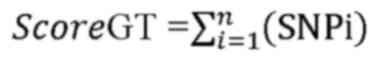Introduction
Child abuse is a pervasive social and public health
issue with profound and long-lasting effects on the psychological,
physiological and neurological well-being of an individual
(1). Exposure to maltreatment
during childhood has been linked to an increased risk of developing
mental health disorders, including depression, anxiety and
post-traumatic stress disorder (2),
as well as physiological conditions, such as cardiovascular
diseases and immune dysregulation (3). While environmental and psychosocial
factors play a crucial role in the consequences of child abuse,
emerging evidence suggests that genetic and epigenetic mechanisms
may significantly influence the susceptibility of an individual to
adversity and their ability to cope with trauma (4).
Genetic factors, including polymorphisms in
stress-related genes, have been found to be associated with
variations in the response of an individual to trauma, potentially
predisposing some individuals to more severe psychological outcomes
(5). Additionally, epigenetic
modifications, such as DNA methylation and histone modifications,
can alter gene expression without changing the DNA sequence,
influencing how individuals respond to early-life stressors
(6). Studies have indicated that
childhood adversity can lead to persistent epigenetic changes that
affect stress response systems, such as the
hypothalamic-pituitary-adrenal (HPA) axis, and contribute to an
increased risk of developing psychiatric disorders later in life
(7,8).
Despite growing interest in this field, there is
still limited research applying bioinformatics approaches to
systematically identify genetic and epigenetic mechanisms
associated with child abuse (9).
The present study aimed to bridge this gap by conducting a
bioinformatics analysis of genetic variations and epigenetic
modifications linked to child abuse. By integrating data from
genome-wide association studies (GWAS) and epigenetic databases,
the present study aimed to identify potential biomarkers that may
enhance the current understanding of the biological underpinnings
of child abuse-related psychopathology (10). The findings presented herein may
contribute to the development of early risk assessment tools and
targeted interventions for affected individuals (11).
Data and methods
Dataset collection
The key terms ‘Child abuse’, ‘Child sexual abuse’,
‘Child maltreatment’, ‘Child neglect’, ‘Child physical abuse’ and
‘Child sexual abuse’ were entered into the MEDLINE and PubMed
databases without date restrictions. English-language publications
related to these terms were searched, transformed from the PubMed
online database (https://pubmed.ncbi.nlm.nih.gov/) and merged
(downloaded and merged) into Medline format. PubMed is a search
engine for life sciences and biomedical articles as it contains
abstracts and references, but often also full articles with open
access. It is part of the NCBI (National Center for Biotechnology
Information) (12).
Data filtering and pre-analysis
In this step, key terms related to abuse,
maltreatment and neglect were extracted. The process was carried
out in the bioinformatics environment of MATLAB Bioinformatics
Toolbox using regular expression and the corresponding PUBMED ID,
during which the data was evaluated in order to extract and store
the results. Using bioinformatics techniques, publications from
different datasets were filtered to remove publications that appear
more than once. This was performed in order to create the complete
set of unique publications.
Extraction of single nucleotide
polymorphisms (SNPs) and data annotation
SNPs associated with child abuse were extracted and
annotated from various genomic databases. The identified SNPs were
located in genes previously implicated in stress response and
neurodevelopment. Additionally, non-coding regions, including long
intergenic non-coding (LINC) RNAs and provisional gene identifiers
(LOC), were analyzed.
LINC RNA genes represent a category of long
non-coding RNAs that do not overlap with protein-coding genes and
are involved in gene regulation. LOC genes serve as provisional
gene identifiers assigned in genomic databases when their precise
function or official gene symbol has yet to be determined.
For annotation, databases such as dbSNP (https://www.ncbi.nlm.nih.gov/snp/), GWAS Catalog
(https://www.ebi.ac.uk/gwas/) and Kyoto
Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg/) were utilized to classify
the SNPs based on their genomic location, functional impact, and
association with biological pathways.
Semantic analysis and gene targets
classification
In this step, the annotated gene targets were scored
through semantic analysis in order to identify and isolate the most
direct and likely targets associated with child abuse. Thus, a
scoring function was created in which we can differentiate the
‘related SNPs’ as well as the ‘strongly related SNPs’, which were
ranked and rated in terms of the frequency of occurrence of
polymorphisms in the articles studied and the frequency of
occurrence of polymorphisms based on each genetic target.
The score genetic targets were as follows:
where i is the number of corresponding SNPs per
genetic target and n is the total number of the SNPs per genetic
target. Scoring function was calculated as follows:
Related_Genetic Targets: Score <3, strongly related genetic
target: score ≥3.
All SNPs were identified and classified into these
two main categories, strongly related genetic targets and related
genetic targets based on their correlation rate with the particular
terms studied.
Genetic targets, disease, biological
pathway analysis and dark DNA matters in adversity genes
From group A ‘Strongly related Genetic Target’ of
genetic targets with high correlation, their related SNPs were
analyzed in order to collect all disease ontologies. A
bioinformatics algorithm was then used to identify the disease
ontologies that occur with higher frequency. This precise result of
the most frequently appearing key terms was captured and visualized
as a word cloud in Fig. 1.
This word cloud visualizes the most frequently
occurring terms related to child abuse in the dataset. Larger words
indicate higher frequency within the dataset, highlighting the
central themes related to child maltreatment and neglect.
In the same manner, using genetic targets, genes,
pseudogenes, primates, characterized regions of dark DNA matter,
such as long non coding RNAs and microRNAs (miRNAs/miRs),
categories of biological pathways were discovered using the
platform Reactome, in order to extract beneficial information
regarding the major biological pathways of the key genetic
targets.
The concept of ‘adversity genes’, as described by
Levine et al (13) in 2017,
is relevant to this analysis. Adversity genes refer to genetic
elements that exhibit differential expression in response to
environmental stressors, including childhood maltreatment. These
genes, which include FKBP prolyl isomerase 5 (FKBP5), corticotropin
releasing hormone receptor 1 (CRHR1),
catechol-O-methyltransferase (COMT) and several non-coding
RNA regions, play a pivotal role in the stress response of the body
and the development of stress-related disorders (13). Their involvement in neuroendocrine
signaling, epigenetic modulation, and immune system regulation
further supports the hypothesis that adverse childhood experiences
leave a lasting imprint at the genetic and epigenetic levels.
Epigenetic target analysis
All suspected SNPs potentially linked to epigenetics
were studied through publications in order to understand the
contribution of epigenetics to this subject. To obtain this, a
search for specific polymorphisms corresponding to the genetic
targets was performed and re-evaluated for their epigenetic
contribution to this subject.
The suspected epigenetic polymorphisms were defined
as polymorphisms corresponding to non-coding regions such as long
non-coding RNAs (lncRNAs), LINCs RNAs, miRs and LOCs. The following
terms were used:
LINC RNAs. A type of lncRNA that does not
overlap protein-coding genes and plays a role in gene regulation,
chromatin remodeling, and cellular processes.
LOC (locus). A placeholder name for genes or
genomic regions that have been identified but are not yet fully
characterized or named. These regions may contain functional
elements, including non-coding RNAs or pseudogenes.
GWAS. A research approach that involves
scanning complete sets of DNA (genomes) from multiple individuals
to identify genetic variants associated with specific traits or
diseases.
Epigenetics. The study of changes in gene
expression that do not involve alterations in the DNA sequence, but
are influenced by environmental and developmental factors, such as
DNA methylation and histone modifications.
DNA methylation. A biochemical process that
adds a methyl group to DNA, often silencing gene expression and
playing a key role in regulating development and disease.
Histone modification. Chemical changes to
histone proteins that affect the structure of chromatin and
influence gene expression.
SNP. A variation in a single nucleotide at a
specific position in the genome that may be associated with genetic
predispositions to diseases or traits.
HPA axis. A major neuroendocrine system that
regulates stress responses, metabolism, immune function, and mood
through the release of hormones such as cortisol.
Results
Dataset collection
A systematic data mining and semantic analysis
approach was employed to identify genes, variants and SNPs
associated with child abuse, maltreatment and neglect. A total of
201,121 publications containing key terms, such as ‘Child abuse’,
‘Child sexual abuse’, ‘Child maltreatment’, ‘Child neglect’ and
‘Child physical abuse’ were retrieved from the MEDLINE and PubMed
databases. The extracted data were filtered and processed to
generate a dataset of genetic factors relevant to child abuse. The
key words used in this analysis are closely related to the research
subject and represent the most frequently occurring terms in the
relevant literature (Table I).
 | Table IKey words used and directly related to
the types of child abuse. |
Table I
Key words used and directly related to
the types of child abuse.
| Key words | Definition |
|---|
| Child abuse | Physical, emotional,
or sexual harm or potential harm inflicted on a child by an adult
or another child. |
| • Child physical
abuse | |
| • Child emotional
abuse | |
| • Child sexual
abuse | |
| Child neglect | Failure to provide
basic needs such as food, shelter, medical care, education, and
supervision to a child. This can include neglecting a child's
health, safety, or emotional well-being. |
| Child
maltreatment | A broad term that
encompasses both child abuse and neglect, as well as any other form
of harmful behavior towards a child. Maltreatment is any form of
harm or mistreatment that a child experiences at the hands of a
caregiver or authority figure. |
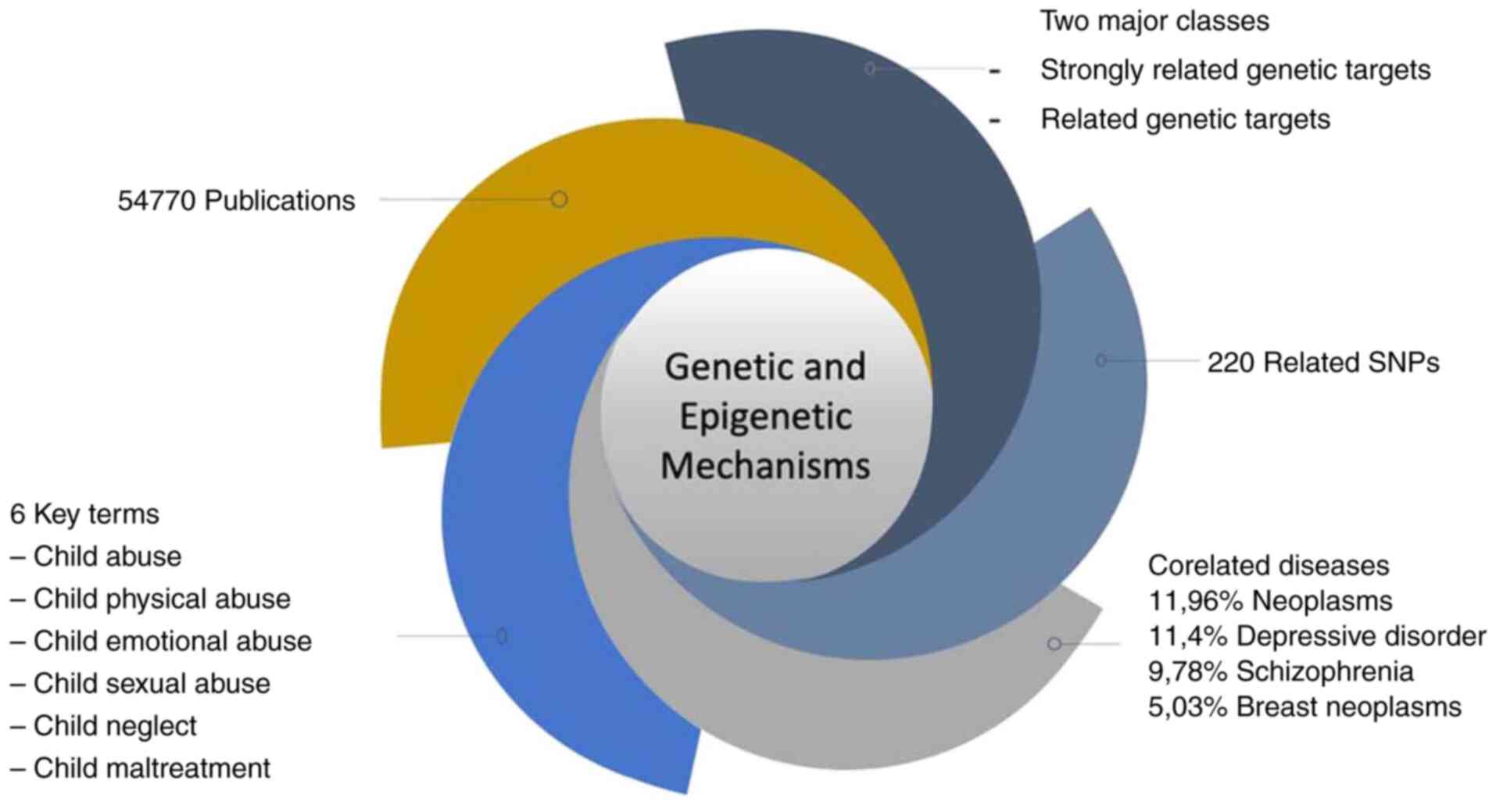
A visual representation of the identified key terms
is presented in Fig. 1, which
illustrates the frequency of key terms related to child abuse
within the dataset. This visualization highlights the most commonly
occurring terminology in the literature, providing insight into the
breadth and focus of existing research. Additionally, a word cloud
was generated to visually represent the most prominent terms
extracted from the dataset (Fig.
2).
Data filtering and pre-analysis
A total of 529 abuse-related keywords were
identified within the dataset, including ‘Child sexual abuse’,
‘Child emotional abuse’ and ‘Child neglect’. Semantic analysis was
applied to refine the dataset, ensuring the inclusion of only the
most relevant terms. The frequency of these key terms was analyzed,
and the most frequently occurring words were identified (Table II). The table provides an overview
of the most frequently occurring key terms within the dataset,
demonstrating the breadth of terminology used in the literature
related to child abuse.
| Table IIList of the most frequently shown key
terms describing child abuse within the dataset (frequency
>150). |
Table II
List of the most frequently shown key
terms describing child abuse within the dataset (frequency
>150).
| Name | Frequency |
|---|
| Child abuse | 563 |
| Legal approach | 360 |
| Population | 305 |
| Child
maltreatment | 295 |
| Child sexual
abuse | 289 |
| Sexual abuse | 249 |
| Adolescents | 242 |
| Behavior | 234 |
| Age factors | 218 |
| Youth | 215 |
| Professional patient
relationship | 205 |
| Developed
countries | 200 |
| Americas | 190 |
| Children | 183 |
| North America | 171 |
| Genetics and
reproduction | 171 |
| Trauma | 161 |
| Depression | 160 |
| Economic
factors | 152 |
A visual representation of the identified key terms
was created in the form of a word cloud (Fig. 2). The figure illustrates the most
common key terms extracted from the dataset using semantic
analysis. Words that appear more frequently in the relevant
literature are depicted in larger fonts, illustrating the
conceptual emphasis of the dataset.
Extraction of SNPs and data
annotation
A total of 209 SNPs and 104 genetic targets,
including genes, pseudogenes and transcription factors, were
identified and extracted from online databases. The SNPs associated
with child abuse were annotated using MATLAB algorithms,
incorporating genomic information from dbSNP, GWAS Catalog and
KEGG. The genes most frequently associated with child abuse were
classified according to their functional role in stress response
and neurodevelopment. The identified genetic targets and SNPs
related to child abuse are listed in Table III. This table compiles the key
genetic targets identified in association with child abuse. It
includes genes, transcription factors, pseudogenes and non-coding
RNAs, highlighting their role in neurodevelopmental and
stress-related biological processes.
| Table IIIList of genetic targets and SNPs
extracted and associated with child abuse. |
Table III
List of genetic targets and SNPs
extracted and associated with child abuse.
| Name | Total unique
SNPS/overall | SNPs | Type of SNPs
(functional consequence) |
|---|
| FKBP5 | 15//87 | rs3800373 |
genic_downstream_transcript_variant,intron_variant,3_prime_UTR_variant |
| | | rs1360780 | intron_variant |
| | | rs4713916 |
intron_variant,genic_upstream_transcript_variant |
| | | rs3777747 | intron_variant |
| | | rs2766533 |
genic_upstream_transcript_variant,intron_variant |
| | | rs9296158 | intron_variant |
| | | rs737054 | intron_variant |
| | | rs9470080 | intron_variant |
| | | rs7771727 |
intron_variant,genic_downstream_transcript_variant |
| | | rs4713902 | intron_variant |
| | | rs9394309 | intron_variant |
| | | rs9470079 | intron_variant |
| | | rs3798347 | intron_variant |
| | | rs10947563 | intron_variant |
| | | rs7748266 | intron_variant |
| | | rs947008 | intron_variant |
| | | rs1360870 | |
|
LINC02210-CRHR1 | 11//47 | rs7209436 | intron_variant |
| | | rs4792887 | intron_variant |
| | | rs110402 | intron_variant |
| | | rs17689882 | intron_variant |
| | | rs242924 | intron_variant |
| | | rs2664008 | intron_variant |
| | | rs12944712 | intron_variant |
| | | rs9900679 | intron_variant |
| | | rs1876831 | intron_variant |
| | | rs9900679 | intron_variant |
| | | rs16940665 |
synonymous_variant,coding_sequence_variant |
| CRHR1 | 10 | rs7209436 | intron_variant |
| | | rs4792887 | intron_variant |
| | | rs110402 | intron_variant |
| | | rs17689882 | intron_variant |
| | | rs242924 | intron_variant |
| | | rs2664008 | intron_variant |
| | | rs12944712 | intron_variant |
| | | rs9900679 | intron_variant |
| | | rs1876831 | intron_variant |
| | | rs16940665 |
synonymous_variant,coding_sequence_variant |
| LOC112267956 | 7 | rs9296158 | intron_variant |
| | | rs1360780 | intron_variant |
| | | rs3777747 | intron_variant |
| | | rs737054 | intron_variant |
| | | rs4713902 | intron_variant |
| | | rs3798347 | intron_variant |
| | | rs7748266 | intron_variant |
| COMT | 6 | rs165599 |
genic_downstream_transcript_variant,3_prime_UTR_variant,intron_variant |
| | | rs5993882 |
genic_upstream_transcript_variant,upstream_transcript_variant,intron_variant |
| | | rs737866 |
2KB_upstream_variant,genic_upstream_transcript_variant,upstream_transcript_variant,intron_variant |
| | | rs4680 |
2KB_upstream_variant,coding_sequence_variant,upstream_transcript_variant,missense_variant |
| | | rs6267 |
2KB_upstream_variant,coding_sequence_variant,upstream_transcript_variant,missense_variant |
| | | rs4633 |
2KB_upstream_variant,coding_sequence_variant,upstream_transcript_variant,synonymous_variant |
| | | rs4818 |
2KB_upstream_variant,coding_sequence_variant,upstream_transcript_variant,synonymous_variant |
| OXTR | 6 | rs2268498 |
2KB_upstream_variant,upstream_transcript_variant |
| | | rs1042778 |
3_prime_UTR_variant,intron_variant |
| | | rs53576 | intron_variant |
| | | rs2254298 | intron_variant |
| | | rs237895 | intron_variant |
| | | rs237885 | intron_variant |
| | | rs237987 | |
| IL1B | 6 | rs16944 |
2KB_upstream_variant,upstream_transcript_variant |
| | | rs1143623 |
2KB_upstream_variant,upstream_transcript_variant |
| | | rs1143627 |
2KB_upstream_variant,upstream_transcript_variant |
| | | rs1143643 | intron_variant |
| | | rs1143633 | intron_variant |
| | | rs1143634 |
synonymous_variant,coding_sequence_variant |
| HTR2A | 5 | rs7997012 | intron_variant |
| | | rs6561333 | intron_variant |
| | | rs1885884 |
non_coding_transcript_variant,intron_variant |
| | | rs9316235 | intron_variant |
| | | rs6313 |
synonymous_variant,coding_sequence_variant,intron_variant |
| CRHR2 | 5 | rs2190242 | intron_variant |
| | | rs2284217 | intron_variant |
| | | rs2014663 | intron_variant |
| | | rs4722999 |
intron_variant,3_prime_UTR_variant |
| | | rs12701020 |
non_coding_transcript_variant,intron_variant |
| FOXP2 | 5 | rs7783012 | intron_variant |
| | | rs10262462 | intron_variant |
| | | rs1456031 |
genic_downstream_transcript_variant,intron_variant |
| | | rs2396753 | intron_variant |
| | | rs2253478 |
intron_variant,genic_upstream_transcript_variant |
| IL19 | 5 | rs1800896 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| | | rs1800871 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| | | rs1800872 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| | | rs1800890 |
genic_upstream_transcript_variant,intron_variant |
| | | rs6676671 |
intron_variant,genic_upstream_transcript_variant |
| GABRA2 | 5 | rs279826 | intron_variant |
| | | rs11503014 |
5_prime_UTR_variant,intron_variant |
| | | rs279858 |
missense_variant,synonymous_variant,coding_sequence_variant |
| | | rs211034 | intron_variant |
| | | rs211035 |
missense_variant,intron_variant,coding_sequence_variant |
| NOS1AP | 4 | rs4145621 |
genic_upstream_transcript_variant,intron_variant |
| | | rs6680461 |
intron_variant,genic_upstream_transcript_variant |
| | | rs3751284 |
genic_upstream_transcript_variant,missense_variant,coding_sequence_variant,synonymous_variant |
| | | rs348624 |
synonymous_variant,coding_sequence_variant |
| NR3C2 | 4 | rs17581262 | intron_variant |
| | | rs5522 |
missense_variant,non_coding_transcript_variant,coding_sequence_variant |
| | | rs5534 |
non_coding_transcript_variant,3_prime_UTR_variant,genic_downstream_transcript_variant |
| | | rs2070951 |
non_coding_transcript_variant,5_prime_UTR_variant |
| IL1RN | 4 | rs9005 |
3_prime_UTR_variant |
| | | rs4251961 |
intron_variant,upstream_transcript_variant,genic_upstream_transcript_variant |
| | | rs315952 |
missense_variant,synonymous_variant,coding_sequence_variant |
| | | rs419598 |
synonymous_variant,coding_sequence_variant |
| GRN | 3 | rs3859268 | intron_variant |
| | | rs2879096 | intron_variant |
| | | rs3785817 | intron_variant |
| SLC6A4 | 3 | rs25531 |
2KB_upstream_variant,intron_variant,genic_upstream_transcript_variant,upstream_transcript_variant |
| | | rs3813034 |
3_prime_UTR_variant |
| | | rs1042173 |
3_prime_UTR_variant |
| LOC105371801 | 3 | rs17689882 | intron_variant |
| | | rs16940665 |
synonymous_variant,coding_sequence_variant |
| | | rs1876831 | intron_variant |
| LOC101929309 | 3 | rs3800373 |
genic_downstream_transcript_variant,intron_variant,3_prime_UTR_variant |
| | | rs6910300 |
intron_variant,genic_downstream_transcript_variant |
| | | rs7771727 |
intron_variant,genic_downstream_transcript_variant |
| IL10 | 3 | rs1800896 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| | | rs1800871 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| | | rs1800872 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,2KB_upstream_variant |
| IL6R | 3 | rs4845617 |
5_prime_UTR_variant,genic_upstream_transcript_variant,intron_variant |
| | | rs4537545 |
intron_variant,genic_downstream_transcript_variant |
| | | rs2228145 |
missense_variant,coding_sequence_variant,intron_variant,genic_downstream_transcript_variant |
| IFNG | 3 | rs1861494 | intron_variant |
| | | rs2069718 | intron_variant |
| | | rs2430561 | intron_variant |
| CRHBP | 3 | rs7728378 | intron_variant |
| | | rs6453267 | intron_variant |
| | | rs10474485 |
genic_downstream_transcript_variant,intron_variant |
| SLC6A2 | 3 | rs1814270 | intron_variant |
| | | rs2242446 |
upstream_transcript_variant,intron_variant,5_prime_UTR_variant,2KB_upstream_variantgenic_upstream_transcript_variant |
| | | rs5569 |
missense_variant,synonymous_variant,coding_sequence_variant |
| NR3C1 | 3 | rs12655166 |
intron_variant,genic_upstream_transcript_variant |
| | | rs10482672 | intron_variant |
| | | rs6198 |
non_coding_transcript_variant,3_prime_UTR_variant,genic_downstream_transcript_variant |
| CRP | 3 | rs3093059 |
upstream_transcript_variant,2KB_upstream_variant |
| | | rs1417938 | intron_variant |
| | | rs1130864 |
intron_variant,3_prime_UTR_variant |
| | | rs2794520 | |
| | | rs3093077 | |
The distribution of genetic targets and evidence
found in the present study is visually presented in Fig. 3. This figure presents a graphical
representation of the most frequently occurring genetic targets and
non-coding regions associated with child abuse. The size of each
genetic term corresponds to its relative frequency within the
dataset, highlighting key genes, such as FKBP5, CRHR1 and COMT.
Semantic analysis and gene targets
classification
A semantic analysis of the extracted SNPs revealed a
genomic map of child abuse-related genetic targets. The genes,
FKBP5, CRHR1, LINC02210-CRHR1 and COMT, were identified as the most
frequently occurring targets. These genes are associated with
stress response regulation, emotional resilience and neuroendocrine
signaling pathways.
FKBP5. The FKBP5 gene is a part of the
immunophilin proteins, which play a role in immune regulation and
functions as a co-chaperone in glucocorticoid receptor activity in
response to stressors, making it one of the most frequently
encountered genes in studies of people who have undergone stress,
and in particular, in children who have been abused (14). In addition to epigenetic
modifications and other environmental factors, it appears that
FKBP5 may modulate GR susceptibility by delaying or reducing its
transcriptional activity (15).
CHRH1. The CRHR1 gene has been extensively
studied due to its implication for sensitized reactivity in
stressful conditions (16). Through
these findings, a significant interaction of CHRH1 with childhood
abuse and trauma and history of suicide attempts emerges. In the
similar direction, in another study on 235 HPA axis SNPs, a trend
of the rs2664008 polymorphism of the CRHR1 gene, early childhood
abuse and suicide attempts in bipolar patients was indicated
(17).
COMT. Variations in some genes, including the
COMT gene, are known to be associated with susceptibility to stress
and some mental disorders. The association between stressful events
and genes is known to activate the mechanism of depression
development (5).
Variations in these genes are associated with stress
sensitivity and depressive cognitive biases. The interaction
between genes and stressful events in childhood is considered to be
a mechanism that plays a role in the development of depression and
therefore helps to proactively identify symptoms of depression or
other diseases through genetic susceptibility (5).
Genetic targets, disease, biological
pathway analysis and dark DNA matters in adversity genes
The analysis of genetic targets revealed that the
most common disorders associated with child abuse include
neoplasms, depressive disorders, schizophrenia, neurodegenerative
diseases and autoimmune conditions. These disorders share key
biological pathways involved in the stress response,
neurodevelopment and immune function. The most frequently occurring
disease ontologies linked to child abuse are summarized in Table IV. The table categorizes the
primary disease ontologies identified in the dataset, emphasizing
conditions frequently associated with child abuse, such as
neuropsychiatric disorders, metabolic syndromes, and immune-related
pathologies.
| Table IVList of the most frequently shown
ontologies describing child abuse within the dataset. |
Table IV
List of the most frequently shown
ontologies describing child abuse within the dataset.
| Disease
ontology | Count |
|---|
| Neoplasms | 4,052 |
| Depressive
disorder | 3,864 |
| Schizophrenia | 3,316 |
| Pain | 2,122 |
| Breast
neoplasms | 1,707 |
| Colorectal
neoplasms | 1,235 |
| Stress
disorders_post_traumatic | 1,098 |
|
Hyperhomocysteinemia | 1,061 |
A visual representation of the key disease
associations with child abuse is provided in Fig. 4, illustrating the most prominent
biological ontologies derived from the analysis. A word cloud
displaying the most frequently identified biological ontologies
related to child abuse is presented in Fig. 4. Terms related to neurodevelopmental
processes, stress response and immune system regulation appear
prominently, emphasizing their significance in the dataset.
Further overrepresentation analysis of genetic
targets demonstrated marked enrichment in biological pathways
related to HPA axis regulation, dopaminergic signaling and synaptic
plasticity. These pathways are crucial in the adaptation of the
body to stress and have been implicated in mental health disorders
commonly observed in individuals with a history of child abuse. The
identified pathways are summarized in Table V. This table details the biological
pathways enriched in the dataset, focusing on molecular mechanisms
linked to stress response, neurodevelopment and immune regulation.
The pathways were identified using overrepresentation analysis.
| Table VBiological ontology mechanisms that
occurred in the search and are associated with child abuse. |
Table V
Biological ontology mechanisms that
occurred in the search and are associated with child abuse.
| Mental
disorders | Cancer | Metabolic
disorders | Neurodegenerative
diseases | Autoimmune
diseases | Other |
|---|
| Depressive
Disorder | Neoplasms | Hypertension | Alzheimer
disease | Arthritis
Rheumatoid | Pain |
| Schizophrenia | Breast
Neoplasms | Obesity | Parkinson
disease | Lupus
Erythematosus |
Hyperhomocysteinemia |
| | | | | Systemic | |
| Stress Disorders
Post Traumatic | Colorectal
Neoplasms | Diabetes
Mellitus | | | Wounds And
Injuries |
| Anxiety
Disorders | Lung Neoplasms | Hypotension | | | Asthma |
| Mental
Disorders | Stomach
Neoplasms |
DiabetesMellitus_Type2 | | | Periodontitis |
| Autistic
Disorder | Carcinoma Non Small
Cell Lung | | | | Infections |
| Depressive Disorder
Major | Prostatic
Neoplasms | | | | Attention Deficit
Disorder |
| | | | | | With
Hyperactivity |
| Bipolar
Disorder | Esophageal
Neoplasms | | | | Alcoholism |
| Pain Insensitivity
Congenital | | | | | Sepsis |
| Psychoses Substance
Induced | | | | | Radiation
Pneumonitis |
| Anxiety | | | | | Tuberculosis |
| Dyskinesia Drug
Induced | | | | | Hepatitis B |
| Substance Related
Disorders | | | | | Headache Disorders
Secondary |
| Adjustment
Disorders | | | | | Pulmonary Disease
Chronic |
| | | | | | Obstructive |
| | | | | | Irritable Bowel
Syndrome |
| | | | | | Fibrosis |
| | | | | | Cognition
Disorders |
| | | | | | Cerebral
Infarction |
| | | | | | Weight Loss |
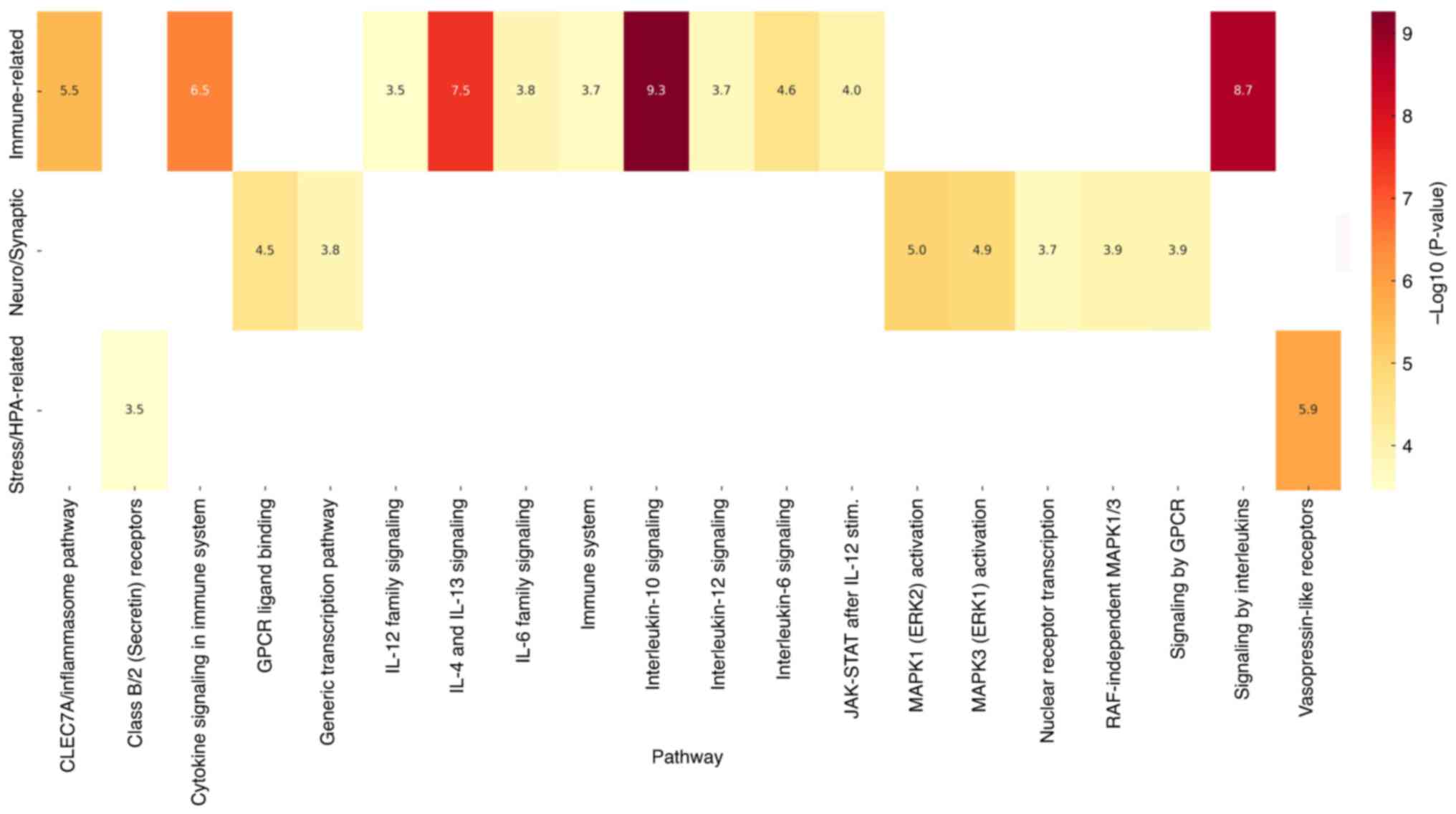
A graphical representation of these enriched
pathways is provided in Fig. 5,
highlighting the statistical significance of biological processes
associated with child abuse-related genetic targets. This heatmap
visually represents the statistical significance (P-values) of the
biological pathways linked to the most frequently occurring genes
in child abuse, grouped by functional categories such as immune
signaling, neurodevelopment, and stress-related mechanisms.
Different colors correspond to varying levels of statistical
significance, with darker shades representing lower P-values and
greater relevance. The exact P-values are presented numerically
within each cell of the heatmap, and statistical significance is
shown using the -log10(P-value) scale, allowing for both
visual and quantitative interpretation.
Table VI provides a
detailed breakdown of the most frequently identified biological
pathways and their associated genetic targets. It highlights key
signaling mechanisms implicated in the biological response to
childhood adversity. The most frequently occurring biological
pathways associated with adversity genes are detailed in Table VI.
| Table VIBiological pathways of the common
genetic targets and the corresponding genes most frequently
involved in child abuse. |
Table VI
Biological pathways of the common
genetic targets and the corresponding genes most frequently
involved in child abuse.
| Pathway | Genetic target | Corresponding
genes |
|---|
| Immune system | • Innate immune
system | - C-type lectin
receptors |
| | | - Toll like
receptor cascades |
| | • Cytokine
signaling in immune system | - Signaling by
interleukins |
| Signal
transduction | • Signaling by
GPCR | - GPCR ligand
binding |
| | | - GPCR downstream
signaling |
| | • MAPK family
signaling cascades | |
| | • Signaling by
receptor tyrosine kinases | |
| Gene expression
(transcription) | • RNA polymerase II
transcription | - Generic
transcription pathway |
| Cellular responses
to stimuli | • Cellular
responses to stress | |
| Programmed cell
death | • Regulated
necrosis | |
| Disease | • Infectious
disease | - Parasitic
infection pathways |
| | | - Bacterial
infection pathways |
| | • Diseases of
immune system | - Diseases
associated with the TLR signaling cascade |
| | • Disorders of
developmental biology | |
| | • Disorders of
transmembrane transporters | |
| Developmental
biology | | |
| Neuronal
system | | |
Epigenetic target analysis
An analysis of epigenetic targets identified key
non-coding RNAs (LINC, LOC and miR) that may mediate the effects of
child abuse. miR-195, implicated in breast cancer, was found to be
epigenetically regulated, while LOC105369506 and LOC100287329 were
associated with stress-related epigenetic modifications (18). These findings suggest that
epigenetic alterations in non-coding genomic regions may contribute
to the long-term consequences of childhood adversity. The
epigenetically significant genetic targets related to child abuse
are compiled in Table VII.
| Table VIIThe ‘usual suspects’ genetic targets
epigenetically associated with child abuse. |
Table VII
The ‘usual suspects’ genetic targets
epigenetically associated with child abuse.
| Genetic target | SNPs | Type of SNPs | | Epigenetic
association with child abuse | Epigenetic
link |
|---|
|
LINC02210-CRHR1 | rs7209436 | intron_variant | locus | x | |
| | rs4792887 | intron_variant | | | |
| | rs110402 | intron_variant | | | |
| | rs17689882 | intron_variant | | | |
| | rs242924 | intron_variant | | | |
| | rs2664008 | intron_variant | | | |
| | rs12944712 | intron_variant | | | |
| | rs9900679 | intron_variant | | | |
| | rs1876831 | intron_variant | | | |
| | rs9900679 | intron_variant | | | |
| | rs16940665 |
synonymous_variant,coding_sequence_variant | | | |
| LOC112267956 | rs9296158 | intron_variant | locus | x | |
| | rs1360780 | intron_variant | | | |
| | rs3777747 | intron_variant | | | |
| | rs737054 | intron_variant | | | |
| | rs4713902 | intron_variant | | | |
| | rs3798347 | intron_variant | | | |
| | rs7748266 | intron_variant | | | |
| LOC105371801 | rs17689882 | intron_variant | locus | x | |
| | rs16940665 |
synonymous_variant,coding_sequence_variant | | | |
| | rs1876831 | intron_variant | | | |
| LOC101929309 | rs3800373 |
genic_downstream_transcript_variant,intron_variant,3_prime_UTR_variant | ncRNA | x | |
| | rs6910300 |
intron_variant,genic_down | | | |
| | |
stream_transcript_variant | | | |
| | rs7771727 |
intron_variant,genic_down | | | |
| | |
stream_transcript_variant | | | |
| miR-4761 | rs4680 |
2KB_upstream_variant,coding_sequence_variant,upstream_transcript_variant,missense_variant | short non-coding
RNA | √ | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7016268/ |
| LOC107986321 | rs6857715 |
genic_upstream_transcript_variant,intron_variant,upstream_transcript_variant,non_coding_transcript_variant | | x | |
| LOC105371720 | rs25531 |
2KB_upstream_variant,intron_variant,genic_upstream_transcript_variant,upstream_transcript_variant | ncRNA | x | |
| LOC107986777 | rs3037354 |
intron_variant,2KB_upstream_variant,upstream_transcript_variant,genic_upstream_transcript_variant | ncRNA | x | |
| LOC105377387 | rs34043524 | intron_variant | RNA, long
non-coding | x | |
| LOC105370115 | rs1886797 | intron_variant | RNA, long
non-coding | x | |
| LOC105377864 | rs6296 |
intron_variant,genic_upstream_transcript_variant,5_prime_UTR_variant,coding_sequence_variant,synonymous | | x | |
| LOC100287329 | rs1041981 |
coding_sequence_variant,upstream_transcript_variant,2KB_upstream_variant,missense_variant | RNA, long
non-coding | √ | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7232649/ |
| LOC105369506 | rs11215217 | intron_variant | RNA, long
non-coding | √ | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6689282/ |
miR-195 is a type of miRNA that has been implicated
in breast cancer. miRNAs are small RNA molecules that regulate gene
expression by binding to messenger RNA (mRNA) molecules and
preventing their translation into proteins. Research has
demonstrated that the expression of miR-195 is downregulated, or
less active, in breast cancer cells compared to normal breast
tissue. This suggests that it may play a role in the development or
progression of breast cancer. The expression of miR-4761 is
regulated by histone modifications in breast cancer cells,
suggesting that epigenetic changes may contribute to its
dysregulation in breast cancer (18).
LOC105369506 is a gene that has been studied in
relation to various behaviors and conditions, and research has
suggested that certain epigenetic modifications can play a role in
the development of antisocial behavior and the effects of child
abuse. For example, studies have found that individuals who have
experienced abuse or neglect during childhood may have differences
in epigenetic marks on genes related to stress response and
emotional regulation, which may increase their risk for antisocial
behavior later in life (19-21).
LOC100287329 is a gene that is also known as
miR-548AA2. It is a miRNA gene located on chromosome 14 in humans.
Abuse, whether physical, emotional, or sexual, can have a
significant impact on the health of an individual. There is
evidence to suggest that exposure to stress and trauma can lead to
epigenetic changes that may contribute to the development of MS and
other diseases (22).
Discussion
The findings of the present study highlight the
genetic and epigenetic factors associated with child abuse,
providing new insight into the biological underpinnings of
stress-related disorders. Through systematic data mining and
bioinformatics analysis, the present study identified 209 SNPs and
104 genetic targets, including FKBP5, CRHR1 and COMT, that are
strongly associated with childhood maltreatment. The results also
revealed that LINC RNAs, provisional gene identifiers (LOC) and
miRNAs contribute to the molecular effects of early-life
adversity.
A key finding of the present study was the prominent
role of FKBP5 in child abuse-related pathways. This gene, which is
involved in glucocorticoid receptor regulation (14,23,24),
was one of the most frequently identified genetic targets in the
dataset. The presence of CRHR1, a key regulator of the HPA axis,
further supports the hypothesis that childhood stress alters
neuroendocrine responses. The identification of COMT, which
influences dopamine metabolism, aligns with increasing evidence
that childhood trauma affects cognitive and emotional processing.
Notably, the present study extends prior knowledge (24-26)
by demonstrating that these genetic markers are not only
statistically overrepresented in the child abuse dataset, but also
frequently co-occur with stress-related SNPs.
Beyond individual genes, the study mapped the
biological pathways most significantly associated with child abuse.
Overrepresentation analysis revealed that child abuse-related genes
are enriched in pathways linked to neuroinflammation, oxidative
stress, and immune system dysregulation. These findings suggest
that the physiological impact of childhood adversity extends beyond
neurological effects to include systemic alterations that may
predispose individuals to chronic diseases, including autoimmune
disorders and metabolic conditions.
A particularly novel contribution of the present
study is the identification of epigenetic markers associated with
childhood trauma. The analysis uncovered miR-4761, LOC105369506 and
LOC100287329 as key regulatory elements that may mediate the
effects of abuse at the molecular level. The detection of these
epigenetic factors underscores the role of non-coding genomic
elements in shaping individual susceptibility to trauma-related
disorders. Unlike previous studies, which have focused primarily on
protein-coding genes, this study expands the scope of genetic
investigation by incorporating non-coding RNA elements, providing a
more comprehensive understanding of the genomic response to
adversity.
The concept of adversity genes, first introduced by
Levine et al (13), is
reinforced by these findings. Their study demonstrated that genes
exhibiting differential expression in response to childhood
maltreatment are frequently involved in stress adaptation,
neurodevelopment and immune regulation (13). While previous research has suggested
that adversity genes contribute to vulnerability in trauma-exposed
individuals, the present study provides a direct
bioinformatics-based validation of their significance. The results
confirm that these genes are consistently overrepresented in child
abuse datasets, further solidifying their relevance in
understanding the biological consequences of early-life stress.
These findings have critical clinical implications.
The identification of genetic and epigenetic biomarkers associated
with child abuse may pave the way for risk prediction models that
could help identify individuals at increased risk of developing
psychiatric or stress-related disorders. Additionally, these
insights may contribute to the development of precision medicine
approaches, where genetic screening informs targeted interventions
for trauma survivors. From a forensic perspective, the biological
evidence linking specific genetic variants to child abuse may also
have applications in legal contexts, offering new tools for
assessing the long-term consequences of maltreatment.
While the present study provides notable
contributions to the field, certain limitations need to be
acknowledged. Genetic predisposition alone does not determine
individual outcomes, as environmental and social factors play a
critical role in shaping resilience. Moreover, epigenetic
modifications are dynamic, necessitating further longitudinal
studies to assess their stability over time. Future research is
thus required to focus on validating these findings through
experimental approaches, including gene expression studies and
methylation analyses in trauma-exposed populations. Additionally,
integrating machine learning algorithms with bioinformatics
pipelines may enhance predictive models for assessing genetic risk
factors in child abuse cases.
In conclusion, the present study presents original
evidence of the genetic and epigenetic alterations associated with
child abuse. By identifying specific SNPs, gene targets, and
regulatory elements linked to early-life adversity, these findings
contribute to a growing understanding of how childhood trauma
becomes biologically embedded. The integration of genetic,
epigenetic, and pathway analysis provides a comprehensive framework
for future research, with implications for both clinical practice
and social policy. As the field progresses, these insights may help
shape personalized intervention strategies aimed at mitigating the
long-term impact of childhood maltreatment.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ED and DV conceived and designed the study. ED
performed the data collection, data analysis and interpretation. LP
contributed to the development of the bioinformatics methodology
and figure processing. EE and GPC performed critical revisions and
were involved in the analysis of the on the genetic data and its
biological relevance, providing expert guidance. DV supervised the
project and provided overall coordination and critical manuscript
review. ED and LP drafted the manuscript. All authors contributed
to the interpretation of results and manuscript preparation. All
authors have read and approved the final manuscript. ED and DV
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
GPC is the Editor in Chief of the journal, and DV
and EE are Editors of the journal. However, they had no personal
involvement in the reviewing process, or any influence in terms of
adjudicating on the final decision, for this article. The other
authors declare that they have no competing interests.
References
|
1
|
Pervanidou P, Makris G, Chrousos G and
Agorastos A: Early life stress and pediatric posttraumatic stress
disorder. Brain Sci. 10(169)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
De Bellis MD and Keshavan MS: Sex
differences in brain maturation in maltreatment-related pediatric
posttraumatic stress disorder. Neurosci Biobehav Rev. 27:103–117.
2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gilbert R, Widom CS, Browne K, Fergusson
D, Webb E and Janson S: Burden and consequences of child
maltreatment in high-income countries. Lancet. 373:68–81.
2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Damaskopoulou E, Chrousos GP, Eliopoulos E
and Vlachakis D: Mechanisms of epigenetic inheritance in children
following exposure to abuse. EMBnet j. 26(e995)2021.
|
|
5
|
Vrijsen JN, van Oostrom I, Arias-Vásquez
A, Franke B, Becker ES and Speckens A: Association between genes,
stressful childhood events and processing bias in depression
vulnerable individuals. Genes Brain Behav. 13:508–516.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tyrka AR, Price LH, Marsit C, Walters OC
and Carpenter LL: Childhood adversity and epigenetic modulation of
the leukocyte glucocorticoid receptor: Preliminary findings in
healthy adults. PLoS One. 7(e30148)2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gershon A, Sudheimer K, Tirouvanziam R,
Williams LM and O'Hara R: The long-term impact of early adversity
on late-life psychiatric disorders. Curr Psychiatry Rep.
15(352)2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kino T and Chrousos GP:
Acetylation-mediated epigenetic regulation of glucocorticoid
receptor activity: Circadian rhythm-associated alterations of
glucocorticoid actions in target tissues. Mol Cell Endocrinol.
336:23–30. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Dalvie S, Maihofer AX, Coleman JRI,
Bradley B, Breen G, Brick LA, Chen CY, Choi KW, Duncan LE, Guffanti
G, et al: Genomic influences on self-reported childhood
maltreatment. Transl Psychiatry. 10(38)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ruisch IH, Dietrich A, Glennon JC,
Buitelaar JK and Hoekstra PJ: Interplay between genome-wide
implicated genetic variants and environmental factors related to
childhood antisocial behavior in the UK ALSPAC cohort. Eur Arch
Psychiatry Clin Neurosci. 269:741–752. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Damaskopoulou E, Papakonstantinou E,
Bacopoulou F, Eliopoulos E, Chrousos G and Vlachakis D: Child
abuse: Past, present and future (Review). World Acad Sci J.
5(4)2022.
|
|
12
|
NCBI Resource Coordinators. Database
resources of the national center for biotechnology information.
Nucleic Acids Res. 44:D7–D19. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Levine ME, Crimmins EM, Weir DR and Cole
SW: contemporaneous social environment and the architecture of
late-life gene expression profiles. Am J Epidemiol. 186:503–509.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zannas AS, Wiechmann T, Gassen NC and
Binder EB: Gene-stress-epigenetic regulation of FKBP5: Clinical and
translational implications. Neuropsychopharmacol. 41:261–274.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wochnik GM, Rüegg J, Abel GA, Schmidt U,
Holsboer F and Rein T: FK506-binding Proteins 51 and 52
differentially regulate dynein interaction and nuclear
translocation of the glucocorticoid receptor in mammalian cells. J
Biol Chem. 280:4609–4616. 2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ray LA, Sehl M, Bujarski S, Hutchison K,
Blaine S and Enoch MA: The CRHR1 gene, trauma exposure, and
alcoholism risk: A test of GxE effects: CRHR1, trauma exposure, and
alcoholism. Genes Brain Behav. 12:361–369. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Breen ME, Seifuddin F, Zandi PP, Potash JB
and Willour VL: Investigating the role of early childhood abuse and
HPA axis genes in suicide attempters with bipolar disorder.
Psychiatr Genet. 25:106–111. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Singh R, Yadav V, Kumar S and Saini N:
MicroRNA-195 inhibits proliferation, invasion and metastasis in
breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1.
Sci Rep. 5(17454)2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cecil CAM, Zhang Y and Nolte T: Childhood
maltreatment and DNA methylation: A systematic review. Neurosci
Biobehav Rev. 112:392–409. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Labonté B, Suderman M, Maussion G, Navaro
L, Yerko V, Mahar I, Bureau A, Mechawar N, Szyf M, Meaney MJ and
Turecki G: Genome-wide epigenetic regulation by early-life trauma.
Arch Gen Psychiatry. 69:722–731. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ma DQ, Rabionet R, Konidari I, Jaworski J,
Cukier HN, Wright HH, Abramson RK, Gilbert JR, Cuccaro ML,
Pericak-Vance MA and Martin ER: Association and gene-gene
interaction of SLC6A4 and ITGB3 in autism. Am J Med Genet B
Neuropsychiatr Genet. 153:477–483. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rehan W, Antfolk J, Johansson A, Aminoff
M, Sandnabba NK, Westberg L and Santtila P: Gene-environment
correlation between the dopamine transporter gene (DAT1)
polymorphism and childhood experiences of abuse. J Interpers
Violence. 33:2059–2072. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Klengel T, Mehta D, Anacker C, Rex-Haffner
M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS,
Bradley B, et al: Allele-specific FKBP5 DNA demethylation mediates
gene-childhood trauma interactions. Nat Neurosci. 16:33–41.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Binder EB: The role of FKBP5, a
co-chaperone of the glucocorticoid receptor in the pathogenesis and
therapy of affective and anxiety disorders.
Psychoneuroendocrinology. 34:S186–S195. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Heim C and Binder EB: Current research
trends in early life stress and depression: Review of human studies
on sensitive periods, gene-environment interactions, and
epigenetics. Exp Neurol. 233:102–111. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Monteiro P and Feng G: learning from
animal models of obsessive-compulsive disorder. Biol Psychiatry.
79:7–16. 2016.PubMed/NCBI View Article : Google Scholar
|