Introduction
Bardet-Biedl syndrome (BBS; Online Mendelian
Inheritance in Man #615987) is a rare autosomal recessive disorder
that involves multiple systems, with clinical manifestations that
may include obesity, mental retardation, retinal dystrophy,
hypogenitalism, renal malformations and polydactyly (1). To date, 26 genes have been reported
to be associated with BBS, and these BBS genes have crucial roles
in both the composition and function of the cilia (2). The BBS1, BBS2, BBS4, BBS5, BBS7,
BBS8, BBS9 and BBS18 proteins are components of the BBSome that
functions as an adaptor for intraflagellar transport molecules, and
the BBS6, BBS10 and BBS12 proteins form the chaperonin-like complex
that has an important role in BBSome assembly (3). The mutations of the genes coding for
components of the BBSome are most frequently (57%) identified in
the patients with BBS, and these are followed by the group of genes
that encode chaperonin-like proteins (30%) (2). BSS10 was identified by
Stoetzel et al (4) in 2006
and mutations in this gene account for 21% of BBS cases. A total of
115 variants distributed across the whole BBS10 gene have
been reported in the Human Gene Mutation Database (HGMD). All types
of mutations have been described; missense variants are the most
frequent, followed by small deletions. Among them, the c.271dupT
(p.Cys91LeufsX5) variant is the most common pathogenic variant in
the BBS10 gene, accounting for up to 48% of cases with a
BBS10 mutation (5). In the
present study, a novel compound heterozygous variants in
BBS10 was detected in a fetus with hyperechoic kidneys and
severe cardiac malformation.
Case report
A 36-year-old G2P0A1 female was referred to the
Prenatal Diagnosis Center of Boai Hospital of Zhongshan Affiliated
to Southern Medical University (Zhongshan, China) in November 2018
for prenatal diagnosis at 25 weeks of gestation due to a fetus with
multiple anomalies. The patient's previous pregnancy was terminated
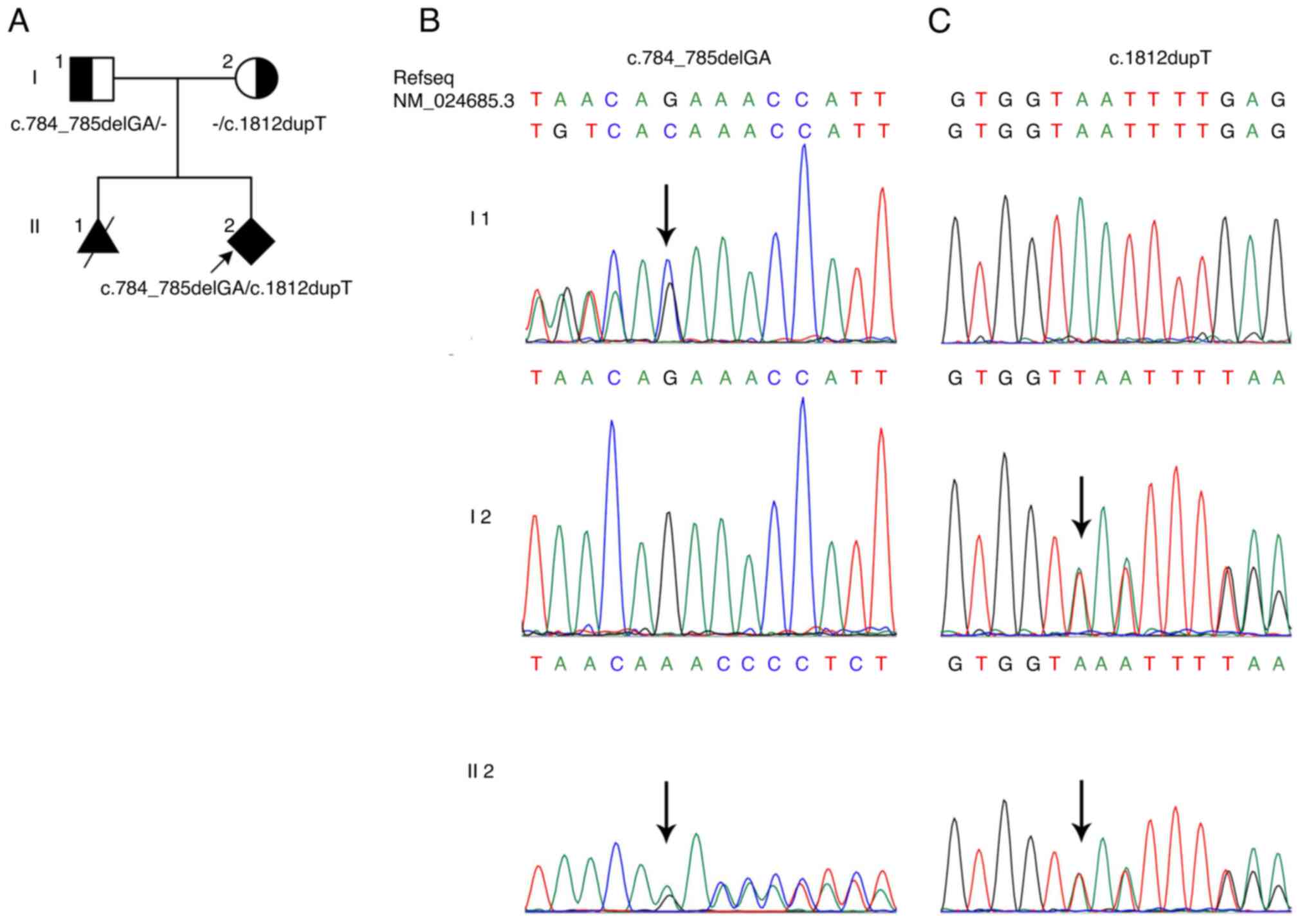
due to similar fetal anomalies, as indicated in the pedigree chart
in Fig. 1. During the patient's
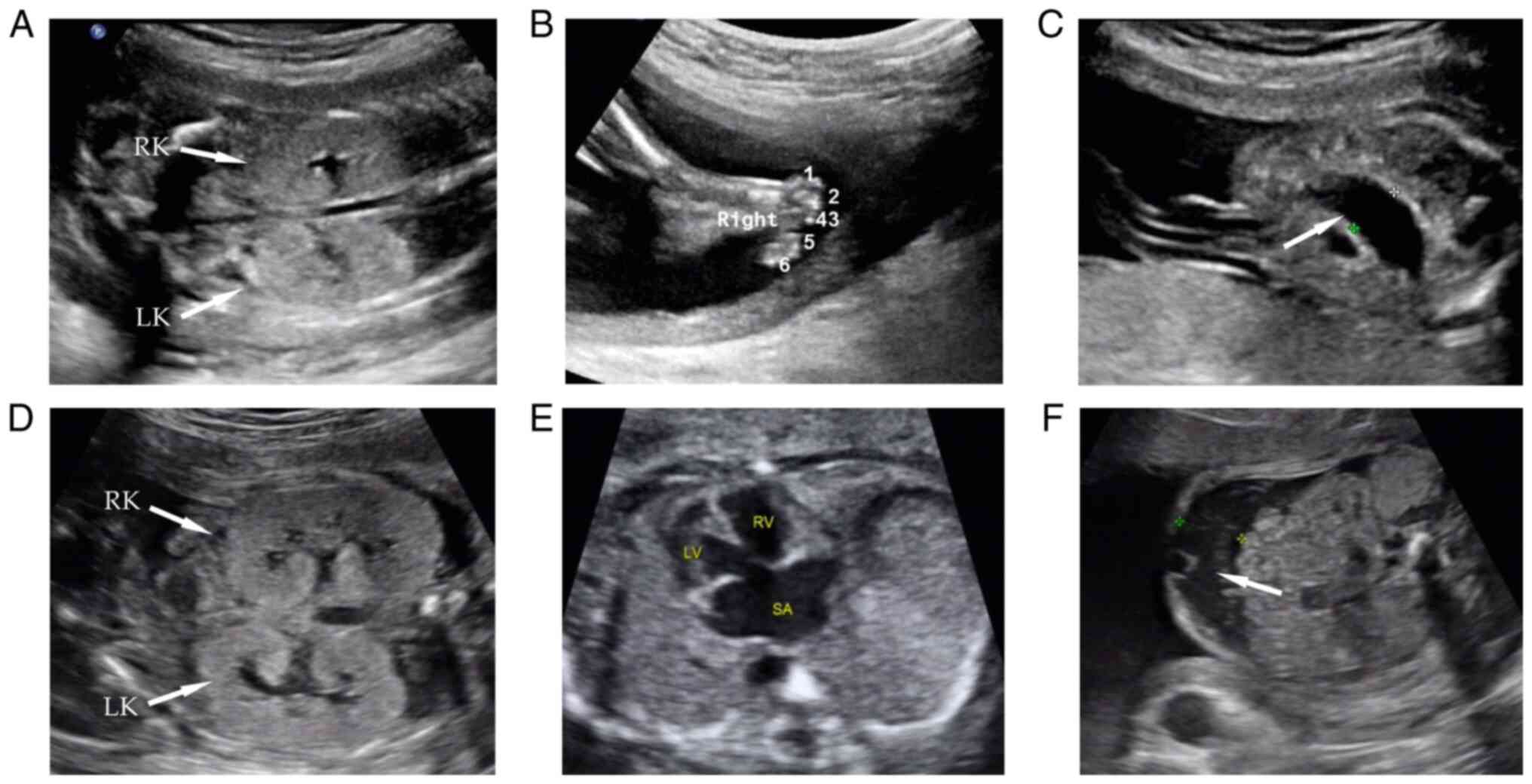
first pregnancy, the 23-week sonography indicated bilateral
enlarged hyperechogenic kidneys, polydactyly of both feet, bowel
dilation and multiple abdominal calcifications (Fig. 2A-C). The pregnancy was terminated
without any genetic analysis. During the second pregnancy, the
first-trimester ultrasound was normal. The 25-week sonography
indicated bilateral enlarged hyperechogenic kidneys, a ventricular
septal defect combined with a single atrium, persistent left
superior vena cava and seroperitoneum (Fig. 2D-F). Considering similar phenotypes
of the kidneys in both fetuses, accompanied by polydactyly, cardiac
malformations and abdominal abnormalities, BBS was suspected in
this family. The parents were non-consanguineous and healthy,
without any family history of congenital malformations.
Genetic testing and analysis were performed with
umbilical cord blood in the second fetus. G-banding normal
chromosomes (46, XX) was found in the second fetus. A genomic
microarray of the umbilical cord blood using an Affymetrix CytoScan
750K array (Thermo Fisher Scientific, Inc.) revealed no
abnormalities. Whole-exome sequencing was performed with umbilical
cord blood on the MGIseq-2000 platform (MGI Tech Co., Ltd.),
revealing that the fetus had two novel compound heterozygous
BBS10 variants, a 2-bp deletion frameshift variant
(NM_024685.3:c.784_785delGA, p.Glu262Asnfs* 41) and a nonsense
variant (NM_024685.3:c.1812dupT, p.Asn605*). The two variants were
also confirmed by Sanger sequencing with an ABI3730 DNA Analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.), i.e.
c.784_785delGA in the father and c.1812dupT in the mother (Fig. 1B and C). These two variants may be the reason
for the disease in this family. Whole-exome sequencing and fetal
ultrasound results were consistent with the diagnostic results of
BBS. After detailed genetic counselling, the couple decided to
terminate the pregnancy at 28 weeks. After induced labor, no
unusual fetal face or polydactyly were observed.
The two variants detected in the present study have
not been reported in any of the reference population gene
databases, including the International Genome Sample Resource
(http://www.internationalgenome.org;
accessed March 11, 2022), Genome Aggregation Database (http://www.gnomad-sg.org/; accessed March 11, 2022),
ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/; accessed March
11, 2022) and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php;
accessed March 11, 2022), which are used to screen pathogenic
variants reported in published studies. BBS10 (NM_024685.3)
c.784_785delGA causes a frameshift. This frameshift variant is
located in the last exon of the BBS10 gene, which is not
predicted to undergo nonsense-mediated decay (NMD) (6). However, the truncated protein loses
the partial apical domain, the C-terminal equatorial domain and the
intermediate domain including In3, which is important for the
protein's function (7). According
to the American College of Medical Genetics and Genomics (ACMG)
(2015) guidelines (8), the
evidence of the variant included PSV1-strong, PM2 and PP4. The
c.784_785delGA variant is classified as likely pathogenic. The
BBS10 mutation (NM_024685.3) c.1812dupT produces a
termination codon. Similarly, this nonsense variant mutation is
also located in the last exon of BBS10 and is not predicted
to undergo NMD. As the role of the lost region on the protein
function is unknown, PVS1 evidence cannot be used (6). MutationTaster (https://www.mutationtaster.org/) predicted the variant
to be ‘disease-causing; probability, 0.999’. According to the ACMG
(2015) guidelines (8), variant
evidence includes PM2, PP3, PM3 and PP4. The c.1812dupT variant is
also classified as a likely pathogenic variant.
The amino acid sequences of the BBS10 protein
(GenBank accession no. NP_078961.3) were obtained from the GenBank
database. Modeling analysis was performed using the homology
modeling program, SWISS-MODEL (http://swissmodel.expasy.org) to visualize the
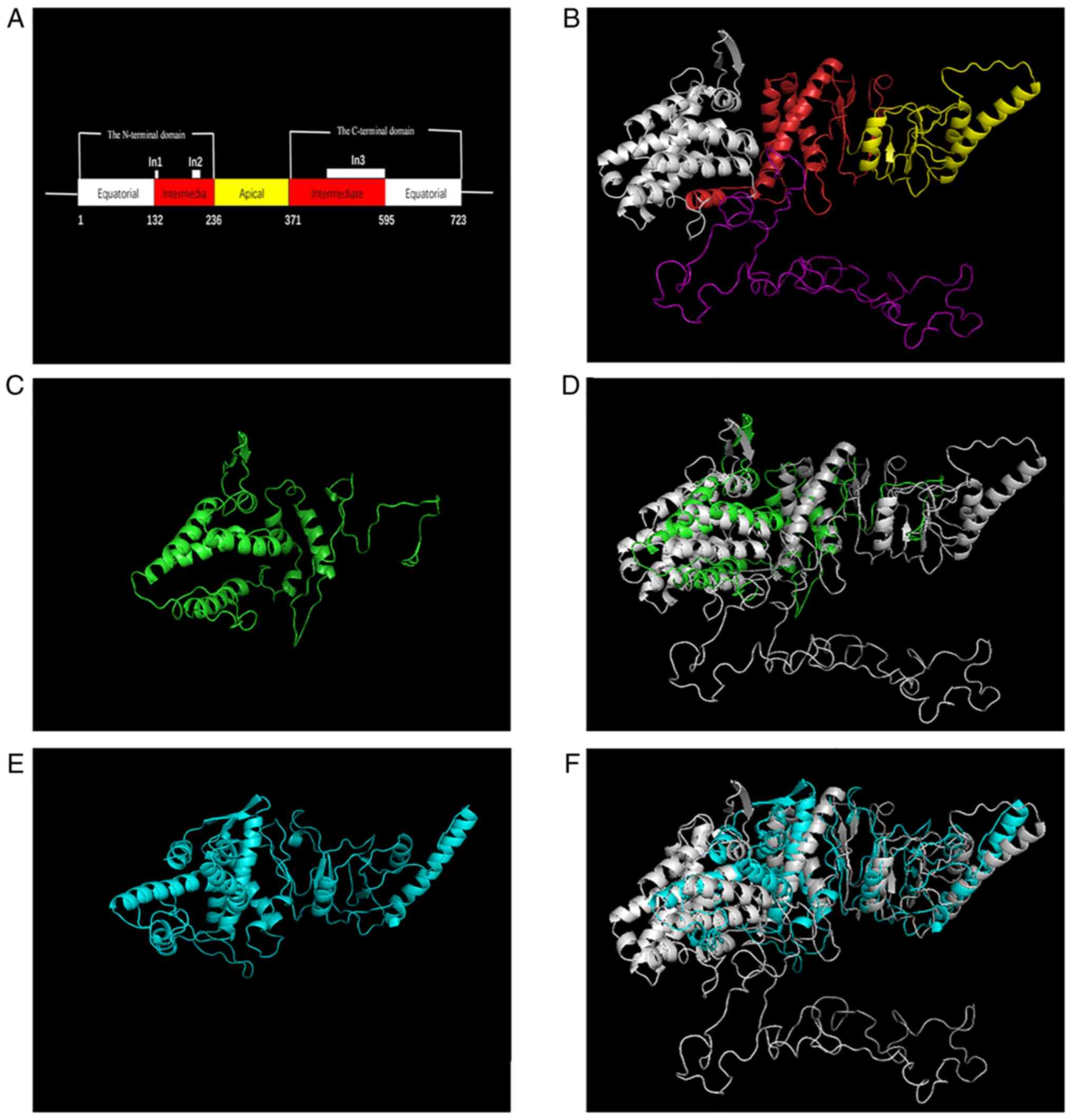
wild-type BBS10 structure with the 5GW4 template. Based on the
structure of wild-type BBS10, the impact of the variant on the
structure of the resulting truncated protein was analyzed according
to SWISS-MODEL (Fig. 3). In
addition, conformations of the wild-type BBS10 protein and the
mutated BBS10 proteins were superimposed by PyMOL (9). The mutated BBS10 proteins had clearly
different structures from the wild-type protein (Fig. 3D and F). Therefore, these compound heterozygous
mutations were predicted to affect BBS10 function as well as
interactions with other molecules.
Discussion
BBS is a rare autosomal recessive inherited
ciliopathy (10). Ciliopathies are
a group of diseases with variants in ciliary-associated genes
(11). To date, >950
cilia-associated genes have been discovered (12). BBS10 is structurally similar
to type II chaperonins and is involved in protein folding (7). Variants in BBS10 have been
reported in patients with a wide range of clinical presentations,
including obesity and rod-cone dystrophy, as well as renal and
polydactyly malformations (13).
Most symptoms of BBS appear postnatally, but kidney abnormalities,
polydactyly and heart structural abnormalities may be detected
antenatally. In 2019, Mary et al (14) reported 45 fetuses with BBS, with
the most prevalent gene mutations in BBS1 (29%),
BBS10 (20%) and BBS12 (18%). That study found no
significant differences in phenotypes among fetal BBS cases with
different mutations, and renal abnormalities (91%) and polydactyly
(82%) were the most common ultrasound manifestations in BBS
fetuses. In 2010, Emmanuelli et al (15) performed a retrospective analysis of
17 fetuses with hyperechogenic kidneys and found two cases that
were diagnosed as BBS. Therefore, for fetuses identified to have
prenatal renal abnormalities, detailed ultrasound scans of the
fetal hands, feet and heart should be performed.
In the present case, enlarged and hyperechogenic
kidneys, polydactyly of the feet and cardiac malformations on
prenatal ultrasonography appeared in one family, suggesting a
diagnosis of BBS. Combined with the clinical manifestations, family
history and genetic testing results, the second fetus was diagnosed
with BBS. However, no fetal specimen of the first pregnancy was
obtained, and thus, it was not possible to clarify whether these
BBS10 variants also existed in the first fetus. BBS is a
possible diagnosis for the first fetus according to the clinical
symptoms. The clinical manifestations of both pregnancies were also
not exactly the same: The first fetus had polydactyly and the
second fetus had a ventricular septal defect combined with single
atrium and persistent left superior vena cava. These differences
are presumed to be due to the diversity of clinical manifestations
of BBS. The first fetus exhibited bowel dilation and multiple
abdominal calcifications, indicating that fetal intestinal
obstruction caused bowel dilation and peritonitis. The second fetus
displayed seroperitoneum, indicating that the presence of
intestinal obstruction resulted in intestinal perforation and
exudation. The abovementioned ultrasonic findings of the abdomen in
the present case were the first reported in a BBS fetuse, which
enriched the antenatal phenotypes of BBS. Two BBS fetuses with
malrotation of the midgut determined by autopsy have been reported,
and one case with BBS10 gene variants (14). In the case of the present study,
the above-mentioned findings of the abdomen may be associated with
malrotation of the midgut, but an autopsy was not performed.
Therefore, the prenatal characteristics of the fetal ultrasound
lacked specificity and were not sufficient to diagnose BBS. The
application of whole-exome sequencing may clarify the diagnosis and
help identify pathogenic genes and variants in the fetus.
In the present study, whole-exome sequencing was
used to detect compound heterozygous variations in the BBS10
gene in the second fetus: Paternal c.784_785delGA (p.Glu262Asnfs*
41) and maternal c.1812dupT (p.Asn605*). BBS10 is located at
12q21.2, containing 2 exons and encoding a large protein of 723
amino acids. BBS10 encodes a vertebrate-specific
chaperonin-like protein. The BBS10 protein regulates the formation
of the BBS-chaperonin complex, which has three chaperonin
functional domains: The equatorial domain, the intermediate domain
and the apical domain (Fig. 3A).
The intermediate domain contains three specific insertions (the
flexible protrusion region specific to group II chaperonins): Two
small insertions, In1 (amino acids at positions 128-139) and In2
(amino acids at positions 187-212), and one large insertion, In3
(amino acids at positions 426-596); these are responsible for ATP
binding and hydrolysis. Both variants found in the BBS10
gene in the present study were novel, not reported in the
literature or databases.
One of the two variants (c.784_785delGA) causes a
frameshift that terminates in the protein in the apical domain,
with partial deletion of the apical domain, the C-terminal
equatorial domain and the intermediate domain including In3, which
is consistent with the predicted structure of the protein. The
other variant (c.1812dupT) causes termination in the C-terminal
equatorial domain with an unknown function, but the region is
conserved in BBS10 homologues by analyzing the conservation using
MutationTaster (https://www.mutationtaster.org/). The predicted
structure of the mutant BBS10 protein (c.1812dupT) indicated that
the normal structure of In3 in the C-terminal equatorial domain was
not able to form. This indicates that the C-terminal equatorial
domain may have an important role in the formation of the normal
structure of In3, but this requires further experimental
confirmation. According to the ACMG (2015) guidelines, these two
variants were classified as likely pathogenic.
In conclusion, whole-exome sequencing provided a
definitive diagnosis for a fetus with multiple malformations. A
total of two variants in BBS10 were determined as likely
pathogenic, which provided a basis for genetic consultation,
pre-implantation genetic diagnosis and prenatal diagnosis for the
pedigree.
Acknowledgements
Not applicable.
Funding
Funding: This work was funded by the Zhongshan Medical
Scientific Research Project (grant no. 2019J164) and by the Project
of Research for Public Benefit and Basic Research of Zhongshan City
(Medical and Health) (grant no. 2020B1057).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to concerns regarding
participant/patient anonymity, but are available from the
corresponding author on reasonable request.
Authors' contributions
XD, TM and YG performed genetic diagnoses and
drafted and critically revised the manuscript. XD, ZL, DW, YX, HL,
PY and LL clinically diagnosed patients, collected and analysed
samples and performed imaging tests. All authors have read and
approved the final manuscript. XD and YG confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the Ethics Committee of
Zhongshan Boai Hospital Affiliated to Southern Medical University
(Zhongshan, China) and with the 1964 Helsinki declaration and its
later amendments or comparable ethical standards. The parents
provided written informed consent for genetic testing.
Patient consent for publication
The parents provided written informed consent for
the publication of their data in this study.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Khan SA, Muhammad N, Khan MA, Kamal A,
Rehman ZU and Khan S: Genetics of human Bardet-Biedl syndrome, an
updates. Clin Genet. 90:3–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Focșa IO, Budișteanu M, Burloiu C, Khan S,
Sadeghpour A, Bohîlțea LC, Davis EE and Bălgrădean M: A case of
Bardet-Biedl syndrome caused by a recurrent variant in BBS12: A
case report. Biomed Rep. 15(103)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Forsythe E, Kenny J, Bacchelli C and
Beales PL: Managing Bardet-Biedl syndrome-now and in the future.
Front Pediatr. 6(23)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Stoetzel C, Laurier V, Davis EE, Muller J,
Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, et al:
BBS10 encodes a vertebrate-specific chaperonin-like protein and is
a major BBS locus. Nat Genet. 38:521–524. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Muller J, Stoetzel C, Vincent MC, Leitch
CC, Laurier V, Danse JM, Hellé S, Marion V, Bennouna-Greene V,
Vicaire S, et al: Identification of 28 novel mutations in the
Bardet-Biedl syndrome genes: The burden of private mutations in an
extensively heterogeneous disease. Hum Genet. 127:583–593.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Abou Tayoun AN, Pesaran T, DiStefano MT,
Oza A, Rehm HL, Biesecker LG and Harrison SM: ClinGen Sequence
Variant Interpretation Working Group (ClinGen SVI). Recommendations
for interpreting the loss of function PVS1 ACMG/AMP variant
criterion. Human mutation. 39:1517–1524. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Alvarez-Satta M, Castro-Sanchez S and
Valverde D: Bardet-Biedl syndrome as a chaperonopathy: Dissecting
the major role of Chaperonin-Like BBS proteins (BBS6-BBS10-BBS12).
Front Mol Biosci. 4(55)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
The PyMOL Molecular Graphics System. In:
Delano Scientific. San Carlos, CA, 2002.
|
|
10
|
Tsang SH, Aycinena ARP and Sharma T:
Ciliopathy: Bardet-Biedl syndrome. Adv Exp Med Biol. 1085:171–174.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
McConnachie DJ, Stow JL and Mallett AJ:
Ciliopathies and the kidney: A review. Am J Kidney Dis. 77:410–419.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
van Dam TJP, Kennedy J, van der Lee R, de
Vrieze E, Wunderlich KA, Rix S, Dougherty GW, Lambacher NJ, Li C,
Jensen VL, et al: CiliaCarta: An integrated and validated
compendium of ciliary genes. PLoS One. 14(e0216705)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Janssen S, Ramaswami G, Davis EE, Hurd T,
Airik R, Kasanuki JM, Van Der Kraak L, Allen SJ, Beales PL,
Katsanis N, et al: Mutation analysis in Bardet-Biedl syndrome by
DNA pooling and massively parallel resequencing in 105 individuals.
Hum Genet. 129:79–90. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mary L, Chennen K, Stoetzel C, Antin M,
Leuvrey A, Nourisson E, Alanio-Detton E, Antal MC, Attié-Bitach T,
Bouvagnet P, et al: Bardet-Biedl syndrome: Antenatal presentation
of forty-five fetuses with biallelic pathogenic variants in known
Bardet-Biedl syndrome genes. Clin Genet. 95:384–397.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Emmanuelli V, Lahoche-Manucci A,
Holder-Espinasse M, Devisme L, Vaast P, Dieux-Coeslier A,
Dehennault M, Petit S, Besson R and Houfflin-Debarge V: Prenatal
diagnosis of hyperechogenic kidneys: A study of 17 cases. J Gynecol
Obstet Biol Reprod (Paris). 39:637–646. 2010.PubMed/NCBI View Article : Google Scholar : (In French).
|