Introduction
Bone defects caused by high-energy trauma, bone
tumors, limb deformities and bone infections can lead to poor and
delayed bone healing, and nonunion (1,2).
Although bone tissue can be completely regenerated in the body with
time, bone defects that are beyond the critical length range of
self-repair experience difficulty in self-repair and reconstruction
to restore normal bone length and stiffness (3). Bone is a highly vascularized, unique,
complex and dynamic tissue with regenerative properties that can
repair itself and restore its original structural integrity after
injury or destruction, although bone damage can lead to a host of
additional problems, such as ruptured blood vessels, hematoma
formation and nerve damage, as it is accompanied by local tissue
hypoxia with the release of systemic cytokines stimulating the
formation of fresh blood vessels in the bone defect site (4). Angiogenesis is a complex process
involving various cell interactions, growth factor stimulation and
biomechanics. Angiogenesis originates from the production of matrix
metalloproteinases in endothelial cells, which degrade the basement
membrane to promote endothelial cell migration (5). Endothelial cell migration is a key
component of the angiogenic response, and endothelial cells move
toward areas with high concentrations of VEGF and other growth
factors through the proteolytic basement membrane (6). When endothelial cells relocate into
tissue, they reproduce and differentiate to construct new blood
vessels. The regeneration of bone is based on the interaction
between osteogenesis and angiogenesis, largely depending on
angiogenesis, which serves a crucial role in bone repair and
remodeling. Angiogenesis refers to the reproduction and migration
of endothelial cells to the sites of original blood vessels to form
a new crisscross vascular network, continuously providing
sufficient nutrients, cytokines, neurotransmitters and oxygen for
bone cells, bone tissue and the inner and outer membranes of bone
(7). Therefore, understanding how
to effectively promote angiogenesis in bone defects may serve an
important role in bone repair.
Monocyte chemoattractant protein-1, also known as CC
motif ligand 2 (CCL2), an effective chemokine of monocytes, is a
member of the CC chemokine family and was the first human chemokine
to be discovered. It is produced or induced by multiple types of
cells in the body in response to oxidative stress. It serves a
valuable role in various pathophysiological processes, such as
inducing macrophages, immune stress, recruitment, inflammatory
response (8), angiogenesis, cell
proliferation, migration and wound repair (9). It also stimulates directional or
nondirectional cell migration (10). Although CCL2 has been reported to
promote angiogenesis, the detailed mechanism of CCL2 has yet to be
fully elucidated. In addition, examining the underlying mechanism
by which CCL2 promotes angiogenesis will yield novel functional
benefits to promote rapid reconstruction and repair of bone
defects. Therefore, the present study investigated the mechanism of
CCL2 in angiogenesis for bone defect repair and remodeling using
cell and animal model experiments.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
obtained from ScienCell Research Laboratories, Inc (cat. no. 8000).
HUVECs were grown in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin and streptomycin (Beijing Solarbio Science &
Technology Co., Ltd.), and cultured in a cell incubator at 95%
humidity at 37˚C with 5% carbon dioxide. Ethics approval for the
use of HUVECs was not required according to our institute's
guidelines (http://www.gdmuah.com/info/1851/8954.htm).
Antibodies and reagents
CCL2 (cat. no. M1203-03S) was purchased from United
States Biological. Antibodies against rho-associated
coiled-coil-containing protein kinase (Rock)1 (cat. no. #4035),
N-cadherin (cat. no. #13116), c-Myc (cat. no. #18583),
phosphorylated (p-)ERK1/2 (cat. no. #4370), p-AKT (cat. no.
#13038), AKT (cat. no. #4691), Wnt5a/b (cat. no. #2530),
low-density lipoprotein receptor related protein 6 (LRP-6; cat. no.
#3395), β-catenin (cat. no. #8480), p-PI3K (cat. no. #17366), PI3K
(cat. no. #4257), GAPDH (cat. no. #5174) and α-tubulin (cat. no.
#2125) and the secondary antibodies goat anti-rabbit IgG (cat. no.
#7074) and anti-mouse IgG (cat. no. #7076) were purchased from Cell
Signaling Technology, Inc. VEGFR2 (cat. no. ab134191) and MMP9
(cat. no. ab228402) antibodies were obtained from Abcam. Antibodies
against Rock2 (cat. no. sc-398519), ERK1/2 (cat. no. sc-514302) and
VEGF (cat. no. sc-57496) were obtained from Santa Cruz
Biotechnology, Inc.
Cell proliferation assay
HUVECs (2x103 cells per well) were seeded
into 96-well plates in ~100 µl total medium, incubated at 37˚C for
24 h, and then treated with various concentrations (0, 25, 50, 75,
100 and 150 ng/ml) of CCL2 at 37˚C for 0-96 h. Following addition
of 10 µl Cell Counting Kit-8 (Beyotime Institute of Biotechnology)
solution per well and incubation at 37˚C for 2 h, the OD value was
determined by a microplate reader (MK3; Thermo Fisher Scientific,
Inc.) at OD 450 nm.
Transwell migration assay
The fresh serum-free DMEM (100 µl) containing
2x104 HUVECs and various concentrations (0, 50, and 100
ng/ml) of CCL2 was added to the upper chamber. The lower chamber
was filled with 500 µl of medium containing 10% FBS and cells were
incubated at 37˚C for 24 h. Subsequently, cells that remained in
the upper chamber without successfully migrating were wiped away.
Cells on the membrane were fixed in methanol at room temperature
for 15 min and stained with crystal violet staining solution at
37˚C for 15 min. The cells were imaged in six random microscopic
fields (magnification, x100) under a light microscope (Olympus
Corporation). Migrated cells (% of control)=(migrated cells treated
with 50 or 100 ng/ml CCL2﹣migrated cells treated with 0 ng/ml
CCL2)/migrated cells treated with 0 ng/ml CCL2 x100.
Wound healing assay
HUVECs were cultured for 24 h in 6-well plates with
lines at the bottom of the plate. When the cell fusion rate
approached 100%, a 100-µl pipette tip was used to draw a line
perpendicular to the mark line in the culture plate, and PBS was
used to clean the culture well twice. Cells that had come loose
were removed, 2 ml serum-free medium with various concentrations of
CCL2 (0, 50 and 100 ng/ml) was added and cells were placed back in
the incubator for further culture. Subsequently, the migration
distance of HUVECs imaged under a light microscope (Olympus
Corporation) at 0 and 24 h was compared, the migration area was
calculated and the effect of different treatments on cell migration
was analyzed. Migration area (%)=(migration area at 0 h﹣migration
area at 24 h)/migration area at 0 h x100.
Tube formation assay
HUVECs (1.5x104) were cultured with
different doses of CCL2 (0, 25, 50 and 100 ng/ml) in DMEM in
Matrigel-precoated (37˚C; 30 min) angiogenesis slides at 37˚C with
5% CO2. After 4 h of incubation, tube formation was
observed under a light microscope (Olympus Corporation) and images
were captured for five randomly selected microscopic fields and
tube formation was analyzed using ImageJ software (v1.8.0; National
Institutes of Health).
Western blotting
Total protein was collected with an appropriate
amount of protein RIPA lysis buffer (PMSF:RIPA=1:100; Beyotime
Institute of Biotechnology). BCA protein assay kit (Beyotime
Institute of Biotechnology) was used to measure protein
concentration and each sample adjusted to the same concentration.
Proteins (20 µg/lane) were separated by SDS-PAGE on 10 and 12%
separation gels. After gel electrophoresis (100 V; 2 h), membrane
transfer (250 V; 2.5 h) and blocking (5% skimmed milk; room
temperature; 2 h), the 0.2 µm pore size PVDF membranes (Merck KGaA,
Darmstadt, Germany) were incubated with primary antibodies (diluted
1:1,000) overnight at 4˚C. Subsequently, the membranes were washed
with TBS with 0.1% Tween-20 (three times; 10 min) before addition
of the secondary antibody (diluted 1:3,000) at room temperature for
1 h. Finally, the immunoreactive membranes were imaged using a
Chemiluminescent and Fluorescent Imaging System (Tanon 5200; Tanon
Science and Technology Co., Ltd.), and the gray levels of the
protein bands were determined using ImageJ software (v1.8.0,
National Institutes of Health).
Establishment of bone defects in a
Sprague Dawley (SD) rat model
All animal experiments were approved by the
Laboratory Animal Ethics Committee of Guangdong Medical University
(Zhanjiang, China; ID Number: GDY1902126; date: 25.05.2019). The SD
rats (Guangdong Medical Laboratory Animal Center) were maintained
at 2 or 3 rats/cage under a 12-h light/dark cycle with adequate
water and standard food, at temperatures of 22-24˚C and relative
humidity of ~45%. Depending on the types of intervention,
12-week-old male SD rats (n=32) weighing 350-400 g were selected
and randomly divided into four groups: Group A (simple bone defect
not filled in), Group B [poly (lactic-co-glycolic acid)/tricalcium
phosphate (PLGA/TCP) porous scaffold; 5x5x5 mm3 filled
in], Group C (PLGA/TCP porous scaffold + Gelma hydrogel filled in)
and Group D (PLGA/TCP porous scaffold + Gelma hydrogel + 1 µg CCL2
filled in). The number of rats in each group was eight. The rats
were anesthetized by intraperitoneal injection with pentobarbital
sodium at a concentration of 1% at a dose of 30 mg/kg. The
appropriate degree of anesthesia was assessed by the
characteristics of stable breathing, sluggish corneal reflex,
generalized muscle relaxation and loss of skin pinch response.
After group assignment, the left femur of each rat was subjected to
3 mm midline osteotomy under anesthesia, and the bone defect area
was filled or not filled according to the experimental group
allocation. Furthermore, the left femur was fixed using an external
fixator (Fig. S1). The bone
defects establishment of each SD rat lasted ~40-60 min. The
specific criteria for SD rat sacrifice were that rats were close to
succumbing, with persistent dyspnea, inability to ingest food,
significant loss of appetite, weight loss of >20% (the maximum
percentage of body weight loss observed in the present study was
~10%), severe ulceration, heavy bleeding and limb paralysis.
Penicillin sodium (80,000 units/kg/d;) was intramuscularly injected
after surgery to prevent infection for 1 week and the surgical
incision was disinfected with iodophor every day. The rats were
monitored daily for surgical incision, mental status, diet, body
weight, activity and excretion. Subsequently, each group was
observed for 4 weeks, at which point half of the specimens were
collected, and 8 weeks, at which point the other half of the
specimens were collected. The SD rats were sacrificed with 10%
sodium pentobarbital at a dose of 200 mg/kg by intraperitoneal
injection. Mortality was verified by confirming the arrest of
breathing, the disappearance of heartbeat and light reflex, and the
dilation of pupils in SD rats. Finally, the femurs of the left leg
were removed and fixed in 10% neutral formalin solution for 48 h
and then stored in 75% ethanol.
Immunohistochemistry
After 10% formalin fixation at 4˚C for 48 h and EDTA
solution (Beijing Solarbio Science & Technology Co., Ltd.)
decalcification (2 months), bone tissue was dehydrated with
gradient ethanol and xylene (75% ethanol for 1 h; 85% ethanol for 1
h; 95% ethanol for 1 h; 100% ethanol for 1 h; xylene I for 30 min;
xylene II for 1 h) at room temperature. Subsequently, the bone
tissue was embedded in paraffin (65˚C). After tissue sectioning (4
µm), bone callus sections were hydrated, blocked with 5% BSA
(Beyotime Institute of Biotechnology) for 1 h, and incubated with
the VEGF primary antibody (diluted 1:200) at 4˚C overnight. After
subsequent secondary antibody (diluted 1:200) incubation (room
temperature; 30 min), 3,3'-diaminobenzidine dyeing solution (5 min)
and hematoxylin (2 sec) were added for staining at room
temperature. The images were captured under a light microscope
(Olympus Corporation).
Statistical analysis
All experimental results in the present study were
obtained from experiments repeated in at least three replicates.
All data are expressed as the mean ± standard deviation. ImageJ
(National Institutes of Health), SPSS Statistics 19.0 software (IBM
Corp.) and GraphPad Prism 8.0 software (GraphPad Software, Inc.)
were used to analyze the data and create the graphs, respectively.
Unpaired Student's t-test was used for comparisons between groups.
Statistical differences between ≥3 groups were determined using
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
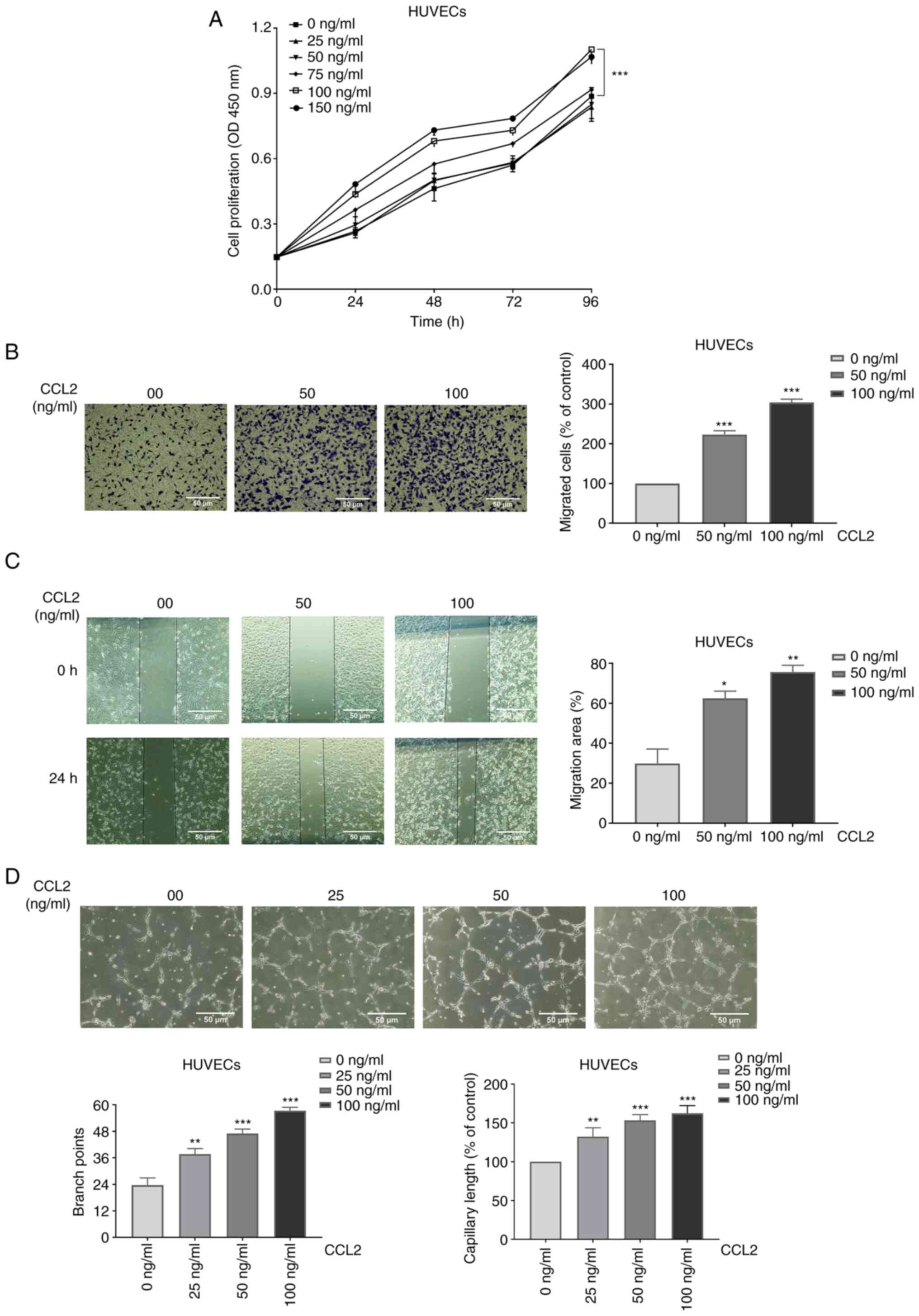
CCL2 promotes proliferation of
HUVECs
Cell proliferation assays revealed that CCL2
increased HUVEC proliferation in a concentration- and
time-dependent manner (Fig.
1A).
CCL2 boosts the migration of
HUVECs
The migration of HUVECs was measured using Transwell
migration assay and wound healing assay. After treatment with
different doses of CCL2 (0, 50 and 100 ng/ml) for 24 h, the cells
that pierced the polycarbonate membrane and the coalescing wound
areas were observed under a microscope at a magnification of x100.
The results of the cell migration assay revealed that the numbers
of migrated cells (% of control) of HUVECs were 100, 223±7 and
304±6% respectively (Fig. 1B). As
shown in Fig. 1C, the migration
area of HUVECs was compared among different treatment conditions.
In addition, the results demonstrated that the migration areas of
HUVECs were 30±5, 62±3, and 76±2%, respectively. The experiments
revealed that CCL2 boosted the migration of HUVECs.
CCL2 promotes tube formation in
HUVECs
One of the most essential steps in the processes of
angiogenesis is endothelial cell proliferation, and the
regeneration of new blood vessels is required for bone tissue
growth. Vessel formation serves a key role in tissue rebuilding
(11). HUVECs exposed to different
concentrations of CCL2 were cultured in Matrigel, and then the
construction of capillary-like structures was imaged by microscopy
at a magnification of x100. Tube formation was assessed based on
the number of branch points and capillary length indexes. Following
treatment with different doses of CCL2 (0, 25, 50 and 100 ng/ml)
for 4 h, the branch points of tube formation were 23±3, 38±2, 47±2
and 57±2. In addition, the capillary lengths (100% of control) of
tube formation were 100, 132±9, 153±6, and 162±9% respectively
(Fig. 1D). The results revealed
that CCL2 at a suitable concentration had a positive effect on tube
formation.
CCL2 promotes the expression levels of
proteins related to proliferation, migration and vascularization in
HUVECs
A number of studies have reported that Rock1, Rock2,
N-cadherin and c-Myc are closely associated with proliferation and
migration, and VEGFR2 is associated with vascularization (12-15).
According to the results of western blotting, HUVECs treated with
CCL2 exhibited marked expression of these markers (Fig. 2A). Therefore, we hypothesized that
CCL2 serves a significant role in the proliferation, migration and
vascularization of HUVECs.
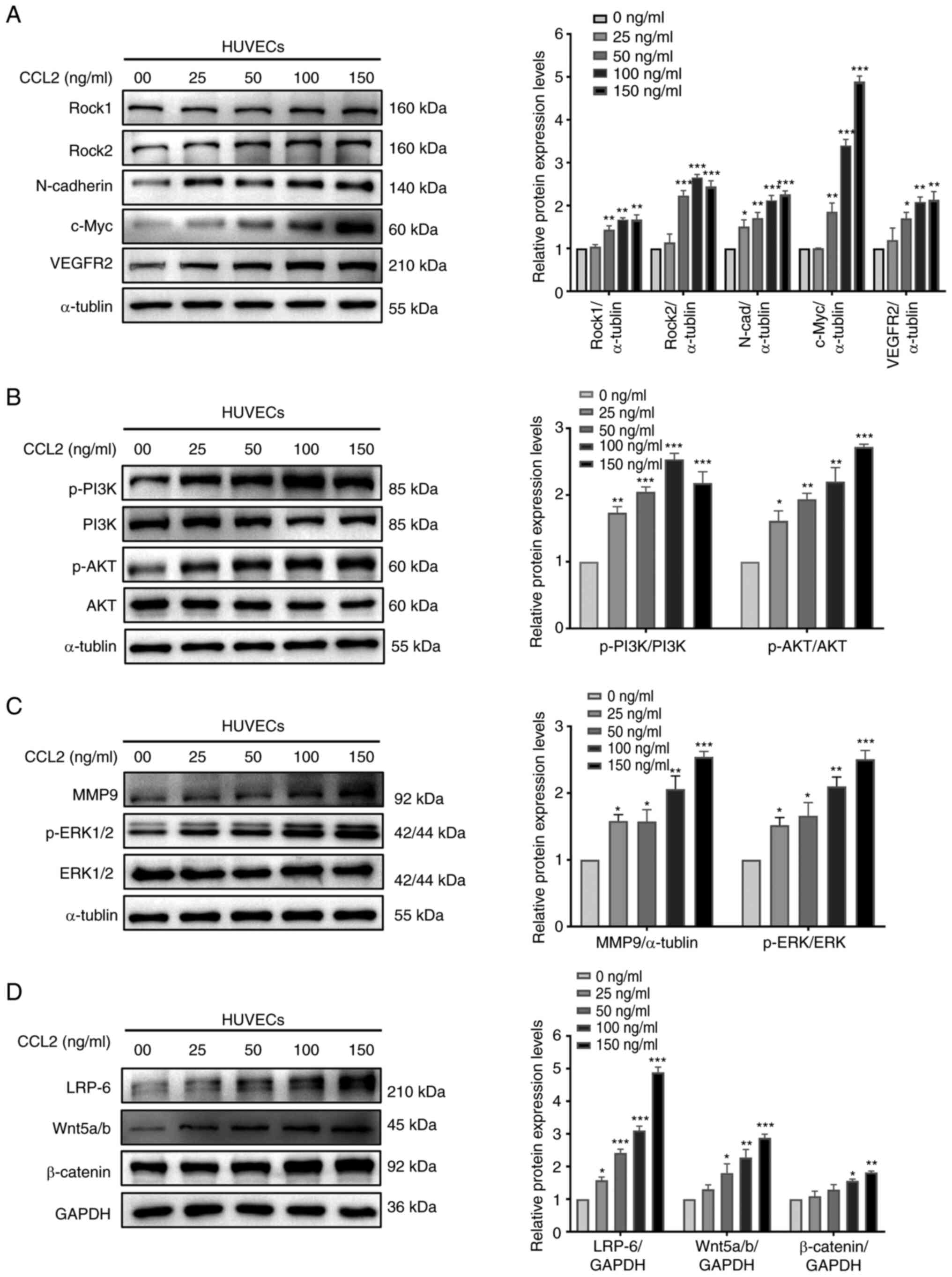 | Figure 2CCL2 promotes proliferation,
migration and angiogenesis via the PI3K/AKT, MAPK/ERK1/2 and
Wnt/β-catenin signaling pathways in HUVECs. (A) Western blot
analysis of Rock1, Rock2, N-cadherin, c-Myc, VEGFR2 and α-tubulin
in HUVECs treated with CCL2 at the indicated doses for 48 h. (B)
Western blot analysis of p-PI3K,PI3K, p-AKT, AKT and α-tubulin in
HUVECs treated with CCL2 at the indicated doses for 48 h. (C)
Western blot analysis of p-ERK1/2, ERK1/2, MMP9 and α-tubulin in
HUVECs treated with CCL2 at the indicated doses for 48 h. (D)
Western blot analysis of LRP-6, Wnt5a/b, β-catenin and GAPDH in
HUVECs treated with CCL2 at the indicated doses for 48 h. Gray
analysis of protein bands was performed using ImageJ.
*P<0.05, **P<0.01,
***P<0.001 vs. control group. CCL2, CC motif ligand
2; HUVECs, human umbilical vein endothelial cells; Rock,
rho-associated coiled-coil-containing protein kinase; p-,
phosphorylated; LRP-6, low-density lipoprotein receptor related
protein 6. |
CCL2 upregulates the PI3K/AKT, ERK1/2
and β-catenin signaling pathways to promote the proliferation and
migration of HUVECs
The PI3K/AKT, ERK1/2 and β-catenin signaling
pathways are related to cell proliferation and migration (16-18).
To gain further insights into the molecular mechanism whereby CCL2
alters HUVEC behavior, the present study investigated the effect of
CCL2 on the PI3K/AKT, ERK1/2 and β-catenin signaling pathways.
Western blotting revealed that CCL2 treatment of HUVECs increased
the levels of p-AKT, p-PI3K, p-ERK1/2, MMP9, Wnt5a/b, β-catenin and
LRP-6 (Fig. 2B-D). Therefore, it
was hypothesized that CCL2 may exert a positive regulatory effect
on proliferation and migration in HUVECs by inducing these
signaling pathways.
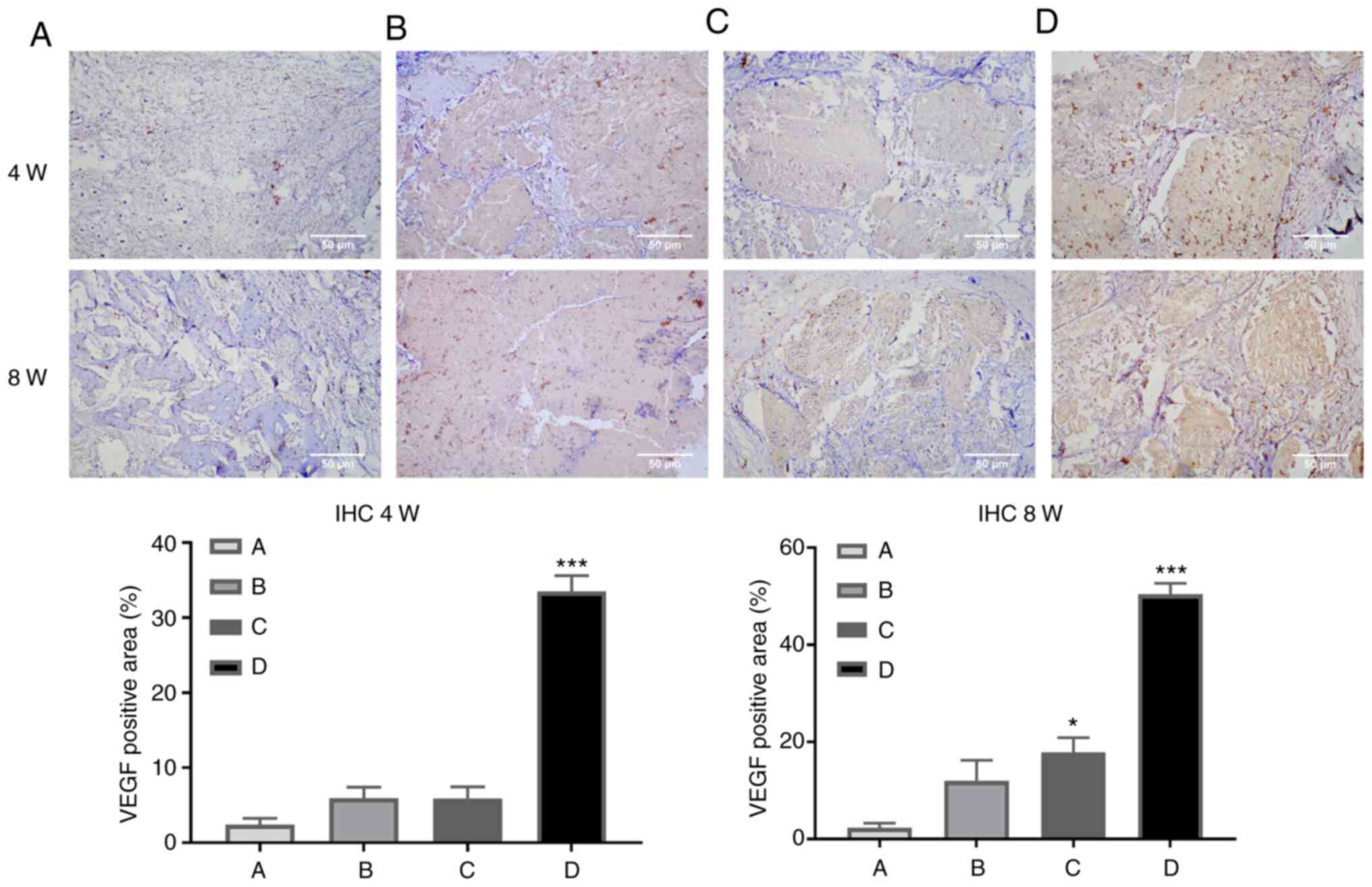
Immunohistochemistry
Femoral samples collected at 4 and 8 weeks after the
bone defect operation underwent EDTA decalcification and
immunohistochemical staining. The results revealed that there was
an increase in the expression area of VEGF in CCL2-treated groups
(Fig. 3).
Discussion
Large bone defects caused by trauma, bone tumors,
limb deformity and bone infection are major clinical challenges in
orthopedics and have become a focus of research in the field of
orthopedics (2,19). In the context of bone defects,
angiogenesis and bone defect reconstruction are closely related in
space and time and achieving adequate vascular development within
regenerating bone tissue remains a significant challenge (20). The formation of blood vessels
regulates the function of osteoblasts during bone regeneration and
is an important regulator of bone regeneration (21). This interconnection between
osteogenesis and angiogenesis is called ‘angiogenesis-osteogenesis
coupling’, which promotes the repair and reconstruction of bone
defects (22,23). VEGF, which is involved in the
possible mechanisms of angiogenesis and osteogenic coupling, serves
an important role between angiogenesis and bone remodeling
(14,24). VEGF binds to three types of
receptor tyrosine kinases in mammals, VEGFR1, VEGFR2 and
VEGFR3(25), which mediate
endothelial cell regeneration, angiogenesis and regulation of
vascular permeability (26). The
VEGF family, including VEGFA-D serve an influential role in
angiogenesis and development, especially VEGFA, which is the major
factor in angiogenesis and works primarily through VEGFR2(27). In present study, the tube formation
assay revealed that CCL2 promoted angiogenesis in HUVECs. High
expression levels of the angiogenesis-associated protein VEGFR2
following CCL2 treatment also suggested that CCL2 promoted
angiogenesis. The results of immunohistochemical staining of the
collected femoral specimens demonstrated that VEGF expression was
markedly increased in the group supplemented with CCL2, which also
confirmed that CCL2 induced angiogenesis associated with bone
defect repair and reconstruction. These results demonstrated that
CCL2 promoted vascular formation in HUVECs, and drove angiogenesis
and osteogenic coupling-related protein VEGF expression.
Cadherin may activate some transduction pathways,
including the Wnt/β-catenin pathway, which serves a crucial role in
the growth and development of cells and tissues (28). N-cadherin is a type of
transmembrane glycoprotein that mediates the adhesion between
endothelial cells and pericytes and is closely related to the
formation and maintenance of blood vessels (29). N-cadherin promotes cell migration,
mobility and polarity through intracellular signaling at cell-cell
junctions (30,31). Rock is known as an effector switch
that regulates a range of cell biological processes, including cell
adhesion, proliferation, migration and gene expression (32,33).
The most typical downstream receptors of ras homolog family member
A are the serine/threonine kinases Rock1 and Rock2, which are two
subtypes of the Rock gene (34).
Rock1 participates in regulation of the signaling pathways of cell
migration (35) and promotes
formation of the actin network (36). Rock1 may mediate the recruitment
and adhesion of circulating leukocytes and inflammatory cells to
sites of vascular injury to form a new intima (37). Similar to Rock1, Rock2 is a key
gene for biological functions related to the transfer process,
including the destruction of adhesion, remodeling of the actin
cytoskeleton, enhancement of cell activity and regulation of
signaling pathways (38), such as
cell proliferation and migration. A study reported that c-Myc
regulates the expression of thousands of downstream genes related
to the regulation of cell proliferation, migration and apoptosis,
affecting biological metabolism, transcriptional expression,
protein synthesis and cell cycle regulation (39). Furthermore, as a key regulator of
tissue growth, angiogenesis and the expression of angiogenic
regulatory factors, c-Myc participates in the dominant regulation
of the angiogenic network architecture. By contrast, c-Myc
deficiency decreases the expression of VEGF (40), which affects the construction of
vascular tissue. The results of the present study revealed that
CCL2 treatment promoted the proliferation and migration of HUVECs
in a concentration-dependent manner as demonstrated by the
increased expression of the proliferation- and migration-related
proteins Rock1, Rock2, N-cad and c-Myc in HUVECs. These results
demonstrated that CCL2 promoted proliferation and migration in
HUVECs.
Although various physiological and pathological
functions of angiogenesis in bone remodeling have been extensively
studied, little is known about the detailed mechanisms by which
CCL2-activated signaling pathways are involved in the
proliferation, migration and vascularization of vascular
endothelial cells. AKT is a specific serine/threonine protein
kinase downstream of the PI3K signaling pathway that regulates key
cellular processes such as glucose metabolism, energy
transformation, cell proliferation, cell growth and cell death
(41,42). It also increases the secretion of
VEGF and the phosphorylation of endothelial nitric oxide synthase,
which promotes vasodilation and angiogenesis to induce growth
factors, blocking apoptosis and increasing the cell survival rate
(43). Phosphorylation of AKT
promotes cell proliferation, migration and angiogenesis, which can
help cells adapt to hypoxia and acidosis (44). In addition, ERK is a member of the
MAPK family, and is the key molecule for transferring signals from
cell surface receptors to the nucleus (45) and is known for its promotion of
proliferation and differentiation (46). The ERK signaling pathway in
endothelial cells mediates various cellular processes, such as
proliferation, migration, survival and differentiation (47). p-ERK promotes the expression of
MMP9, which is one of the Zn2+-dependent endopeptidases
downstream of ERK signal transduction (48). MMP9 can degrade denatured collagen
and lytic extracellular matrix to facilitate remodeling of the
extracellular matrix (49), which
is significant in a variety of biological and molecular processes,
including tissue repair, wound healing, cell differentiation and
metastasis (50). Wnt5a is one of
the Wnt family members whose signaling pathways regulate most cell
growth and development processes, including cell proliferation,
migration and survival (51).
Wnt5a is involved in the regulation of angiogenesis by activating
the Wnt/β-catenin signaling pathway. Upregulation of Wnt5a/b
promotes the accumulation of β-catenin and mobilizes the classic
Wnt signaling pathway to promote the expression of downstream
target genes, such as vascular endothelial cadherin and MMP9, which
are implicated in accelerating angiogenesis (52). In the classic Wnt/β-catenin
signaling pathway, the connection of Wnt ligands to frizzled
transmembrane receptors and LRP-6 to form protein complexes,
ensures cell survival. It is also associated with the promotion of
bone formation (53). The present
study revealed that CCL2 treatment enhanced the levels of p-PI3K,
p-AKT, p-ERK1/2, MMP9, LRP-6, Wnt5a/b and β-catenin in HUVECs.
Regrettably, the lack of evaluation for ERK, PI3K, and β-catenin
levels in tissue is a limitation of the present study. In summary,
these results suggested that CCL2 served a positive regulatory role
in the proliferation, migration and angiogenesis of HUVECs by
upregulating the PI3K/AKT, MAPK/ERK1/2/MMP9 and Wnt/β-catenin
signaling pathways.
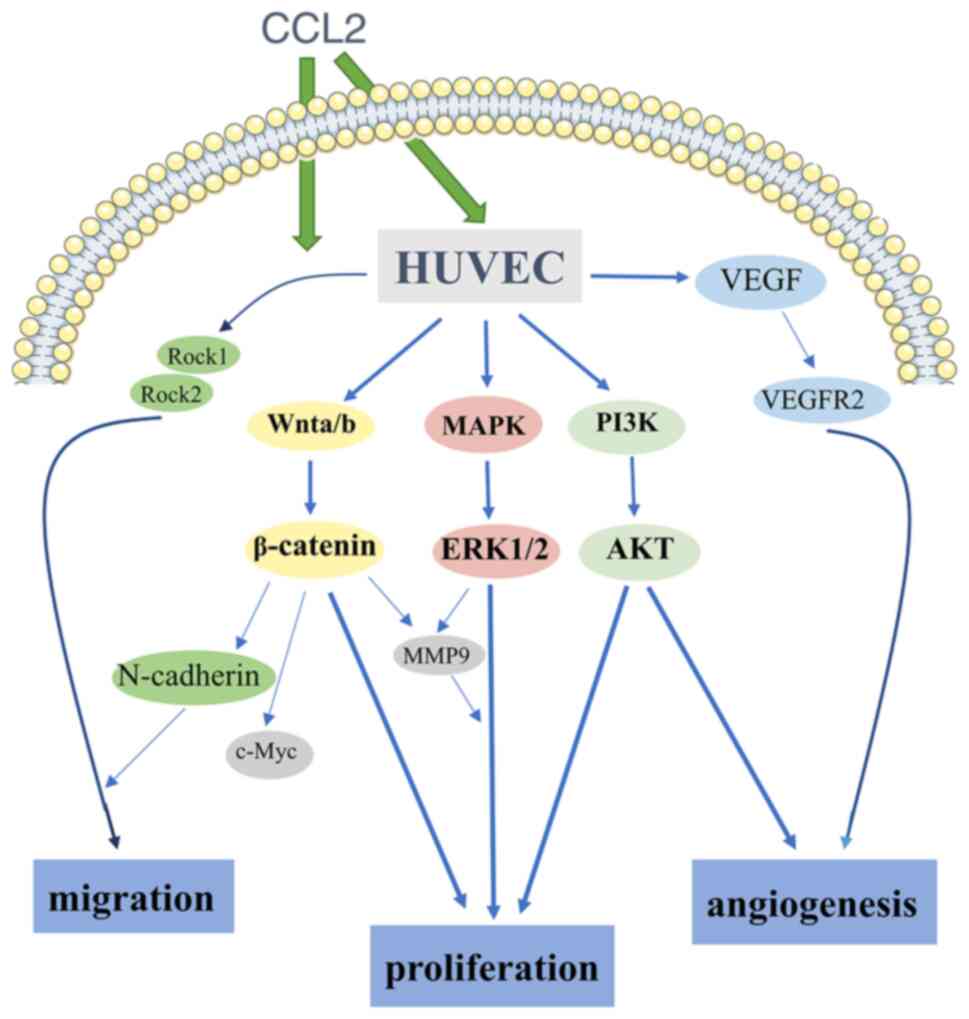
In conclusion, to the best of the authors'
knowledge, the present study was the first to demonstrate that CCL2
promoted proliferation, migration and angiogenesis in HUVECs. In
addition, the present study investigated the high expression of
molecular markers linked to these functions induced by CCL2,
including Rock1, Rock2, N-cadherin and c-Myc. Next, using reliable
and sufficient methods, it was demonstrated that CCL2 promoted
proliferation, migration and angiogenesis by activating the
PI3K/AKT, MAPK/ERK1/2/MMP9 and Wnt/β-catenin signaling pathways. In
addition, the SD rat bone defect model confirmed that CCL2 promoted
the expression of the angiogenesis-osteogenesis coupling associated
protein VEGF in bone defect reconstruction. Due to its mechanism of
inducing angiogenesis (Fig. 4),
CCL2 may be a novel angiogenesis-osteogenesis coupling agent for
bone defect repair and reconstruction.
Supplementary Material
Detailed procedure for establishing
bone defects in SD rat models. (A) General view of the external
fixation brace used. (B) Anesthesia, skin preparation, disinfection
and towel laying for SD rats. (C) An incision of about 2 cm on the
left lateral thigh. (D) The skin and fascia were incised layer by
layer. The femoral stem was bluntly separated, exposed, and propped
open with a small spreader. (E) Using the retractor holes as anchor
points, the electric rotor drilled four holes at low speed. The
external fixation device was attached to the four screws. (F and G)
The bone was cut with a wire saw 3 mm between the two screws in the
middle. (H) The external fixation device was adjusted in groups B,
C, and D to align the broken end of the femur with the composite
brace. Group A left the bone defect area open. (I) The muscle, deep
and superficial fascia, and skin were sutured layer by layer. The
wound was disinfected with iodophor after suturing. SD, Sprague
Dawley rats.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Guangdong (grant no. 2020A1515010003), Peaking Plan
for the reconstruction of the high-level hospital at Affiliated
Hospital of Guangdong Medical University (grant no.
20501DFY20190168) and Zhanjiang Science and Technology Bureau
(grant no. 200513174547221).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BW and SL conceived the current study. ZP, HW, XP
and HP performed the experiments. ZP, HP, XP, QT and HW analyzed
the results. ZP, HP, and QT drafted the manuscript. HW, BW and SL
and HP revised the manuscript. HP and ZP confirm the authenticity
of all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments are approved by the
Laboratory Animal Ethics Committee of Guangdong Medical University.
(ID Number: GDY1902126; date: 25.05.2019).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Freeman F, Burdis R and Kelly DJ: Printing
new bones: From print-and-implant devices to bioprinted bone organ
precursors. Trends Mol Med. 27:700–711. 2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Arealis G and Nikolaou VJ: Bone printing:
New frontiers in the treatment of bone defects. Injury 46 Suppl.
8:S20–S22. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tateishi-Yuyama E, Matsubara H, Murohara
T, Ikeda U, Shintani S, Masaki H, Amano K, Kishimoto Y, Yoshimoto
K, Akashi H, et al: Therapeutic angiogenesis for patients with limb
ischaemia by autologous transplantation of bone-marrow cells: A
pilot study and a randomised controlled trial. Lancet. 360:427–435.
2002.PubMed/NCBI View Article : Google Scholar
|
|
4
|
McLaughlin KI, Milne TJ, Zafar S,
Zanicotti DG, Cullinan MP, Seymour GJ and Coates DE: The in vitro
effect of VEGF receptor inhibition on primary alveolar osteoblast
nodule formation. Aust Dent J. 65:196–204. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Moses MA: The regulation of
neovascularization of matrix metalloproteinases and their
inhibitors. Stem Cells. 15:180–189. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: Structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Herzog C, Haun RS, Shah SV and Kaushal GP:
Proteolytic processing and inactivation of CCL2/MCP-1 by meprins.
Biochem Biophys Rep. 8:146–150. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Whelan DS, Caplice NM and Clover AJP:
Mesenchymal stromal cell derived CCL2 is required for accelerated
wound healing. Sci Rep. 10(2642)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tu MM, Abdel-Hafiz HA, Jones RT, Jean A,
Hoff KJ, Duex JE, Chauca-Diaz A, Costello JC, Dancik GM, Tamburini
BAJ, et al: Inhibition of the CCL2 receptor, CCR2, enhances tumor
response to immune checkpoint therapy. Commun Biol.
3(720)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Anada T, Pan CC, Stahl AM, Mori S, Fukuda
J, Suzuki O and Yang Y: Vascularized Bone-mimetic hydrogel
constructs by 3D bioprinting to promote osteogenesis and
angiogenesis. Int J Mol Sci. 20(1096)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cao Z, Hao Z, Xin M, Yu L, Wang L, Zhang
Y, Zhang X and Guo X: Endogenous and exogenous galectin-3 promote
the adhesion of tumor cells with low expression of MUC1 to HUVECs
through upregulation of N-cadherin and CD44. Lab Invest.
98:1642–1656. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Farrell AS and Sears RC: MYC degradation.
Cold Spring Harb Perspect Med. 4(a014365)2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hu K and Olsen BR: Osteoblast-derived VEGF
regulates osteoblast differentiation and bone formation during bone
repair. J Clin Invest. 126:509–526. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Loirand GJPr: Rho kinases in health and
disease: From basic science to translational research. Pharmacol
Rev. 67:1074–1095. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pai SG, Carneiro BA, Mota JM, Costa R,
Leite CA, Barroso-Sousa R, Kaplan JB, Chae YK and Giles FJ:
Wnt/beta-catenin pathway: Modulating anticancer immune response. J
Hematol Oncol. 10(101)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pompura SL and Dominguez-Villar M: The
PI3K/AKT signaling pathway in regulatory T-cell development,
stability, and function. J Leukoc Biol 2018 (Epub ahead of
print).
|
|
18
|
Roskoski R Jr: ERK1/2 MAP kinases:
Structure, function, and regulation. Pharmacol Res. 66:105–143.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lu H, Liu Y, Guo J, Wu H, Wang J and Wu G:
Biomaterials with antibacterial and osteoinductive properties to
repair infected bone defects. Int J Mol Sci. 17(334)2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Stahl A and Yang YP: Regenerative
approaches for the treatment of large bone defects. Tissue Eng Part
B Rev. 27:539–547. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhu S, Bennett S, Kuek V, Xiang C, Xu H,
Rosen V and Xu J: Endothelial cells produce angiocrine factors to
regulate bone and cartilage via versatile mechanisms. Theranostics.
10:5957–5965. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kusumbe AP, Ramasamy SK and Adams RH:
Coupling of angiogenesis and osteogenesis by a specific vessel
subtype in bone. Nature. 507:323–328. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhu S, Yao F, Qiu H, Zhang G, Xu H and Xu
J: Coupling factors and exosomal packaging microRNAs involved in
the regulation of bone remodelling. Biol Rev Camb Philos Soc.
93:469–480. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Street J, Winter D, Wang JH, Wakai A,
McGuinness A and Redmond HP: Is human fracture hematoma inherently
angiogenic? Clin Orthop Relat Res. 378:224–237. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Stuttfeld E and Ballmer-Hofer K: Structure
and function of VEGF receptors. IUBMB Life. 61:915–922.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Simons M, Gordon E and Claesson-Welsh L:
Mechanisms and regulation of endothelial VEGF receptor signalling.
Nat Rev Mol Cell Biol. 17:611–625. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487.
2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Blaschuk OW: N-cadherin antagonists as
oncology therapeutics. Philos Trans R Soc Lond B Biol Sci.
370(20140039)2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sabatini PJ, Zhang M, Silverman-Gavrila R,
Bendeck MP and Langille BL: Homotypic and endothelial cell
adhesions via N-cadherin determine polarity and regulate migration
of vascular smooth muscle cells. Circ Res. 103:405–412.
2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tomita K, van Bokhoven A, van Leenders GJ,
Ruijter ET, Jansen CF, Bussemakers MJ and Schalken JA: Cadherin
switching in human prostate cancer progression. Cancer Res.
60:3650–3654. 2000.PubMed/NCBI
|
|
32
|
Heasman SJ and Ridley AJ: Mammalian Rho
GTPases: New insights into their functions from in vivo studies.
Nat Rev Mol Cell Biol. 9:690–701. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Dong M, Yan BP, Liao JK, Lam YY, Yip GW
and Yu CM: Rho-kinase inhibition: A novel therapeutic target for
the treatment of cardiovascular diseases. Drug Discov Today.
15:622–629. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lock FE and Hotchin NA: Distinct roles for
ROCK1 and ROCK2 in the regulation of keratinocyte differentiation.
PLoS One. 4(e8190)2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wu YJ, Tang Y, Li ZF, Li Z, Zhao Y, Wu ZJ
and Su Q: Expression and significance of Rac1, Pak1 and Rock1 in
gastric carcinoma. Asia Pac J Clin Oncol. 10:e33–e39.
2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lou Z, Billadeau DD, Savoy DN, Schoon RA
and Leibson PJ: A role for a RhoA/ROCK/LIM-kinase pathway in the
regulation of cytotoxic lymphocytes. J Immunol. 167:5749–5757.
2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Noma K, Rikitake Y, Oyama N, Yan G,
Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, et al: ROCK1
mediates leukocyte recruitment and neointima formation following
vascular injury. J Clin Invest. 118:1632–1644. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
38
|
Lock FE, Ryan KR, Poulter NS, Parsons M
and Hotchin NA: Differential regulation of adhesion complex
turnover by ROCK1 and ROCK2. PLoS One. 7(e31423)2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Baudino TA, McKay C, Pendeville-Samain H,
Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN and Cleveland
JL: c-Myc is essential for vasculogenesis and angiogenesis during
development and tumor progression. Genes Dev. 16:2530–2543.
2002.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Peiris TH, Ramirez D, Barghouth PG and
Oviedo NJ: The Akt signaling pathway is required for tissue
maintenance and regeneration in planarians. BMC Dev Biol.
16(7)2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Abeyrathna P and Su Y: The critical role
of Akt in cardiovascular function. Vascul Pharmacol. 74:38–48.
2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Radisavljevic Z: AKT as locus of cancer
phenotype. J Cell Biochem. 116:1–5. 2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhai H, Pan T, Yang H, Wang H and Wang Y:
Cadmium induces A549 cell migration and invasion by activating ERK.
Exp Ther Med. 18:1793–1799. 2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Gough NR: Focus issue: Recruiting players
for a game of ERK. Sci Signal. 4(eg9)2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Liu P and Zhong TP: MAPK/ERK signalling is
required for zebrafish cardiac regeneration. Biotechnol Lett.
39:1069–1077. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin F, Chengyao X, Qingchang L, Qianze D,
Enhua W and Yan W: CRKL promotes lung cancer cell invasion through
ERK-MMP9 pathway. Mol Carcinog 54 Suppl. 1:E35–E44. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Gillard JA, Reed MW, Buttle D, Cross SS
and Brown NJ: Matrix metalloproteinase activity and
immunohistochemical profile of matrix metalloproteinase-2 and -9
and tissue inhibitor of metalloproteinase-1 during human dermal
wound healing. Wound Repair Regen. 12:295–304. 2004.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Raeeszadeh-Sarmazdeh M, Do LD and Hritz
BG: Metalloproteinases and their inhibitors: Potential for the
development of new therapeutics. Cells. 9(1313)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Shi YN, Zhu N, Liu C, Wu HT, Gui Y, Liao
DF and Qin L: Wnt5a and its signaling pathway in angiogenesis. Clin
Chim Acta. 471:263–269. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yao L, Sun B, Zhao X, Zhao X, Gu Q, Dong
X, Zheng Y, Sun J, Cheng R, Qi H and An J: Overexpression of Wnt5a
promotes angiogenesis in NSCLC. Biomed Res Int.
2014(832562)2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Silvério KG, Davidson KC, James RG, Adams
AM, Foster BL, Nociti FH Jr, Somerman MJ and Moon RT: Wnt/β-catenin
pathway regulates bone morphogenetic protein (BMP2)-mediated
differentiation of dental follicle cells. J Periodontal Res.
47:309–319. 2012.PubMed/NCBI View Article : Google Scholar
|