Introduction
Waldenström's macroglobulinemia (WM), also
recognized as lymphoplasmacytic lymphoma (LPL), represents a rare
mature B-cell proliferative disorder, accounting for <2% of all
non-Hodgkin lymphomas (1,2). WM is composed of small B lymphocytes,
plasma cell-like lymphocytes and plasma cells, which produce a
large amount of monoclonal immunoglobulin M (IgM) in the bone
marrow, leading to increased blood viscosity and potentially
causing various symptoms and complications (3,4). The
diagnosis of WM usually requires a combination of clinical
symptoms, hematological tests and bone marrow biochemical analysis
(5,6). The common symptoms of WM are anemia,
thrombocytopenia, elevated serum IgM levels and increased blood
viscosity. The treatment options for WM include chimeric antigen
receptor-T cell therapy, chemotherapy, targeted therapy [such as
Bruton's tyrosine kinase (BTK) inhibitors, BCL-2 inhibitors],
immunomodulators and monoclonal antibodies (7-9).
The common therapeutic drugs for WM are chemotherapy drugs (such as
bortezomib, cyclophosphamide), immunomodulators (such as rituximab)
and proteasome inhibitors (such as thalidomide) (10). The median age at diagnosis for WM
is ~71 years with the median survival associated with this disease
being >10 years (11,12). Patients diagnosed with WM have a
heightened risk of developing amyloidosis, including the light
chain amyloidosis (AL) type driven by light chain proteins and the
transthyretin amyloidosis (ATTR) type precipitated by abnormal
transthyretin protein synthesis (13,14).
Notably, 10% of amyloidosis cases associated with WM present as the
AL type, implicating multiple organ systems and leading to
significant morbidity (13). This
comprehensive study explores and reviews the diagnosis and
treatment methods for a patient with cardiac AL amyloidosis
secondary to WM.
Case report
Basic information
A 67-year-old male patient presented to Luohu
People's Hospital of Shenzhen (Shenzhen, China) in August 2021 with
the chief complaints of chest tightness, nocturnal dyspnea and a
paroxysmal cough persisting for half a month. The patient had no
family history of hematologic disorders or amyloidosis. Past
medical history included hypertension (controlled) and no
psychosocial risk factors. Between January 2019 and July 2021, the
patient had sought medical attention at an external hospital
(Shenzhen People's Hospital, Shenzhen, China) due to chest
tightness and nocturnal dyspnea and was diagnosed with ‘chronic
heart failure’. Prior interventions for heart failure in 2019
included diuretics, spironolactone tablets, furosemide tablets and
sacubitril valsartan sodium tablets for 2 years, which provided
transient symptom relief but no improvement in cardiac function.
The patient suffered from the symptoms of chest tightness and
orthopnoea at night, accompanied by paroxysmal cough, as well as
numbness in the toes.
The patient also had symptoms such as difficulty
breathing, edema, fatigue and atrial fibrillation in chronic heart
failure. However, the patient's symptoms of chronic heart failure
were different from those caused by traditional coronary heart
disease and hypertension. The patient presented with the following:
i) Postural hypotension and diastolic dysfunction; ii) special
cardiac signs: a) Thickened ventricular wall but relatively
preserved systolic function (ultrasound showed ‘granular’
myocardial echoes); b) low voltage on electrocardiogram (related to
myocardial amyloid infiltration); iii) multiple systemic
involvement manifestations (systemic deposition of amyloid
protein): Peripheral neuropathy; numbness and pain in hands and
feet; renal dysfunction and elevated creatinine levels; iv) poor
response to traditional heart failure treatment: According to
disease feedback, diuretics have limited efficacy in treating
current patients Beta blockers and angiotensin-converting enzyme
inhibitors are also not suitable as they can exacerbate
hypotension. The main pathological manifestation of traditional
chronic heart failure is myocardial systolic and diastolic
dysfunction (13). Cardiac
amyloidosis is an infiltrative cardiomyopathy and mainly
characterized by amyloid deposition leading to myocardial stiffness
and affecting diastolic function (14). Therefore, examinations for cardiac
amyloidosis were necessary.
Laboratory tests
The blood routine examination indicated a white
blood cell count of 5.68x109/l, platelet count of
184x109/l and hemoglobin concentration of 136 g/l, all
within normal ranges. Cardiac enzyme tests revealed cardiac stress
with a creatine kinase level of 48 U/l and an N-terminal pro-brain
natriuretic peptide II level of 8,288 pg/ml. Cardiac infarction
markers indicated myocardial injury with a troponin T level of
0.154 ng/ml and a myoglobin level of 96 ng/ml. The renal function
panel showed a creatinine level of 256 µmol/l, a uric acid level of
521 µmol/l and a β2-microglobulin concentration of 14.7 mg/l,
indicating impaired renal function. The Ig profile showed abnormal
levels with results of IgG 19.5 g/l, IgA 0.60 g/l and IgM 9.98 g/l
(Table I). Serum protein
electrophoresis (Fig. S1;
Data SI; Table SI) revealed albumin (ALB) at
49.2%, β1 globulin (β1G) decreased to 4.5% and β2G at 19.2%.
Immunofixation electrophoresis (Data
SI; Table SII) showed
positive IgM-λ type M protein, an erythrocyte sedimentation rate of
59.0 mm/h, negative Bence Jones protein and a free light chain
ratio (κ/λ ratio) of 0.26, with free κ and λ light chains at 132
and 507 mg/l, respectively. The urine analysis showed no protein
and the 24-h urine protein measurement was 93.6 mg, which was
within the normal range. The electrocardiogram showed low voltage
in the limb leads, suggesting cardiac abnormalities (Fig. S2). Genetic mutation detection
(Fig. S3; Data S1; Table SIII) was tested by droplet digital
polymerase chain reaction based on droplet separation and
single-molecule amplification techniques, which identified the
presence of MYD88 innate immune signal transduction adaptor (MYD88)
with positive L265P mutation with a mutation frequency of 2.89%.
Noteworthy abnormalities included cardiac stress, myocardial
injury, impaired renal function, abnormal Ig levels, positive IgM-λ
type M protein, significant proteinuria and low voltage in the limb
leads.
 | Table ILaboratory test results of the
patient. |
Table I
Laboratory test results of the
patient.
| Examination
items | Test values | Normal range |
|---|
| Blood cell count,
x109/l | 5.68 | 3.5-9.5 |
| Platelet count,
x109/l | 184 | 125-350 |
| Hemoglobin
concentration, g/l | 136 | 130-175 |
| N-terminal
pro-brain natriuretic peptide II, pg/ml | 8,288 | <125 |
| Troponin T level,
ng/ml | 0.154 | <0.014 |
| Myoglobin level,
ng/ml | 96 | 28-72 |
| Creatinine level,
µmol/l | 256 | 57-111 |
| Uric acid level,
µmol/l | 521 | 208-428 |
| Creatine kinase
level, U/l | 48 | 50-310 |
| β2-microglobulin
concentration, mg/l | 14.7 | 0.8-2.2 |
| Immunoglobulin IgG,
g/l | 19.5 | 8.6-17.4 |
| Immunoglobulin IgA,
g/l | 0.60 | 1.0-4.2 |
| Immunoglobulin IgM,
g/l | 9.98 | 0.3-2.2 |
| Erythrocyte
sedimentation rate, mm/h | 59.0 | <43.5 |
| Serum light chain
ratio (κ/λ) | 0.26 | 0.31-1.56 |
| Serum light chains
κ, mg/l | 132 | 6.7-22.4 |
| Serum light chains
λ, mg/l | 507 | 8.3-27.0 |
| 24-h urine protein,
mg | 93.6 | <150 |
Pathological examination
The echocardiography images (Fig. 1A and B) revealed left ventricular posterior
wall hypertrophy with a thickness of 15.6 mm and interventricular
septum of bilateral atrial enlargement with a thickness of 13.3 mm,
which were both higher than normal values (both <12 mm). The
thickened myocardium exhibited scattered sparkling granular echoes
and a small amount of pericardial effusion. The heart valves were
thick and rough and regurgitated. Fig.
1B shows that the left ventricular fractional shortening ratio
was 22%, which is lower than the normal value (25-35%). The
broadened main pulmonary artery diameter resulted in high pulmonary
artery pressure. Fig. 1C shows
that the left ventricular ejection fraction value of cardiac
function was 44.3%, which is lower than the normal value (>50%).
Fig. 1D shows that the left
ventricular wall motion amplitude was slightly lower than normal,
with a peak value of 61.6 cm/sec (normal range, 67-109 cm/sec). The
aforementioned findings suggested that the left ventricular
diastolic and systolic function were reduced, resulting in mild
heart failure.
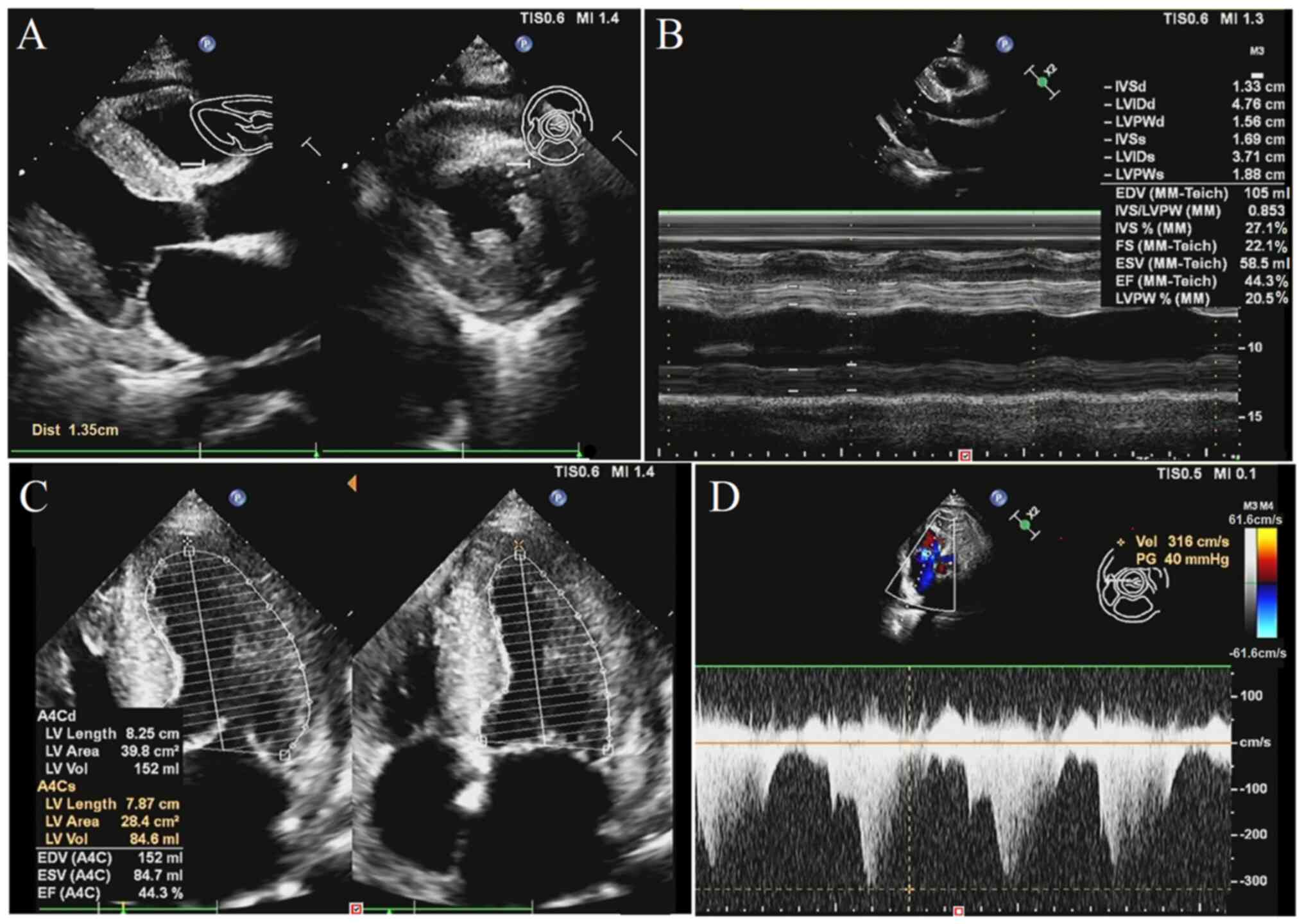 | Figure 1Echocardiography images. (A) LV (left
ventricular) posterior wall hypertrophy and thickened bilateral
atrial. (B) Various cardiac parameters with ultrasonography
measurements. (C) Low LV ejection fraction value with decreasing
blood-pumping function. (D) Decrease of LV systolic function.
Cardiac echocardiography demonstrating LV hypertrophy (16 mm),
sparkling granular echoes (amyloid deposition) and pericardial
effusion. LV, left ventricle; IVSd, interventricular septum
thickness in diastole; LVIDd, left ventricular internal diameter in
diastole; LVPWd, left ventricular posterior wall thickness in
diastole; IVSs, interventricular septum thickness in systole;
LVIDs, left ventricular internal diameter in systole; LVPWs, left
ventricular posterior wall thickness in systole; EDV, end-diastolic
volume; MM-Teich, M-mode Teichholz formula; IVS, interventricular
septum thickness; LVPW, left ventricular posterior wall thickness;
FS, fractional shortening; ESV, end-systolic volume; EF, ejection
fraction; LV, left ventricular; A4C, apical four-chamber view;
A4Cd/A4Cs, apical four-chamber view in diastole/systole; Vol,
volume; TIS, thermal index for soft tissue; MI, mechanical index;
Dist, distance. |
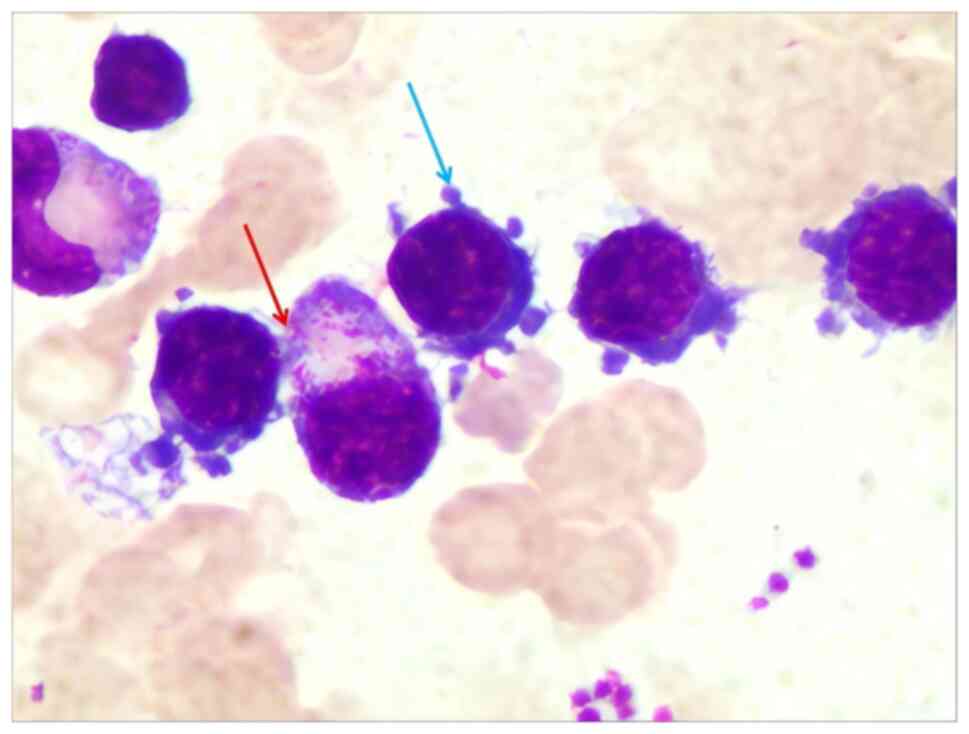
The bone marrow morphology (Data SI; Table SIV) revealed active proliferation
with an increased proportion of lymphocytes, as shown in Fig. 2. Certain lymphocytes exhibited
plasma-like differentiation, while lymphocytes and plasma-like
lymphocytes exhibited focal aggregation. The lymphocytes were
predominantly mature, with ~10% identified as abnormal lymphocytes.
The abnormal lymphocytes had a small size, dense nuclear chromatin
and a small cell mass, and part of the cell cytoplasm exhibited
blue, foam-like, plasma cell-like changes or irregular cytoplasm
edges with burrs. The proportion and morphology of erythrocytes
were normal. Platelets were distributed in single or small clusters
and were easily visible with a small number of abnormal platelets.
The proportion of plasma cells was about 4%, which were mature
plasma cells.
The flow cytometry of bone marrow (Data SI; Table SV) results showed that lymphocytes
constituted 13.72% of nucleated cells (normal range, 10-20%), with
T lymphocytes comprising 49.69% (normal range, 60-80%). The
CD4+/CD8+ ratio was 0.37 (normal range, 1.4-2.0), reflecting a
decreased ratio, yet no discernible phenotypic abnormalities were
apparent. Among the lymphocytes, the proportion of CD19+ B cells
was 32.78% (normal range, 5-15%; Fig.
S4), exhibiting a phenotype characterized by CD19+, CD20+,
Lambda++, Kappa-, CD5-, CD10-, CD3-, GD4-, CD8-, CD2- and CD7-.
Granulocytes accounted for 68.96% (normal range, 50-60%), with a
proportion within the normal range, and no notable abnormalities
were observed in the proportion and phenotype of granulocytes at
various stages. The CD34+ and CD117+ marrow cells had a low
proportion of 0.47% (normal value, <1%), and no apparent
phenotypic abnormalities were observed. Notably, abnormal mature B
cells, displaying immunophenotypic abnormalities, made up ~4.94%
(normal value, 0%) of nucleated cells. B-cell lymphoma excluding
CD5- and CD10-could not be conclusively ruled out, necessitating
further investigation to establish a definitive diagnosis.
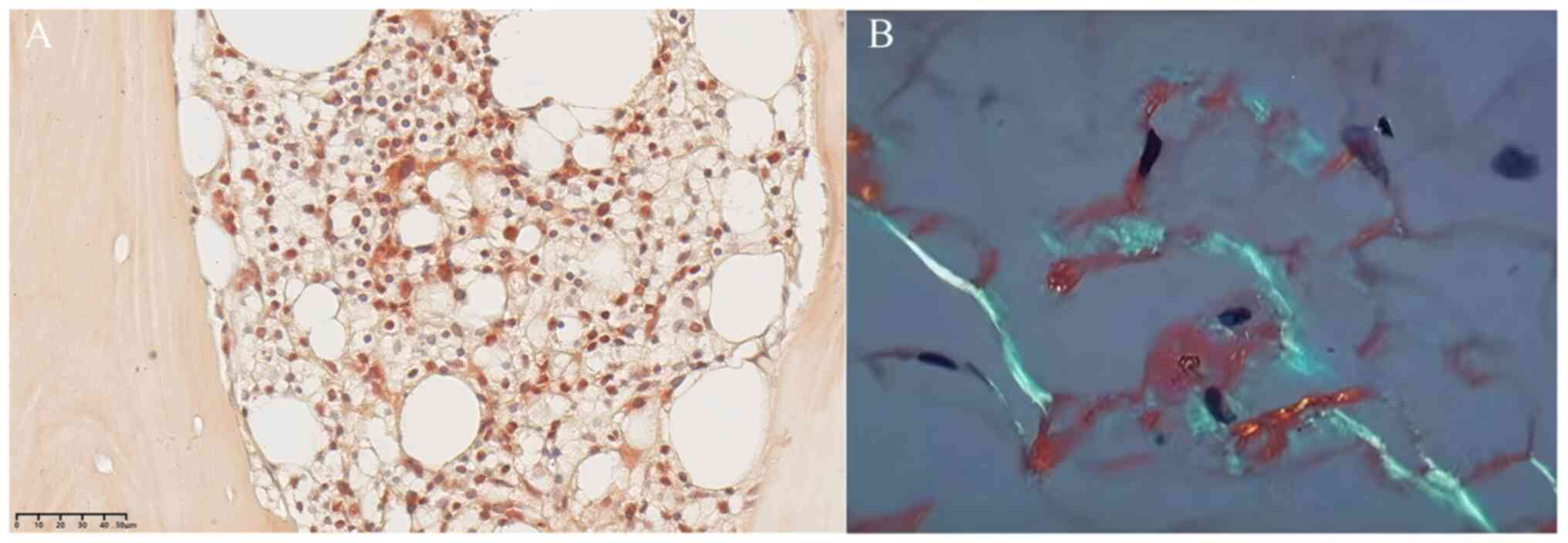
Fig. 3A shows the
bone marrow biopsy with immunohistochemistry and Congo red staining
(Data S1), which was completed
through biopsy of the posterior superior iliac spine. The results
indicate that the proportion of red bone marrow decreased, the
proportion of granulocytes, red cells and megakaryocytes in the red
pulp was normal, and the morphology of each cell was relatively
mature, with a few scattered plasma cells. In typical amyloidosis
cases, amyloid substance is an amorphous extracellular eosinophilic
substance, which is commonly identified by the normal Congo red
staining method because with other staining, it is difficult to
distinguish colors. The Congo red staining was positive, displaying
in orange red, which indicated the existence of amyloid substance.
Amyloid protein mass spectrometry was performed on abdominal fat
biopsy (Congo red-positive) with 84% sensitivity (15), which is a recommendation surrogate
biopsy tissue for most patients, and confirming AL type with
predominant Ig λ C2 spectra (12%). Cardiac tissue biopsy was not
feasible due to the patient's unstable condition. Fig. 3B shows the biopsy of abdominal wall
fat tissue with Congo red staining; an apple green color
originating from amyloid substance with double refraction was
visible under a polarized light microscope, which was transformed
through the amyloid substance. Therefore, the Congo red staining
with orange red and apple green color transformation both confirmed
cardiac amyloidosis in this case.
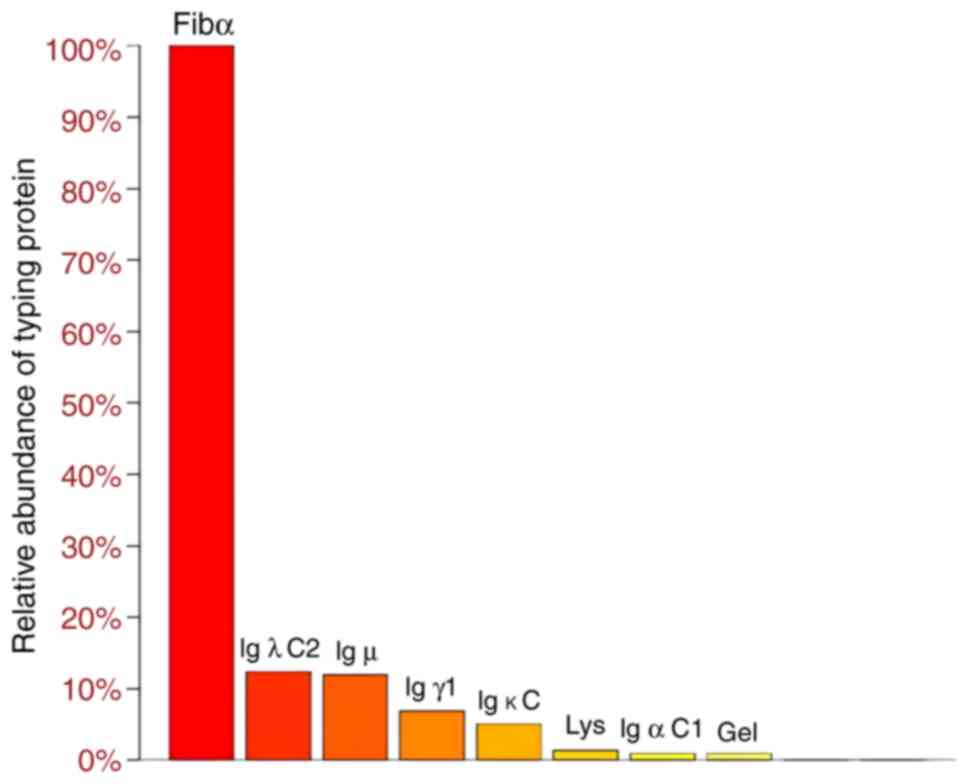
The amyloid protein mass spectrometry spectrum of
abdominal adipose tissue was used to determine the amyloidosis type
(Data S1), as shown in Fig. 4. Fibrinogen α chain (Fibα), the Ig
light chain constant (Igλ C2), the Ig heavy chains Igµ, Igγ1 and
Igα C1, Ig light chain κ C, lysozyme (lys) and gelsolin (gel) were
detected. Among the currently known sub-typing proteins in Fig. 4, Fibα has the highest relative
abundance in spectra, but Fibα<β+γ indicates that Fibα comes
from blood contamination (α 218, β 294, γ 188) and is not a
sedimentary protein (16).
Compared with heavy chains, light chains (especially λ type) are
more likely to form amyloid fibrils due to their genetic sequence
and high secretion characteristics (13). Thus, light chains are more prone to
deposit in the cardiac position, leading to cardiac amyloidosis.
Excluding Fibα, among other typing proteins, Igλ C2 had the highest
relative abundance in the spectrum, κ and λ values are
significantly higher than the normal range (Table I), indicating that the main type is
AL amyloidosis. Therefore, based on the positive Congo red staining
and abdominal adipose tissue mass spectrometry analysis, the
diagnosis was cardiac amyloidosis of the AL type.
Diagnosis and treatment process
Based on the comprehensive characteristics of the
case and auxiliary examination results, the diagnosis aligns with
the criteria for LPL/WM, IgM-λ type, high-risk, accompanied by
cardiac AL amyloidosis. The patient's prior diagnosis of chronic
heart failure made in 2019 was attributed to non-specific causes,
and no amyloidosis or hematologic malignancy was suspected until
2021, which highlights the importance of considering amyloidosis in
refractory heart failure. The diagnosis of WM was confirmed through
abnormal IgM (9.98 g/l) levels, λ-free light chains (507 mg/l), the
existence of small B lymphocytes, plasma cell-like lymphocytes,
plasma cells, M protein and MYD88 with L265P mutation. The cardiac
amyloidosis was identified through cardiac biomarkers (N-terminal
pro-B-type natriuretic peptide: 8,288 pg/ml), cardiac
echocardiography (sparkling echoes), positive Congo red staining of
bone marrow and apple green color transformation of abdominal fat
biopsy, and mass spectrometry peaks of Ig λ C2 and µ heavy chain as
the dominant amyloid protein with AL type.
In August 2021, the patient initiated the BR
regimen, which consisted of Bendamustine (0.15 g intravenously on
days 2-3) and Rituximab (0.6 g intravenously on day 1), following a
28-day chemotherapy cycle. A second course of the BR regimen was
administered in September 2021, maintaining the same regimen and
dosage as the initial course. The disease was subsequently
evaluated as stable disease. In October 2021, the patient underwent
a third-line treatment with the BRD regimen. This regimen included
subcutaneous injections of 2 mg bortezomib on days 1, 8, 15 and 22,
intravenous infusion of 0.5 g rituximab on the same days, and
intravenous infusion of 10 mg dexamethasone on days 1, 8, 15 and
22, with treatments administered once a week within a 28-day
chemotherapy cycle. After the treatment, the patient was determined
to show a partial response (PR). The patient adhered to treatment
with no adverse events and at the six-month follow-up, the patient
showed sustained hematological PR but progressive diastolic
dysfunction on echocardiography. The patient voluntarily left the
hospital due to economic difficulties. The PR was mainly based on
hematological criteria: >50% reduction in serum IgM (from 9.98
to 4.79 g/l), >50% reduction in the difference of serum free
light chain (dFLC) and a decrease of cardiac injury markers
(troponin T: 0.119 ng/ml). Cardiac function (ejection faction: 50%)
remained stable but did not improve due to irreversible amyloid
deposition. The follow-up was terminated as the patient did not
return to the hospital for treatment after discharge.
Discussion
Cardiac AL amyloidosis secondary to WM presents
diverse clinical manifestations and treatment complexities.
According to the Chinese Society of Clinical Oncology, the
‘Diagnosis and Treatment of LPL/WM Chinese Expert Consensus (2022
version)’ outlines the diagnostic criteria for WM (17). The diagnostic markers of WM are
small B lymphocytes, plasma cell-like lymphocytes and plasma cells,
as well as high concentrations of IgM. The diagnostic markers of
amyloidosis are positive Congo red staining of the fat tissue
biopsy and apple green color transformation. The diagnosis requires
exclusion of other lymphomas and the presence of the MYD88 with
L265P mutation, found in >90% of WM cases (18). Herein, the genetic mutation
detection of this case identified the presence of MYD88 with
positive L265P mutation type.
At present, the specific mechanisms of WM are not
fully understood, but studies have confirmed that genetic
alterations may occur. García-Sanz et al (19) studied the clonality of tumor
populations in WM and how clonal complexity can evolve and impact
disease progression. Single-cell transcriptomics analysis also
indicated that MYD88 could be an early event in tumorigenesis
(20). In addition, X-box binding
protein 1 (XBP1)-endoplasmic reticulum to nucleus signaling 1α
(ERN1α) may play a role in the pathogenesis of WM because about 80%
of patients have high XBP1 splicing mRNA expression, with 80%
showing high mRNA ERN1α expression (21). The WM tumor progression is closely
related to the bone marrow microenvironment and cytokines (22).
A study that included 20 years of data (1988 to
2007) from the Surveillance, Epidemiology and End Results (SEER)
program showed that the median age at diagnosis for WM was 73
years, with an overall age-adjusted incidence rate of 0.38/100,000
per year, increasing to 2.85 among patients older than 80 years
(23). Risk factors for WM
prognosis include age (>65 years), elevated β-2 microglobulin,
organomegaly, anemia, thrombocytopenia, low ALB, high serum
monoclonal protein and high serum free light chain concentration.
Recent research indicates that the median age at diagnosis for WM
is ~71 years (12). Previous
studies indicated that the median survival of patients with WM
under the age of 70 is in excess of 10 years, that of patients aged
70-79 years is ~7 years and the median survival of patients aged 80
years or above is nearly 4 years (8,12).
The incidence rate of WM is ~24,000 individuals every year
globally, and the male-to-female ratio is 3.2:1. One study based on
5,784 patients with WM in the SEER database indicated that the
median overall survival (OS) improved from 6 to 8 years from 1991
to 2010(24). Another study
determined that the median survival after treatment initiation was
87 months among 587 patients with WM (25).
The median diagnostic age of cardiac amyloidosis was
determined to be >60 years (26). The median survival of cardiac ATTR
and AL amyloidosis was determined to be 2-5 years and the OS at 2
years was 63% for AL and 98% for ATTR from diagnosis, indicating
that the prognosis of cardiac ATTR amyloidosis is better than that
of cardiac AL amyloidosis (27).
The median survival of patients with AL amyloidosis associated with
WM was reported to be in the range of 49-78 months, which was not
significantly different compared with patients without IgM, as
survival is independently affected by cardiac involvement (28-30).
WM may lead to various complications, including
cardiac amyloidosis, and the heart is easily affected by
amyloidosis secondary to WM. Among patients with WM in SEER
database, most subjects were found to present with symptoms related
to tumor burden, while only ~10% of patients concurrently suffer
from AL amyloidosis (12,13). Cardiac amyloidosis is an uncommon
cardiopathy and characterized by the deposition of insoluble
amyloid proteins within cardiac tissues. The mechanism of
amyloidosis secondary to WM is that abnormal production and
deposition of monoclonal IgM may cause cardiac amyloidosis,
affecting cardiac structure and function and resulting in heart
failure (13). The definition, the
diagnostic criteria, criteria for initiation of therapy and a new
classification scheme of amyloidosis and WM were established in
2004(31). A study of 50 patients
with serum IgM monoclonal gammopathy showed that 53% of the
patients died of cardiac amyloidosis, while 32, 14 and 10% of
patients also had renal, liver and lung amyloidosis, respectively
(32). IgM-related amyloidosis
accounts for only 6-10% in patients with various Ig-related
amyloidoses, and 54% of cases among them were associated with
underlying non-Hodgkin's lymphoma, including WM (33,34).
A test of the MYD88 mutation status is necessary for the evaluation
of IgM-related AL, as these mutations are distinct features of WM
(35). Gustine et al
(36) studied consecutive patients
from 2006 to 2022; all patients were diagnosed with WM-AL
amyloidosis through the criteria of the presence of IgM
paraprotein, bone marrow infiltration by lymphoplasmacytic lymphoma
and positive Congo red staining of a biopsy specimen. The median OS
was 7.3 years in 49 patients with WM-AL amyloidosis. Patients with
WM with cardiac amyloidosis may be misdiagnosed as either AL or
ATTR amyloidosis by immunohistochemistry and can be correctly
identified by mass spectrometry (37). The reported patients with WM-ATTR
amyloidosis had a lower dFLC than patients with AL. In the case of
the present study, a delay in diagnosis occurred due to overlapping
symptoms with heart failure and the rarity of WM-associated AL
amyloidosis.
WM secondary to cardiac amyloidosis is controlled
and treated through chemotherapy, immunotherapy and targeted
therapy, and a high level of autologous stem cell transplantation.
The management of cardiac symptoms requires specific interventions
for heart failure, arrhythmias and conduction abnormalities.
First-line treatment options for symptomatic patients with WM
include Rituximab-chemotherapy and BTK inhibitors, such as
ibrutinib, either alone or combined with rituximab and zanubrutinib
(13,36). For patients with WM-AL amyloidosis,
the most common salvage therapy method is a bortezomib- and/or
bendamustine-based regimen (36).
BR is preferred for rapid tumor reduction or high tumor burden,
showing potential for longer progression-free survival. The choice
of BTK inhibitors should consider the MYD88 L265P mutation, as the
wild-type shows the best response and the mutated type has a poor
response and may benefit from added rituximab (38). The patient of the present study
carried the MYD88 L265P mutation, which was treated with the BR and
BRD regimen rather than BTK inhibitors. Compared with Bendamustine,
proteasome inhibitors such as bortezomib are not first-line
medications but may be used in resistant cases to target and kill
tumor cells. The Bendamustine is more expensive than bortezomib,
and thus, the patient preferred the BRD regimen; however, both the
BR and BRD regimens failed to achieve the expected effect with only
a PR. AL amyloidosis is one of the most dangerous disorders related
to the M protein, which is better treated before irreversible organ
impairment has occurred (28,39).
In the present study, the patient with WM-AL amyloidosis failed to
respond to the BR and BRD regimens, presumably because he was
diagnosed with chronic heart failure at an external hospital in
2019, but was diagnosed as WM accompanied by cardiac AL amyloidosis
in 2021. The irreversible organ impairment caused by WM-AL
amyloidosis may have occurred before therapy at our hospital.
Numerous specific cases of WM accompanied by cardiac
amyloidosis have been reported in the literature. A 70-year-old
male was diagnosed as WM with AL amyloidosis, was then started on
bortezomib, cyclophosphamide, dexamethasone and rituximab, and
after three cycles of therapy, IgM had decreased to 1,500 mg/dl
with improved performance status (13). A 52-year-old male was diagnosed
with WM accompanied by AL amyloidosis and the rituximab and
bortezomib-based chemotherapy was ineffective (40). A 76-year-old African American male
with chronic kidney disease was diagnosed with WM accompanied by
ATTR amyloidosis, and after 12 months of tafamidis-based
chemotherapy, cardiac and hematological conditions were stable
(41). An 80-year-old female
hospitalized for chronic dyspnea and cough was diagnosed with λ
light chain and µ heavy chain type amyloidosis; the patient was not
suited for chemotherapy or immunotherapy due to severe heart
failure and hypotension (42). The
previous study demonstrated that WM-AL amyloidosis can also be
treated with the standard WM regimens, such as bortezomib,
dexamethasone and rituximab or bendamustine and rituximab (36). High-dose melphalan and stem cell
transplantation (HDM/SCT) should be a new option for patients with
WM-AL amyloidosis, as HDM/SCT-based therapy can prolong survival to
>20 years in typical AL amyloidosis (43).
In conclusion, a case was diagnosed with WM
accompanied by cardiac AL amyloidosis based on the presence of
abnormal IgM paraprotein levels and the high Ig λ C2 spectrum in
the amyloid protein mass spectrometry. The novelty of this case
lies in the rare cardiac AL amyloidosis secondary to WM,
particularly the diagnostic challenges and the limited treatment
response due to irreversible cardiac damage. The BR and BRD-based
therapy only yielded a PR and the symptom of chronic heart failure
due to irreversible organ impairment had occurred before treatment
at our hospital. This result indicates that WM secondary to cardiac
amyloidosis needs to be diagnosed and treated as early as possible
by inspecting IgM levels, amyloid protein mass spectrometry,
genetic mutation analysis and pathological examination of tissues
before the irreversible organ impairment at the site of the heart.
In addition, the mechanisms, treatment methods and survival of WM,
ATTR or AL amyloidosis and WM-AL amyloidosis were reviewed in this
paper. The complexities of WM and its secondary complications such
as cardiac amyloidosis highlight the need for ongoing research and
collaboration. Integrating advanced diagnostic techniques and
personalized treatment strategies will improve the management of
this disease and increase patient quality of life.
Supplementary Material
Supplementary methods
Test result of serum protein
electrophoresis. The peak on the left represents ALB protein and
the peak on the right represents γ globulin. The appearance of
steep ‘peaks’ in the right elecrophoretic bands indicates the
presence of abnormally increased immunoglobulin. Ref. Conc.,
reference concentration΄ ALB, albumin.
Low voltage in the limb leads in the
electrocardiogram.
Gene mutation detection by droplet
digital polymerase chain reaction. The blue dots represent the
MYD88 mutant type. The green dots represent MYD88 wild-type (no
mutation). Orange dots represent a mixed state of mutant and
wild-type (such as chimeric mutations or low abundance mutations).
The black dots represent the background noise.
Flow cytometry results. The antibody
labeling method is the five color method. The antibodies include
CD19-PC5.5 for recognizing B cells, and CD20-APC for recognizing
mature B cells. The gated area includes CD19+ and CD20+ cells (pink
area), representing a subset of B cells that simultaneously express
CD19 and CD20. The green area on the left represents CD19 low
expression or negative cells. The 4.94% is the proportion of CD19+
and CD20+ in the total lymphocytes based on upstream lymphocyte
gating. The 32.78% represents the proportion of CD19+ and CD20+ in
CD19+ cells in this case.
Test results of serum protein
electrophoresis.
Test results of immunofixation
electrophoresis.
Test results of genetic mutation
detection.
Test results of bone marrow histology
morphology.
Test results of bone marrow histology
morphology.
Acknowledgements
The authors thank Miss Shuaiyang Wang
(technologist-in-charge) and Miss Hongmei Mo (associate senior
technologist) of the Clinical Laboratory of Luohu People's Hospital
of Shenzhen (Shenzhen, China), and Miss Huizhi Guo (attending
physician) of the Department of Ultrasound of Luohu People's
Hospital of Shenzhen for providing the examination reports of the
current patient.
Funding
Funding: This research was funded by Sanming Project of Medicine
in Shenzhen (grant no. SZSM202301035).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FD was responsible for the case diagnosis and
treatment, as well as writing and analysis of the manuscript. AG
contributed to the data analysis and collection. LZ analyzed
patient data, DL provided treatment advice, JC collected image
data, HX collected pathological data, WL collected ultrasound data,
JL collected flow cytometry of bone marrow data and YL collected
the amyloid protein mass spectrometry spectrum data. HC was
responsible for study conception and revisions to the manuscript.
All authors have read and approved the final manuscript. FD and HC
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
This study received approval from the Shenzhen Luohu
People's Hospital Research Ethics Committee (grant no.
2024-LHQRMYY-KYLL-26). The protocol and informed consent form were
reviewed and approved on May 12, 2024, with a subsequent simplified
review conducted on May 23, 2024. The study adheres to ethical
standards as outlined by the committee. The patient provided
written informed consent for participation.
Patient consent for publication
The patient provided written informed consent for
publication of their data, including case information and
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Olszewski AJ, Treon SP and Castillo JJ:
Evolution of management and outcomes in Waldenström
macroglobulinemia: A population-based analysis. Oncologist.
21:1377–1386. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vijay A and Gertz MA: Waldenström
macroglobulinemia. Blood. 109:5096–5103. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zheng L, Zeng Z, Lin J and Chen J: IgM
multiple myeloma presenting with bleeding tendency: A case report
with immunophenotype analysis. Oncol Lett. 2:55–57. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang Q, Liu Q, Liang H and Gao W: Biclonal
lymphoplasmacytic lymphoma/Waldenström macroglobulinemia associated
with POEMS syndrome: A case report and literature review. Oncol
Lett. 25(97)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Dimopoulos MA, Kyle RA, Anagnostopoulos A
and Treon SP: Diagnosis and management of Waldenstrom's
macroglobulinemia. J Clin Oncol. 23:1564–1577. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dimopoulos MA, Merlini G, Leblond V,
Anagnostopoulos A and Alexanian R: How we treat Waldenstrom's
macroglobulinemia. Haematologica. 90:117–125. 2005.PubMed/NCBI
|
|
8
|
Oza A and Rajkumar SV: Waldenstrom
macroglobulinemia: Prognosis and management. Blood Cancer J.
5(e391)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Grunenberg A and Buske C: How to manage
waldenström's macroglobulinemia in 2024. Cancer Treat Rev.
125(102715)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Castillo JJ and Treon SP: What is new in
the treatment of Waldenstrom macroglobulinemia? Leukemia.
33:2555–2562. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hodge LS and Ansell SM: Waldenström's
macroglobulinemia: Treatment approaches for newly diagnosed and
relapsed disease. Transfus Apher Sci. 49:19–23. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gertz MA: Waldenström macroglobulinemia:
2023 Update on diagnosis, risk stratification, and management. Am J
Hematol. 98:348–358. 2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lu R and Richards T: A focus on
Waldenström macroglobulinemia and AL amyloidosis. J Adv Pract
Oncol. 13 (Suppl 4):S45–S56. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Merlini G, Sarosiek S, Benevolo G, Cao X,
Dimopoulos M, Garcia-Sanz R, Gatt ME, Fernandez de Larrea C,
San-Miguel J, Treon SP and Minnema MC: Report of consensus panel 6
from the 11 th international workshop on Waldenström's
macroglobulinemia on management of Waldenström's macroglobulinemia
related amyloidosis. Semin Hematol. 60:113–117. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rubin J and Maurer MS: Cardiac
amyloidosis: Overlooked, underappreciated, and treatable. Annu Rev
Med. 71:203–219. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sethi S, Vrana JA, Theis JD, Leung N,
Sethi A, Nasr SH, Fervenza FC, Cornell LD, Fidler ME and Dogan A:
Laser microdissection and mass spectrometry-based proteomics aids
the diagnosis and typing of renal amyloidosis. Kidney Int.
82:226–234. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yi S and Li J: Chinese expert consensus on
diagnosis and treatment of lymphatic plasma cell
lymphoma/fahrenheit megaglobulinemia (2022 edition). Chin J
Hematol. 43:624–630. 2022.(In Chinese).
|
|
18
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J
Med. 367:826–833. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
García-Sanz R, García-Álvarez M, Medina A,
Askari E, González-Calle V, Casanova M, de la Torre-Loizaga I,
Escalante-Barrigón F, Bastos-Boente M, Bárez A, et al: Clonal
architecture and evolutionary history of Waldenström's
macroglobulinemia at the single-cell level. Dis Model Mech.
16(dmm050227)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Qiu Y, Wang XS, Yao Y, Si YM, Wang XZ, Jia
MN, Zhou DB, Yu J, Cao XX and Li J: Single-cell transcriptome
analysis reveals stem cell-like subsets in the progression of
Waldenström's macroglobulinemia. Exp Hematol Oncol.
12(18)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Leleu X, Hunter ZR, Xu L, Roccaro AM,
Moreau AS, Santos DD, Hatjiharissi E, Bakthavachalam V, Adamia S,
Ho AW, et al: Expression of regulatory genes for lymphoplasmacytic
cell differentiation in Waldenstrom macroglobulinemia. Br J
Haematol. 145:59–63. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Boutilier AJ, Huang L and Elsawa SF:
Waldenström macroglobulinemia: Mechanisms of disease progression
and current therapies. Int J Mol Sci. 23(11145)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang H, Chen Y, Li F, Delasalle K, Wang J,
Alexanian R, Kwak L, Rustveld L, Du XL and Wang M: Temporal and
geographic variations of Waldenstrom macroglobulinemia incidence: A
large population-based study. Cancer. 118:3793–3800.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Castillo JJ, Olszewski AJ, Kanan S, Meid
K, Hunter ZR and Treon SP: Overall survival and competing risks of
death in patients with Waldenström macroglobulinaemia: An analysis
of the surveillance, epidemiology and end results database. Br J
Haematol. 169:81–89. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Morel P, Duhamel A, Gobbi P, Dimopoulos
MA, Dhodapkar MV, McCoy J, Crowley J, Ocio EM, Garcia-Sanz R, Treon
SP, et al: International prognostic scoring system for Waldenstrom
macroglobulinemia. Blood. 113:4163–4170. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Aimo A, Merlo M, Porcari A, Georgiopoulos
G, Pagura L, Vergaro G, Sinagra G, Emdin M and Rapezz C: Redefining
the epidemiology of cardiac amyloidosis. A systematic review and
meta-analysis of screening studies. Eur J Heart Fail. 24:2342–2351.
2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rapezzi C, Merlini G, Quarta CC, Riva L,
Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E,
et al: Systemic cardiac amyloidoses: Disease profiles and clinical
courses of the 3 main types. Circulation. 120:1203–1212.
2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Palladini G and Merlini G: Diagnostic
challenges of amyloidosis in Waldenström macroglobulinemia. Clin
Lymphoma Myeloma Leuk. 13:244–246. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wechalekar AD, Lachmann HJ, Goodman HJ,
Bradwell A, Hawkins PN and Gillmore JD: AL amyloidosis associated
with IgM paraproteinemia: Clinical profile and treatment outcome.
Blood. 112:4009–4016. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Palladini G, Russo P, Bosoni T, Sarais G,
Lavatelli F, Foli A, Bragotti LZ, Perfetti V, Obici L, Bergesio F,
et al: AL amyloidosis associated with IgM monoclonal protein: A
distinct clinical entity. Clin Lymphoma Myeloma. 9:80–83.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gertz MA, Merlini G and Treon SP:
Amyloidosis and Waldenström's Macroglobulinemia. Hematology Am Soc
Hematol Educ Program. 2004:257–282. 2004.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gertz MA, Kyle RA and Noel P: Primary
systemic amyloidosis: A rare complication of immunoglobulin M
monoclonal gammopathies and Waldenström's macroglobulinemia. J Clin
Oncol. 11:914–920. 1993.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Bou Zerdan M, Valent J, Diacovo MJ, Theil
K and Chaulagain CP: Utility of Bruton's tyrosine kinase inhibitors
in light chain amyloidosis caused by lymphoplasmacytic lymphoma
(Waldenström's macroglobulinemia). Adv Adv Hematol.
2022(1182384)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zanwar S, Abeykoon JP, Ansell SM, Gertz
MA, Dispenzieri A, Muchtar E, Sidana S, Tandon N, Rajkumar SV,
Dingli D, et al: Primary systemic amyloidosis in patients with
Waldenstrom macroglobulinemia. Leukemia. 33:790–794.
2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sidana S, Larson DP, Greipp PT, He R,
McPhail ED, Dispenzieri A, Murray DL, Dasari S, Ansell SM, Muchtar
E, et al: IgM AL amyloidosis: Delineating disease biology and
outcomes with clinical, genomic and bone marrow morphological
features. Leukemia. 34:1373–1382. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gustine JN, Szalat RE, Staron A, Joshi T,
Mendelson L, Sloan JM and Sanchorawala V: Light chain amyloidosis
associated with Waldenstrom macroglobulinemia: Treatment and
survival outcomes. Haematologica. 108:1680–1684. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Singh A, Geller HI, Alexander KM, Padera
RF, Mitchell RN, Dorbala S, Castillo JJ and Falk RH: True, true
unrelated? Coexistence of Waldenström macroglobulinemia and cardiac
transthyretin amyloidosis. Haematologica. 103:e374–e376.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
National Comprehensive Cancer Network
(NCCN). Waldenström macroglobulinemia/lymphoplasmacytic lymphoma.
Version 1.2022, 2024.
|
|
39
|
Merlini G and Palladini G: Amyloidosis: Is
a cure possible? Ann Oncol. 19 (Suppl 4):iv63–iv66. 2008.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Charles Lobo A, Bhat V, Anandram S, Devi A
M S, Rao SS, Vinister GV, Lobo V and Reuben Ross C: Rare
complication of a rare malignancy: Case report of cardiac
amyloidosis secondary to waldenstrom macroglobulinemia. Qatar Med
J. 2022(7)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Qian X, Bakhshi H, Biswas R, Gattani R and
Kennedy JL: Concurrent waldenstrom macroglobulinemia and mutant
transthyretin cardiac amyloidosis. J Community Hosp Intern Med
Perspect. 12:95–99. 2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ho VV, O'Sullivan JW, Collins WJ, Ozdalga
E, Bell CF, Shah ND, Krishnam MS, Ozawa MG and Witteles RM:
Constrictive pericarditis revealing rare case of ALH amyloidosis
with underlying lymphoplasmacytic lymphoma (waldenstrom
macroglobulinemia). JACC Case Rep. 4:271–275. 2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gustine JN, Staron A, Szalat RE, Mendelson
LM, Joshi T, Ruberg FL, Siddiqi O, Gopal DM, Edwards CV, Havasi A,
et al: Predictors of hematologic response and survival with stem
cell transplantation in AL amyloidosis: A 25-year longitudinal
study. Am J Hematol. 97:1189–1199. 2022.PubMed/NCBI View Article : Google Scholar
|