Introduction
Von Hippel-Lindau (VHL) syndrome is an inherited
multisystem disorder caused by mutations in the VHL tumor
suppressor gene located at chromosome 3p25-26(1). The incidence of VHL disease is ~1 in
36,000 live births with a penetrance >90% (2). This syndrome is characterized by a
predisposition to develop both benign and malignant tumors in
multiple organs, including hemangioblastomas of the brain/spinal
cord/retina, renal cell carcinomas (RCCs), pheochromocytomas
(PhCs), pancreatic neuroendocrine tumors and endolymphatic sac
tumors (ELSTs).
The VHL gene serves a pivotal role in regulating
critical cellular processes. The VHL protein plays a central role
in cellular oxygen sensing and hypoxic adaptation through the
erythropoietin-VHL-hypoxia-inducible factor (HIF) signaling axis,
while also participating in multiple HIF-independent pathways
(3). Notably, deficient VHL gene
function results in dysregulated homeostasis of HIFs, causing
pathological accumulation of these transcription factors that
ultimately drive transcriptional programs, which promote neoplastic
proliferation and angiogenesis (4). Current pharmacotherapeutic research
primarily targets HIF cascade proteins and functional restoration
of mutated VHL proteins (2).
VHL syndrome exhibits marked heterogeneity in
clinical manifestations with a variable age of onset, thus
mandating the lifelong clinical monitoring of patients. The
combination of early genetic diagnosis and periodic tumor
surveillance is pivotal for optimizing therapeutic outcomes in
affected individuals (5). The
present study retrospectively reviewed the clinical data of four
patients diagnosed with VHL syndrome at Hebei General Hospital
(Shijiazhuang, China), aiming to summarize their clinical features
and therapeutic management.
Case report
Case 1
A 34-year-old man presented with posterior neck pain
persisting for 14 weeks, which had worsened over the preceding
week. The patient was admitted to Hebei General Hospital in October
2010. A neurological examination revealed clear consciousness and
intact cranial nerve function. Muscle strength was graded 4+ in the
right limbs and grade 4 in the left limbs (6). The medical history of the patient
included bilateral adrenal PhCs diagnosed in 2005, for which they
underwent laparoscopic pheochromocytoma resection to manage
hypertension. There was no history of infectious diseases or
hereditary conditions. Cranial magnetic resonance imaging (MRI)
demonstrated a space-occupying lesion in the left cerebellar
hemisphere, along with multiple areas of abnormal enhancement in
the cerebellar vermis, right cerebellar tonsils and right
cerebellar hemisphere (data not shown). The findings were
consistent with various hemangioblastomas. The patient underwent
intracranial tumor resection on October 2010, with postoperative
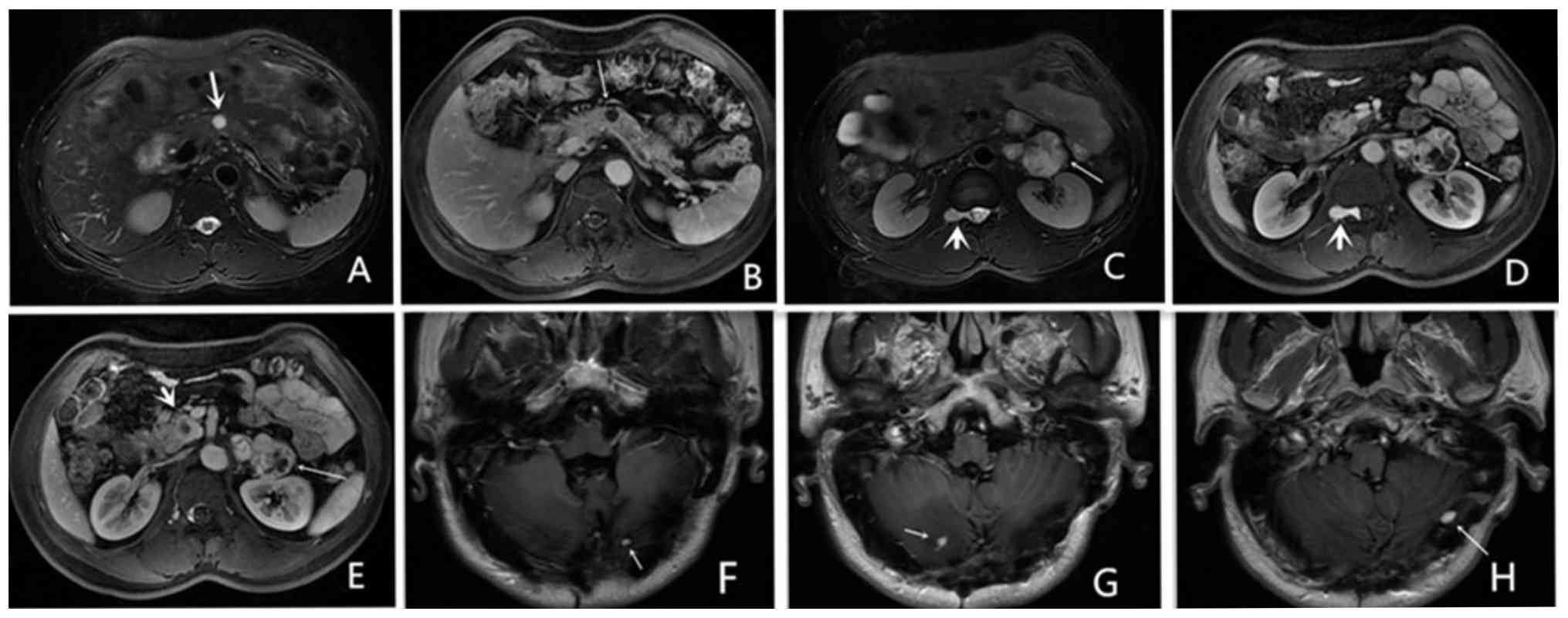
pathology confirming hemangioblastoma (Fig. 1A and C-L) and the following pathological
results: Hematoxylin and eosin staining showed that the tumor
tissue exhibited nested and alveolar growth patterns, composed of
large tumor cells with vacuolated, lipid-rich cytoplasm. Marked
nuclear pleomorphism was observed, featuring atypical nuclei with
hyperchromasia. Mitotic figures were rare. The tumor cell nests
were surrounded by abundant capillary networks involving
predominantly thin-walled vessels, with some showing characteristic
highly branched ‘staghorn’ morphology. Immunohistochemical findings
were as follows: CD34(+++; strong diffuse positivity); solute
carrier family 2 facilitated glucose transporter member 1(+);
vimentin(+++; strong diffuse positivity); epithelial membrane
antigen(-), effectively ruling out angiomatous meningioma; Ki-67
(<5% positive), supporting the diagnosis of hemangioblastoma;
glial fibrillary acidic protein(-), excluding diffuse astrocytoma;
pan-cytokeratin(-); CD10(-); and paired box protein Pax-8(-),
excluding metastatic carcinoma and metastatic renal cell carcinoma
(7). The patient's symptoms
improved after surgery and they were subsequently discharged.
In March 2014, the patient was re-admitted to the
Department of Neurosurgery due to gait instability persisting for
>2 months. Neurological examination revealed no notable
deficits. Cranial MRI demonstrated multiple small nodules within
the cerebellum and a cystic-solid mass in the cerebellar vermis,
exerting pressure on the fourth ventricle and resulting in mild
supratentorial hydrocephalus (data not shown). Intracranial surgery
was performed 7 days after readmission, and postoperative
histopathology confirmed a capillary hemangioblastoma (Fig. 1B). Tissue sections were cut from
formalin-fixed, paraffin-embedded tissue blocks at a 4-µm
thickness. Sections were deparaffinized in xylene and rehydrated
through graded ethanol. Heat-induced epitope retrieval was
performed in citrate buffer (pH 6.0) for 5 min. Sections were
treated with 3% hydrogen peroxide in methanol for 30 min to quench
endogenous peroxidase activity. Sections were incubated overnight
at 4˚C in a humidified chamber with anti-HIF-1α (1:200 dilution;
GeneTex, Inc.) and anti-PKM2 (1:400 dilution; Cell Signaling
Technology, Inc.). After washing with PBS, immunostaining was
performed using the SP detection kit (Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.) according to the manufacturer's
instructions. Nuclei were counterstained with hematoxylin. Parallel
staining omitting primary antibodies was included for each
experiment. The patient was discharged in a stable condition
following clinical improvement.
Abdominal ultrasound revealed a hypoechoic area in
the pancreas (data not shown), whereas the adrenal glands and
kidneys appeared normal. A pancreatic neuroendocrine tumor
measuring 2.1x2.4 cm was identified in January 2018. Surgical
intervention was deferred as there was no marked tumor growth
during follow-up. Intracranial surgery was performed 4 days after
admission and a postoperative histopathological examination was
completed. Tissues were fixed in 10% neutral buffered formalin at
room temperature (20-25˚C) for 24 h before being sectioned to 4-µm
thick. Hematoxylin and eosin staining was performed using
hematoxylin for 5 min at room temperature and using eosin for 2 min
at room temperature. Results were examined using an Olympus BX53
microscope (Olympus Corporation) and confirmed the diagnosis of
hemangioblastoma (Fig. 1A)
(8). The patient experienced
symptomatic improvement following the procedure, owing to a
clinically notable resolution of gait disturbance.
Follow-up evaluations showed that the patient
underwent a pancreatic and adrenal MRI in October 2018. Imaging
revealed multiple abnormal signal intensities within the pancreas
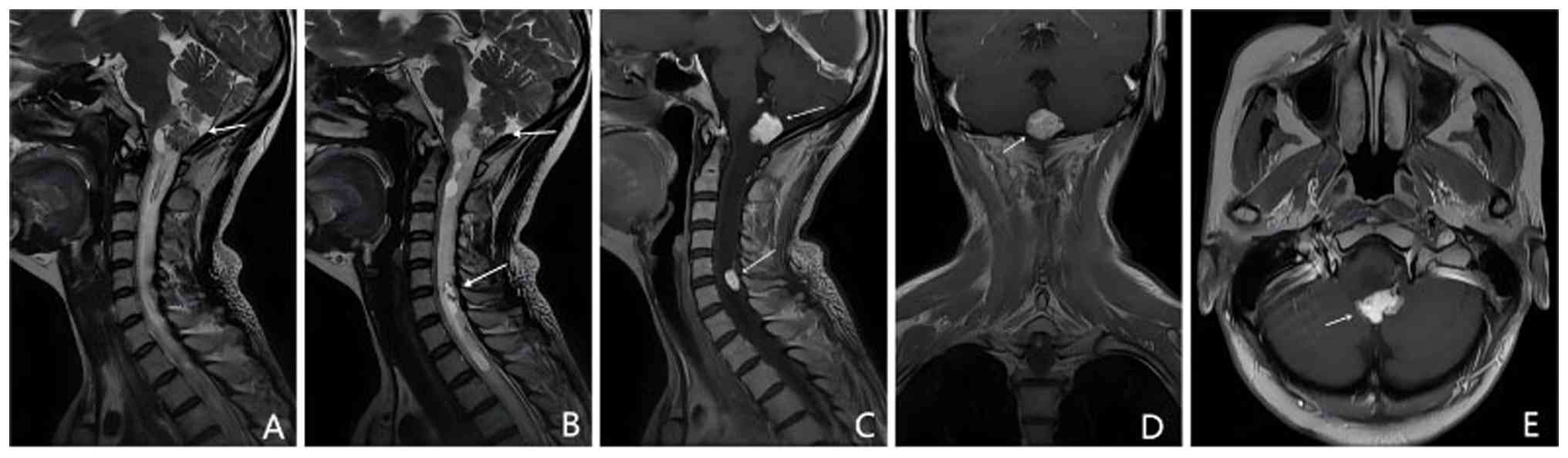
(Fig. 2A and B), suggestive of neuroendocrine tumors.
An additional lesion in the pancreatic body was consistent with a
pancreatic cyst. Multiple signal abnormalities in the left adrenal
gland indicated PhC (Fig. 2C-E),
and an abnormal signal intensity at the lower pole of the right
kidney was consistent with a renal cyst. A contrast-enhanced
cranial MRI performed in February 2019, showed nodular enhancement
in both cerebellar hemispheres, raising concern for recurrence of
VHL-associated lesions (Fig.
2F-H). Based on the presence of cerebellar hemangioblastomas,
pancreatic neuroendocrine tumors, adrenal PhC and a renal cyst, a
diagnosis of VHL syndrome was strongly suspected. As of May 2025,
patient 1 was alive; their parents and son were also alive at this
time and showed no signs of VHL syndrome-related symptoms.
Case 2
A 45-year-old woman presented with a 2-year history
of paresthesia in the left hand and left lower limb, which had
progressively worsened over the past 6 months. The patient was
admitted to Hebei General Hospital in October 2019. Neurological
examination revealed a positive Hoffmann's sign on the left side,
with no other significant abnormalities.
In 2012, the patient underwent a
choledochojejunostomy due to bile duct obstruction caused by
multiple pancreatic cysts. The patient had also experienced
elevated blood glucose levels for >2 years. There was no
reported family history of infectious or hereditary diseases. A
contrast-enhanced brain and cervical spine MRI revealed abnormal
signal intensities in the cerebellar vermis, dorsal medulla
oblongata and dorsal spinal cord at the C5-6 level (Fig. 3A and B). Numerous tortuous flow-void vascular
structures were observed, consistent with multiple central nervous
system hemangioblastoma (CNS-H) tumors (Fig. 3). Abdominal ultrasound demonstrated
pancreatic thickening with multiple anechoic cystic areas and
calcifications (data not shown). A diagnosis of VHL syndrome was
considered based on the presence of numerous CNS-H tumors found in
the cerebellum, brainstem and spinal cord, in conjunction with a
history of pancreatic cysts. As of May 2025, the patient was lost
to follow-up. No health status information is available regarding
the parents or children of the patient.
Case 3
A 30-year-old woman was admitted to Hebei General
Hospital in May 2018, with a complaint of a fever persisting for
>20 days. Neurological examination revealed no evident
abnormalities. The medical history of the patient included surgical
resection of brain tumors in 2012 and 2014, with postoperative
pathology confirming hemangioblastomas. There was no reported
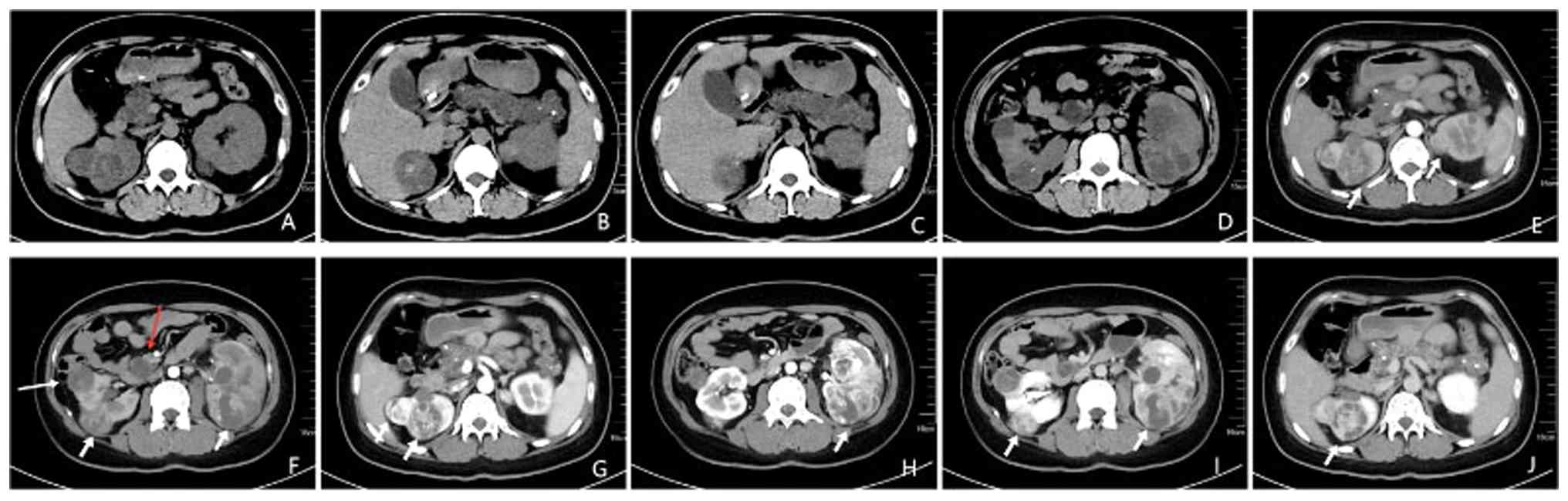
family history of infectious or hereditary diseases. Renal computed
tomography (CT) revealed bilateral renal masses suggestive of RCC,
cysts in the upper poles of both kidneys, multiple pancreatic cysts
and a hemangioblastoma at the L2-3 vertebral level (Fig. 4). In this case, the largest of the
bilateral renal cell carcinomas was found to be 9.3x5.4x6.8 cm in
2018. During a follow-up examination in July 2019 after treatment,
the tumor lesion was found to be significantly smaller than before.
Based on the presence of CNS-H, RCC, renal cysts and pancreatic
cysts, a diagnosis of VHL syndrome was suspected. As of May 2025,
the patient was confirmed deceased. It was also reported that the
father of the patient had succumbed (without a known history of VHL
syndrome), while their mother, who was suspected to have VHL
syndrome, was alive at this time. The patient had a son who showed
no evidence of VHL-related manifestations.
Case 4
A 50-year-old woman with a 12-year history of
intermittent headaches and dizziness was admitted to Hebei General
Hospital in March 2019. Neurological examination revealed no
notable abnormalities. The medical history of the patient included
brain tumor resection 12 years earlier, with postoperative
pathology confirming hemangioblastoma. In April 2018, the patient
experienced tumor recurrence and underwent a second surgical
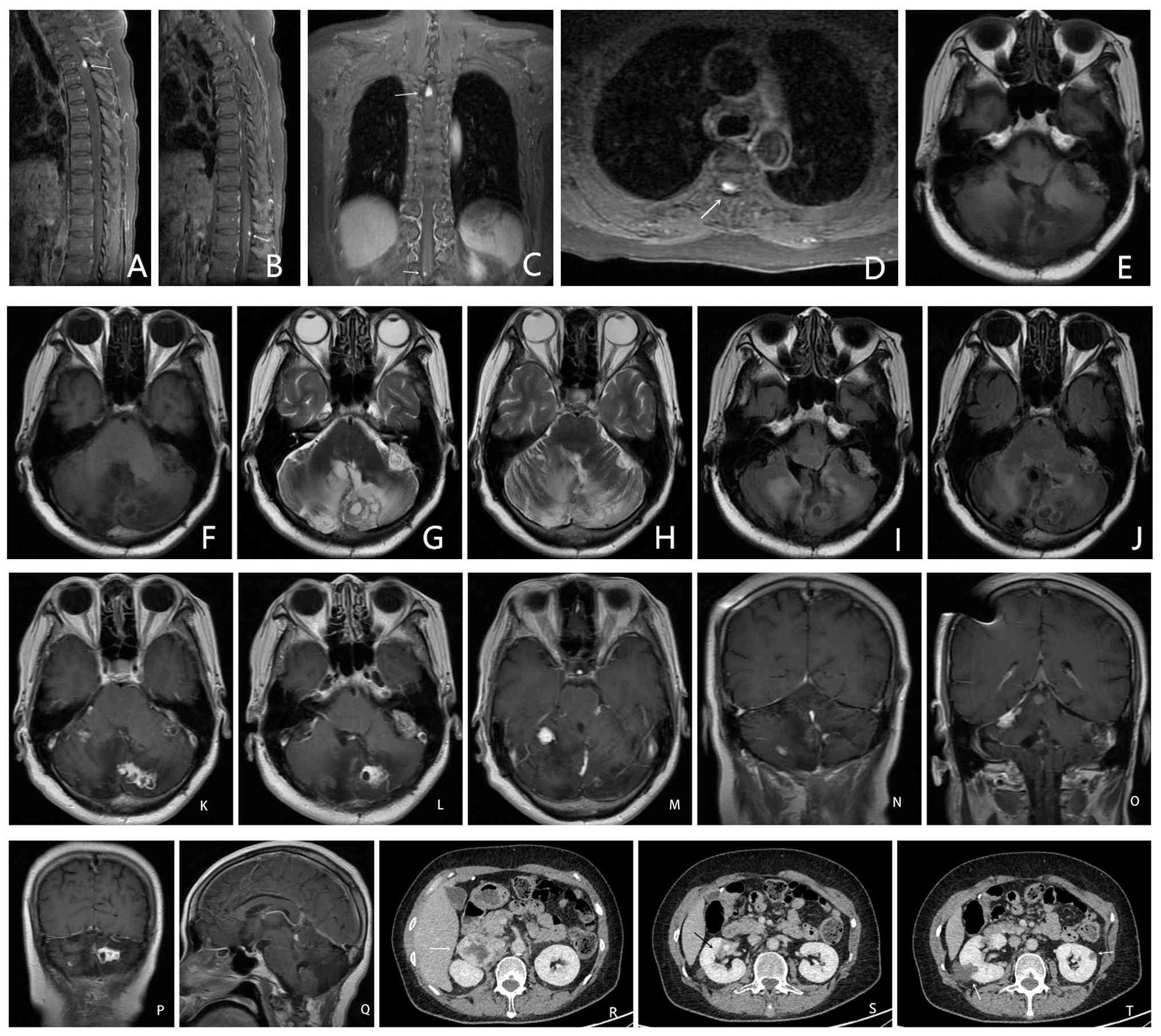
resection (Fig. 5A-Q), which again
confirmed hemangioblastoma. There was no family history of
infectious or hereditary diseases. Contrast-enhanced chest and
abdominal CT scans revealed a pancreatic mass, a cyst in the
pancreatic tail, bilateral adrenal PhC (Fig. 5R) and radiologic features highly
suggestive of bilateral RCC (Fig.
5S and T).
Thoracic spine MRI (Fig. 5A-C) with contrast showed abnormal
enhancement of the posterior spinal cord at the T3 level and the
T12-L1 intervertebral space, raising suspicion for meningeal
metastases of hemangioblastoma. A diagnosis of VHL syndrome was
established based on the presence of multiple hemangioblastomas in
the cerebellum and thoracic spinal cord, along with RCC, pancreatic
cysts and adrenal PhC. This patient was diagnosed with a
non-functioning pheochromocytoma due to the absence of hypertensive
manifestations, and therefore no specific treatment was
administered. As of May 2025, the patient was in good health. Both
parents and two daughters were physically healthy with no clinical
manifestations related to VHL syndrome at this time.
Literature review
VHL syndrome is a rare autosomal dominant disorder
characterized by the development of tumors and cysts across
multiple organ systems. The syndrome results from mutations in the
VHL gene, which is a tumor suppressor gene, located on chromosome
3p25-p26(1). The estimated
incidence of VHL syndrome ranges from 1 in 36,000-45,500
individuals (9). Mutations in the
VHL gene lead to dysregulation of the HIF pathways, resulting in
the upregulation of vascular endothelial growth factor (VEGF) and
platelet-derived growth factor-β, both of which are implicated in
tumorigenesis (10). Genetic
alterations in the VHL gene can be identified in 39-75% of familial
cases, whereas 25-61% of cases occur sporadically, without a family
history (11). The present study
diverges from previous research by not only cataloging tumor
heterogeneity across patients with VHL syndrome, but also
longitudinally tracking treatment response statistics for each
therapeutic intervention in individual patients, with a median
follow-up period of ~8 years.
Clinical diagnosis and
classification
VHL syndrome is characterized by the development of
tumors across multiple organ systems, specifically CNS-H, retinal
hemangioblastoma (RH), RCC, PhC and ELST, among others. Diagnosis
is based on both clinical assessment and genetic analysis. The most
widely accepted clinical diagnostic criteria, proposed by Maher
et al (12), include: i)
The presence of any one of the characteristic tumor types in a
patient with a positive family history; ii) in the absence of a
family history, a diagnosis may be established when a patient
presents with either two or more hemangioblastomas, or a single
hemangioblastoma in combination with a related visceral tumor.
Genetic testing [sequence analysis and gene-targeted
deletion/duplication analysis (1)]
remains the definitive method for diagnosis (13). Genetic testing should be promptly
pursued to confirm the diagnosis in patients with a strong clinical
suspicion but inconclusive findings.
Classification
VHL syndrome is classified internationally into two
types (14), based on the presence
or absence of PhC (Table I).
 | Table IClassification of VHL syndrome. |
Table I
Classification of VHL syndrome.
| Type |
Characteristics |
|---|
| Type 1 | No
pheochromocytoma |
|
1A | Renal cancer (case
3) |
|
1B | No renal cancer
(case 2) |
| Type 2 |
Pheochromocytoma |
|
2A | No renal cancer +
tumors of other organs (case 1) |
|
2B | Renal cancer (case
4) |
|
2C | Only
pheochromocytoma |
Pathological features of VHL syndrome
in organs
VHL syndrome demonstrates diverse clinical
manifestations and can involve multiple organs, either concurrently
or sequentially over time. The interval between the onset of
lesions in different organs may range from several years to
decades. For example, in the cases described in the present study,
the intervals were 5 years in case 1, 7 years in case 2, 6 years in
case 3 and 11 years in case 4 (Table
II).
| Table IIClinical data and affected
organs. |
Table II
Clinical data and affected
organs.
| | CNS
hemangioblastoma | |
|---|
| Case | Sex | Age at first onset,
years | Chief
complaint | Head | Spinal cord | Retina | Pancreas | Kidney |
|---|
| 1 | Male | 29 | Posterior neck
pain | Cerebellum | L1 | No | Pancreatic
neuroendocrine tumor, pancreatic cyst | Left adrenal
pheochromocytoma, right renal cysts |
| 2 | Female | 38 | Paresthesia in the
left hand and left lower limb | Medulla
oblongata | C5-6 | No | Multiple pancreatic
cysts | No |
| 3 | Female | 24 | Fever | Cerebellar
hemisphere and medulla oblongata | L2-3 | No | Multiple pancreatic
cysts | Bilateral renal
cysts, bilateral RCC |
| 4 | Female | 38 | Intermittent
headache and dizziness | Cerebellar
hemisphere and vermis | T3, T12-L1 | No | Pancreatic
cyst | Bilateral RCC,
bilateral adrenal pheochromocytoma |
CNS-H
CNS-H is the most frequent tumor associated with VHL
syndrome and represents the leading cause of mortality in affected
patients. Onset typically occurs between 20 and 30 years of age.
CNS-H predominantly arises in the cerebellum (44-72%), spinal cord
(13-50%) and brainstem (10-25%), with rare involvement of the optic
pathway, choroid plexus, anterior pituitary and infundibulum (~1%)
(15). Clinical manifestations are
variable and may include headache, dizziness, balance disturbances,
limb pain, weakness, numbness and aspiration pneumonia (16). Early symptoms are often
nonspecific, making differential diagnosis challenging; for
instance, in case 2, the patient underwent ulnar nerve
decompression due to an initial misdiagnosis of hand numbness
attributed to ulnar nerve compression. Surgical resection remains
the standard treatment for large or symptomatic CNS-H tumors
(17). Radiotherapy is
increasingly employed for lesions that are small, deep-seated,
multifocal or otherwise unsuitable for surgery (18). Recurrence is common following
initial treatment, necessitating regular monitoring. Given that
5-38% of CNS-H cases are linked to VHL syndrome (19), thorough family history assessment
and prompt screening of other organ systems are critical upon CNS-H
detection. Genetic testing facilitates early diagnosis and
management of VHL syndrome when clinically indicated.
In the present study, cases 1, 3 and 4 underwent
surgical removal of intracranial tumors at initial presentation;
however, tumor recurrence was observed 4, 2 and 11 years
postoperatively, respectively. Case 2 did not receive surgery due
to the complexity of the lesion.
RH
RH is the second most prevalent tumor linked to VHL
syndrome worldwide, occurring in ~73% of cases (20). However, the incidence among Chinese
patients is markedly lower, at ~22% (21), possibly reflecting variations
associated with ethnicity or genetics. RH is usually diagnosed at
~25 years of age, with most patients presenting between 10 and 40
years of age (22). Bilateral
involvement occurs in 50-60% of cases (23). Early-stage RH is generally
asymptomatic, but as the disease advances, patients may experience
visual deterioration, potentially leading to blindness.
Consequently, early detection and timely intervention are vital to
preserve vision. The primary treatment method involves tumor
ablation through laser photocoagulation or cryotherapy (24). When ablative therapies are
ineffective or unsuitable, intravitreal injections of anti-VEGF
agents serve as an alternative option (25).
Pancreatic disease
Pancreatic lesions develop in ~60% of patients with
VHL syndrome, including simple cysts, serous cystadenomas and
neuroendocrine tumors (26). Among
these, multiple cysts are the most common, with a subset developing
into pancreatic neuroendocrine tumors (27). At initial diagnosis, pancreatic
lesions constitute the only abdominal finding in ~12% of patients
with VHL syndrome (28).
Clinically, >90% of patients remain asymptomatic; however,
hyperglycemia may arise if insulin secretion is disrupted. Tumors
that compress the pancreatic or bile ducts can lead to obstructive
jaundice and gastrointestinal symptoms, such as diarrhea and
constipation. Simple cysts and serous cystadenomas generally have a
benign course and warrant regular surveillance. Neuroendocrine
tumors, which carry a risk of metastasis, require management
tailored to tumor size, growth dynamics and specific gene mutations
(29).
In case 2, a choledochojejunostomy was performed to
relieve bile duct obstruction which was caused by compression from
pancreatic cysts. The patient developed elevated blood glucose
levels 2 years after the cysts were identified, suggesting a
potential link between cyst compression and hyperglycemia. For
patients with VHL syndrome, regular blood sugar monitoring is
advisable following the detection of pancreatic cysts. If
hyperglycemia occurs and other causes are excluded, addressing the
pancreatic cyst may help achieve improved glucose control. In case
1, a pancreatic neuroendocrine tumor measuring 2.1x2.4 cm was
identified in January 2018. Surgical intervention was deferred as
there was no marked tumor growth during follow-up, however close
observation was maintained. The remaining two cases had multiple
asymptomatic pancreatic cysts that did not require specific
treatment, although continued monitoring was recommended.
Kidney lesions
Approximately two-thirds of patients with VHL
syndrome develop renal cysts or RCC. Although renal cysts are
benign, they carry the risk of progressing to cancer, necessitating
careful surveillance (12). RCC is
the second most common cause of mortality among patients with VHL
syndrome and typically manifests between the ages of 20 and 50
years. It often presents as multiple bilateral lesions, which grow
slowly and metastasize late (30).
Early disease stages are usually asymptomatic, while advanced
stages may cause symptoms such as lower back pain and hematuria.
For tumors <3 cm, regular monitoring is advised (31). The diagnosis primarily relies on
imaging modalities including ultrasonography, CT and MRI (4). Notably, 68Ga-NY104 PET/CT has
demonstrated marked diagnostic value for imaging metastatic RCC,
while its application in primary RCC requires further clinical
validation (32). Tumors >3 cm
are generally managed with nephron-sparing surgery. Breakthroughs
in targeted therapies, particularly HIF and VEGF inhibitors, have
provided novel therapeutic options for advanced or metastatic RCC
(33).
In case 1, the patient had a solitary renal cyst
without any clinical symptoms; therefore, no treatment was
administered. Case 3 was diagnosed with bilateral RCC in 2018, with
the largest tumor measuring 9.3x5.4x6.8 cm. Following treatment, a
marked reduction in tumor size was observed during the follow-up
examination in July 2019. Case 4 presented with kidney cancer
involving a tumor <3 cm in diameter, and thus, no further
treatment was deemed necessary.
PhC
PhC occurs in 10-20% of patients with VHL syndrome,
with >90% of cases arising in the adrenal glands. Rare cases
involve locations such as the carotid sinus, vagus nerve or
abdominal aorta (34). The average
age of onset is 34 years. Adrenal PhC can be unilateral or
bilateral, with ~44% of cases affecting both adrenal glands. These
tumors may be either functional or non-functional. Functional
pheochromocytomas demonstrate overproduction of catecholamines
(epinephrine, norepinephrine and dopamine), clinically presenting
with characteristic symptom clusters: Sudden-onset high blood
pressure, rapid heartbeat, profuse sweating, severe headaches and
shaking tremor. Hypertension is the most common clinical
manifestation (4). Surgical
removal remains the primary treatment for PhC, although there is a
recurrence risk, underscoring the importance of regular follow-up
(35).
Case 1 underwent surgery for bilateral adrenal PhC
in 2005 and initially recovered well; however, a recurrence of the
left adrenal PhC was detected in 2018. Histologically, bilateral
PhC associated with VHL syndrome is indistinguishable from solitary
PhC. In young patients with hypertension and bilateral adrenal PhC,
VHL syndrome should be suspected. In case 4, PhC was considered
non-functional due to the absence of hypertension, thus no
treatment was initiated. However, close surveillance was
maintained, with plans to address adrenal PhC before any other
surgical interventions to reduce intraoperative risks.
Other lesions of VHL syndrome
Bilateral epididymal papillary cystadenomas occur in
approximately one-half of all male patients with VHL syndrome,
usually presenting as painless scrotal masses. These lesions
typically do not impact fertility. Female patients may develop
cystadenomas of the broad ligament, which are generally
asymptomatic and do not require specific treatment (2).
None of the four cases in the current study showed
involvement of the reproductive system.
Discussion
VHL syndrome is a rare disorder affecting multiple
organs, and its diagnosis can be challenging due to the variability
in which organs are involved and the order of their appearance.
When VHL syndrome is clinically suspected, a thorough examination
and detailed family history review are essential. Screening and
monitoring family members, especially offspring, is recommended
after diagnosis. Advances in the study of the VHL gene have
introduced new diagnostic tools and treatment options. Genetic
testing allows for the identification of specific mutations and
helps to assess the risk of tumor development across various
organs, facilitating focused surveillance. Currently, surgery
remains the primary treatment, although tumor recurrence after
surgery is common. Insights into VHL gene mutations affecting the
VEGF signaling pathway have spurred progress in targeted drug
therapies, such as belzutifan (HIF-2 inhibitor) (36), tivozanib (VEGF receptor tyrosine
kinase inhibitor) (37) and
bevacizumab (anti-VEGF monoclonal antibody) (38).
With ongoing advancements in molecular biology, gene
therapy presents a promising option for managing VHL syndrome in
the future. The critical research trajectory in this disease domain
hinges on establishing a translational pipeline where clinicians
can: i) Systematically correlate clinical-radiographic phenotypes
with genetic suspicion indices: Germline VHL mutations are
classified into distinct subtypes, each associated with specific
clinical manifestations of the syndrome (4); and ii) implement tiered molecular
diagnostics as per the American College of Medical Genetics and
Genomics guidelines (39). The
primary clinical implication of the present study lies in its
capacity to alert clinicians to consider VHL syndrome, a heritable
syndrome of tumor predisposition, in the differential diagnosis
when evaluating patients with multisystem tumors, particularly
those demonstrating atypical organ involvement patterns.
The primary limitations of the current study
include: A restricted cohort size (n=4), and the absence of novel
clinical manifestations or previously undocumented organ
involvement patterns in VHL syndrome. We subsequently aim to
establish a longitudinal VHL registry to systematically expand the
sample size and comprehensively characterize its phenotypic
evolution, with the ultimate goal of identifying potential
genotype-phenotype associations and emerging disease patterns.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SD prepared the initial draft of the manuscript and
participated in patient data analysis and medical history
follow-up. HC handled data collection for the study, providing the
patient's clinical history and medical records, and participated in
both data acquisition and analysis. RD contributed to revising and
refining the manuscript, and participated in both data analysis and
interpretation. YB and DJ contributed to data acquisition and data
analysis, and the evaluation of treatment recommendations. TP and
YW contributed to data analysis and interpretation. SD and HC
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All studies involving human participants were
reviewed and approved by the Hebei General Hospital Ethics
Committee (Shijiazhuang, China; approval no. 2024-LW-150) and were
conducted following the ethical principles outlined in the 1964
Declaration of Helsinki and its subsequent amendments.
Patient consent for publication
Patient identities were anonymized throughout data
collection and analysis. All personal identifiers were either
removed or encrypted, including names, addresses, social security
numbers and hospital ID numbers. The data were then aggregated and
analyzed to ensure that individual patients could not be
re-identified. A waiver of patient consent for publication was
approved by the Hebei General Hospital Ethics Committee.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
van Leeuwaarde RS, Ahmad S, van
Nesselrooij B, Zandee W and Giles RH: Von Hippel-Lindau syndrome.
In: GeneReviews®. Adam MP, Feldman J and Mirzaa GM
(eds). University of Washington, Seattle, WA, 2025.
|
|
2
|
Gläsker S, Neumann HPH, Koch CA, Vortmeyer
A, Feingold KR, Ahmed SF, Anawalt B, Blackman MR, Boyce A and
Chrousos G (eds): Endotext, Von Hippel-Lindau Disease. MDText.com, South Dartmouth, MA, 2018.
|
|
3
|
Hudler P and Urbancic M: The role of VHL
in the development of von Hippel-Lindau disease and erythrocytosis.
Genes (Basel). 13(362)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Harbi E and Aschner M: Von Hippel-Lindau
syndrome: Clinical features, genetic foundations, and management
strategies. Mol Biol Rep. 52(281)2025.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ohh M, Taber CC, Ferens FG and Tarade D:
Hypoxia-inducible factor underlies von Hippel-Lindau disease
stigmata. Elife. 11(e80774)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Florence JM, Pandya S, King WM, Robison
JD, Baty J, Miller JP, Schierbecker J and Signore LC: Intrarater
reliability of manual muscle test (medical research council scale)
grades in Duchenne's muscular dystrophy. Phys Ther. 72:115–126.
1992.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hofman FM and Taylor CR:
Immunohistochemistry. Curr Protoc Immunol. 103:21.4.1–21.4.26.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hao Z, Wang Y, Li J, Liu W, Zhao W and
Wang J: Expression of HIF-1α/PKM2 axis correlates to biological and
clinical significance in papillary thyroid carcinoma. Medicine
(Baltimore). 102(e33232)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wong M, Chu YH, Tan HL, Bessho H, Ngeow J,
Tang T and Tan MH: Clinical and molecular characteristics of East
Asian patients with von Hippel-Lindau syndrome. Chin J Cancer.
35(79)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nielsen SM, Rhodes L, Blanco I, Chung WK,
Eng C, Maher ER, Richard S and Giles RH: Von Hippel-Lindau disease:
Genetics and role of genetic counseling in a multiple neoplasia
syndrome. J Clin Oncol. 34:2172–2181. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Stolle C, Glenn G, Zbar B, Humphrey JS,
Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R and
Linehan WM: Improved detection of germline mutations in the von
Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 12:417–423.
1998.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Maher ER, Nrumann HP and Richard S: von
Hippel-Lindau disease: A clinical and scientific review. Eur J Hum
Gene. 19:617–623. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nordstrom-O'Brien M, van der Luijt RB, van
Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van
Brussel A, Voest EE and Giles RH: Genetic analysis of von
Hippel-Lindau disease. Hum Mutat. 31:521–537. 2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Crespigio J, Berbel LCL, Dias MA, Berbel
RF, Pereira SS, Pignatelli D and Mazzuco TL: Von Hippel-Lindau
disease: A single gene, several hereditary tumors. J Endocrinol
Invest. 41:21–31. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Weil RJ, Lonser RR, DeVroom HL, Wanebo JE
and Oldfield EH: Surgical management of brainstem hemangioblastomas
in patients with von Hippel-Lindau disease. J Neurosurg. 98:95–105.
2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Panayi C, Antoun N and Sandford R: Bulbar
dysfunction and aspiration pneumonia due to a brainstem
haemangioblastoma: An unusual complication of von Hippel-Lindau
disease. BMJ Case Rep. 13(2016:bcr2016217076)2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ammerman JM, Lonser RR, Dambrosia J,
Butman JA and Oldfield EH: Long-term natural history of
hemangioblastomas in patients with von Hippel-Lindau disease:
Implications for treatment. J Neurosurg. 105:248–255.
2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Aronow ME, Wiley HE, Gaudric A, Krivosic
V, Gorin MB, Shields CL, Shields JA, Jonasch EW, Singh AD and Chew
EY: Von Hippel-Lindau disease: Update on pathogenesis and systemic
aspects. Retina. 39:2243–2253. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gläsker S: Central nervous system
manifestations in VHL: Genetics, pathology and clinical phenotypic
features. Fam Cancer. 4:37–42. 2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ong KR, Woodward ER, Killick P, Lim C,
Macdonald F and Maher ER: Genotype-phenotype correlations in von
Hippel-Lindau disease. Hum Mutat. 28:143–149. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang JY, Peng SH, Ning XH, Li T, Liu SJ,
Liu JY, Hong BA, Qi NN, Peng X, Zhou BW, et al: Shorter telomere
length increases age-related tumor risks in von Hippel-Lindau
disease patients. CaIlcer Med. 6:2131–2141. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chang JH, Spraul CW, Lynn ML, Drack A and
Grossniklaus HE: The two-stage mutation model in retinal
hemangioblastoma. Ophthalmic Genet. 19:123–130. 1998.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tsang SH and Sharma T: Von Hippel-Lindau
disease. Adv Exp Med Biol. 1085:201–203. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hajjaj A, van Overdam KA, Gishti O, Ramdas
WD and Kiliç E: Efficacy and safety of current treatment options
for peripheral retinal haemangioblastomas: A systematic review.
Acta Ophthalmol. 100:e38–e46. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Papastefanou VP, Pilli S, Stinghe A,
Lotery AJ and Cohen VML: Photodynamic therapy for retinal capillary
hemangioma. Eye (Lond). 27:438–442. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gao HL, Jin KZ, Wang XH, Chen ZY, Lu RQ,
Huang D and Yu XR: Clinicopathological conference: Multiple
pancreatic cysts with intermittent abdominal discomfort. Fudan Univ
J Med Sci. 44:528–531. 2017.(In Chinese).
|
|
27
|
Laks S, van Leeuwaarde R, Patel D, Keutgen
XM, Hammel P, Nilubol N, Links TP, Halfdanarson TR, Daniels AB and
Tirosh A: Pancreatic Manifestations Recommendations Development
Subcommittee of the VHL Alliance. Management recommendations for
pancreatic manifestations of von Hippel-Lindau disease. Cancer.
128:435–446. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Keutgen XM, Hammel P, Choyke PL, Libutti
SK, Jonasch E and Kebebew E: Evaluation and management of
pancreatic lesions in patients with von Hippel-Lindau disease. Nat
Rev Clin Oncol. 13:537–549. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zwolak A, Świrska J, Tywanek E, Dudzińska
M, Tarach JS and Matyjaszek-Matuszek B: Pancreatic neuroendocrine
tumours in patients with von Hippel-Lindau disease. Endokrynol Pol.
71:256–259. 2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chittiboina P and Lonster RR: Von
Hippel-Lindau disease. Handb Clin Neurol. 132:139–156.
2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kim E and Zschiedrich S: Renal cell
carcinoma in von Hippel-Lindau disease from tumor genetics to novel
therapeutic strategies. Front Pediatr. 6(16)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhu W, Zheng G, Yan X, Liu M, Li X, Cheng
Y, Bai C, Zhang Y and Huo L: Diagnostic efficacy of
[68Ga]Ga-NY104 PET/CT to identify clear cell renal cell
carcinoma. Eur J Nucl Med Mol Imaging. 51:4127–4133.
2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Choueiri TK and Kaelin WG Jr: Targeting
the HIF2-VEGF axis in renal cell carcinoma. Nat Med. 26:1519–1530.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Walther MM, Reiter R, Keiser HR, Choyke
PL, Venzon D, Hurley K, Gnarra JR, Reynolds JC, Glenn GM, Zbar B
and Linehan WM: Clinical and genetic characterization of
pheochromocytoma in von Hippel-Lindau families: Comparison with
sporadic pheochromocytoma gives insight into natural history of
pheochromocytoma. J Urol. 162:659–664. 1999.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Aufforth RD, Ramakant P, Sadowski SM,
Mehta A, Trebska-McGowan K, Nilubol N, Pacak K and Kebebew E:
Pheochromocytoma screening initiation and frequency in von
Hippel-Lindau syndrome. J Clin Endocrinol Metab. 100:4498–4504.
2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Jonasch E, Donskov F, Iliopoulos O,
Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie
JK, Welsh SJ, et al: Belzutifan for renal cell carcinoma in von
Hippel-Lindau disease. N Engl J Med. 385:2036–2046. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jacob A, Shook J and Hutson TE: Tivozanib,
a highly potent and selective inhibitor of VEGF receptor tyrosine
kinases, for the treatment of metastatic renal cell carcinoma.
Future Oncol. 16:2147–2164. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hrisomalos FN, Maturi RK and Pata V:
Long-term use of intravitreal bevacizumab (avastin) for the
treatment of von hippel-lindau associated retinal
hemangioblastomas. Open Ophthalmol J. 4:66–69. 2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|