Introduction
Tuberous sclerosis complex (TSC) is a hereditary
intractable disease characterized by various neoplastic lesions and
psychoneurotic symptoms. Currently, mTOR inhibitors (rapamycin and
its derivatives) are used to treat TSC due to the fact that loss of
the causative gene (TSC1 or TSC2) (1,2)
results in the increased activity of mTOR kinase complex 1 (mTORC1)
in the pathological process of TSC (3-8).
While therapeutic benefits have been obtained to some extent in the
reduction of neoplastic lesions (9,10),
the lack of complete tumor eradication (9), the side effects associated with
long-term drug administration (such as interstitial pneumonia,
severe stomatitis and immunosuppression) (11), and the sporadic occurrence of
unresponsive cases has led to the urgent need to develop new
therapeutic options.
Numerous studies have attempted to identify new drug
targets for TSC in order to improve treatment. For example,
rapamycin has been shown to induce feedback activation of
pro-oncogenic kinases upstream of mTORC1, such as Akt and
mitogen-activated protein kinase (MAPK), in TSC gene-deficient
tumor cells (12-14).
These kinases promote cell proliferation in rapamycin-treated TSC
gene-deficient tumor cells (15).
Studies have shown that endoplasmic (ER) stress is involved in the
mechanism underlying negative feedback regulation by
hyper-activated mTORC1(16).
Autophagy is activated by rapamycin independent of such feedback
regulation and might support the proliferation of TSC
gene-deficient tumor cells by providing free amino acids (17,18).
These rapamycin-responsive pathways are thought to be candidate for
drug targets. The single intervention of these pathways, along with
certain rapamycin combination treatments for TSC gene-deficient
tumors have been tested using cellular and animal models (15,18-20).
While some of these interventions were effective, none have been
applied in practical scenarios.
Many reports in the literature have addressed the
dysregulation of mTORC1-independent signaling pathways in TSC gene
deficiency. Because the protein complex of TSC1/TSC2 gene products
(hamartin/tuberin) directly regulates the GTP-binding protein Rheb
as a GTPase-activating protein (GAP), researchers have attempted to
identify new pathways downstream of Rheb (21,22).
For example, the contribution of PAK2 has already been demonstrated
(23). In terms of
neuropsychiatric disorders, previous research identified that the
GDP-binding form of Rheb downregulates the expression levels of
syntenin in an mTORC1-independent manner and helps to maintain the
appropriate formation of the spine (24,25).
The contribution of syntenin-related pathways in TSC-related
tumorigenesis has yet to be reported. There are many other reports
relating to mTORC1- and Rheb-independent pathways, although these
findings have yet to be applied clinically.
In addition, heat shock protein (HSP)-related
pathways have been implicated in the development and progression of
cancers (26). The expression
levels of HSP, especially HSP70, HSP90, and Hspb1 (HSP25/27), have
been identified as markers of the stage and prognosis of various
cancers (26,27). These HSPs are thought to represent
therapeutic targets for the treatment of cancer (26). Research has identified an
interaction between hamartin and HSP70; this interaction was found
to regulate apoptosis in TSC (28). Other research has shown that
harmatin acts as a chaperone for HSP90(29). However, the significance of these
functions in the pathogenesis of TSC has yet to be fully
elucidated. Furthermore, the relationship between Hspb1 and
TSC-related tumorigenesis and therapy has yet to be defined.
In an attempt to identify key pathways for the
development of new drugs, we analyzed rapamycin-induced changes in
Tsc2-deficient tumor cells from an animal model of TSC
(30-32).
In this study, we focused on the pathways related to Hspb1 and
found identified their role in the proliferation of
Tsc2-deficient cells under rapamycin treatment.
Materials and methods
Cell culture
A Tsc2-deficient kidney tumor cell line,
MKOC1-277 (MKOC) was established in our laboratory from the
Tsc2 knockout mouse and, together with its derivatives, had
been used in various researches (30-35).
MKOC cells were cultured in RPMI 1640 medium supplemented with 10%
fetal bovine serum (FBS), 100 U/ml of penicillin and 100 µg/ml of
streptomycin. Rapamycin (Sigma-Aldrich, Darmstadt, Germany)
treatment was performed with dimethylsulfoxide (DMSO) as a vehicle.
HeLa, COS7 and Plat-E cells (COSMO BIO, Tokyo, Japan) were cultured
in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 1
µg/ml of puromycin, 100 U/ml of penicillin and 100 µg/ml of
streptomycin. In the case of Plat-E cells, 10 µg/ml of blasticidin
was also added. All cell cultures were performed at 37˚C with 5%
CO2. SB203580 was purchased from Sigma.
Cell proliferation assays
Cell proliferation was assessed with the
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) assay system (Roche, Basel, Switzerland) in accordance with
the manufacturer's guidelines using 96-well plates. To 200 µl of
culture medium, XTT reagent (50 µl) was added to each well and
incubated for 4 h. Measurement of the absorbance at 450 nm
(reference wavelength of 650 nm) was performed with a microplate
reader (Benchmark Plus-microplate Spectrophotometer; Bio-Rad,
Hercules, United States).
Western blot analysis
Cell lysates were prepared in Laemmli's sample
buffer for sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE). Protein concentration was determination
by DC Protein Assays (Bio-Rad). Equal amounts of proteins were
separated by SDS-PAGE using standard methodology and proteins were
transferred to nylon membranes (Millipore, Burlington, United
States). For blocking and antibody reactions, we used 1% skimmed
milk/Tris buffered saline supplemented with 0.05% of Tween 20
(TBST). Anti-mouse or anti-rabbit Envision HRP-conjugate (DAKO,
K4001 or K4003, respectively) was used as secondary antibodies.
SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher
Scientific, Waltham, United States) or Immobilon Forte (Millipore)
was used for the development of signals. A Bio-Rad ChemiDoc MP
imaging system was used for the detection and analysis of signals.
We used several different primary antibodies: anti-HSP27
(#sc13132), -pHSP27(S82) (#sc166693), -Hsp70 (#sc24, #sc59569), and
Hsp90 (#sc13119); these antibodies were purchased from Santa Cruz
Biotechnology (Dallas, United States). We also used
anti-pS6(S235/S236) (#2211), -S6 (#2217), -tuberin (#3612), and
-ß-actin (#4970) antibodies from Cell Signaling Technology
(Danvers, United States) and anti-α-tubulin (B-5-1-2, #T5168) from
Sigma.
RNA interference (RNAi)
Transfection of siRNA was performed with
Lipofectamine RNAi MAX (Thermo Fisher Scientific) according to the
manufacturer's protocol. Cells were treated with 10 nM siRNA for
the Hspb1 gene (5'-CGGAGGAGCUCACAGUGAATT-3' and
5'-UUCACUGUGAGCUCCUCCGGA-3') (36)
or control RNA (Thermo Fisher Scientific, #4390843) for 48-72 h. To
analyze cell proliferation, rapamycin treatment was started one day
after siRNA transfection to avoid severe toxicity.
Plasmid construction
The full-length cDNA of mouse Hspb1 was
amplified using primers mHsp27-F,
5'-AAGAATTCCCAGTGCTTCTAGATCCTCA-3' (forward), and mHsp27-R,
5'-AACTCGAGGCAATGGCTATGGGAGATAG-3' (reverse). The amplified
fragment was digested with EcoRI and XhoI and
subcloned into the pBabe-puro vector (pBabe-puro was a gift from
Hartmut Land & Jay Morgenstern & Bob Weinberg (Addgene
plasmid #1764 ; http://n2t.net/addgene:1764 ; RRID:Addgene_1764))
(37). The vector had been
modified to contain the XhoI recognition sequence by
introducing synthetic oligonucleotides to the cloning site. The
resulting Hspb1 expression vector was designated as pBabe-mHSP27.
To generate cDNA for the Ser86-to-Asp86 (S86D) Hspb1 mutant, PCRs
were carried out with specific primers: mHsp27-F and H27ASPR1,
5'-ACCCCGCTGTCGAGCTGT-3' (reverse), H27ASPF1,
5'-ACAGCTCGACAGCGGGGT-3' (forward), and mHsp27-R, using
pBabe-mHSP27 as a template. Then, the two amplified fragments were
mixed and a second PCR was performed using mHsp27-F and mHsp27-R to
amplify the full-length cDNA. Similarly, cDNA for the
Ser86-to-Ala86 (S86A) Hspb1 mutant (pBabe-mHsp27-S86A) was
amplified using the following primers: mHsp27F, H27ALAR1,
5'-ACCCCGCTTGCGAGCTGT-3' (reverse), H27ALAF1,
5'-ACAGCTCGCAAGCGGGGT-3' (forward), and mHsp27R. The resulting full
length mutant cDNA fragments were cloned into the pBabe-puro vector
to generate pBabe-mHSP27-S86D and -S86A. The accuracy of the cDNA
sequence was checked by the dideoxy-sequencing method using an ABI
310 sequencer (Thermo Fisher Scientific).
Establishment of stable cell
lines
Plat-E cells (pre-cultured without puromycin or
blasticidin) were transfected with the vector for wild-type, S86A
and S86D Hspb1, or empty vector using FuGENE6 transfection reagent
(Promega, Wisconsin, United States) according to the manufacturer's
protocol. Next, 48 h after transfection, we collected the culture
media which was filtered through a 0.45-µm filter. Then, we added
polybrene to a final concentration of 8 µg/ml. MKOC cells were
infected with supernatant and the selection of stably transduced
cells with 0.6 µg/ml of puromycin was started 48 h after infection.
Established cell lines were maintained in the presence of puromycin
in the medium.
Statistical analysis
Statistical analyses were performed with Microsoft
Excel software (Microsoft, Redmond, United States), and Statistical
analysis program for pharmaceutical data (Osaka university, Osaka,
Japan; http://www.gen-info.osaka-u.ac.jp/MEPHAS/tukey-e.html).
The significance of difference was examined by unpaired Student's
t-test for two groups comparison, and one-way ANOVA followed by
Tukey's post hoc test for multiple groups comparison. A p value
less than 0.05 was considered statistically significant.
Results
Hspb1 expression was upregulated by
rapamycin treatment in Tsc2-deficient tumor cells
To analyze the levels of Hspb1 protein, we treated
MKOC cells with rapamycin (20 nM) for 24 and 48 h and then
performed western blotting. Rapamycin suppressed the proliferation
of Tsc2-deficient cells by approximately 35% (P<0.001)
(Fig. 1A). In this condition, we
found that the amount of Hspb1 protein was increased by 24 h
rapamycin treatment (Fig. 1B). The
phosphorylation of Hspb1 on Ser86 (corresponding to Ser82 in human
HSPB1), the main phosphorylation site that regulates
oligomerization, was also upregulated, concomitant with an increase
in the total amount of protein (Fig.
1B). Furthermore, rapamycin treatment did not result in the
increased expression of HSP70 and Hsp90 (Fig. 1C), thus revealing that each HSP
responded differently to rapamycin. To ascertain whether these
phenomena occurred generally in cultured cells, we investigated the
effects of rapamycin on HSP expression in routine HeLa and COS7
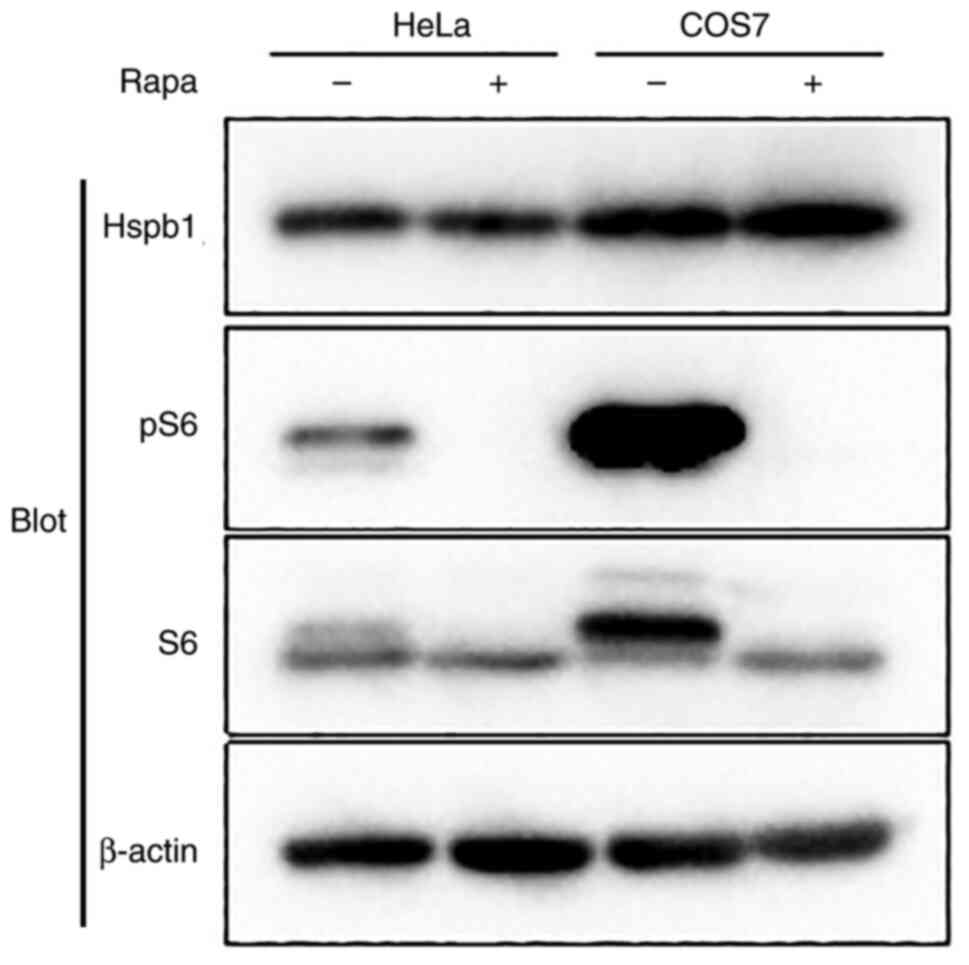
cells. No induction of Hspb1 expression was observed in either HeLa
or COS7 cells (Fig. 2). These
results indicated that the suppression of mTORC1 by rapamycin
causes the upregulation of Hspb1 expression in
Tsc2-deficient cells.
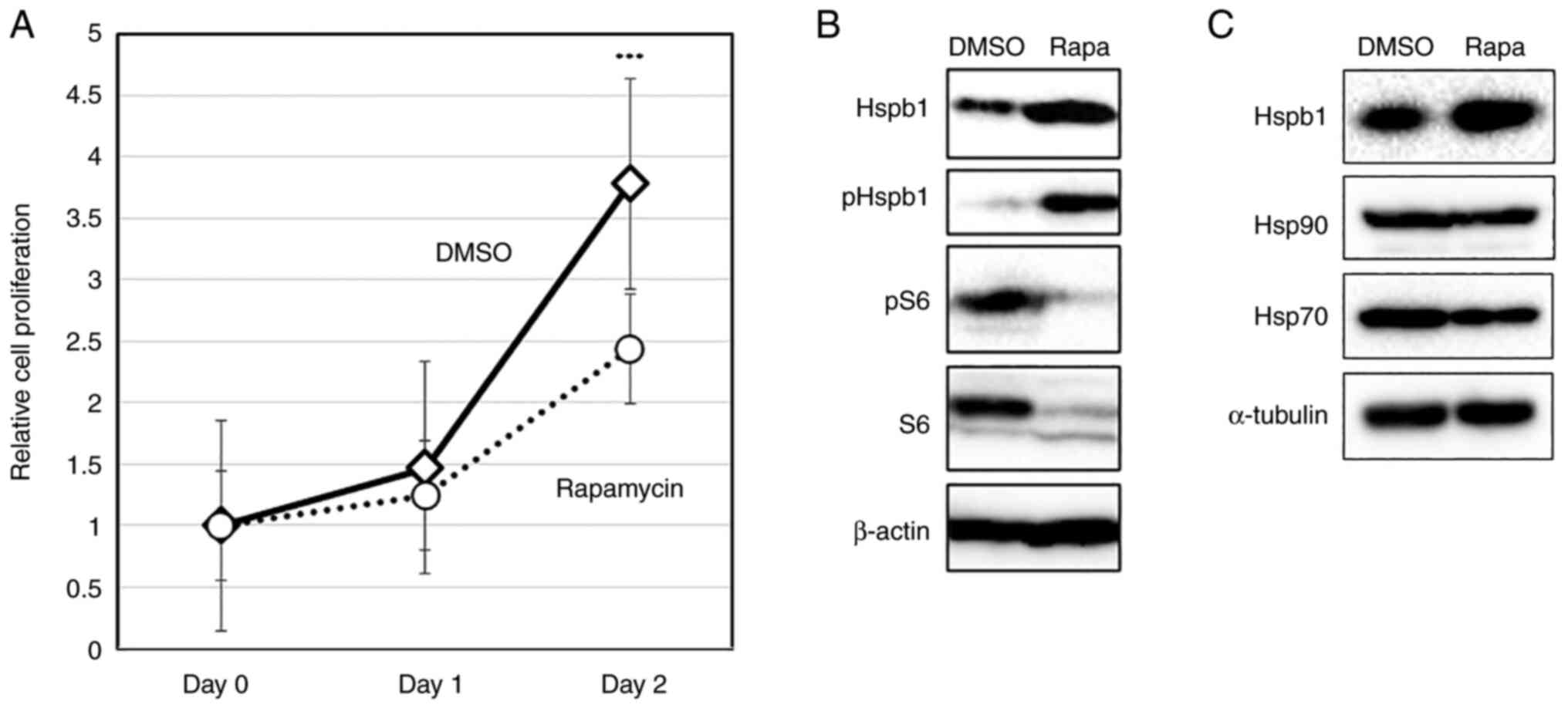 | Figure 1Analysis of Hspb1 expression in TSC
complex subunit 2-deficient kidney tumor cells (MKOC). (A) MKOC
cells were treated with 20 nM Rapa or vehicle only (DMSO) for 48 h.
Cell proliferation was then analyzed by XTT assays. Data are
representative of three independent experiments. Each experiment
was performed with three independent culture wells. The data are
presented as the mean + SD. ***P<0.001 compared with
Rapa. (B) MKOC cells were treated with 20 nM Rapa or vehicle only
(DMSO) for 24 h. Total cell lysates were analyzed by western
blotting with the indicated antibodies (Hspb1, pHspb1, pS6, S6 and
βactin). (C) MKOC cells were treated with 20 nM Rapa or vehicle
only (DMSO) for 24 h. Total cell lysates were analyzed by western
blotting with the indicated antibodies (Hspb1, Hsp90, Hsp70 and
α-tubulin). Hsp90, heat shock protein 90; Hsp70, heat shock protein
70; Hspb1, heat shock protein family B (small) member 1; MKOC,
MKOC1-277; p, phosphorylated; Rapa, rapamycin. |
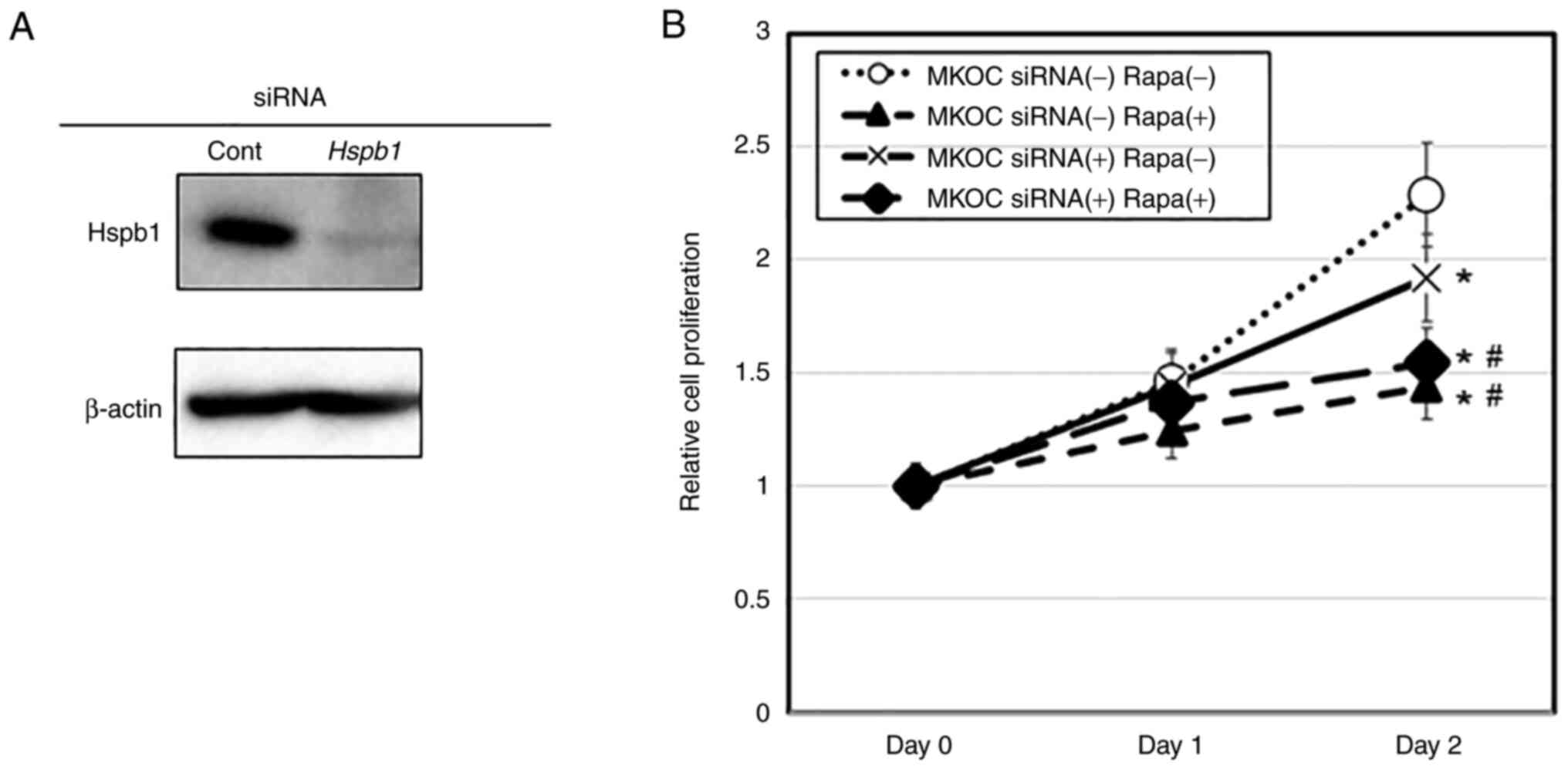
SiRNA for Hspb1 suppressed cell
proliferation
To investigate whether Hspb1 exerted a positive or
negative role in cell proliferation under rapamycin treatment, we
used RNAi to suppress the expression of Hspb1. The efficacy of
siRNA for the Hspb1 gene was confirmed by western blotting
analysis (Fig. 3A). Hspb1
siRNA treatment significantly inhibited the proliferation of MKOC
cells under normal culture condition (P<0.05) (Fig. 3B). Under the rapamycin treatment,
however, the extent of inhibition was similar when compared between
control RNA and Hspb1 siRNA conditions (P<0.05 between
DMSO and rapamycin without siRNAs; no significance between control
and Hspb1 siRNA under the rapamycin-treated condition)
(Fig. 3B), thus suggesting that
rapamycin-induced growth regulation may involve the suppression of
Hspb1-related pathways.
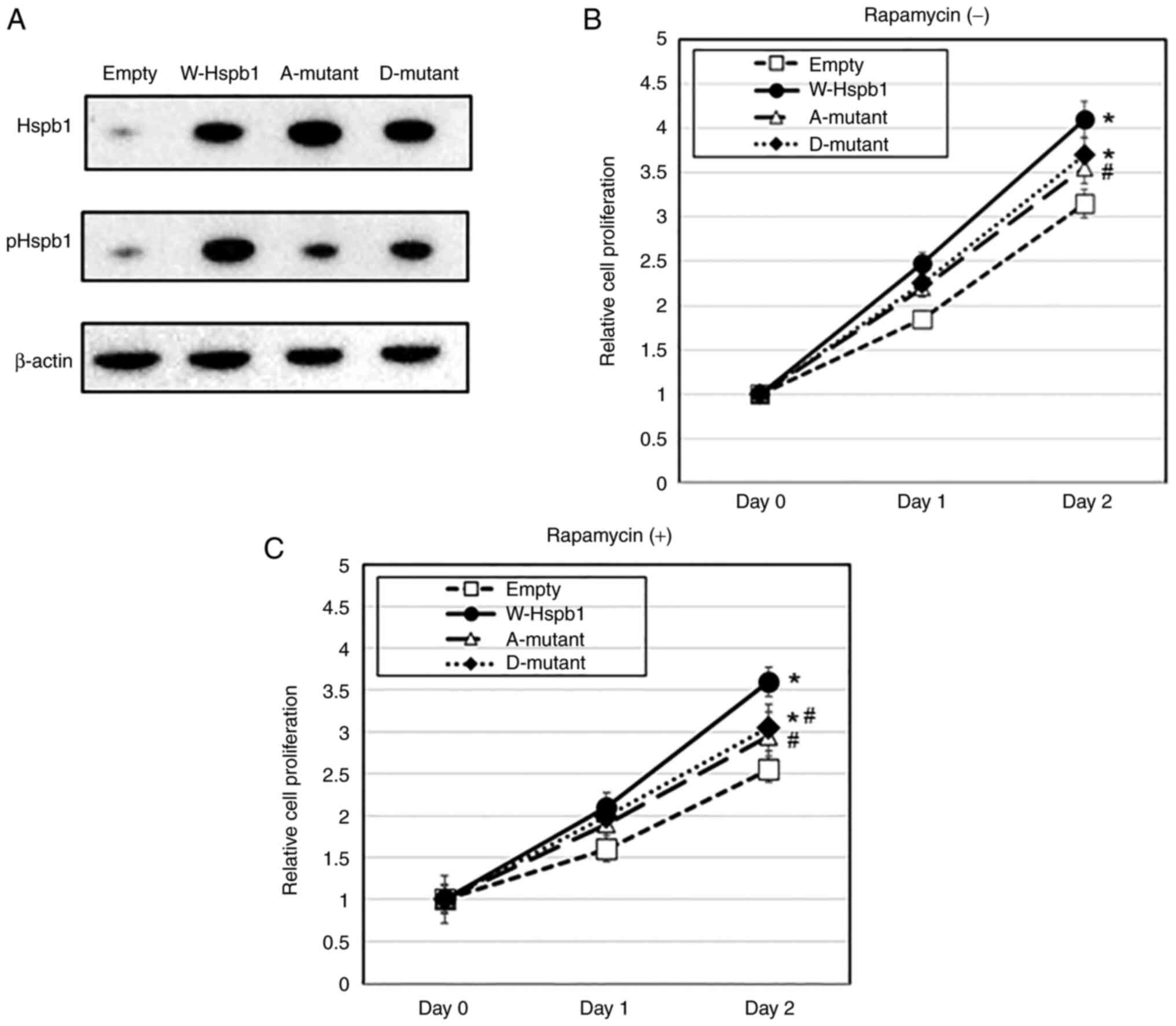
The forced expression of Hspb1
enhanced proliferation under rapamycin treatment
Next, we analyzed the effects of the forced
expression of Hspb1 on cell proliferation. We established MKOC
cells stably expressing Hspb1 and control cells (Fig. 4A). When cultured without rapamycin
for 48 h, the proliferation of HSP27-expressing cells was promoted
when compared with control cells (P<0.05) (Fig. 4B). Under the action of rapamycin,
there was also proliferation promoting activity in the wild-type
Hspb1 (P<0.05) (Fig. 4B). These
results suggested that the function of Hspb1 promoted proliferation
in Tsc2-deficient cells even in the presence of
rapamycin.
We also assayed the effect of Ser86 phosphorylation
in Hspb1 upon cell proliferation by the forced expression of a
phosphorylation-mimetic S86D mutant (D-mutant) and resistant S86A
mutant (A-mutant) Hspb1 in MKOC cells (Fig. 4A). Western blotting revealed a
reduction in pHspb1 antibody binding in D-mutant and A-mutant
cases, thus suggesting that site-directed mutagenesis had been
successfully accomplished. In the proliferation assays, both with
and without rapamycin treatment, there was a reduction of
proliferation-promoting activity in the A-mutant case (P<0.05
vs. wild-type case both with and without rapamycin) when compared
with the wild-type case, thus suggesting that phosphorylation on
S86 might be a positive regulator of proliferation-promoting
activity in Hspb1 (Fig. 4A and
B). However, there was no obvious
difference in cell proliferation when compared between the A- and
D-mutant (Fig. 4A and B). From these observations, it was
difficult to conclude that Ser86 phosphorylation on Hspb1 affects
cell proliferation.
Discussion
In this study, we focused on the stress-response
pathways involving Hspb1 that have not been explored in the
pathogenesis and treatment of TSC. We confirmed that Hsp27 was not
increased by rapamycin treatment in other cells, a phenomenon that
may be related to Tsc2 mutation, thus, we decided to further
analyze Hsp27. We found that the Hspb1 induced by rapamycin
promoted cell proliferation in Tsc2-deficient tumor cells in
culture.
There was no significant difference in proliferation
inhibition at 24 h, but there was a difference in Hspb1 expression.
Although the mechanism is unknown at present, we hypothesize that
Hspb1 expression fluctuations occur quickly in response to
rapamycin the network of proliferation inhibitory mechanisms
(Fig. S1), and then actual cell
proliferation suppression appears as a phenotype.
Previous studies have shown that the resistance to
anti-cancer drugs in many cancers is associated with higher
expression levels of Hspb1(26).
The findings of the present study suggest that Hspb1 might be
responsible, at least in part, for tumor remnants then TSC is
treated with rapamycin (9). In
siRNA experiments, no effect of suppression of Hspb1 was
observed in addition to the action of rapamycin, but in forced
expression experiments, a proliferation-promoting effect of
increased Hspb1 expression appeared under the rapamycin treatment
condition. In such a condition, it is presumed that the
proliferation-promoting pathway that can be supported by Hspb1
expression was sufficiently suppressed, and that further
suppression of Hspb1 by siRNA did not show any effect. In
the case of forced expression experiments, it is presumed that the
suppression of proliferation by rapamycin could be partly recovered
by increased amount of Hspb1. The regulatory mechanisms of
proliferation exerted by Hspb1 under rapamycin treatment remain
unclear. Nonetheless, the combined treatment of TSC gene-deficient
tumors with a drug targeting Hspb1-related pathway might be more
effective when compared with the single use of rapamycin. It might
be possible that the effect of Hspb1 suppression will appear at the
concentration of rapamycin-related drugs acting during
treatment.
In this study, we revealed the functional importance
of Hspb1 in the proliferation of Tsc2-deficient cells. The
mechanistic effect of Hspb1 has been implicated in the regulation
of ferroptosis, a mechanism of cell death that is associated with
iron metabolism (38,39). In Hspb1-suppressed cells, it
is possible that a proliferation suppressive mechanism related to
ferroptosis might be induced.
In the present study, we also observed elevated
levels of phosphorylation. However, the forced expression of
phosphorylation-mimicking and non-phosphorylated mutants both
showed slight attenuation of the proliferation-promoting effect. At
present, the significance of phosphorylation remains unclear but
should be investigated in the future.
A recent study found a possibility that
Hspb1-related pathway might be a chemotherapeutic target of
TSC-related neuropsychiatric disorders (40). To the best of our knowledge, the
present study is the first to report the function of Hspb1 in the
proliferation of TSC-related tumors. Further experiments are needed
to determine whether other types of TSC gene-deficient cells,
especially human-origin cells, show identical phenotypes.
In conclusion, we identified Hspb1 as a key molecule
in the proliferation-supporting mechanism that occurs under
rapamycin treatment. Intervention of the Hspb1-related pathway may
increase the efficacy of rapamycin in the treatment of TSC. It is
now necessary to perform in vitro as well as in vivo
studies with animal models to clarify the importance of Hspb1 in
the pathogenesis and treatment of TSC.
Supplementary Material
Model of rapamycin-induced changes in
Hspb1-related pathways. Tsc, tuberous sclerosis complex; Hspb1,
heat shock protein family B (small) member 1.
Acknowledgements
The authors would like to thank Dr Akira Orimo
(Department of Molecular Pathogenesis, Juntendo University Graduate
School of Medicine, Tokyo, Japan) for valuable advice and
discussion.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TKi, OH and TKo designed the study. TKi, KN, TT, YS
and TKo performed the experiments. TKi and TKo prepared the
figures. TKi and TKo prepared the manuscript. TKi and TKo confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Henske EP, Jóźwiak S, Kingswood JC,
Sampson JR and Thiele EA: Thiele: Tuberous sclerosis complex. Nat
Rev Dis Primers. 2(16035)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Northrup H, Aronow ME, Bebin EM, Bissler
J, Darling TN, de Vries PJ, Frost MD, Fuchs Z, Gosnell ES, Gupta N,
et al: Updated international tuberous sclerosis complex diagnostic
criteria and surveillance and management recommendations. Pediatr
Neurol. 123:50–66. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Inoki K, Li Y, Zhu T, Wu J and Guan KL:
TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signalling. Nat Cell Biol. 4:648–657. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Gao X, Zhang Y, Arrazola P, Hino O,
Kobayashi T, Yeung RS, Ru B and Pan D: Tsc tumor suppressor
proteins antagonize amino-acid-TOR signaling. Nat Cell Biol.
4:699–704. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Kwiatkowski DJ, Zhang H, Bandura JL,
Heiberger KM, Glogauer M, el-Hashemite N and Onda H: A mouse model
ot TSC1 reveals sex-dependent lethality from liver hemangiomas, and
up-regulation of p70S6 kinase activity in Tsc1 null cells.
Hum Mol Genet. 11:525–534. 2002.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tee AR, Fingar DC, Manning BD, Kwiatkowski
DJ, Cantley LC and Blenis J: Tuberous sclerosis complex-1 and -2
gene products function together to inhibit mammalian target of
rapamycin (mTOR)-mediated downatream signaling. Proc Natl Acad Sci
USA. 99:13751–13756. 2002.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kenerson HL, Aicher LD, True LD and Yeung
RS: Activated mammalian target of rapamycin pathway in the
pathogenesis of tuberous sclerosis complex renal tumors. Cancer
Res. 62:5645–5650. 2002.PubMed/NCBI
|
|
8
|
El-Hashemite N, Zhang H, Henske EP and
Kwiatkowski DJ: Mutation in TSC2 and activation of mammalian
target of rapamycin signaling pathway in renal angiomyolipoma.
Lancet. 361:1348–1349. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bissler JJ, McCormack FX, Young LR, Elwing
JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J,
et al: Sirolimus for angiomyolipoma in tuberous sclerosis complex
or lymphangioleiomyomatosis. N Engl J Med. 358:140–151.
2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Krueger DA, Care MM, Holland K, Agricola
K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T and Franz
DN: Everolimus for subependymal giant-cell astrocytomas in tuberous
sclerosis. N Engl J Med. 363:1801–1811. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Davies M, Saxena A and Kingswood JC:
Management of everolimus-associated adverse events in patients with
tuberous sclerosis complex: A practical guide. Orphanet J Rare Dis.
12(35)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Manning BD, Logsdon MN, Lipovsky AI,
Abbott D, Kwiatkowski DJ and Cantley LC: Feedback inhibition of Akt
signaling limits the growth of tumors lacking Tsc2. Genes Dev.
19:1773–1778. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wan X, Harkavy B, Shen N, Grohar P and
Helman LJ: Rapamycin induces feedback activation of Akt signaling
through an IGF-1R-dependent mechanism. Oncogene. 26:1932–1940.
2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Carracedo A, Baselga J and Pandolfi PP:
Deconstructing feedback-signaling networks to improve anticancer
therapy with mTORC1 inhibitors. Cell Cycle. 7:3805–3809.
2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mi R, Ma J, Zhang D, Li L and Zhang H:
Efficacy of combined inhibition of mTOR and ERK/MAPK pathways in
treating a tuberous sclerosis complex cell model. J Genet Genomics.
36:355–361. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ozcan U, Ozcan L, Yilmaz E, Düvel K, Sahin
M, Manning BD and Hotamisligil GS: Loss of the tuberous sclerosis
complex tumor suppressors truggers the unfolded protein response to
regulate insulin signaling and apoptosis. Mol Cell. 29:541–551.
2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yu J, Parkhitko AA and Henske EP:
Mammalian target of rapamycin signaling and autophagy: Roles in
lymphangioleiomyomatosis therapy. Proc Am Thorac Soc. 7:48–53.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Parkhitko A, Myachina F, Morrison TA,
Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski
DJ, Yu JJ and Henske EP: Tumorigenesis in tuberous sclerosis
complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent.
Proc Natl Acad Sci USA. 108:12455–12460. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pollizzi K, Malinowska-Kolodziej I, Stumm
M, Lane H and Kwiatkowski D: Equivalent benefit of mTORC1 blockade
and combined PI3K-mTOR blockade in a mouse model of tuberous
sclerosis. Mol Cancer. 8(38)2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Siroky BJ, Yin H, Babcock JT, Lu L,
Hellmann AR, Dixon BP, Quilliam LA and Bissler JJ: Human
TSC-associated renal angiomyolipoma cells are hypersensitive to ER
stress. Am J Physiol Renal Physiol. 303:F831–F844. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Armijo ME, Campos T, Fuentes-Villalobos F,
Palma ME, Pincheira R and Castro AF: Rheb signaling and
tumorigenesis: mTORC1 and new horizons. Int J Cancer.
138:1815–1823. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gau CL, Kato-Stankiewicz J, Jiang C,
Miyamoto S, Guo L and Tamanoi F: Farnesyltransferase inhibitors
reverse altered growth and distribution of actin filaments in
Tsc-deficient cells via inhibition of both rapamycin-sensitive and
-insensitive pathways. Mol Cancer Ther. 4:918–926. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Alves MM, Fuhler GM, Queiroz KC, Scholma
J, Goorden S, Anink J, Spek CA, Hoogeveen-Westerveld M, Bruno MJ,
Nellist M, et al: PAK2 is an effector of TSC1/2 signaling
independent of mTOR and a potential therapeutic target for tuberous
sclerosis complex. Sci Rep. 5(14534)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sugiura H, Yasuda S, Katsurabayashi S,
Kawano H, Endo K, Takasaki K, Iwasaki K, Ichikawa M, Kobayashi T,
Hino O and Yamagata K: Rheb activation disrupts spine synapse
formation through accumulation of syntenin in tuberous sclerosis
complex. Nat Commun. 6(6842)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sugiura H, Shimada T, Moriya-Ito K, Goto
JI, Fujiwara H, Ishii R, Shitara H, Taya C, Fujii S, Kobayashi T,
et al: A farnesyltransferase inhibitor restores cognitive deficits
in Tsc2+/- mice through inhibition of Rheb1. J Neurosci.
23:2598–2612. 2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wu J, Liu T, Rios Z, Mei Q, Lin X and Cao
S: Heat shock proteins and cancer. Trends Pharmacol Sci.
38:226–256. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dimas DT, Perlepe CD, Sergentanis TN,
Misitzis I, Kontzoglou K, Patsouris E, Kouraklis G, Psaltopoulou T
and Nonni A: The prognostic significance of Hsp70/Hsp90 expression
in breast cancer: A systematic review. Anticancer Res.
38:1551–1562. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Inoue H, Uyama T, Suzuki T, Kazami M, Hino
O, Kobayashi T, Kobayashi K, Tadokoro T and Yamamoto Y:
Phosphorylated hamartin-Hsp70 complex regulates apoptosis via
mitochondrial localization. Biochem Biophys Res Commun.
391:1148–1153. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Woodford MR, Sager RA, Marris E, Dunn DM,
Blanden AR, Murphy RL, Rensing N, Shapiro O, Panaretou B, Prodromou
C, et al: Tumor suppressor Tsc1 is a new HSP90 co-chaperone that
facilitates folding of kinase and non-kinase clients. EMBO J.
36:3650–3665. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kobayashi T, Minowa O, Kuno J, Mitani H,
Hino O and Noda T: Renal carcinogenesis, hepatic hemangiomatosis,
and embryonic lethality caused by a germ-line Tsc2 mutation
in mice. Cancer Res. 59:1206–1211. 1999.PubMed/NCBI
|
|
31
|
Piao X, Kobayashi T, Wang L, Shiono M,
Takagi Y, Sun G, Abe M, Hagiwara Y, Zhang D, Okimoto K, et al:
Regulation of folliculin (the BHD gene product) phosphorylation by
Tsc2-mTOR pathway. Biochem Biophys Res Commun. 389:16–21.
2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Goncharova EA, Goncharov DA, Fehrenbach M,
Khavin I, Ducka B, Hino O, Colby TV, Merrilees MJ, Haczku A,
Albelda SM and Krymskaya VP: Prevention of alveolar destruction and
airspace enlargement in a mouse model of pulmonary
lymphangioleiomyomatosis (LAM). Sci Transl Med.
4(154ra134)2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Okura H, Kobayashi T, Koike M, Ohsawa M,
Zhang D, Arai H, Uchiyama Y and Hino O: Tuberin activates and
controls the distribution of Rac1 via association with p62 and
ubiquitin through the mTORC1 signaling pathway. Int J Oncol.
43:447–456. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sato T, Akasu H, Shimono W, Matsu C,
Fujiwara Y, Shibagaki Y, Heard JJ, Tamanoi F and Hattori S: Rheb
protein binds CAD (Carbamoyl-phosphate synthetase 2, aspartate
transcarbamoylase, and dihydroorotase) protein in a GTP- and
effector domain-dependent manner and influences its cellular
localization and Carbamoyl-phosphate synthetase (CPSase) activity.
J Biol Chem. 290:1096–1105. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Liu HJ, Lizotte PH, Du H, Speranza MC, Lam
HC, Vaughan S, Alesi N, Wong KK, Freeman GJ, Sharpe AH and Henske
EP: TSC2-deficient tumors have evidence of T cell exhaustion and
respond anti-PD-1/anti-CTLA-4 immunotherapy. JCI Insight.
3(e98674)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang HX, Yang Y, Guo H, Hou DD, Zheng S,
Hong YX, Cai YF, Huo W, Qi RQ, Zhang L, et al: HSPB1 deficiency
sensitizes melanoma cells to hyperthermia induced cell death.
Oncotarget. 7:67449–67462. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Morgenstern JP and Land H: Advanced
mammalian gene transfer: High titre retroviral vectors with
multiple drug selection markers and a complementary helper-free
packaging cell line. Nucleic Acids Res. 18:3587–3596.
1990.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X,
Wang H, Cao L and Tang D: HSPB1 as a novel regulator of ferroptotic
cancer cell death. Oncogene. 34:5617–5625. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Liu CC, Li HH, Lin JH, Chiang MC, Hsu TW,
Li AF, Yen DH, Hsu HS and Hung SC: Esophageal cancer stem-like
cells resist ferroptosis-induced cell death by active Hsp27-GPX4
pathway. Biomolecules. 12(48)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Di Nardo A, Lenoël I, Winden KD, Rühmkorf
A, Modi ME, Barrett L, Ercan-Herbst E, Venugopal P, Behne R, Lopes
CAM, et al: Phenotypic screen with TSC-deficient neurons reveals
heat-shock machinery as a druggable pathway for mTORC1 and reduced
cilia. Cell Rep. 31(107780)2020.PubMed/NCBI View Article : Google Scholar
|