Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a
hematological malignancy with marked heterogeneity. A variety of
genetic mutations or chromosomal variations lead to the malignant
transformation of T-cell progenitors and define different subtypes
(1,2). In Europe, the USA and Japan, T-ALL
accounts for 10-15% of pediatric ALL cases and 20-25% of adult ALL
cases. Since the introduction of intensive chemotherapy, the
remission rate for T-ALL is increasing, and the prognosis is
gradually improving. Unfortunately, T-ALL relapse remains a
significant concern (1,3,4).
Highly aggressive relapsed T-ALL has a poor prognosis and is
generally resistant to chemotherapy (5-7).
In the face of these clinical challenges, researchers are committed
to identifying more specific therapeutic targets and developing
more effective and less toxic anti-leukemia drugs or drug
combinations.
The Janus kinase/signal transducer and activator of
transcription (JAK/STAT) signaling pathway is a signaling hub that
mediates the intracellular signaling of a variety of cytokines and
growth factors. Its aberrant activation is associated with a
variety of hematological malignancies (8-10).
Inhibition of the JAK/STAT pathway holds promise for the treatment
of a range of diseases, and several inhibitors of the JAK/STAT
pathway are currently being tested in clinical trials (8). Most STAT3 small-molecule inhibitors
block STAT3 phosphorylation and/or dimerization. Several STAT3
inhibitors, including STX-0119, STA-21, LLL-3, LLL12, XZH-5, and
OPB-31121, exhibit antitumor properties, but few have been approved
for clinical trials or clinical use (11-16).
BP-1-102, an orally available inhibitor of STAT3
phosphorylation, blocks STAT3 phosphorylation and dimerization by
directly interfering with the binding of phosphorylated tyrosine
residue (pTyr) of the upstream molecule to the Src homology 2 (SH2)
domain of STAT3. BP-1-102 reduces phosphorylated STAT3 levels in
cells and subsequently inhibits the expression of its downstream
proteins c-Myc, Bcl-xL, Cyclin D1, Survivin and VEGF, thereby
suppressing the growth, survival, migration and invasion of tumor
cells (17). BP-1-102 exerts
antitumor effects in several solid tumor cell lines, including
breast cancer, non-small cell lung cancer and gastric
adenocarcinoma (17-19).
Although the majority of ALL is B-ALL, there is a
greater need for targeted therapies for T-ALL. Our preliminary
results have demonstrated high expression of the JAK2/STAT3/c-Myc
pathway in T-ALL cell lines and samples (20,21).
Given that STAT3 mutations have been identified in T-cell
malignancies (22), it is
worthwhile to investigate whether BP-1-102 could be used to treat
T-ALL. In this study, BP-1-102 was administered to T-ALL cell lines
(MOLT4 and CUTLL1), and its effects on cell proliferation, colony
formation, apoptosis, cell cycle distribution as well as the
JAK2/STAT3/c-Myc signaling pathway were examined. The findings
revealed that BP-1-102 can inhibit the JAK2/STAT3/c-Myc signaling
pathway in T-ALL cells, exerting various antitumor effects.
Therefore, BP-1-102 represents a promising targeted antitumor
inhibitor.
Materials and methods
Cell culture
MOLT-4 is a cell line of adult T-cell acute
lymphoblastic leukemia. CUTLL1 is a cell line of childhood T-cell
lymphoblastic lymphoma. Cells were cultured in Roswell Park
Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine
serum (FBS). All cells were incubated in an incubator (Thermo
Scientific, Waltham, MA, USA) containing 95% air and 5%
CO2 at 37˚C.
Reagents and Antibodies
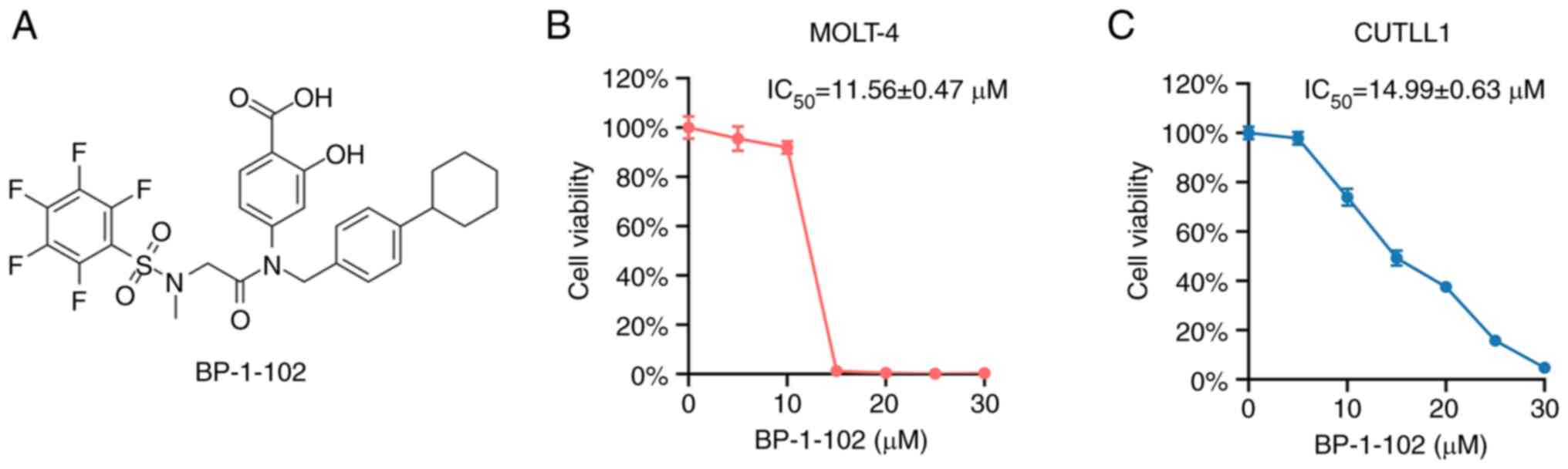
The STAT3-specific inhibitor BP-1-102 was purchased
from MedChemExpress (MCE). The chemical structure is shown in
Fig. 1A. Antibodies against JAK2
(#3230), STAT3 (#4904), p-STAT3 (Tyr705) (#9145), c-Myc (#5605),
Bcl-2 (#2870), Bcl-xL (#2764), Cyclin D1 (#55506) and β-actin
(#3700) were purchased from Cell Signaling Technology (CST).
Cell viability assay
The experimental groups were treated with the
indicated concentrations of BP-1-102, and the control group was
treated with an equal volume of dimethylsulfoxide (DMSO) vehicle
for 48 h. Cells were seeded at 105 µl per well in 96-well plates in
quadruplicate at a density of 1x105/ml. Ten microliters
of Cell Counting Kit-8 (CCK-8) reagent (NCM Biotech, C6005) was
added, and the cells were incubated for 3 h at 37˚C. The optical
density at 450 nm (OD450) was measured using a spectrophotometer
(Thermo Electron, Waltham, MA, USA). Relative cell viability and
half-maximal inhibitory concentration (IC50) were
calculated.
Cell colony formation assay
Cells were mixed at a density of 4,000/ml in
semisolid culture with 30% serum and indicated concentrations of
the drug. The semisolid culture system was seeded at 1 mL per dish
in 35-mm dishes in triplicate. Petri dishes were incubated for 12
days in a humidified incubator at 37 ˚C. Colonies with more than 50
cells were counted under a microscope (ZEISS, Axio Vert. A1) and
photographed.
Cell apoptosis assay
Cells were seeded in 6-well plates in triplicate at
a density of 4x105/ml and treated with 0, 15, or 20 µM
BP-1-102 for 24 h, ensuring the same concentration of DMSO in every
group. Cells were subsequently stained with Annexin V and propidium
iodide (PI) dye following the instructions of the Annexin-V-FITC
kit (4A Biotech Co., Ltd., Beijing, China). The apoptosis signal
was detected by flow cytometry (Cytek, NL-CLC) and processed with
FlowJo 10.5.3 software.
Inverted light microscopy
Cells were seeded in 6-well plates at a density of
4x105/ml and treated with 15 µM BP-1-102 or an equal
volume of DMSO vehicle. After incubation for 6 h, the cells were
observed and photographed using an inverted light microscope
(ZEISS, Axio Vert. A1) to search for subcellular structures, such
as apoptotic bodies.
Transmission electron microscopy
Cells were seeded in 6-well plates at a density of
4x105/ml and treated with 15 µM BP-1-102 or an equal
volume of DMSO vehicle for 6 h. Cells were then fixed in precooled
2.5% glutaraldehyde solution for 24 h. After dehydration,
permeation, embedding, sectioning and staining, the samples were
prepared. The samples were observed and photographed by a
transmission electron microscope (Hitachi TEM system, HT7800).
Cell cycle assay
Cells were seeded in 6-well plates in triplicate at
a density of 4x105/ml and treated with 10 µM BP-1-102 or
an equal volume of DMSO vehicle for 24 h. Cells were collected and
stained with PI dye (4A Biotech Co., Ltd., Beijing, China) after
being fixed with ethanol solution. The PI signal was detected by
flow cytometry (Cytek, NL-CLC), and cell cycle distribution was
processed with ModFit LT 3.1 software.
Western blotting
Cells were seeded in 6-well plates at a density of
1x106/ml and treated with 15 or 20 µM BP-1-102 or an
equal volume of DMSO vehicle for 6 h. Total protein was extracted
by Radio Immunoprecipitation Assay (RIPA) lysis buffer (with
protease inhibitor, phosphatase inhibitor and phenylmethanesulfonyl
fluoride added) using a sonicator (Sonics, Newtown, CT, USA). The
protein concentration was determined using the Bicinchoninic Acid
(BCA) assay (KeyGEN Biotech, KGP902).
Twenty-five micrograms of the cell lysate was
subjected to sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred to polyvinylidene
fluoride (PVDF) membranes (Millipore, Boston, MA, USA). The
membranes were sequentially incubated with 5% skim milk, primary
antibodies and horseradish peroxidase (HRP)-conjugated secondary
antibodies (Proteintech, Chicago, IL, USA). Proteins were
visualized using an HRP-based chemiluminescence kit (Monad,
PW30601S) and Gel Imaging System (Bio-Rad, Alfred Nobel Drive,
Hercules, CA, USA). All experiments were repeated thrice. These
bands were quantified by densitometry with Scion Image software
(ImageJ v1.53s).
Statistical methods
GraphPad Prism 8.0 (GraphPad Software, San Diego,
CA, USA) was applied for statistical analysis. Unpaired Student
t-test was used to compare normally distributed data from two
groups. Dunnett's test was used for multiple comparisons of each
drug-treated group versus the control group. Nonlinear regression
was used to fit the dose-viability curves and calculate their
IC50 values. Data are expressed as the mean ± standard
deviation. P<0.05 was considered statistically significant.
Results
BP-1-102 inhibited cell
proliferation
To test the antitumor effect of BP-1-102, we first
performed a CCK-8 assay to examine the effect of BP-1-102 on T-ALL
cell proliferation. We treated the T-ALL cell line with a gradient
concentration of the drug for 48 h before the CCK-8 assay was
performed. The results showed that BP-1-102 inhibited cell
proliferation, and the inhibitory effect was enhanced as the drug
concentration increased (Fig. 1B
and C). The half-maximal
inhibitory concentration (IC50) of BP-1-102 in the
MOLT-4 cell line was 11.56±0.47 µM (Fig. 1B). The half-maximal inhibitory
concentration (IC50) of BP-1-102 in the CUTLL1 cell line
was 14.99±0.63 µM (Fig. 1C). The
cell viability of MOLT-4 was slightly lower than that of CUTLL1 due
to cell line based differences.
BP-1-102 inhibited cell colony
formation
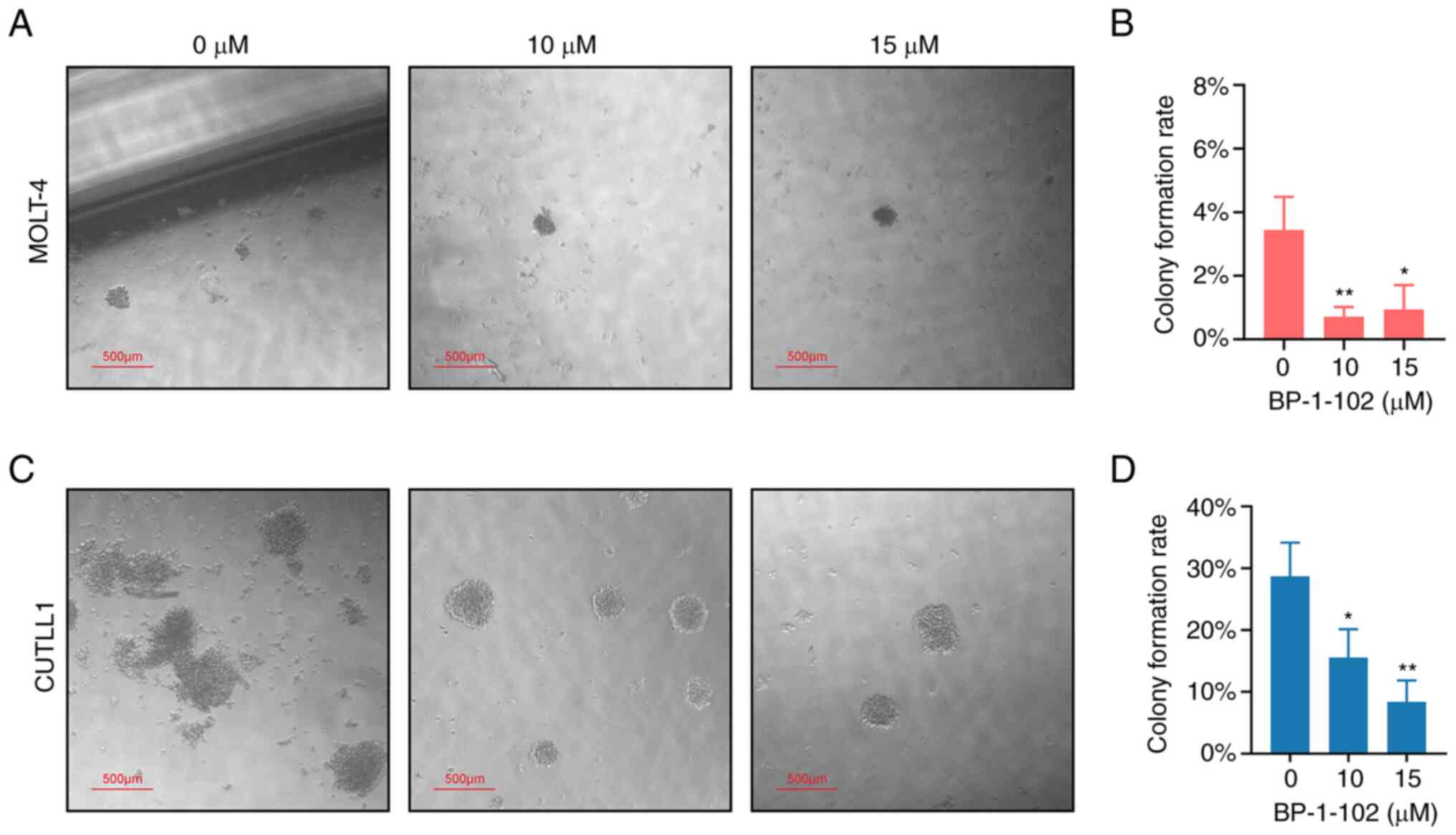
In addition to cell proliferation, we also examined
the effect of BP-1-102 on the colony formation ability of T-ALL
cell lines. We cultured T-ALL cells with methylcellulose semisolid
medium containing the indicated drug concentration for 12 days and
counted the number of colonies. The results showed that BP-1-102
inhibited the colony formation ability of T-ALL cells (Fig. 2). The colony formation rate of
MOLT-4 was much lower than that of CUTLL1, and the size of its
colonies was much smaller (Fig.
2). A significant decrease in the colony formation rate of the
10 and 15 µM groups of MOLT-4 was observed by counting the number
of colonies under a microscope (Fig.
2B). The colony formation capacity of CUTLL1 was higher with
dense colonies noted in the control group (Fig. 2C). A gradient decrease in colony
formation rate of CUTLL1 was observed in the 10 and 15 µM groups
(Fig. 2D).
BP-1-102 induced cell apoptosis
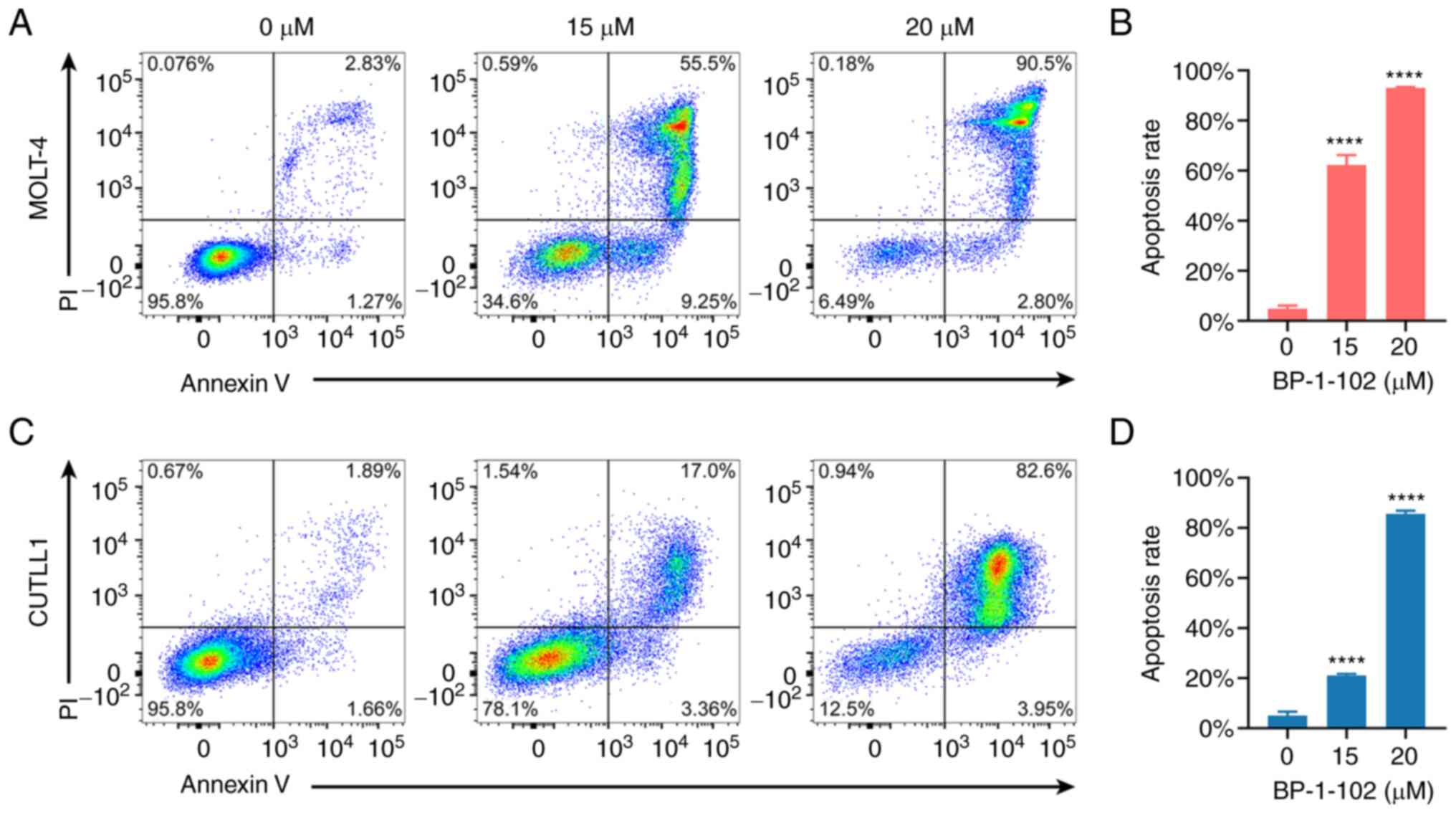
To explore the mechanism of the antitumor effect of
BP-1-102, we examined the apoptosis-inducing effect of BP-1-102 in
T-ALL cell lines by flow cytometry. After incubating the T-ALL cell
line with BP-1-102 for 24 h, the cells were labeled with Annexin V
and PI dye, and the fluorescence intensity of each cell was
measured by flow cytometry. The results showed that BP-1-102
induced apoptosis in T-ALL cells and the apoptosis-inducing effect
was enhanced with increasing drug concentration (Fig. 3). Significant MOLT-4 and CUTLL1
cell apoptosis were noted in both the 15 and 20 µM groups; in
particular, the 20 µM group showed very high apoptosis rates of
93.00±0.32% and 85.66±1.00% in the two cell lines, respectively
(Fig. 3B, D). The vast majority of
apoptosis in MOLT-4 and CUTLL1 cells exhibited late apoptosis
(Fig. 3A and C). Considering that 20 µM BP-1-102 could
lead to such a high apoptosis rate, we concluded that among the
various cytotoxic and antitumor effects of BP-1-102, the induction
of apoptosis represented its most dominant role.
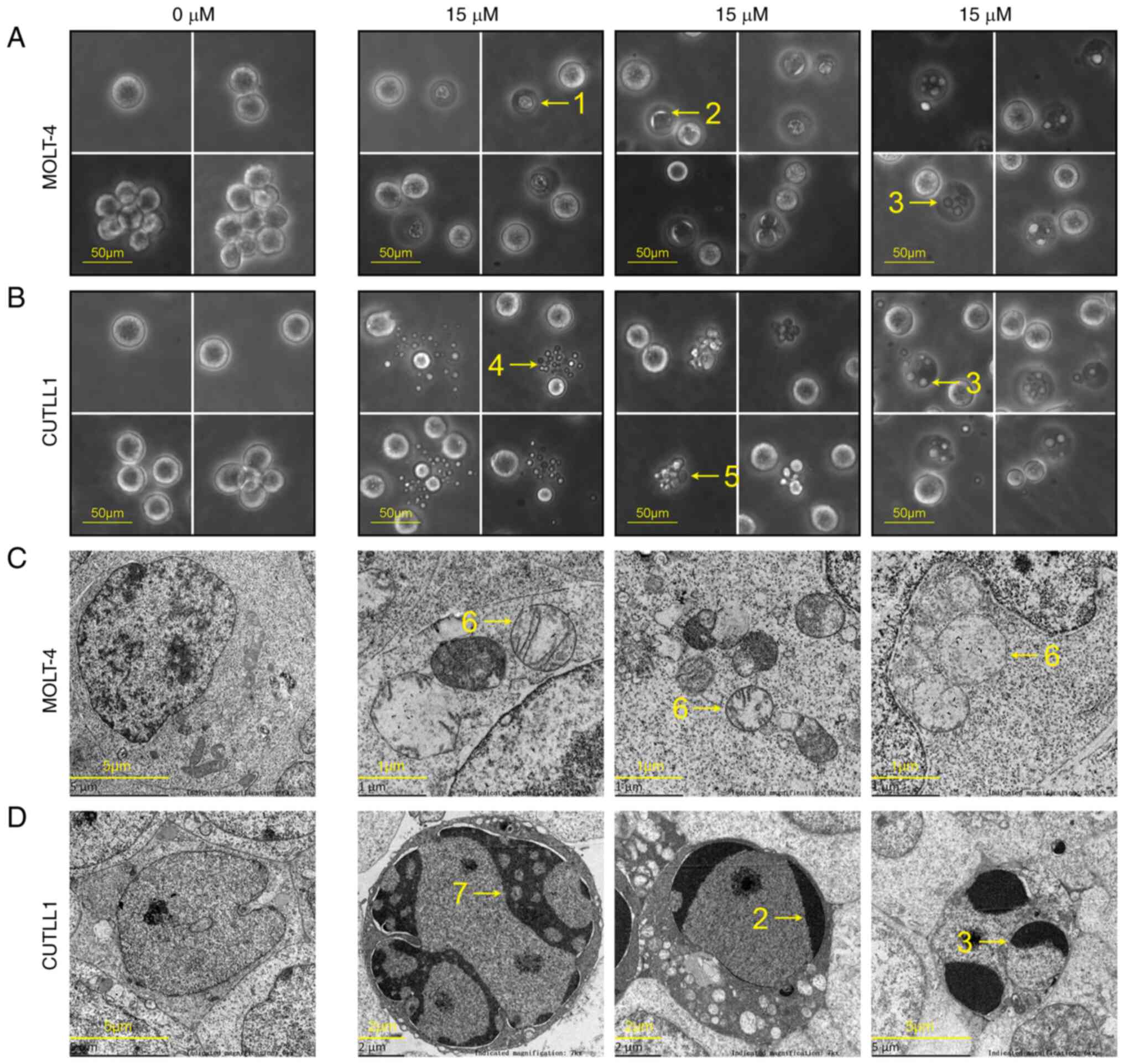
To demonstrate that apoptosis occurred in T-ALL
cells after BP-1-102 treatment, we incubated the cells with 15 µM
BP-1-102 for 6 h and then observed cell morphology using inverted
light microscopy and transmission electron microscopy (Fig. 4). Healthy cells in the control
group were round and full under a light microscope, and healthy
organelles, such as mitochondria, were visible under a transmission
electron microscope. In contrast, cells in the drug-treated group
showed subcellular structural features of apoptosis and necrosis:
apoptotic bodies, cell shrinkage, swollen mitochondria,
crescent-shaped chromatin, petal-like chromatin, pyknosis
(condensed nuclei) and karyorrhexis (fragmented nuclei).
Morphological results showed that apoptosis occurred in the
drug-treated cells (Fig. 4).
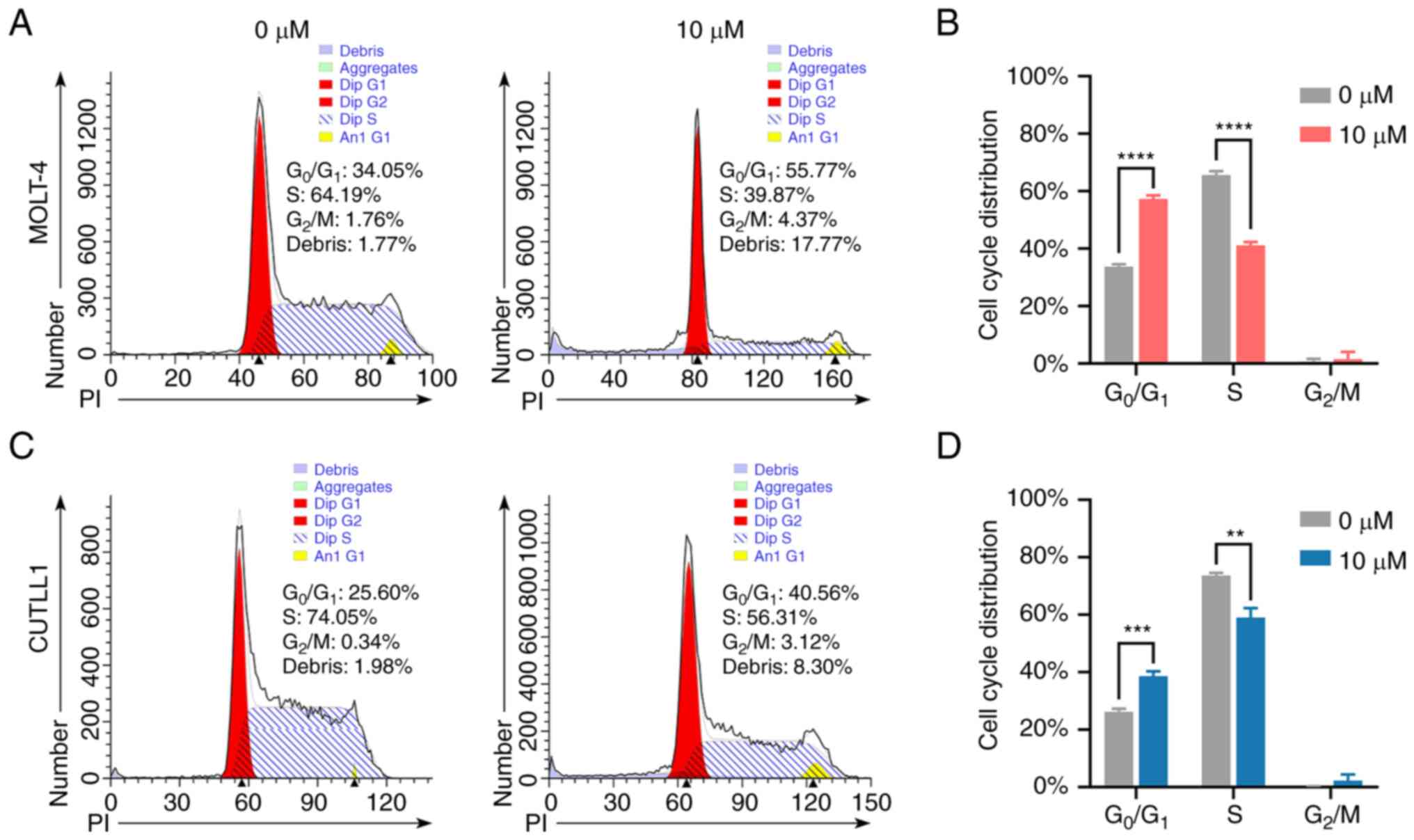
BP-1-102 caused cell cycle arrest at
the G0/G1 phase
To explore the mechanism of the antitumor effect of
BP-1-102, we examined the effect of BP-1-102 on the cell cycle
distribution of T-ALL cell lines by flow cytometry. MOLT-4 and
CUTLL1 cells were treated with 10 µM BP-1-102 or an equal volume of
DMSO for 24 h, fixed in ethanol and then stained with PI dye. The
PI fluorescence intensity of each cell was measured by flow
cytometry. The results showed that BP-1-102 blocked the cell cycle
at the G0/G1 phase (Fig. 5). The proportion of
G0/G1 phase cells in MOLT-4 increased from
33.67±0.74% to 57.27±1.06%, and the proportion of S phase cells
decreased from 65.59±1.11% to 41.16±0.92% (Fig. 5A and B). The percentage of
G0/G1 phase cells in CUTLL1 increased from
26.24%±0.84% to 38.60±1.40%, and the percentage of S phase cells
decreased from 73.64±0.76% to 59.07±2.64% (Fig. 5C and D). The altered cell cycle distribution
increased the proportion of resting cells, decreased the proportion
of proliferating cells and slowed the proliferation of the cell
population as a whole. In addition, the results showed that there
was no sub-G1 peak in the drug-treated group, but a higher
percentage of cellular debris.
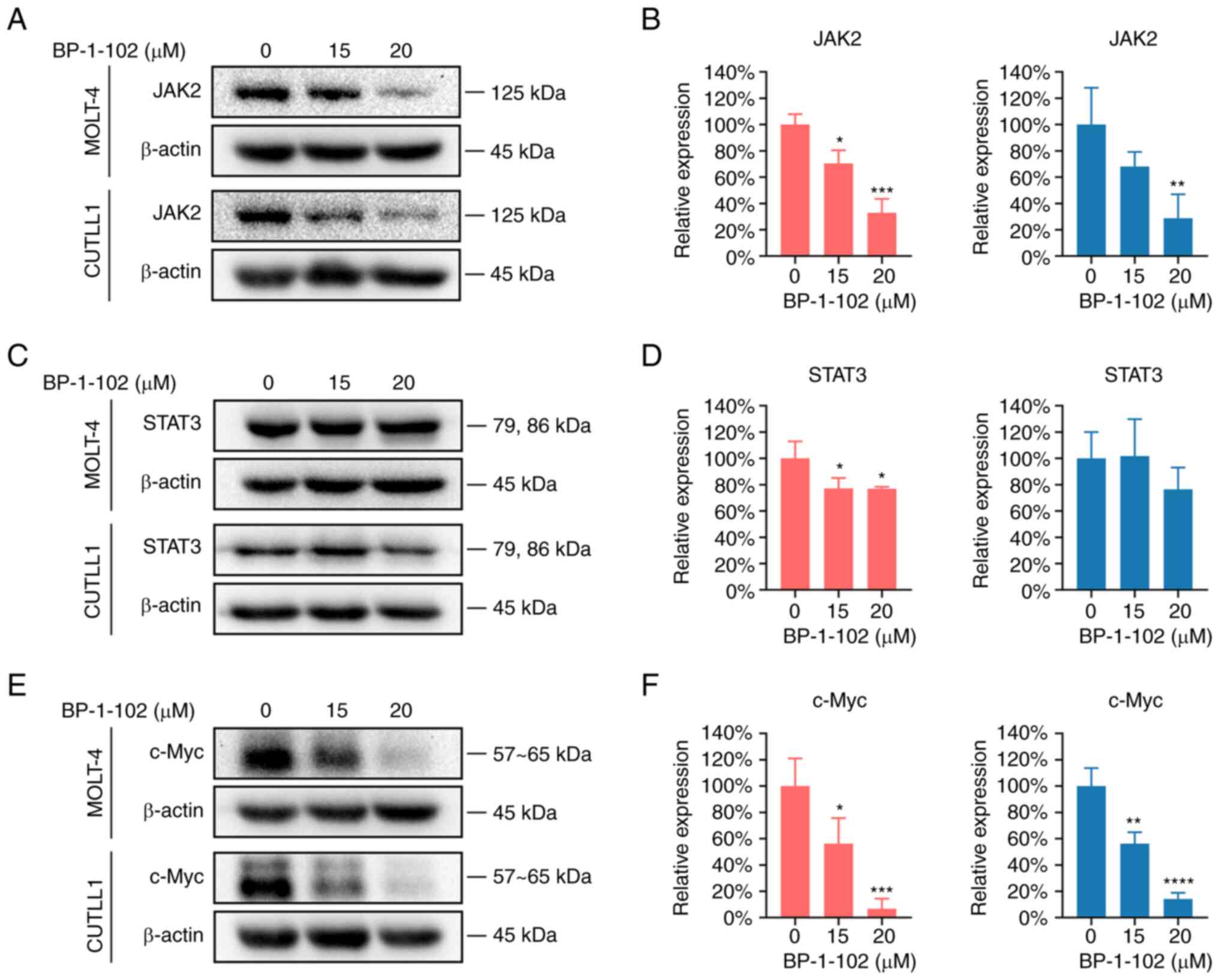
BP-1-102 suppressed the
JAK2/STAT3/c-Myc signaling pathway
To investigate the mechanisms by which BP-1-102
exerted the various effects described above, the expression levels
of the factors involved in the JAK2-STAT3 pathway and several
downstream proteins were measured by Western blotting. The results
showed that BP-1-102 downregulated the JAK2-STAT3 pathway and
significantly inhibited c-Myc protein expression (Fig. 6).
Although the inhibitory effects of BP-1-102 on cell
proliferation were slightly different between MOLT-4 and CUTLL1
cells, the basal expression levels of the JAK2/STAT3/c-Myc pathway
were high in both cell lines. For both MOLT-4 and CUTLL1 cells,
BP-1-102 significantly inhibited JAK2 expression in a
dose-dependent manner. This finding reflected the interaction
between STAT3 and JAK2 (Fig. 6A
and B). The effect of BP-1-102 on
total STAT3 was not apparent and only differed significantly in the
drug-treated MOLT-4 group (Fig. 6C
and D). Regretfully,
phosphorylated STAT3 was likely minimally expressed in both T-ALL
cell lines and was not detected by Western blotting in either (data
not shown). Perhaps changes in phosphorylated STAT3 levels can be
detected by flow cytometry or enzyme-linked immunosorbent assay
(ELISA) kits.
As a classical oncogene that promotes tumor cell
proliferation, c-Myc is a downstream gene of STAT3. Both the bands
of c-Myc and the histogram of its quantification reflected a
significant reduction in c-Myc expression in the drug-treated
groups, exhibiting a typical stepwise decrease. The expression of
c-Myc in the 20 µM groups was only approximately 10% of that in the
control group (Fig. 6E and
F). The antitumor effects of
BP-1-102 on T-ALL cell lines were likely to result from direct
inhibition of STAT3 phosphorylation and the subsequent indirect
inhibition of c-Myc protein.
Discussion
T-cell acute lymphoblastic leukemia is a highly
aggressive and heterogeneous hematological malignancy. Over the
past decades, a variety of targeted drugs have been developed to
target the pathological process of T-ALL. Unfortunately, none of
the new targeted drugs have been approved as first-line treatment
options. The research and development of new targeted drugs for
T-ALL are translationally important.
The JAK-STAT signaling pathway plays an important
role in leukemia pathogenesis and could be used as a potential
target for drug therapy. Activation of STAT proteins, particularly
STAT5 and STAT3, plays a crucial role in the pathogenesis of
lymphoid and myeloid malignancies (23). Human T-cell lymphotropic virus type
I (HTLV-I) is a cause of adult T-cell leukemia. The JAK-STAT
signaling pathway is constitutively activated in HTLV-I-mediated
malignant T cells (24). STAT3 is
constitutively activated in lymphoblastoid-like cells and Burkitt's
lymphoma cells that are EBV-positive or permanently expressing
interleukin-10(25). Recurrent
STAT3 mutations were identified in 40% of T-cell large granular
lymphocytic leukemia cases. These mutations increase the
transcriptional activity of STAT3 and lead to the upregulation of
STAT3 target genes, such as Bcl-2-like protein 1 (BCL2L1) and JAK2
(26,27). STAT3 mutations were found in 4 of
258 patients with T-cell tumors, and the mutations were confined to
malignant cells. These mutations in STAT3 (Y640F) lead to
constitutive activation of STAT3 and malignant hematopoiesis in
vivo (28). As demonstrated by
Western blotting, Cheng et al. found significantly increased
JAK2 and STAT3 expression in T-ALL patient samples compared with
healthy controls (20). Taken
together, STAT3 emerges as a potential therapeutic target for
T-ALL.
BP-1-102 is a homologue of S3I-201.1066(29) obtained by computer-aided
optimization. As a ligand for the SH2 domain of STAT3, BP-1-102 can
compete with the pTyr of the upstream molecule, subsequently
interfering with the binding of pTyr to the SH2 domain of STAT3
and, as a result, blocking STAT3 phosphorylation and dimerization
(17). We administered gradient
concentrations of BP-1-102 to T-ALL cell lines and examined its
antitumor properties. The results showed that BP-1-102 inhibited
tumor cell proliferation and cell colony formation, induced
apoptosis and blocked the cell cycle at the
G0/G1 phase. The antiproliferative and
apoptosis-inducing effects were dose-dependent. In summary,
BP-1-102 represents a promising targeted antitumor medication that
is expected to be used in clinical trials.
When the apoptosis rates were detected by flow
cytometry, the cells had been treated for 24 h, when late apoptosis
dominated in MOLT-4 and CUTLL1 (Fig.
3). Yet we later found that incubation for 6 h was most
suitable for morphological analysis, when the cells exhibited the
most typical apoptotic morphological features such as apoptotic
bodies (Fig. 4B). Both Bcl-2 and
Bcl-xL are classical anti-apoptotic oncogenes downstream of
STAT3(30). The expression levels
of Bcl-2 and Bcl-xL were slightly reduced by BP-1-102 treatment,
which contributed to apoptosis. Yet such inhibition effects were
weak and poorly reproducible (data not shown).
Cyclin D1 drives cell cycle progression from G0/G1
phase to S phase. Overexpressed Cyclin D1 has oncogenic effects
(31). Xiaoxia Jiang et al
once administered BP-1-102 to gastric cancer cell line AGS cells
and found that Cyclin D1 was downregulated, although the cell cycle
distribution of AGS cells was not affected significantly (18). We hypothesized that BP-1-102
exerted its cell cycle-blocking effects on T-ALL cells by
inhibition of cyclin D1 downstream of STAT3 (Fig. 5). Regrettably, cyclin D1 expression
levels examined by Western blotting were not significantly reduced
(data not shown).
c-Myc is a transcription factor that is highly
expressed in over 70% of cancers. As a very important oncogene with
a broad role, c-Myc is involved in the regulation of proliferation,
differentiation, apoptosis and the cell cycle in many tumors
(32). c-Myc also promotes tumor
angiogenesis, metastasis and chemoresistance (33). c-Myc is a downstream gene regulated
by STAT3. The STAT3/c-Myc axis is involved in the oncogenesis,
progression, metabolism and therapeutic response of a variety of
cancers (34-37).
Haizhi Yu et al. found that c-Myc expression levels were
increased in patients' T-ALL cells as well as Jurkat and DU528
T-ALL cell lines compared with peripheral blood mononuclear cells
(PBMCs) from healthy donors (21).
In the present study, c-Myc protein expression was
completely inhibited in the high-concentration group (Fig. 6). The antiproliferative,
apoptosis-inducing and cell cycle blocking effects of BP-1-102 in
T-ALL were mainly mediated through indirect inhibition of c-Myc
(Fig. 6). The principal mechanism
by which BP-1-102 exerts its antitumor effects in T-ALL cell lines
is the suppression of the JAK2/STAT3/c-Myc oncogenic pathway
(Fig. 6). Considering the driver
role of c-Myc in many types of cancers, BP-1-102 may be applied to
a wide range of other tumors.
The shortcoming of this study should be noted.
Specifically, samples from T-ALL patients have not been studied,
and in vivo studies have not been conducted. Such
experiments may further validate the antitumor effects and safety
of BP-1-102.
BP-1-102 exerts anti-inflammatory effects in
addition to its antitumor effects. Qi-Ying Wu et al
demonstrated that BP-1-102 inhibited JAK2/STAT3/NF-κB pathway
activation in a mouse abdominal aortic aneurysm model, thereby
reducing the expression of proinflammatory cytokines, decreasing
inflammatory cell infiltration, and ultimately impeding the
development and progression of abdominal aortic aneurysms (38). Zhixian Jiang et al.
demonstrated that BP-1-102 suppressed the JAK/STAT3/NF-κB pathway
in intracranial aneurysms, reduced the vascular inflammatory
response, alleviated the symptoms of intracranial aneurysms and
prevented their rupture (39,40).
Chronic inflammation is associated with the
oncogenesis of many tumors, the most typical of which is
hepatocellular carcinoma. Transcription factors NF-κB and STAT3 are
both implicated in the development of hepatocellular carcinoma
(41,42). Regarding the mechanism of
inflammation-induced malignant transformation in cancers, such as
hepatocellular carcinoma, BP-1-102 may provide researchers with new
clues. Necroptosis is a type of pro-inflammatory cell death
(43). Induction of necroptosis
may bypass treatment resistance in cancers and accelerate the
development of novel therapeutic strategies for apoptosis-resistant
leukemia (44). Whether BP-1-102
plays a role in inducing necroptosis in leukemia is worth
investigating in the future.
In summary, we demonstrated that the
JAK2/STAT3/c-Myc pathway is necessary for the survival and
maintenance of T-ALL cells. In T-ALL cell lines, BP-1-102
suppressed the JAK2/STAT3/c-Myc signaling pathway, thus inhibiting
cell proliferation and colony formation, inducing apoptosis and
blocking the cell cycle at the G0/G1 phase.
BP-1-102 is a promising targeted inhibitor that offers an effective
option for targeted therapy in T-ALL. BP-1-102 and its potential
derivatives deserve to be explored in in vivo experiments
and clinical trials.
Acknowledgements
We wish to thank Professor Guangsen Zhang
(Department of Hematology, The Second Xiangya Hospital, Central
South University, Changsha, China) for his patient guidance and
great support. We wish to thank Dr Weiwei Yang (School of Basic
Medical Sciences, Harbin Medical University, Harbin, China) for her
kind help with TEM figure labeling.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant no. 82070175).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CY and ZC conceptualized this study. CY conducted
the experiments. CY, XR, YZ, HZ, CW, ZC and HP were involved in the
data analysis and validation. CY and ZC confirmed the authenticity
of all the raw data. CY and ZC wrote the manuscript. HP provided
project administration and funding. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Belver L and Ferrando A: The genetics and
mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer.
16:494–507. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Girardi T, Vicente C, Cools J and De
Keersmaecker K: The genetics and molecular biology of T-ALL. Blood.
129:1113–1123. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hunger SP and Mullighan CG: Acute
lymphoblastic leukemia in children. N Engl J Med. 373:1541–1552.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hunger SP, Lu X, Devidas M, Camitta BM,
Gaynon PS, Winick NJ, Reaman GH and Carroll WL: Improved survival
for children and adolescents with acute lymphoblastic leukemia
between 1990 and 2005: A report from the children's oncology group.
J Clin Oncol. 30:1663–1669. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bhojwani D and Pui CH: Relapsed childhood
acute lymphoblastic leukaemia. Lancet Oncol. 14:e205–e217.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Reismüller B, Attarbaschi A, Peters C,
Dworzak MN, Pötschger U, Urban C, Fink FM, Meister B, Schmitt K,
Dieckmann K, et al: Long-term outcome of initially homogenously
treated and relapsed childhood acute lymphoblastic leukaemia in
Austria--a population-based report of the Austrian
Berlin-Frankfurt-Münster (BFM) study group. Br J Haematol.
144:559–570. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pocock R, Farah N, Richardson SE and
Mansour MR: Current and emerging therapeutic approaches for T-cell
acute lymphoblastic leukaemia. Br J Haematol. 194:28–43.
2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hu X, Li J, Fu M, Zhao X and Wang W: The
JAK/STAT signaling pathway: From bench to clinic. Signal Transduct
Target Ther. 6(402)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Aittomäki S and Pesu M: Therapeutic
targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol.
114:18–23. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
O'Shea JJ, Schwartz DM, Villarino AV,
Gadina M, McInnes IB and Laurence A: The JAK-STAT pathway: Impact
on human disease and therapeutic intervention. Annu Rev Med.
66:311–328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ashizawa T, Akiyama Y, Miyata H, Iizuka A,
Komiyama M, Kume A, Omiya M, Sugino T, Asai A, Hayashi N, et al:
Effect of the STAT3 inhibitor STX-0119 on the proliferation of a
temozolomide-resistant glioblastoma cell line. Int J Oncol.
45:411–418. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Song H, Wang R, Wang S and Lin J: A
low-molecular-weight compound discovered through virtual database
screening inhibits Stat3 function in breast cancer cells. Proc Natl
Acad Sci USA. 102:4700–4705. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Fuh B, Sobo M, Cen L, Josiah D, Hutzen B,
Cisek K, Bhasin D, Regan N, Lin L, Chan C, et al: LLL-3 inhibits
STAT3 activity, suppresses glioblastoma cell growth and prolongs
survival in a mouse glioblastoma model. Br J Cancer. 100:106–112.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nie Y, Li Y and Hu S: A novel small
inhibitor, LLL12, targets STAT3 in non-small cell lung cancer in
vitro and in vivo. Oncol Lett. 16:5349–5354. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu A, Liu Y, Jin Z, Hu Q, Lin L, Jou D,
Yang J, Xu Z, Wang H, Li C and Lin J: XZH-5 inhibits STAT3
phosphorylation and enhances the cytotoxicity of chemotherapeutic
drugs in human breast and pancreatic cancer cells. PLoS One.
7(e46624)2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kim MJ, Nam HJ, Kim HP, Han SW, Im SA, Kim
TY, Oh DY and Bang YJ: OPB-31121, a novel small molecular
inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an
antitumor activity in gastric cancer cells. Cancer Lett.
335:145–152. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang X, Yue P, Page BD, Li T, Zhao W,
Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT and Turkson J:
Orally bioavailable small-molecule inhibitor of transcription
factor Stat3 regresses human breast and lung cancer xenografts.
Proc Natl Acad Sci USA. 109:9623–9628. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jiang X, Tang J, Wu M, Chen S, Xu Z, Wang
H, Wang H, Yu X, Li Z and Teng L: BP-1-102 exerts an antitumor
effect on the AGS human gastric cancer cell line through modulating
the STAT3 and MAPK signaling pathways. Mol Med Rep. 19:2698–2706.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Belton A, Xian L, Huso T, Koo M, Luo LZ,
Turkson J, Page BD, Gunning PT, Liu G, Huso DL and Resar LM: STAT3
inhibitor has potent antitumor activity in B-lineage acute
lymphoblastic leukemia cells overexpressing the high mobility group
A1 (HMGA1)-STAT3 pathway. Leuk Lymphoma. 57:2681–2684.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cheng Z, Yi Y, Xie S, Yu H, Peng H and
Zhang G: The effect of the JAK2 inhibitor TG101209 against T cell
acute lymphoblastic leukemia (T-ALL) is mediated by inhibition of
JAK-STAT signaling and activation of the crosstalk between
apoptosis and autophagy signaling. Oncotarget. 8:106753–106763.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yu H, Yin Y, Yi Y, Cheng Z, Kuang W, Li R,
Zhong H, Cui Y, Yuan L, Gong F, et al: Targeting lactate
dehydrogenase A (LDHA) exerts antileukemic effects on T-cell acute
lymphoblastic leukemia. Cancer Commun (Lond). 40:501–517.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pencik J, Pham HT, Schmoellerl J, Javaheri
T, Schlederer M, Culig Z, Merkel O, Moriggl R, Grebien F and Kenner
L: JAK-STAT signaling in cancer: From cytokines to non-coding
genome. Cytokine. 87:26–36. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vainchenker W and Constantinescu SN:
JAK/STAT signaling in hematological malignancies. Oncogene.
32:2601–2613. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Migone TS, Lin JX, Cereseto A, Mulloy JC,
O'Shea JJ, Franchini G and Leonard WJ: Constitutively activated
Jak-STAT pathway in T cells transformed with HTLV-I. Science.
269:79–81. 1995.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Weber-Nordt RM, Egen C, Wehinger J, Ludwig
W, Gouilleux-Gruart V, Mertelsmann R and Finke J: Constitutive
activation of STAT proteins in primary lymphoid and myeloid
leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma
cell lines. Blood. 88:809–816. 1996.PubMed/NCBI
|
|
26
|
Ohgami RS, Ma L, Merker JD, Martinez B,
Zehnder JL and Arber DA: STAT3 mutations are frequent in CD30+
T-cell lymphomas and T-cell large granular lymphocytic leukemia.
Leukemia. 27:2244–2247. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Koskela HL, Eldfors S, Ellonen P, van
Adrichem AJ, Kuusanmäki H, Andersson EI, Lagström S, Clemente MJ,
Olson T, Jalkanen SE, et al: Somatic STAT3 mutations in large
granular lymphocytic leukemia. N Engl J Med. 366:1905–1913.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Couronné L, Scourzic L, Pilati C, Della
Valle V, Duffourd Y, Solary E, Vainchenker W, Merlio JP,
Beylot-Barry M, Damm F, et al: STAT3 mutations identified in human
hematologic neoplasms induce myeloid malignancies in a mouse bone
marrow transplantation model. Haematologica. 98:1748–1752.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang X, Yue P, Fletcher S, Zhao W,
Gunning PT and Turkson J: A novel small-molecule disrupts Stat3 SH2
domain-phosphotyrosine interactions and Stat3-dependent tumor
processes. Biochem Pharmacol. 79:1398–1409. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pistritto G, Trisciuoglio D, Ceci C,
Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: Function
and dysfunction of its modulators and targeted therapeutic
strategies. Aging. 8:603–619. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Montalto FI and De Amicis F: Cyclin D1 in
cancer: A molecular connection for cell cycle control, adhesion and
invasion in tumor and stroma. Cells. 9(2648)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Madden SK, de Araujo AD, Gerhardt M,
Fairlie DP and Mason JM: Taking the Myc out of cancer: Toward
therapeutic strategies to directly inhibit c-Myc. Mol Cancer.
20(3)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ala M: Target c-Myc to treat pancreatic
cancer. Cancer Biol Ther. 23:34–50. 2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Mo M, Tong S, Yin H, Jin Z, Zu X and Hu X:
SHCBP1 regulates STAT3/c-Myc signaling activation to promote tumor
progression in penile cancer. Am J Cancer Res. 10:3138–3156.
2020.PubMed/NCBI
|
|
35
|
Gao S, Chen M, Wei W, Zhang X, Zhang M,
Yao Y, Lv Y, Ling T, Wang L and Zou X: Crosstalk of mTOR/PKM2 and
STAT3/c-Myc signaling pathways regulate the energy metabolism and
acidic microenvironment of gastric cancer. J Cell Biochem.
120:1193–1202. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ning R, Chen G, Fang R, Zhang Y, Zhao W
and Qian F: Diosmetin inhibits cell proliferation and promotes
apoptosis through STAT3/c-Myc signaling pathway in human
osteosarcoma cells. Biol Res. 54(40)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhu Y and Feng Y: GRAIL inhibits the
growth, migration and invasion of lung adenocarcinoma cells by
modulating STAT3/C-MYC signaling pathways. J BUON. 26:353–358.
2021.PubMed/NCBI
|
|
38
|
Wu QY, Cheng Z, Zhou YZ, Zhao Y, Li JM,
Zhou XM, Peng HL, Zhang GS, Liao XB and Fu XM: A novel STAT3
inhibitor attenuates angiotensin II-induced abdominal aortic
aneurysm progression in mice through modulating vascular
inflammation and autophagy. Cell Death Dis. 11(131)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jiang Z, Huang J, You L, Zhang J and Li B:
Pharmacological inhibition of STAT3 by BP-1-102 inhibits
intracranial aneurysm formation and rupture in mice through
modulating inflammatory response. Pharmacol Res Perspect.
9(e00704)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jiang Z, Huang J, You L and Zhang J:
Protective effects of BP-1-102 against intracranial
aneurysms-induced impairments in mice. J Drug Target. 29:974–982.
2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Berasain C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
new molecular links. Ann N Y Acad Sci. 1155:206–221.
2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
He G and Karin M: NF-κB and STAT3-key
players in liver inflammation and cancer. Cell Res. 21:159–168.
2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Negroni A, Colantoni E, Cucchiara S and
Stronati L: Necroptosis in intestinal inflammation and cancer: New
concepts and therapeutic perspectives. Biomolecules.
10(1431)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Huang X, Xiao F, Li Y, Qian W, Ding W and
Ye X: Bypassing drug resistance by triggering necroptosis: Recent
advances in mechanisms and its therapeutic exploitation in
leukemia. J Exp Clin Cancer Res. 37(310)2018.PubMed/NCBI View Article : Google Scholar
|