Introduction
Myocardial injury incurred during ischemia is
generally attributed to a depletion of myocyte adenosine
triphosphate (ATP) production. The reperfusion of the ischemic
myocardium is the only recourse to salvaging cardiac function.
Paradoxically, reperfusion itself induces further tissue injury.
Thus, immense efforts have been expended to gain insight into
potential drivers of reperfusion injury, in order to enable the
development of therapeutic strategies for the mitigation of
reperfusion injury (1,2). To this end, in vivo and ex
vivo models of cardiac ischemia/reperfusion (I/R) have been
used. In vivo models involve occlusion and the subsequent
release of a coronary artery, and tracking cardiac injury and
inflammation. This approach more closely simulates the clinical
situation; however, due to input from various resident (myocytes,
fibroblasts, endothelial cells) and infiltrating immune
(neutrophils, monocytes) cells, unravelling the cellular mechanisms
is challenging (3). Frequently,
cell-based models are used, involving the challenge of
cardiomyocytes with hypoxia/reoxygenation (H/R).
Consensus holds that reactive oxygen species (ROS)
play a critical role in reperfusion-induced cardiac injury
(4-6).
Whereas there are several potential sources of ROS in
cardiomyocytes, mitochondrial-derived ROS are particularly
attractive targets. Mitochondria most likely generate ROS during
the normal course of oxidative phosphorylation. The rapid transfer
of electrons along the electron transport chain (ETC) can result in
the ‘leakage’ of electrons that can interact with O2 to
form superoxide. A particularly devastating feature of
mitochondrial ROS production is the feed-forward mechanism by which
ROS-induced ROS release spreads from one mitochondrion to another,
ultimately resulting in myocyte dysfunction and death (7,8).
Regulatory mechanisms are operative, to limit
excessive cellular ROS levels and associated sequelae. At the
transcriptional level, ROS can transactivate genes encoding
enzymatic and non-enzymatic antioxidants (4,5). At
the translational level, an additional layer of regulation is
achieved by microRNAs (miRNAs/miRs) (9-11).
miRNAs are non-coding RNAs that can modulate genetic output of
antioxidants, primarily by transcriptional silencing. Members of
the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and
miR-429) can respond to and regulate cellular ROS levels (9,10).
Two members of this family, miR-200c and miR-141, have been
implicated in various pathologies associated with excessive oxidant
stress and mitochondrial dysfunction within the heart, including
aging and diabetes (11,12). While an increase in miR-200c
driving cardiac I/R or H/R injury has been supported by previous
studies (1,2), the role of miR-141 is rather
ambiguous (13,14). This is contrary to the available
evidence for a ROS/miR-141 pathway being intimately involved in
mitochondrial dysfunction (15-17).
Thus, in the present study, a reductionist approach was applied to
systematically re-examine this issue. The current findings indicate
that the challenge of cardiomyocytes with H/R may result in
mitochondrial dysfunction via a ROS/miR-141 pathway, in line with a
ROS-induced ROS release mechanism.
To address specific targets of miR-141 that may
mediate the H/R-induced mitochondrial dysfunction, the present
study focused on sirtuin-1 (Sirt1) and mitofusin-2 (MFN2). Sirt1
and MFN2 both are intimately involved in mitochondrial function
(18-20)
and exerts cardioprotective effects in I/R models (18,21).
Of note, a Sirt1/MFN2 pathway has been implicated in I/R-induced
mitochondrial dysfunction in hepatocytes (22,23).
Herein, evidence was provided that both Sirt1 and MFN2 have
functional binding sites for miR-141 on their gene transcripts.
Additionally, the blockade of miR-141 can prevent the H/R-induced
downregulation of Sirt1and MFN2. Collectively, the results of the
present study indicate that the ROS/miR-141/Sirt1/MFN2 pathway may
promote myocardial I/R injury by inducing mitochondrial
dysfunction.
Materials and methods
Reagents
Mitochondrial ROS was detected using MitoSOX (cat.
no. M36008; Thermo Fisher Scientific, Inc.) and Mito-Tempo (cat.
no. 1334850995; MilliporeSigma) was used to quench mitochondrial
ROS. Mitochondrial membrane potential was assessed using JC-1 (cat.
no. 40705ES03; Shanghai Yeasen Biotechnology Co., Ltd.).
For reverse transcription-quantitative PCR
(RT-qPCR), the primers used for miRNA were 5'-TAGCCTGCTGGGGTGGAA-3'
(forward), and 5'-TATGGTTTTGACGACTGTGTGAT-3' (reverse). The control
primers (U6) were 5'-CGCGCTTCGGCAGCACATATACT-3' (forward) and
5'-ACGCTTCACGAATTTGCGTGTC-3' (reverse).
For western blotting, the primary antibodies used
were as follows: MFN2 (1:1,000; cat. no. sc-515647; Santa Cruz
Biotechnology, Inc.), Sirt1 (1:1,000; cat. no. sc-74465; Santa Cruz
Biotechnology, Inc.), and β-actin (1:5,000; cat. no. 66009-1-Ig;
ProteinTech Group, Inc.). A horseradish peroxidase-conjugated goat
anti-mouse secondary antibody (1:2,000; cat. no. ab205719; Abcam)
was used for detection.
siRNA transfection
For RNA interference, two approaches were followed:
Small interfering RNA (siRNA) and miRNA. siRNA-Sirt1, as well as a
miR-141-3p (miR-141) mimic and an inhibitor were synthesized by
Shanghai GenePharma Co., Ltd. The target sequence for Sirt1 siRNA
was 5'-CCCUGUAAAGCUUUCAGAA-3' and that of the negative control was
5'-TTCTCCGAACGTGTCACGT-3'. The sequence of miR-141 mimic was
5'-UAACACUGUCUGGUAAAGAUGG-3' and that of the negative control was
5'-UUCUCCGAACGUGUCACGUTT-3'. The sequence of the miR-141 inhibitor
was 5'-CCAUCUUUACCAGACAGUGUUA-3' and that of the negative control
was 5'-CAGUACUUUUGUGUAGUACAA-3'. All the siRNAs and miRNA
mimics/inhibitors were transfected into the HL-1 cells (described
below) using Lipofectamine 2000 reagent (cat. no. 11668019; Thermo
Fisher Scientific, Inc.). Briefly, two sterilized Eppendorf tubes
were prepared for each group of cells. Each tube was filled with
100 µl Opti-MEM (Thermo Fisher Scientific, Inc.). One tube was
filled with 5 µl Lipofectamine 2000, whereas the other was filled
with 100 pmol mimics/inhibitors/negative control or with 75 pmol
siRNAs/negative controls. The two tubes were then evenly mixed and
incubated at room temperature for 20 min. The cells transfected
with siRNA/mimics/inhibitors were incubated in a standard incubator
at 37˚C and 5% CO2 for 48 h before subsequent
experiments.
Hypoxia/reoxygenation (H/R) of HL-1
cells
The HL-1 immortalized murine cardiomyocyte cell line
(cat. no. SCC065; Merck KGaA) was grown in DMEM (cat. no. 11995065;
Gibco; Thermo Fisher Scientific, Inc.) with 10% FBS (cat. no.
16140071; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (cat. no. 10378016; Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C in a standard humidified incubator (95%
air/5% CO2). The H9c2 cell line was obtained from the
American Type Culture Collection (cat. no. CRL-1446) and cultured
in DMEM containing 10% FBS at 37˚C with 5% CO2. To
induce hypoxia, the culture medium was changed to serum- and
glucose-free DMEM, placed into an anaerobic chamber, wherein
O2 was purged with AnaeroPack (Mitsubishi Gas Chemical
Co., Inc.) for 3 h (<0.1% O2, >15%
CO2). Subsequently, the cells were reoxygenated through
replenishment with fresh DMEM and rapidly transferred back to the
standard incubator. Cell and mitochondrial functions were evaluated
at the indicated time points (6 and 12 h) following reoxygenation.
As a control, HL-1 cardiomyocytes were incubated with serum- and
glucose-free DMEM for 3 h under standard normoxic (20%
O2) conditions at 37˚C with 5% CO2. and then
replenished with fresh DMEM and cultured in normal conditions
[normoxia/reoxygenation (N/R)].
Cell injury assays
Cell viability was assessed using four approaches. A
Cell Counting Kit-8 assay (CCK-8; cat. no. C0038; Beyotime
Institute of Biotechnology) was applied for the detection of
metabolic dysfunction of live cells. Cell supernatants were
evaluated for the presence of WST-8 formazan produced by
dehydrogenase activity of live cells. After incubation for 2 h at
37˚C, the optical density values were detected at 450 nm. Cell
apoptosis was measured using an Annexin-V/propidium iodide
apoptosis detection kit (cat. no. 640914; BioLegend, Inc.). In
brief, cells labelled with FITC-labelled Annexin-V, excluding
propidium iodide, were quantified using flow cytometry. Flow
cytometric analyses were performed using BD LSRFortessa X-20 (BD
Biosciences). Flow cytometry data were analyzed by FlowJo V10
(FlowJo, LLC). As an index of cell membrane disruption, the lactate
dehydrogenase (LDH) content in the cell supernatants was measured
using an LDH assay kit (cat. no. A0202; Nanjing Jiancheng
Bioengineering Institute). In addition, the creatine kinase-MB
(CK-MB) levels in the cell supernatants were measured
spectrophotometrically using standard enzyme-linked immunosorbent
assay kits (cat. nos. F3500-A and F2801-B; FANKEL Industrial Co.,
Ltd.) and a microplate reader (800 TS; BioTek Instruments, Inc.)
according to the manufacturer's instructions.
Measurement of mitochondrial ROS
production
Mitochondrial ROS production was detected using
MitoSOX (cat. no. M36008; Thermo Fisher Scientific, Inc.), a
cationic cell-permeable probe that enters the mitochondria, is
oxidized by superoxide, and fluoresces when bound to nucleotides
(e.g., DNA). Cell fluorescence was assessed using a confocal
scanning microscope (LSM800; Zeiss AG). The superoxide dismutase
(SOD) mimetic, Mito-Tempo (cat. no. 1334850995; MilliporeSigma) was
used to quench mitochondrial ROS. ImageJ software (v1.8.0; National
Institutes of Health) was used to quantify the fluorescence
intensity.
Measurement of mitochondrial membrane
potential
The fluorescent probe, JC-1 (cat. no. 40705ES03;
Shanghai Yeasen Biotechnology Co., Ltd.), accumulates in the
mitochondria and exhibits a red-to-green shift in emission, which
is inversely proportional to membrane polarization. The red/green
fluorescence intensity ratio of the HL-1 cells was assessed using a
confocal scanning microscope (LSM800). ImageJ software (v1.8.0) was
used to quantify the fluorescence intensity.
Determination of mitochondrial
function
The mitochondrial oxygen consumption rate (OCR) of
the HL-1 cells was assessed using an extracellular flux analysis
using Seahorse XF Cell Mito Stress kits (Agilent Technologies,
Inc.). The OCR of the HL-1 cells (1x104 cells/well)
under various conditions (e.g., ETC inhibitors) was quantified in a
Seahorse XFp analyzer using accompanying software (Seahorse
Bioscience; Agilent Technologies, Inc.). Various compounds were
used to alter mitochondrial ETC function at 37˚C: oligomycin (1 µM,
27 min) to inhibit complex V, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (1 µM, 27 min) to
uncouple oxidative phosphorylation, and a combination of antimycin
A/rotenone (1 µM/100 nM, 27 min) to inhibit complexes I/III.
Transmission electron microscopy
The HL-1 cells were prefixed in 2.5% glutaraldehyde
(cat. no. P1126; Beijing Solarbio Science & Technology Co.,
Ltd.) overnight at 4˚C, washed with 0.1 M sodium cacodylate buffer
(cat. no. 20840; Sigma-Aldrich; Merck KgaA) and then fixed in 1%
osmium tetroxide (cat. no. 18459; Ted Pella, Inc.) for 24 h at 4˚C.
The cells were then washed with 0.1 M sodium cacodylate buffer
again, dehydrated in an ethanol series, infiltrated with PolyBed
epoxy resin (cat. no. GP2001; Wuhan Servicebio Technology Co.,
Ltd.) and stained with 2% uranyl acetate (Wuhan Servicebio
Technology Co., Ltd.) for 10 min and lead citrate (Wuhan Servicebio
Technology Co., Ltd.) for 5 min at room temperature. Ultrathin
sections were observed under a transmission electron microscope
(HT7800; Hitachi, Ltd.). The extent of mitochondrial and cellular
pathology was scored using a modification of previously published
paradigms (24,25), with 0 assigned to intact
mitochondria and 3 to severely damaged mitochondria (Table SI).
Dual luciferase assay
miRanda (http://www.bioinformatics.com.cn/local_miranda_miRNA_target_prediction_120),
Tarbase (http://microrna.gr/tarbase/) and
TargetScan (www.targetscan.org/vert_72/) were used to predict
target genes of miR-141. MFN2 and Sirt1 were identified as
potential target genes. The wild-type 3'-UTR and the miR-141 ‘seed’
mutant 3'-UTR of MFN2 and Sirt1 were synthesized in vitro
and cloned into the psi-CHECK2 (Hanbio Biotechnology Co., Ltd.).
luciferase reporter plasmid. 293T cells (cat. no. 12022001;
MilliporeSigma). were transfected with psi-CHECK-2 plasmid
containing wild-type or mutant derivatives, along with the miRNA
control or miR-141 mimic using Lipofectamine 2000. After 24 h, the
cells were harvested, lysed, and the ratio of Firefly luciferase to
Renilla luciferase was assessed. The luciferase activity was
detected using the Dual Luciferase Reporter Assay kit (Promega
Corp.). The psi-Check2 vector was used for the dual luciferase
assay. In this vector, the promoter of Firefly luciferase (Fluc) is
sv40, and the promoter of Renilla-luciferase (Rluc) is
HSV-TK. The binding site region were constructed in the 3'UTR
region of Fluc.
RT-qPCR
Total RNA, including miRNA, was extracted from the
cultured cells by using Trizol reagent (cat. no. B511311; Sangon
Biotech Co., Ltd.). The target miRNA was reverse transcribed into
cDNA using the miRNA 1st Strand cDNA synthesis kit (cat. no.
MR101-01; Vazyme Biotech Co. Ltd.). The miRNA expression level was
determined with a miRNA Universal SYBR qPCR master mix (cat. no.
MQ101-01; Vazyme Biotech Co. Ltd.) using the CFX96 Touch Real-Time
PCR Detection System (Bio-Rad Laboratories, Inc.). The cycling
conditions are listed as follows: Pre-denaturation at 95˚C for 5
min, followed by 40 cycles at 95˚C for 10 sec and 60˚C for 30 sec,
and elongation at 95˚C for 15 sec and 60˚C for 60 sec. The relative
fold expression of the target, normalized to the corresponding
control, was calculated by the comparative
2-ΔΔCq method (26).
Western blotting
HL-1 cells were lysed with ice-cold RIPA lysis
buffer (cat. no. P0013E; Beyotime Institute of Biotechnology). BCA
protein assay kit (cat. no. P0012; Beyotime Institute of
Biotechnology) was used for protein quantification. Equal amounts
of protein (20 µg) from each sample were separated using 10%
SDS-PAGE and subsequently electro-transferred on to nitrocellulose
membranes (cat. no. FFN08; Beyotime Institute of Biotechnology).
Following blocking with 5% skim milk powder in 1X Tris-buffered
saline containing 0.1% Tween-20 for 2 h at room temperature, the
blots were probed with primary antibodies (as aforementioned)
overnight at 4˚C. Subsequently, the membrane was incubated with
horseradish peroxidase-conjugated secondary antibody (as
aforementioned) for 1 h at 37˚C. The immunoreactive proteins were
visualized using enhanced chemiluminescence reagent kit (cat. no.
P0018M; Beyotime Institute of Biotechnology)and analyzed using
ImageJ software (v1.8.0). β-actin served as the loading control to
normalize relative protein expression levels.
Statistical analysis
Data are presented as the mean ± standard error of
the mean (SEM). An unpaired Student's t-test was performed for
direct two-group comparisons and one-way ANOVA was performed for
multiple group comparisons followed by the Bonferroni test when
ANOVA found a significant F-value and there was no variance in
homogeneity; otherwise, Tamhane's T2 post hoc test was used. All
statistical analyses were performed using SPSS 13.0 statistical
software (SPSS, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
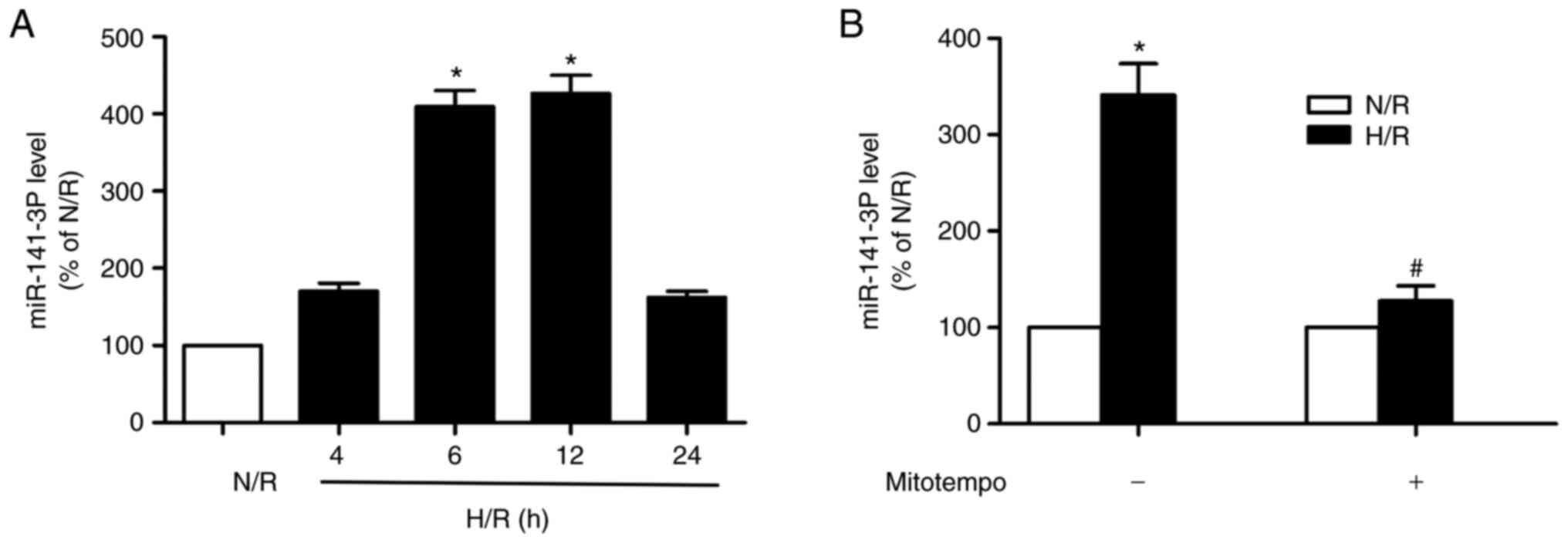
H/R increases miR-141-3p expression in
HL-1 and H9c2 cardiomyocytes and this event is prevented by
quenching mitochondrial superoxide
The challenge of HL-1 cardiomyocytes with H/R
increased miR-141 expression within 6 h following reoxygenation.
The increment in expression remained elevated at 12 h following
reoxygenation and returned to control (normoxia) levels by 24 h
(Fig. 1A). Mitochondrial
superoxide production is implicated in the I/R-induced myocardial
injury (4-6).
Thus, a SOD mimetic (Mitotempo) targeting the mitochondria, was
used to address the role of mitochondrial ROS in the upregulation
of miR-141 expression induced by H/R challenge of the
cardiomyocytes. As depicted in Fig.
1B, the SOD mimetic prevented the increase in miR-141
expression at 6 h following reoxygenation. In addition, similar
results were observed in H9c2 cells (Fig. S1).
miR-141-3p modulates cardiomyocyte
viability following H/R
As shown in Fig. 2
(for HL-1 cells), Fig. S2 (for
HL-1 cells) and Fig. S3 (for H9c2
cells), gain- and loss-of-function approaches (miR-141 mimic and
inhibitor, respectively) were used to systematically evaluate the
role of miR-141 in cardiomyocyte viability following H/R. Firstly,
the dehydrogenase activity of live cells was measured, using a
CCK-8 assay. As depicted in Figs.
2A and S3A, H/R impaired this
index of cell activity. The miR-141 mimic exacerbated the effects
of H/R, while the miR-141 inhibitor exerted protective effects. For
the N/R of the challenged cells, the miR-141 mimic impaired cell
activity, while the miR-141 inhibitor exerted protective effects.
Subsequently, using differential Annexin-V/propidium iodide
staining, the apoptotic cells were detected. H/R increased the
number of cells in early apoptosis; this effect was exacerbated by
miR-141 mimic and ameliorated by miR-141 inhibitor (Figs. 2B and S2). Late apoptotic or necrotic cells
were also detected following H/R, as indicated by LDH (Figs. 2C and S3B) and CK-MB (Figs. 2D and S3C) release into the supernatants.
miR-141 mimic exacerbated the effects of H/R, while miR-141
inhibitor exerted protective effects (Figs. 2C and D, and S3B and C). Establishment of the miR-141 mimic
and inhibitor models was shown in Fig. S4.
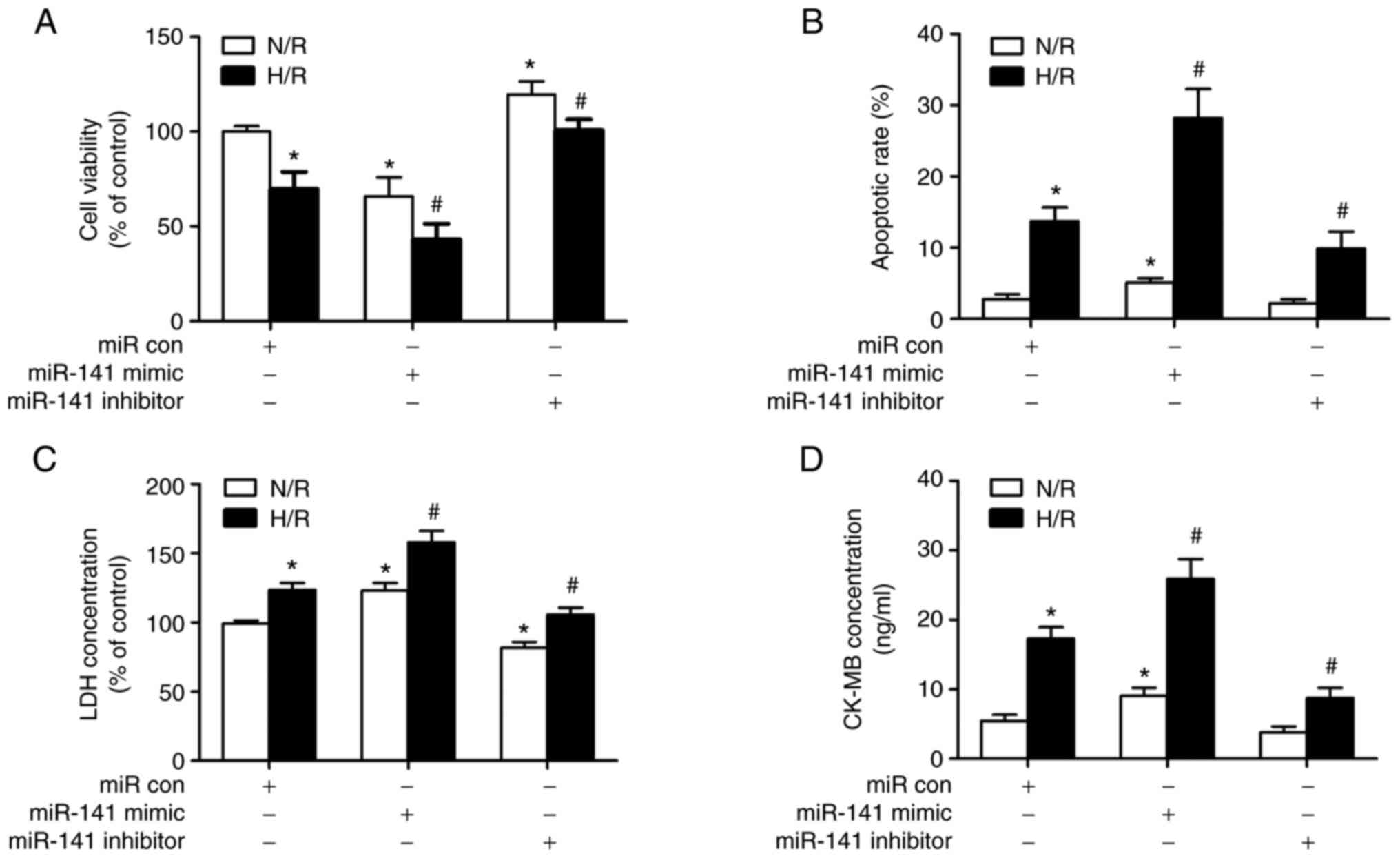 | Figure 2Role of miR-141 in H/R-induced
cardiomyocyte viability and death. HL-1 cardiomyocytes were
transfected with a miR-141 mimic, miR-141 inhibitor, or their
negative controls (miR con). Subsequently, the cells were
challenged with H/R and 12 h following reoxygenation and indices of
viability, apoptosis and plasma membrane disruption were assessed.
(A) The viability of cells was assessed using a CCK-8 assay. (B)
Cell apoptosis was assessed using the FACS analysis of
Annexin-V/propidium iodide (please see Fig. S1 for quadrant depictions). (C and
D) LDH and CK-MB release from cells with ruptured membranes.
Results are presented as the mean ± SEM; n=3. *P<0.05
vs. N/R + miR con, #P<0.05 vs. H/R + miR con. H/R,
hypoxia/reoxygenation; CCK-8, cell counting kit-8 assay; LDH,
lactate dehydrogenase; CK-MB, creatine kinase-MB; SEM, standard
error of the mean; miR con, miR control. |
miR-141-3p modulates the mitochondrial
cardiomyocyte OCR induced by H/R
A metabolic flux analysis was performed to assess
the mitochondrial function of cardiomyocytes challenged with H/R.
Specifically, the OCR of the HL-1 cells was measured during the
pharmacological modulation of the mitochondrial ETC. The challenge
of the cardiomyocytes with H/R reduced their basal OCR; the miR-141
mimic exacerbated (Fig. 3A), while
the inhibitor ameliorated (Fig.
3C) the decrement of basal OCR. The inhibition of ATP synthase
with oligomycin revealed the ATP-linked OCR of HL-1 cells. The OCR
attributed to mitochondrial ATP production was reduced in cells
subjected to H/R; the miR-141 mimic exacerbated (Fig. 3B), while the miR-141 inhibitor
(Fig. 3D) blunted the decrement in
ATP-coupled OCR.
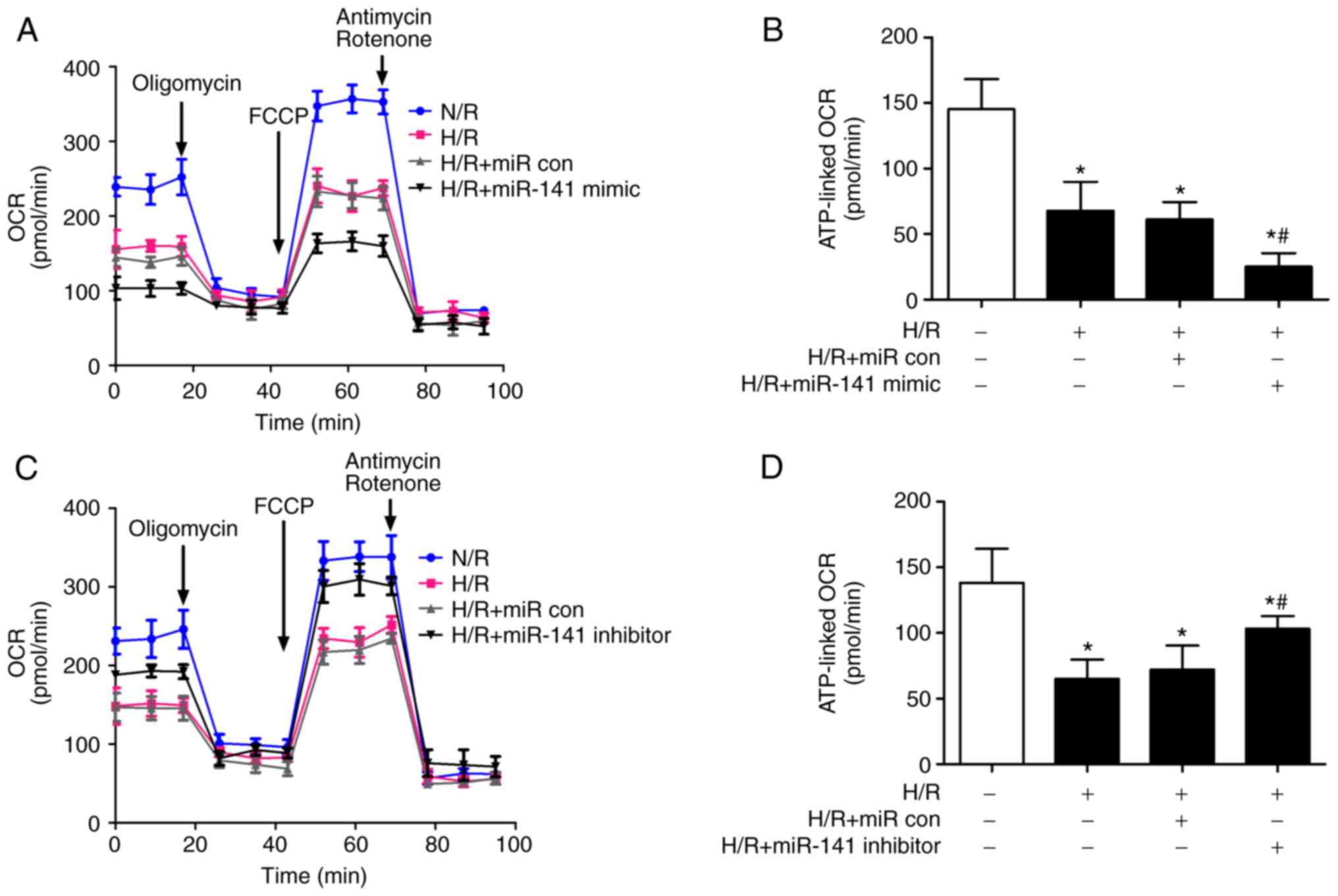 | Figure 3Role of miR-141 in H/R-induced
mitochondrial dysfunction of cardiomyocytes. HL-1 cardiomyocytes
were transfected with a miR-141 mimic, miR-141 inhibitor, or their
negative controls (miR con). Subsequently, the cells were
challenged with H/R and 12 h and OCR was evaluated by extracellular
flux analysis. In brief, after the baseline OCR was obtained,
oligomycin (1 µM, 27 min) was added to obtain ATP-coupled OCR
(baseline-oligomycin). Subsequently, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (1 µM, 27 min) was added
to uncouple oxidative phosphorylation followed by a combination of
antimycin A/rotenone (1 µM/100 nM, 27 min) to inhibit complexes
I/III. (A and B) The complete extracellular flux analyses of
cardiomyocyte OCR challenged with (A) H/R and miR-141 mimic, and
the OCR linked to (B) ATP production. (C and D) The complete
extracellular flux analyses of cardiomyocyte OCR challenged with
(C) H/R and miR-141 inhibitor, and (D) the OCR linked to ATP
production. Bar graphs represent the mean ± SEM; n=3.
*P<0.05 vs. normoxic control (open bars),
#P<0.05 vs. H/R + miR con. miR con, miR control; H/R,
hypoxia/reoxygenation; OCR, reoxygenation mitochondrial oxygen
consumption rate; SEM, standard error of the mean. |
miR-141-3p modulates mitochondrial
superoxide production, membrane potential and damage induced by
H/R
The scanning confocal microscopy of the HL-1 cells
was used to monitor fluorescent markers of mitochondrial superoxide
(mitoSOX) and membrane potential (JC-1). Upon the reoxygenation of
hypoxic cardiomyocytes, there was an increase in mitochondrial
superoxide production; miR-141 mimic exacerbated, while the miR-141
inhibitor prevented the H/R-induced generation of superoxide
(Fig. 4A and B). The challenge of the HL-1 cells with
H/R depolarized the mitochondrial membrane; miR-141 mimic
exacerbated, while the inhibitor prevented membrane depolarization
(Fig. 4C and D).
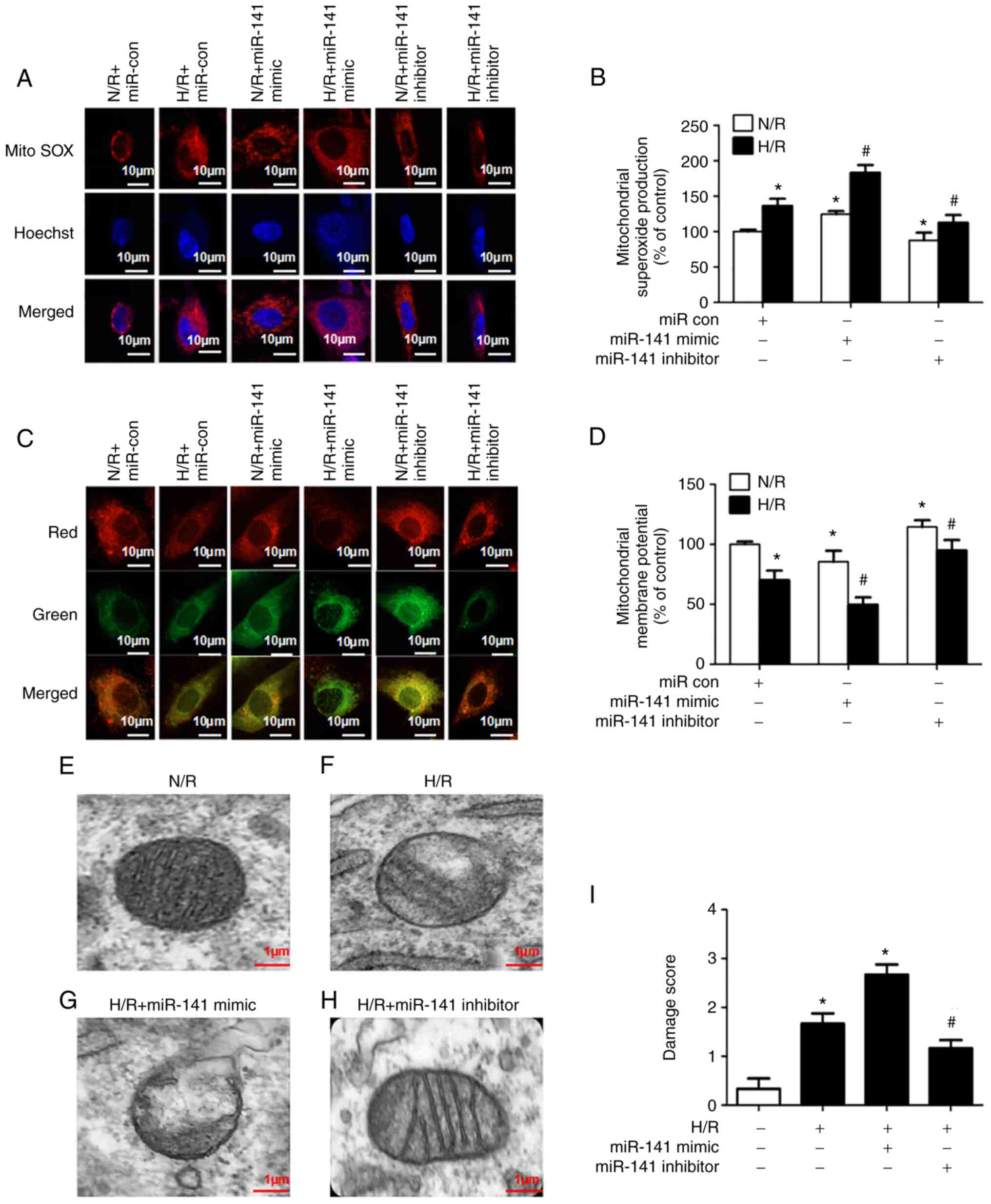 | Figure 4Role of miR-141 in the H/R-induced
increase in mitochondrial superoxide production, decrease in
mitochondrial membrane potential, and ultrastructural derangements.
HL-1 cardiomyocytes were transfected with a miR-141 mimic, miR-141
inhibitor, or their negative controls (miR con). Subsequently, the
cells were challenged with H/R. (A and B) Mitochondrial superoxide
production was assessed using the fluorescence microscopy of
MitoSOX red, 6 h after reoxygenation. An increase in superoxide
production is indicated by an increase in intensity of red
fluorescence. (A) Representative images and (B) quantification
results are presented. (C and D) Mitochondrial membrane potential
was assessed using the fluorescence microscopy of JC-1 6 h after
reoxygenation. Mitochondrial membrane depolarization is indicated
by a shift from red to green fluorescence. (C) Representative
images and (D) quantification results are presented. Bar graphs are
represent the mean ± SEM; n=3. *P<0.05 vs. control
(open bar); #P<0.05 vs. H/R. (E-I) Mitochondrial
morphology was assessed using electron microscopy, 12 h after
reoxygenation. (E-H) Representative images and in (E-H) and (I)
quantification results are presented. The scoring paradigm is
presented in Table SI. In brief,
a value of 0 was assigned to intact mitochondria and 3 to severely
damaged mitochondria. Each bar graph represents scoring of 6
photomicrographs/group by 3 investigators. Bar graphs represent the
mean ± SEM. *P<0.05 vs. control (open bar);
#P<0.05 vs. H/R. H/R, hypoxia/reoxygenation; miR con,
miR control; SEM, standard error of the mean. |
Mitochondrial morphology was examined using electron
microscopy (Fig. 4E-H) and the
results were quantified (Fig. 4I).
The normoxic HL-1 cells exhibited discernable mitochondrial
membranes and cristae (Fig. 4E).
The mitochondria of cells subjected to H/R exhibited signs of
structural derangement; they appeared swollen and many lacked
defined cristae (Fig. 4F). The
miR-141 mimic aggravated the H/R-induced mitochondrial damage,
additionally resulting in cell lysis in some cases (Fig. 4G). By contrast, the miR-141
inhibitor partially protected the cells from H/R-induced
mitochondrial injury (Fig.
4H).
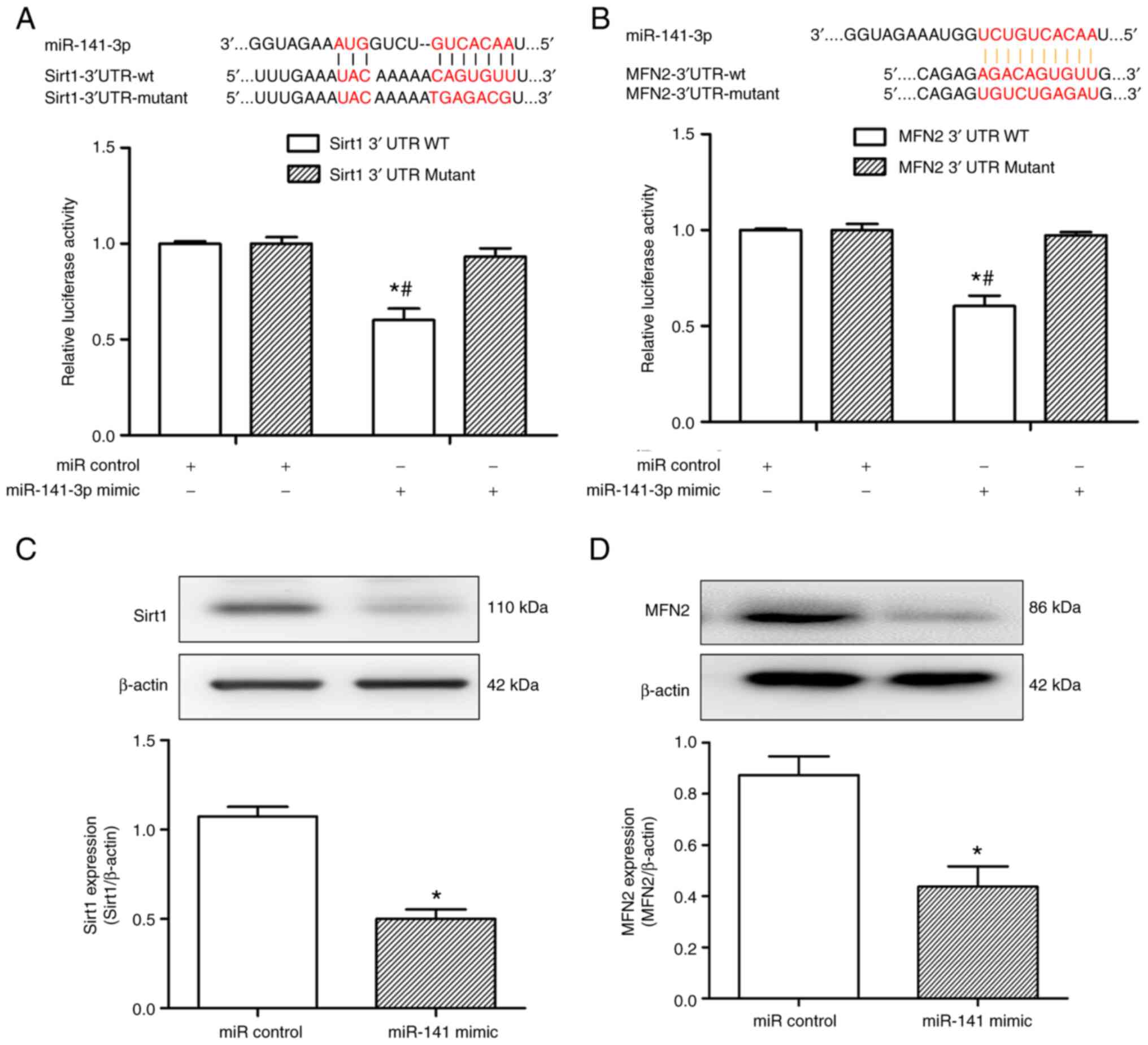
Sirt1 and MFN2 mRNA are targets of
miR-141
Sirt1 and MFN2 have been reported to be depleted in
hepatocytes challenged with H/R (22). miRNA target predictor programs
(miRanda, Tarbase and TargetScan) identified putative binding sites
for miR-141, concurrently on Sirt1 and MFN2 transcripts. To
determine whether miR-141 is able to bind to either the 3'-UTR of
Sirt1 or MFN2, a luciferase assay was performed. As demonstrated in
Fig. 5A and B, co-transfection with miR-141 mimic
decreased the relative (Firefly/Renilla) luciferase activity
of 293T cells transfected with the 3'-UTR of Sirt1 or MFN2, which
was not observed in the control sample. According to the binding
data, the transfection of HL-1 cells with the miR-141 mimic but not
with the miR control, simultaneously decreased Sirt1 and MFN2
protein expression (Fig. 5C and
D). Collectively, these findings
indicate that miR-141 can bind to the 3'-UTRs of both Sirt1 and
MFN2, and decrease their cellular protein levels.
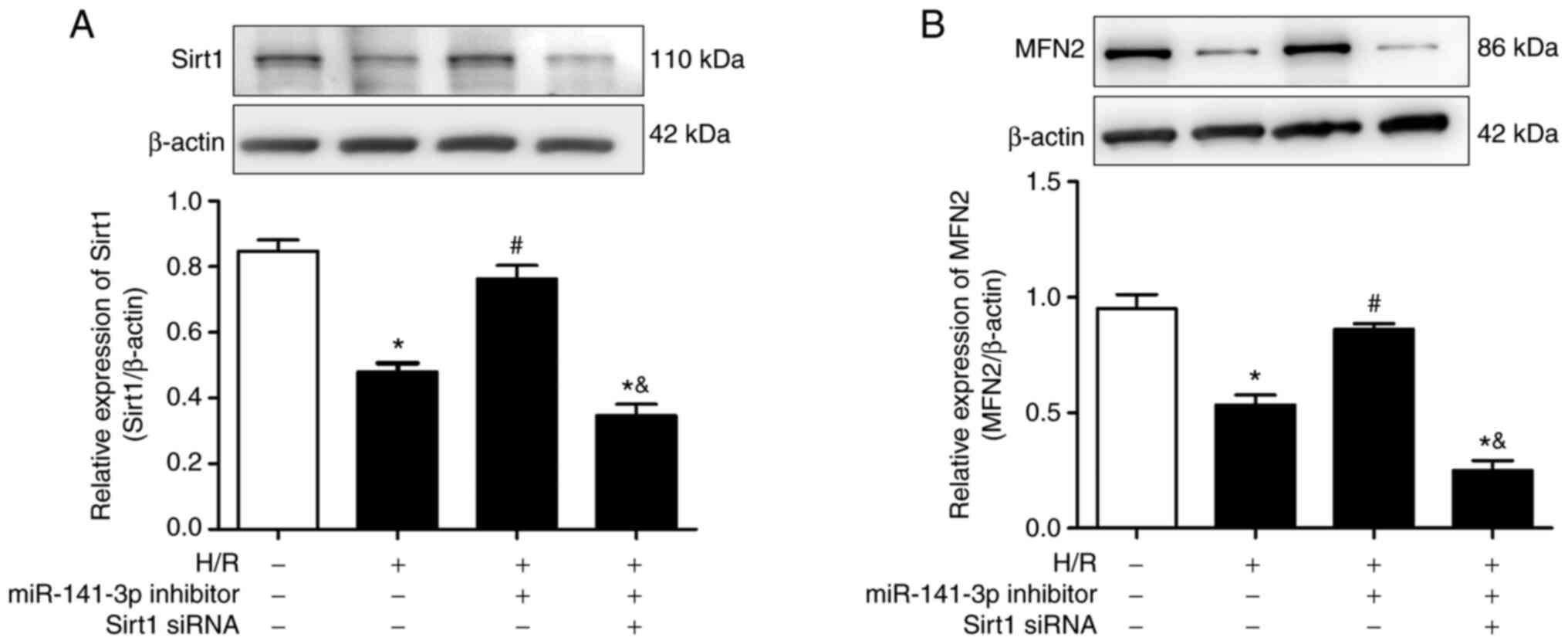
miR-141 inhibitor prevents the
H/R-induced downregulation of Sirt1 and MFN2 in HL-1 cells
The H/R challenge of cardiomyocytes decreased their
expression of Sirt1 and MFN2 protein (Fig. 6A and B). The miR-141 inhibitor prevented the
H/R-induced downregulation of Sirt1 and MFN2. As shown in Fig. S5, Sirt1 siRNA effectively
inhibited the expression of Sirt1 in HL-1 cardiomyocytes.
Furthermore, transfection of the HL-1 cells with siRNA against
Sirt1 reversed the promoting effects of miR-141 inhibitor on MFN2
protein expression (Fig. 6B). The
latter observation is consistent with MFN2 being a downstream
target of Sirt1 (22,23).
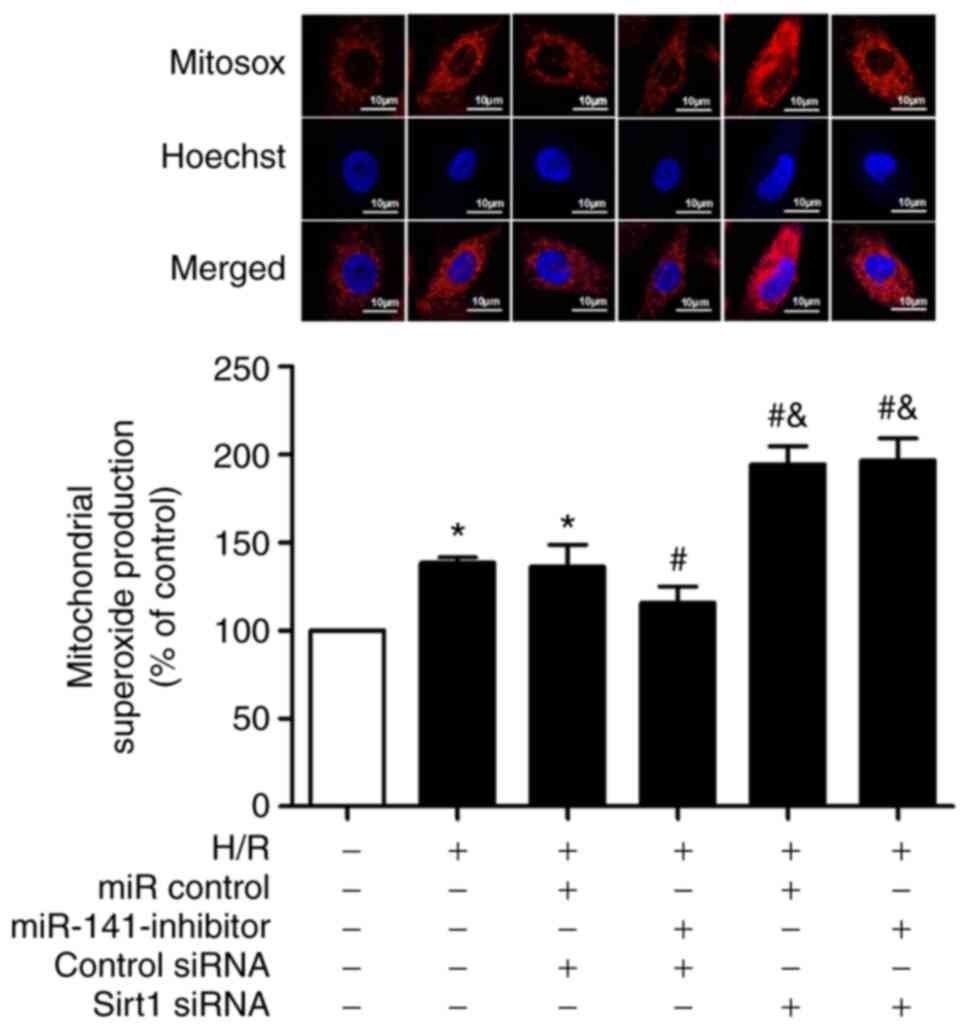
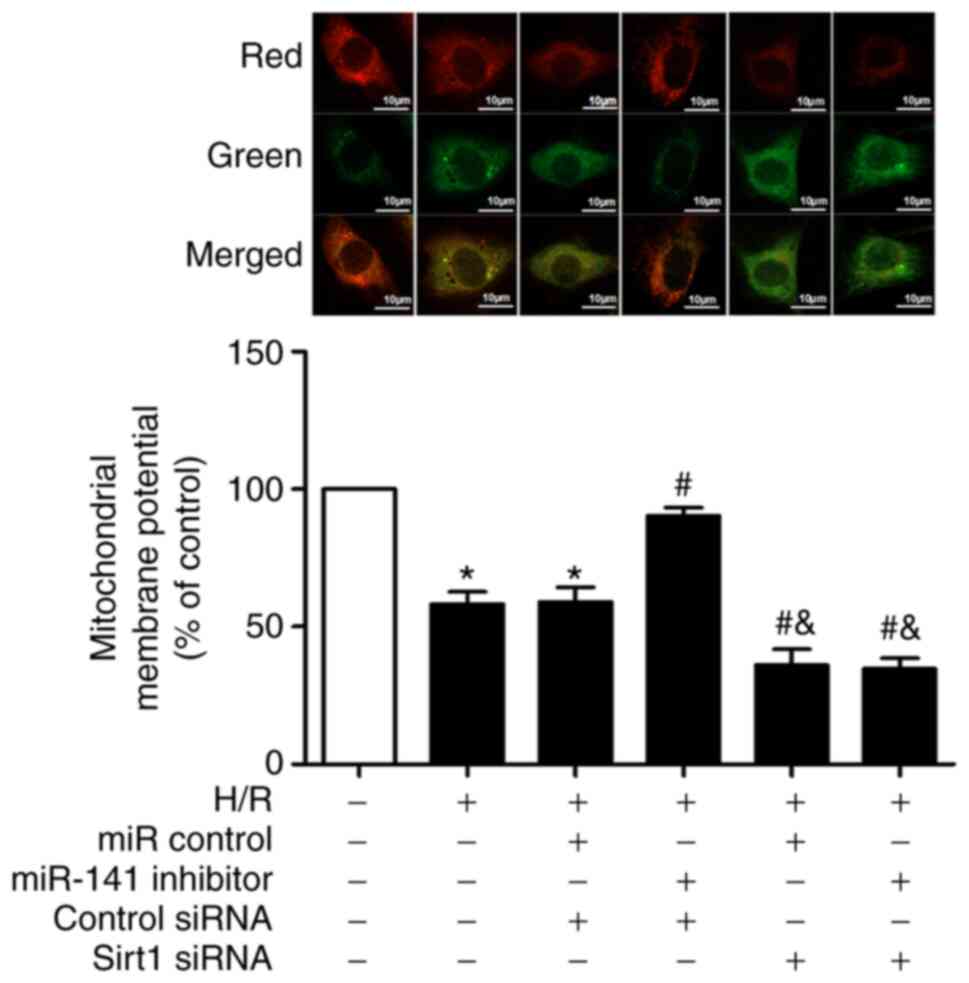
Sirt1 siRNA reverses the protective
effects of miR-141 inhibitor on H/R-induced mitochondrial
superoxide production and membrane potential
As anticipated, the challenge of cardiomyocytes with
H/R increased mitochondrial superoxide production (Fig. 7); an effect significantly prevented
by the miR-141 inhibitor. Moreover, Sirt1 siRNA abolished the
protective effect of the miR-141 inhibitor, and exacerbated the
H/R-induced mitochondrial superoxide production compared with in
the H/R + mir-141-inhibitor + control siRNA group.
Similar results were observed concerning the
H/R-induced changes in mitochondrial membrane potential. The
challenge of cardiomyocytes with H/R depolarized the mitochondrial
membrane (Fig. 8), an effect which
was attenuated by miR-141 inhibitor. Additionally, Sirt1 siRNA
abolished the protective effects of the miR-141 inhibitor, and also
exacerbated the H/R-induced depolarization of the mitochondrial
membrane.
Discussion
I/R-induced myocardial dysfunction and injury are
attributed to enhanced ROS production by cardiomyocytes, a major
source of ROS being the ETC of mitochondria (4-8).
The initial overproduction of ROS generates a self-amplifying wave
of oxidant stress along the ETC that leads to mitochondrial
dysfunction and culminates in cardiomyocyte death and tissue
necrosis (5,7,8).
Transcriptional regulatory mechanisms exist to limit cellular
oxidative stress and associated sequelae; however, the generation
of defensive proteins can be negated at the post-transcriptional
level by non-coding RNAs, miRNAs. miRNAs exert their effects by
binding to complementary mRNA sites, preventing their translation.
miR-141, a member of the miR-200 family, can respond to oxidative
stress (27) and can induce
mitochondrial dysfunction (16)
and cell death (13,14). Thus, the main focus of the present
study was to elucidate the role of miR-141 in the mitochondrial
dysfunction of cardiomyocytes, elicited by an H/R challenge. Three
novel findings-to the best of our knowledge-were presented in the
current study: i) The H/R-induced decrement in mitochondrial oxygen
utilization, increment in ROS production, and membrane
depolarization, as well as cardiomyocyte apoptosis is ameliorated
by miR-141 inhibitor; ii) miR-141 simultaneously targets Sirt1 and
MFN2 mRNA; and iii) the mitochondria-associated protein, MFN2, is a
downstream target of Sirt1.
The results of the present study using a cell-based
model of simulated I/R are in line with mitochondrial ROS-induced
ROS release playing a prominent role in I/R-induced cardiomyopathy
(5,7,8). In
this scenario, upon the reoxygenation of hypoxic cardiomyocytes
(H/R), there is a surge in mitochondrial ROS production, resulting
in an increase in inner membrane permeability and the subsequent
collapse of the membrane potential. The oxidative stress is
propagated from one mitochondrion to another, eventually resulting
in cell death. Herein, the H/R challenge of HL-1 or H9c2
cardiomyocytes resulted in an upregulation of miR-141; an event
inhibited by a SOD-mimetic that accumulates in mitochondria
(Figs. 1 and S1). Furthermore, the H/R-induced
mitochondrial ROS generation, membrane depolarization,
ultrastructural derangements, as well as cardiomyocyte apoptosis
were blunted by an inhibitor of miR-141 (Figs. 2, 4, S2
and S3). In addition, miR-141
also impaired the viability of the N/R-treated cells, while miR-141
inhibitor increased cell viability. The possible reason is that
miR-141 affects cell viability by regulating the expression of
relevant target genes. Collectively, these findings indicated that
subjecting cardiomyocytes to simulated I/R increased mitochondrial
ROS production, subsequently inducing more mitochondrial ROS
production and dysfunction via miR-141.
The specific components of the signaling pathway by
which mitochondrial ROS initially promotes expression of miR-141
are not yet entirely clear. However, the tumor suppressor protein,
p53, is an attractive candidate. An in silico approach
indicates that functional p53 motifs are present on the miR-200
family of genes, including miR-141(28), which can be activated by p53 in
response to oxidant stress in hepatocytes (29). Moreover, the cardiomyocyte
expression of p53 can be increased by an H/R challenge in a
ROS-dependent manner (30).
Further studies are required to systematically evaluate the
signaling pathway leading to miR-141 expression induced by
mitochondrial oxidant stress.
Of note, in the present study, the H/R challenge of
HL-1 cardiomyocytes reduced the mitochondrial oxygen consumption
associated with ATP production. This defect in oxidative
phosphorylation was exaggerated by a miR-141 mimic (Fig. 3A and B), but was attenuated by a miR-141
inhibitor (Fig. 3C and D). These findings are in accordance with
those of a previous study implicating a role for miR-141 in
mitochondrial oxidative phosphorylation (16). Specifically, miR-141 targets
Slc25a3, a mitochondrial phosphate carrier. The Slc25a3 carrier is
important for the delivery of inorganic phosphate to the
mitochondrial matrix for the generation of ATP from ADP. The
overexpression of miR-141 in HL-1 cardiomyocytes reduces Slc25a3
and cellular ATP production (16).
Thus, a potential mechanism by which miR-141 decreased ATP-linked
oxygen uptake in the present study may be by limiting inorganic
phosphate availability for ATP production.
Sirt1 is a cardioprotective protein that promotes
redox homeostasis during oxidant stress among other beneficial
effects, which are exerted through the deacetylation of proteins
with antioxidant activities, including FOXO, and PGC-1α (18,31,32).
Notably, PGC1α is a critical regulator of mitochondrial function
through activation of mitochondrial biogenesis and energy
metabolism (30). Specifically,
Sirt1 can ameliorate cardiomyocyte apoptosis and ROS production
(18). Furthermore, the induction
of Sirt1 reduces cardiomyocyte dysfunction induced by I/R or H/R
(30,33). Of note, miR-141 is expression
increased and targets Sirt1 in a cell-based model of Parkinson's
disease, involving the challenge of neuron-like cells with a toxin
that disrupts the mitochondrial ETC and resulting in cellular
oxidant stress and death (15,17).
Additional evidence in support of the role of Sirt1 in maintaining
mitochondrial hemostasis during oxidative stress is provided by the
results of the present study. In brief, miR-141 regulates the
impact of H/R on mitochondrial structure (Fig. 4E-H) and function (Fig. 3), as well as cell viability
(Figs. 2 and S3). The miR-141 mimic exacerbated the
effects of H/R, while a miR-141 inhibitor was protective.
Additionally, Sirt1 was a target of miR-141 and the silencing of
Sirt1 negated the regulatory function of miR-141 on H/R-induced
mitochondrial ROS production (Fig.
7) and membrane collapse (Fig.
8).
MFN2 is an outer mitochondrial membrane GTPase that
has been implicated in optimal mitochondrial functioning (20). As depicted in Fig. 6, MFN2 is a target of Sirt1.
Previous studies have indicated that MFN2 levels decrease after
H/R, and either stabilizing or enhancing MFN2 levels protects cells
from H/R (21-23).
The protective effect of MFN2 has been attributed to either the
fusion-induced optimization of mitochondrial oxidative
phosphorylation (21) or the
induction of mitophagy to remove defective mitochondria (22,23).
Irrespectively, both events serve to maintain a cellular pool of
optimally functioning mitochondria. In fact, MFN2 maintains
mitochondrial homeostasis in cardiomyocytes via both fusion events
and mitophagy (34). Of note, the
deacetylation of MFN2 by Sirt1 is protective against hepatic I/R or
hepatocyte H/R (22,23). Specifically, Sirt1 deacetylates at
two lysine residues in the C-terminus of MFN2 leading to autophagy
activation (22). In the present
study, the Sirt1/MFN2 pathway was operative in cardiomyocyte H/R,
with miR-141 targeting both components of this pathway (Fig. 5). This observation provides an
explanation for the observation that i) simulated I/R of aged
hepatocytes resulted in the loss of both Sirt1 and MFN2; and ii)
the co-overexpression of Sirt1 and MFN2, and not the overexpression
of either protein alone, mitigated the mitochondrial dysfunction
and hepatocyte death induced by H/R (22).
The H/R challenge of cardiomyocytes reduced MFN2
protein levels, an effect mitigated by the silencing of Sirt1
(Fig. 6B). This observation is in
accordance with the ability of Sirt1 to promote mitochondrial
biogenesis in hepatocytes by induction of the MFN2 gene (35). Whereas the deacetylation of MFN2 by
Sirt1 has been previously reported (22), the mechanisms by which Sirt1
regulates MFN2 protein expression remain unclear. Previous research
has revealed that PGC-1α is a target protein of Sirt1, and that the
overexpression of Sirt1 can cause an increase in PGC-1α protein
expression (36). Furthermore,
PGC1α can regulate Mfn2 transcription by binding to its promoter
region (37). More importantly,
the mitochondrial dysfunction induced by an H/R challenge of H9c2
cardiomyocytes is blunted by Sirt1-induced expression of PGC1α
(30). Furthermore, PGC1α can
stimulate MFN2 mRNA and protein expression in skeletal muscle
(38). Briefly, the ability of
Sirt1 to induce MFN2 protein expression is not unprecedented,
albeit the mechanisms involved have not been systematically
assessed. A possible mechanism through which Sirt1 can regulate
MFN2 expression is that Sirt1 may regulate MFN2 transcription
through its co-transcriptional regulator, PGC1α.
Mitochondria are dynamic organelles that undergo
fission and fusion. MFN2 is a well characterized protein driving
mitochondrial fusion, while dynamin-related protein 1 (DRP1) is a
major regulator of mitochondrial fission (39,40).
The H/R-induced mitochondrial ROS production and structural
derangements noted in the present study are reminiscent of
mitochondrial fission/fragmentation. Of note, a role for the
Sirt1/DRP1 pathway in a cell-based model of hyperglycemia to
simulate diabetes has been previously reported by the authors. In
brief, an H/R challenge of cardiomyocytes pre-conditioned with
glucose (30 mM) decreased Sirt1 levels and activated DRP1, thereby
promoting mitochondrial fragmentation (41). Thus, it is likely that Sirt1 can
modulate mitochondrial dynamics by reciprocal regulation of
regulatory proteins, including MFN2 and DRP1.
A major disadvantage is that the total number of
miRNAs that can target Sirt1 and MFN2 is expanding. For example,
miR-22, -34a, -9, -29c and -92a, as well as miR-141 can target
Sirt1 [(17,42-44),
and the results of the present study], while miR-195, -106b, -93
and -20b, as well as miR-141 can target MFN2 [(45-47),
and the results of the present study]. Specifically, miR-34a,
miR-141 and miR-9 may influence Parkinson's disease pathogenic
processes by targeting Sirt1(17).
Suppression of miR-34a has been reported to provide anti-apoptotic
protection by directly targeting Sirt1 in a myocardial IR injury
model (42). Furthermore, miR-92a
can prevent the migration of H2O2-induced
vascular smooth muscle cells by suppressing the expression of
Sirt1(43) and miR-29c suppresses
liver tumorigenesis by binding to the 3'-untranslated region of
Sirt1 mRNA (44). In addition,
miR-195 may impair mitochondrial function by targeting MFN2 in
breast cancer cells (45), and
miR-106b and miR-93 can control excessive mitophagy and restrain
cell death by targeting MFN2(46).
Furthermore, miR-20b promotes cardiac hypertrophy by directly
targeting MFN2(47). Thus, drawing
firm conclusions regarding the therapeutic potential of any miRNA
is rather difficult. Additionally, the use of a cell-based model of
simulated ischemia/reperfusion further complicates the
translational applicability. Whereas cell-based models are very
useful in uncovering signaling pathways in a given resident cardiac
cell (e.g., cardiomyocyte), contributions from other resident or
infiltrating cells cannot be considered (3). Nonetheless, miR-141 can target both
components of the Sirt1/MFN2 pathway (Fig. 5), which has been implicated in
mitochondrial homeostasis during I/R or H/R [(22,23),
and the results of the present study]. The targeting of both
components of the Sirt1/MFN2 pathway permits an improved degree of
specificity in the modulation of mitochondrial function by miR-141.
This intriguing aspect of miR-141 modulation of the Sirt1/MFN2
pathway warrants further investigation with a view toward potential
therapeutic application.
In conclusion, the challenge of cardiomyocytes with
H/R increases miR-141 expression. miR-141 expression is induced by
mitochondrial ROS production and serves to perpetuate the oxidant
stress, ultimately leading to cell death. The H/R-induced
mitochondrial ROS production and dysfunction are mediated by
miR-141, most likely via the targeting of both components of the
Sirt1/MFN2 pathway.
Supplementary Material
H/R increases miR-141 expression in
H9C2. H9C2 cardiomyocytes were challenged with hypoxia for 3 h and
subsequently reoxygenated (H/R). Cells maintained under normoxic
conditions for relevant durations served as controls (N/R).
Cellular levels of miR-141 were measured using reverse
transcription-quantitative PCR. (A) miR-141 levels in N/R group and
H/R group. (B) Effects of a SOD mimetic (Mitotempo) targeting
mitochondrial superoxide on miR-141 levels assessed at 6 h after
reoxygenation. The cardiomyocytes were pretreated with 20 μM
Mitotempo for 1 h prior to the challenge with H/R or N/R. The
results are expressed as the mean ± standard error of the mean;
n=3. *P<0.01 vs. N/R; #P<0.01 vs. H/R
without Mitotempo. H/R, hypoxia/reoxygenation; N/R,
normoxia/reoxygenation; SOD, superoxide dismutase.
Role of miR-141 in H/R-induced
cardiomyocyte apoptosis. HL-1 cardiomyocytes were transfected with
a miR-141 mimic, miR-141 inhibitor, or miR con. Subsequently, the
cells were challenged with H/R, and 12 h following reoxygenation,
the indices of apoptosis were evaluated using FACS analyses with
Annexin-V/propidium iodide. These figures are from representative
experiments carried out at least three independent tests. The
viable cells, early apoptotic and necrotic/secondary necrotic cells
were represented by the lower left quadrant (Annexin-V-/PI-), lower
right (Annexin-V+/PI-) and upper (Annexin-V+/PI+) quadrant,
respectively. H/R, hypoxia/reoxygenation; miR con, negative
control.
The role of miR-141 in H/R-induced
H9C2 cardiomyocyte viability and death. H9C2 cardiomyocytes were
transfected with a miR-141 mimic, miR-141 inhibitor, or their
negative controls (miR con). Subsequently, the cells were
challenged with H/R and 12 h after reoxygenation indices of
viability and plasma membrane disruption were assessed. (A) The
viability of cells was assessed using a CCK-8 assay. (B and C) LDH
and CK-MB release from cells with ruptured membranes. The results
are presented as the mean ± standard error of the mean; n=3.
*P<0.05 vs. N/R + miR con, #P<0.05 vs.
H/R + miR con. H/R, hypoxia/reoxygenation; miR con, miR control;
CCK-8, cell counting kit-8 assay; LDH, lactate dehydrogenase;
CK-MB, creatine kinase-MB.
Eestablishment of the miR-141 mimic
and inhibitor models. HL-1 cardiomyocytes were transfected with a
miR-141 mimic, miR-141 inhibitor, or their negative controls (miR
con) respectively. Following transfection, cellular levels of
miR-141 were measured using reverse transcription-quantitative PCR.
(A) Transfection with miR-141 mimics significantly increased the
expression of miR-141. (B) Transfection with miR-141 inhibitor
reduced the expression of miR-141. All bar graphs represent the
mean ± standard error of the mean; n=3. *P<0.001 vs.
NC group (for A); *P<0.05 vs. NC group (for B).
Construction of Sirt1 knockdown cell
model. HL-1 cardiomyocytes were transfected with Sirt1 siRNA or
control siRNA. After 24 h, the protein expression of Sirt1 in HL-1
cells with or without Sirt1 siRNA was analyzed using western
blotting. All bar graphs represent the mean ± standard error of the
mean; n=3. *P<0.01 vs. control (open bar).
Scoring paradigm for mitochondrial
ultrastructure.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 82172172) and the Science
and Technology Bureau of Zhengjiang (grant no. SH2019048).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ, HW and TR contributed to the conceptualization
and the design of the present study. HZ, RL and HW performed the
experiments and analyzed the data. YW, YY, PK and KW were
responsible for the acquisition, analysis and interpretation of the
data. HZ and PK contributed to the drafting of the manuscript. RL
and KW confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heusch G: Myocardial ischaemia-reperfusion
injury and cardioprotection in perspective. Nat Rev Cardiol.
17:773–789. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Davidson SM, Adameová A, Barile L,
Cabrera-Fuentes HA, Lazou A, Pagliaro P, Stensløkken KO and
Garcia-Dorado D: EU-CARDIOPROTECTION COST Action (CA16225).
Mitochondrial and mitochondrial-independent pathways of myocardial
cell death during ischaemia and reperfusion injury. J Cell Mol Med.
24:3795–3806. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ren D, Wang X, Ha T, Liu L, Kalbfleisch J,
Gao X, Williams D and Li C: SR-A deficiency reduces myocardial
ischemia/reperfusion injury; involvement of increased microRNA-125b
expression in macrophages. Biochim Biophys Acta. 1832:336–346.
2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866(165768)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen YR and Zweier JL: Cardiac
mitochondria and reactive oxygen species generation. Circ Res.
114:524–537. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Magenta A, Ciarapica R and Capogrossi MC:
The emerging role of miR-200 family in cardiovascular diseases.
Circ Res. 120:1399–1402. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kalinina EV, Ivanova-Radkevich VI and
Chernov NN: Role of MicroRNAs in the regulation of redox-dependent
processes. Biochemistry (Mosc). 84:1233–1246. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Magenta A, Lorde R, Syed SB, Capogrossi
MC, Puca A and Madeddu P: Molecular therapies delaying
cardiovascular aging: Disease- or health-oriented approaches. Vasc
Biol. 2:R45–R58. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Qadir MMF, Klein D, Álvarez-Cubela S,
Domínguez-Bendala J and Pastori RL: The role of MicroRNAs in
diabetes-related oxidative stress. Int J Mol Sci.
20(5423)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yao B, Wan X, Zheng X, Zhong T, Hu J, Zhou
Y, Qin A, Ma Y and Yin D: Critical roles of microRNA-141-3p and
CHD8 in hypoxia/reoxygenation-induced cardiomyocyte apoptosis. Cell
Biosci. 10(20)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Qin Q, Cui L, Zhou Z, Zhang Z, Wang Y and
Zhou C: Inhibition of microRNA-141-3p reduces hypoxia-induced
apoptosis in H9c2 rat cardiomyocytes by activating the
RP105-dependent PI3K/AKT signaling pathway. Med Sci Monit.
25:7016–7025. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zheng Y, Dong L, Liu N, Luo X and He Z:
Mir-141-3p regulates apoptosis and mitochondrial membrane potential
via targeting sirtuin1 in a 1-methyl-4-phenylpyridinium in vitro
model of Parkinson's disease. Biomed Res Int.
2020(7239895)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Baseler WA, Thapa D, Jagannathan R,
Dabkowski ER, Croston TL and Hollander JM: miR-141 as a regulator
of the mitochondrial phosphate carrier (Slc25a3) in the type 1
diabetic heart. Am J Physiol Cell Physiol. 303:C1244–C1251.
2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Delavar MR, Baghi M, Safaeinejad Z,
Kiani-Esfahani A, Ghaedi K and Nasr-Esfahani MH: Differential
expression of miR-34a, miR-141, and miR-9 in MPP+-treated
differentiated PC12 cells as a model of Parkinson's disease. Gene.
662:54–65. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang J, Ren D, Fedorova J, He Z and Li J:
SIRT1/SIRT3 modulates redox homeostasis during ischemia/reperfusion
in the aging heart. Antioxidants (Basel). 9(858)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lee IH: Mechanisms and disease
implications of sirtuin-mediated autophagic regulation. Exp Mol
Med. 51:1–11. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Filadi R, Pendin D and Pizzo P: Mitofusin
2: From functions to disease. Cell Death Dis. 9(330)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Olmedo I, Pino G, Riquelme JA, Aranguiz P,
Díaz MC, López-Crisosto C, Lavandero S, Donoso P, Pedrozo Z and
Sánchez G: Inhibition of the proteasome preserves Mitofusin-2 and
mitochondrial integrity, protecting cardiomyocytes during
ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis.
1866(165659)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chun SK, Lee S, Flores-Toro J, Rebecca YU,
Yang MJ, Go KL, Biel TG, Miney CE, Louis SP, Law BK, et al: Loss of
sirtuin 1 and mitofusin 2 contributes to enhanced
ischemia/reperfusion injury in aged livers. Aging Cell.
17(e12761)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Biel TG, Lee S, Flores-Toro JA, Dean JW,
Go KL, Lee MH, Law BK, Law ME, Dunn WA Jr, Zendejas I, et al:
Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse
livers in a mitofusin 2-dependent manner. Cell Death Differ.
23:279–290. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu Y, Nguyen P, Baris TZ and Poirier MC:
Molecular analysis of mitochondrial compromise in rodent
cardiomyocytes exposed long term to nucleoside reverse
transcriptase inhibitors (NRTIs). Cardiovasc Toxicol. 12:123–134.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Poli G, Guasti D, Rapizzi E, Fucci R, Canu
L, Bandini A, Cini N, Bani D, Mannelli M and Luconi M:
Morphofunctional effects of mitotane on mitochondria in human
adrenocortical cancer cells. Endocr Relat Cancer. 20:537–550.
2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Magenta A, Cencioni C, Fasanaro P,
Zaccagnini G, Greco S, Sarra-Ferraris G, Antonini A, Martelli F and
Capogrossi MC: miR-200c is upregulated by oxidative stress and
induces endothelial cell apoptosis and senescence via ZEB1
inhibition. Cell Death Differ. 18:1628–1639. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tamura M, Sasaki Y, Kobashi K, Takeda K,
Nakagaki T, Idogawa M and Tokino T: CRKL oncogene is downregulated
by p53 through miR-200s. Cancer Sci. 106:1033–1040. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xiao Y, Yan W, Lu L, Wang Y, Lu W, Cao Y
and Cai W: p38/p53/miR-200a-3p feedback loop promotes oxidative
stress-mediated liver cell death. Cell Cycle. 14:1548–1558.
2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang
CN, Tang XL, Lu JQ, Zhu XY and Ni X: Upregulation of microRNA-22
contributes to myocardial ischemia-reperfusion injury by
interfering with the mitochondrial function. Free Radic Biol Med.
96:406–417. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
D'Onofrio N, Servillo L and Balestrieri
ML: SIRT1 and SIRT6 signaling pathways in cardiovascular disease
protection. Antioxid Redox Signal. 28:711–732. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Elibol B and Kilic U: High levels of SIRT1
expression as a protective mechanism against disease-related
conditions. Front Endocrinol (Lausanne). 9(614)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hsu CP, Zhai P, Yamamoto T, Maejima Y,
Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J:
Silent information regulator 1 protects the heart from
ischemia/reperfusion. Circulation. 122:2170–2182. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xiong W, Ma Z, An D, Liu Z, Cai W, Bai Y,
Zhan Q, Lai W, Zeng Q, Ren H and Xu D: Mitofusin 2 participates in
mitophagy and mitochondrial fusion against angiotensin II-induced
cardiomyocyte injury. Front Physiol. 10(411)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mouchiroud L, Houtkooper RH, Moullan N,
Katsyuba E, Ryu D, Cantó C, Mottis A, Jo YS, Viswanathan M,
Schoonjans K, et al: The NAD(+)/sirtuin pathway modulates longevity
through activation of mitochondrial UPR and FOXO signaling. Cell.
154:430–441. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou Z, Ma D, Li P, Wang P, Liu P, Wei D,
Wang J, Qin Z, Fang Q, Wang J, et al: Sirt1 gene confers Adriamycin
resistance in DLBCL via activating the PCG-1α mitochondrial
metabolic pathway. Aging (Albany NY). 12:11364–11385.
2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hu L, Guo Y, Song L, Wen H, Sun N, Wang Y,
Qi B, Liang Q, Geng J, Liu X, et al: Nicotinamide riboside promotes
Mfn2-mediated mitochondrial fusion in diabetic hearts through the
SIRT1-PGC1α-PPARα pathway. Free Radical Biol Med. 183:75–88.
2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Soriano FX, Liesa M, Bach D, Chan DC,
Palacín M and Zorzano A: Evidence for a mitochondrial regulatory
pathway defined by peroxisome proliferator-activated receptor-gamma
coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin
2. Diabetes. 55:1783–1791. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sabouny R and Shutt TE: Reciprocal
regulation of mitochondrial fission and fusion. Trends Biochem Sci.
45:564–577. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu YJ, McIntyre RL, Janssens GE and
Houtkooper RH: Mitochondrial fission and fusion: A dynamic role in
aging and potential target for age-related disease. Mech Ageing
Dev. 186(111212)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Tao A, Xu X, Kvietys P, Kao R, Martin C
and Rui T: Experimental diabetes mellitus exacerbates
ischemia/reperfusion-induced myocardial injury by promoting
mitochondrial fission: Role of down-regulation of myocardial Sirt1
and subsequent Akt/Drp1 interaction. Int J Biochem Cell Biol.
105:94–103. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Fu BC, Lang JL, Zhang DY, Sun L, Chen W,
Liu W, Liu KY, Ma CY, Jiang SL, Li RK and Tian H: Suppression of
miR-34a expression in the myocardium protects against
ischemia-reperfusion injury through SIRT1 protective pathway. Stem
Cells Dev. 26:1270–1282. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Liu P, Su J, Song X and Wang S: miR-92a
regulates the expression levels of matrix metalloproteinase 9 and
tissue inhibitor of metalloproteinase 3 via sirtuin 1 signaling in
hydrogen peroxide-induced vascular smooth muscle cells. Mol Med
Rep. 17:1041–1048. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bae HJ, Noh JH, Kim JK, Eun JW, Jung KH,
Kim MG, Chang YG, Shen Q, Kim SJ, Park WS, et al: MicroRNA-29c
functions as a tumor suppressor by direct targeting oncogenic SIRT1
in hepatocellular carcinoma. Oncogene. 33:2557–2567.
2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Purohit PK, Edwards R, Tokatlidis K and
Saini N: MiR-195 regulates mitochondrial function by targeting
mitofusin-2 in breast cancer cells. RNA Biol. 16:918–929.
2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang C, Nie P, Zhou C, Hu Y, Duan S, Gu
M, Jiang D, Wang Y, Deng Z, Chen J, et al: Oxidative stress-induced
mitophagy is suppressed by the miR-106b-93-25 cluster in a
protective manner. Cell Death Dis. 12(209)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Qiu Y, Cheng R, Liang C, Yao Y, Zhang W,
Zhang J, Zhang M, Li B, Xu C and Zhang R: MicroRNA-20b promotes
cardiac hypertrophy by the inhibition of mitofusin 2-mediated
inter-organelle Ca(2+) cross-talk. Mol Ther Nucleic Acids.
19:1343–1356. 2020.PubMed/NCBI View Article : Google Scholar
|