Introduction
Hearing loss is one of the most common sensory
disorders in humans. Approximately half of hearing loss cases have
a genetic etiology with autosomal dominant, autosomal recessive,
X-linked, or mitochondrial modes of inheritance (1), with autosomal recessive being the
most common. There are two monogenic forms of hearing loss
including syndromic or non-syndromic ones. Approximately 71 genes
have been described for non-syndromic hearing loss (as mentioned in
the hereditary hearing loss homepage, http://hereditaryhearingloss.org/). The gene most
frequently involved in autosomal recessive non-syndromic hearing
loss is GJB2, which encodes the gap junction protein
connexin 26 (Cx26) and is responsible for over half of the cases,
followed by SLC26A4, MYO15A, OTOF,
CDH23 and TMC1 (2).
Over 150 mutations, polymorphisms and unclassified variants have
been identified in the GJB2 gene (http://davinci.crg.es/deafness), some of which are
frequent, while others are extremely rare. These mutations occur at
different frequencies across populations (3), with c.35delG, c.167delT and
c.235delC predominating in Caucasian, Ashkenazi Jewish and East
Asian populations, respectively (4–8).
In addition, Pendred syndrome mutations in SLC26A4 account
for 10% of hereditary hearing loss in most world populations. In
China, almost 50% of patients with nonsyndromic hearing loss carry
the GJB2 or SLC26A4 mutations (8). Identification of these mutations is
of primary interest in genetic counseling. Although a large number
of cases are caused by hotspot mutations of these genes as revealed
by molecular epidemiologic studies, rare mutations may also
contribute to hearing loss.
In this study, we reported the identification of a
novel compound heterozygote with two missense mutations in the
GJB2 gene, and assessed the pathogenic effects of these
mutations based on bioinformatic structural analysis and the
subcellular localization of the compound heterozygous mutant Cx26
protein in HEK293 cells.
Materials and methods
Subjects and clinical examinations
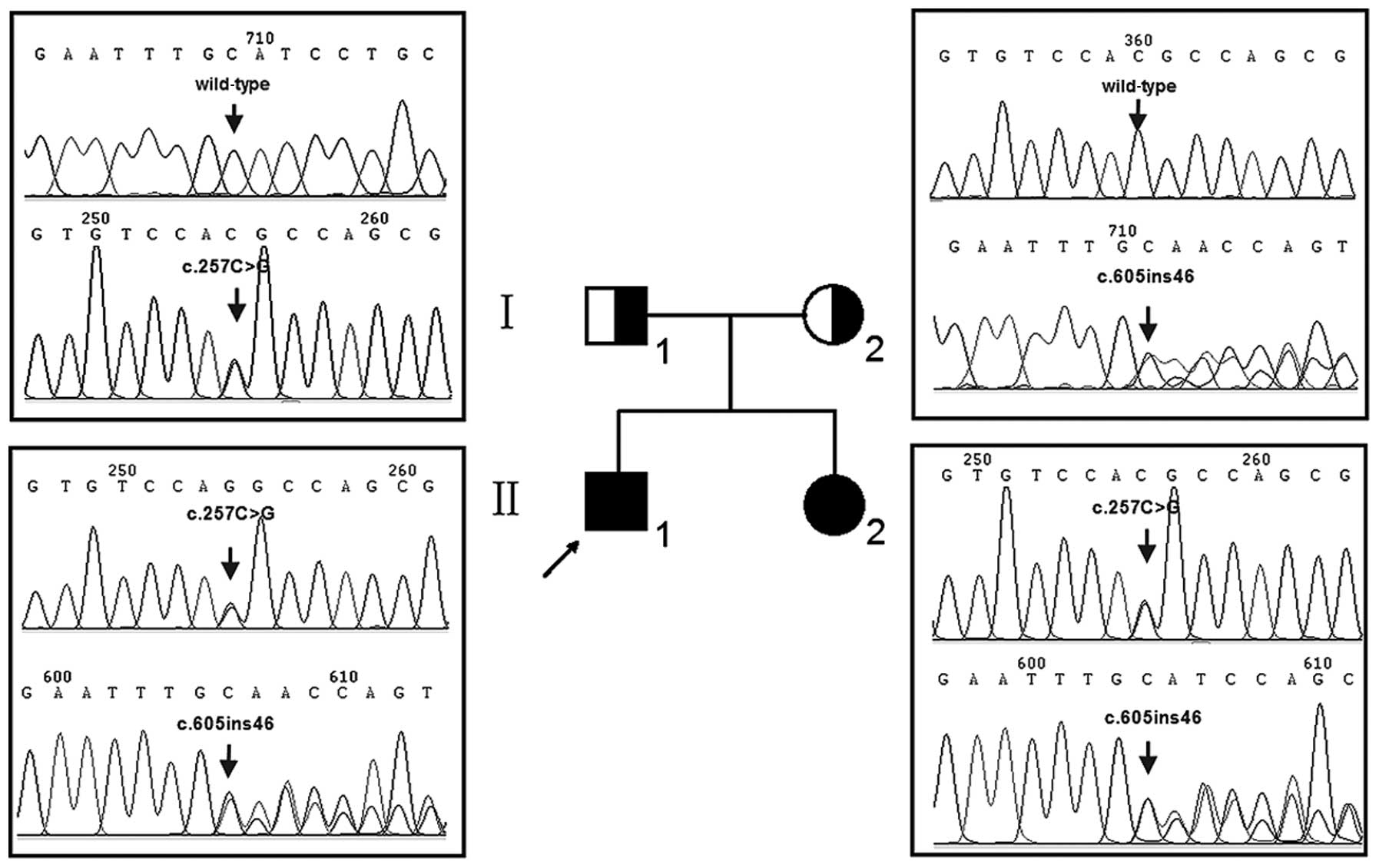
Two siblings (II-1 and II-2) (Fig. 1) of Chinese Han origin suffering
from prelingual hearing loss were referred to our departments for
clinical and molecular evaluation. Informed consent was obtained
from their parents prior to their participation in the study, which
was conducted in accordance with the Ethics Committee of the First
Affiliated Hospital of Nanjing Medical University. A comprehensive
history and physical examination were performed to identify any
syndromic findings, the history of the use of aminoglycosides, and
genetic factors related to hearing loss. Audiological studies
including pure tone audiometry, auditory brainstem response (ABR),
immittance and distortion product otoacoustic emissions (DPOAEs)
were conducted in a soundproof room. The pure-tone average was
calculated from the sum of the audiometric thresholds at 500, 1,000
and 2,000 Hz. The severity of hearing loss was classified into five
grades: normal, <26 decibel (dB); mild, 26–40 dB; moderate,
41–70 dB; severe, 71–90 dB; and profound, >90 dB.
Molecular analysis
Genomic DNA was isolated from EDTA-anticoagulated
blood samples of the two siblings and their parents using Puregene
DNA Isolation kits (Gentra Systems, Minneapolis, MN, USA). Nine
hotspot mutations of deafness genes present in Chinese populations
were screened by using a universal array approach, termed a
multiplex allele-specific PCR-based universal array (ASPUA), as
previously described (9). The
mutations included c.35delG, c.176del16bp, c.235delC and c.299delAT
in the GJB2 gene, c.538C>T in the GJB3 gene,
c.IVS7-2A>G and c. 2168A>G in the SLC26A4 gene, and
m.1555A>G and m.1494C>T in the RNR1 gene of
mitochondrial DNA (mtDNA). The participants were then subjected to
bidirectional sequencing of the coding region of the GJB2
gene to investigate the existence of possible rare or novel
pathogenic mutations (methods are available upon request). Samples
from 400 unrelated Chinese individuals with normal hearing were
collected served as controls.
Computer-assisted model building and
structure-based analysis
3D models of the human wild-type (WT) and mutant
Cx26 proteins were constructed using SWISS-MODEL (Basel,
Switzerland) (10–12). The SWISS-MODEL (http://swissmodel.expasy.org/) is a server for the
automated modeling of 3D protein structures, and the resulting
protein can be visualized and analyzed using visual molecular
dynamics (VMD) 1.9 (http://www.ks.uiuc.edu/Research/vmd/vmd-1.9/). By
comparing the 3D protein structures and Anolea mean force potential
energy of the WT and mutant Cx26 proteins, we evaluated the effect
of GJB2 mutations on the protein structure (13).
Molecular cloning of WT and mutant GJB2
genes
A WT human GJB2 sequence fragment cDNA was
subcloned into the pEGFP-N1 and pmCherry-N1 vectors to construct
Cx26-EGFP and Cx26-mCherry fusion proteins. The mutant GJB2
sequences were obtained from the genomic DNA of the proband
carrying the compound heterozygous mutation (c.257C>G/WT,
c.605ins46/WT). PCR was conducted using the primers that contained
HindIII and BamHI restriction sites: forward,
5′-GCGCAAGCTTTATGGATTGGGGCACGCT-3′ and reverse,
5′-GCGCGGATCCCTAACTGGCTTTTTTGAC-3′. The sequences of
Cx26-c.257C>G and Cx26-c.605ins46 were confirmed by DNA
sequencing.
Immunocytochemical analysis of Cx26 WT
and mutant in HEK293 cells
The HEK293 cell line, which is deficient for gap
junctions, was obtained from the American Type Culture Collection
(ATCC, Manassas, VA, USA). HEK293 cells were grown in the
recommended medium consisting of Dulbecco’s modified Eagle’s
medium, 10% fetal bovine serum (both from Invitrogen, Carlsbad, CA,
USA), and 0.5% penicillin and streptomycin. The plasmids encoding
WT or mutated Cx26 tagged with fluorescent protein markers were
transfected to the cells using X-tremeGENE HP transfection reagent
(Roche Diagnostics, Indianapolis, IN, USA) according to the
manufacturer’s instructions. The intracellular localization of WT
and mutant Cx26 proteins was investigated by immunocytochemical
analysis 48 h after transfection.
Results
Clinical and molecular analysis
The two children suffered from severe to profound
congenital hearing loss that was confirmed by consecutive previous
ABR tests. The patients presented to the Department of
Otolaryngology at the First Affiliated Hospital of Nanjing Medical
University at the ages of 13 and 16 years. Complete medical
histories showed that neither child had any abnormal history during
pregnancy or delivery, or had a history of exposure to
aminoglycosides prior to their deafness. Physical and otoscopic
examination failed to identify any syndromic findings. Unaided
audiometric testing indicated bilaterally symmetric and profound
sensorineural hearing loss in each of the two siblings, with normal
results for their parents. Other audiological examinations,
including immittance, ABR and DPOAEs, revealed cochlear
involvement.
Following molecular screening by ASPUA, hotspot
mutations in GJB2, SLC26A4, GJB3 and mtDNA
RNR1 genes were excluded as causative factors of the hearing
loss of the two children. Subsequent sequence analysis of the
GJB2 gene, however, revealed that both the profoundly deaf
children were compound heterozygotes for a previously unreported
combination of the GJB2 mutations, c.257C>G (p.T86R) and
c.605ins46 (Fig. 1). The parents
were each unaffected heterozygotes, the father a c.257C>G
heterozygote and the mother a c.605ins46 heterozygote. The
c.257C>G and c.605ins46 mutations were not detected in 400
Chinese controls.
Comparison of 3D structure and Anolea
mean force potential of WT and mutant Cx26 proteins
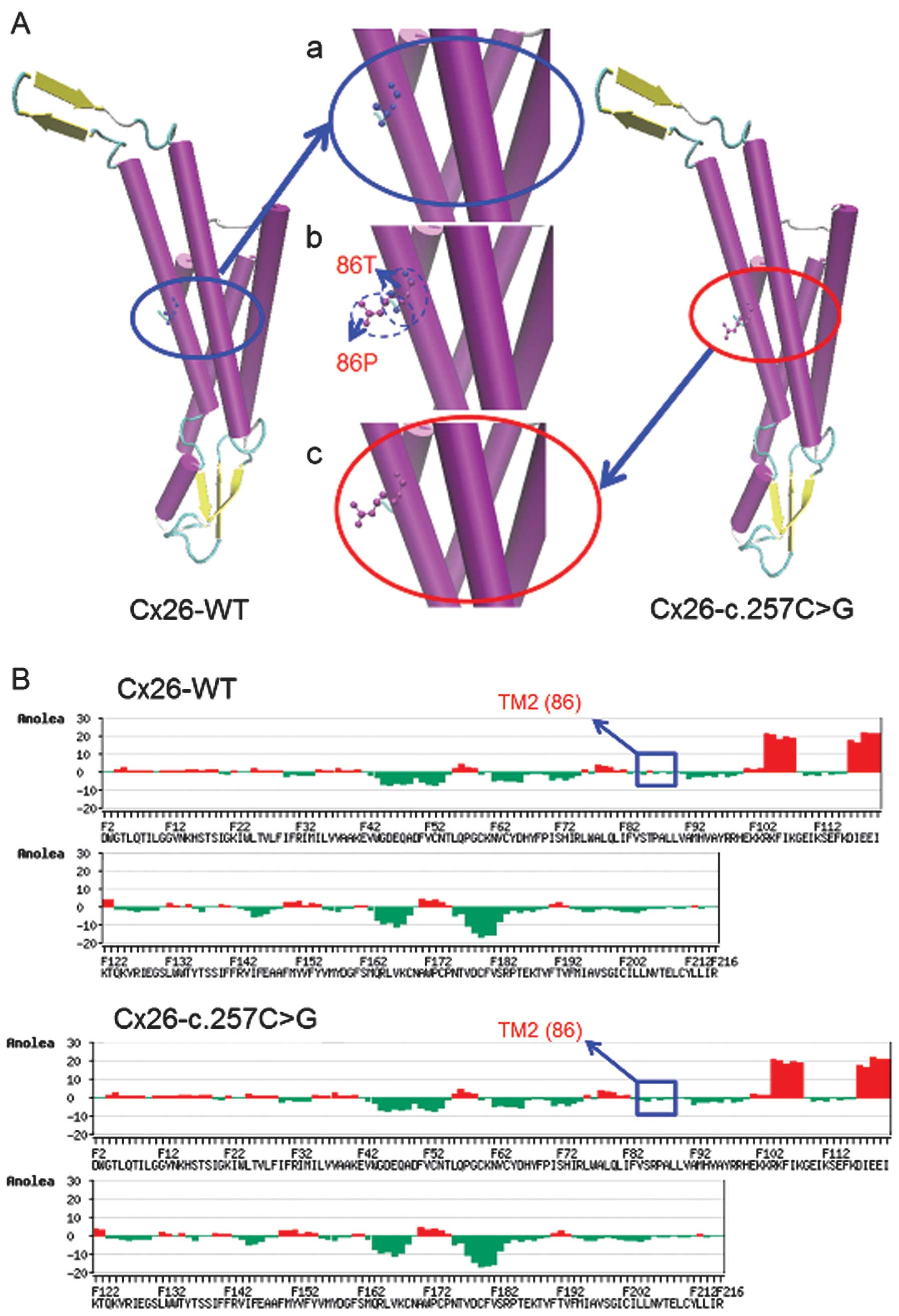
To understand the mechanism of the mutations at a
protein level, 3D models of WT and mutant Cx26 proteins were
constructed for bioinformatic structural analysis. The results
obtained by SWISS-MODEL demonstrated that the c.257C>G mutation
is located in transmembrane domain 2 (TM2) and the c.605ins46
mutation is in the TM4 of Cx26. These mutations may change the Cx26
structure both in transmembrane domains and in the extracellular
loop and may play a significant role in the development of hearing
loss.
Overlapping analysis of the 3D structures of the
Cx26-WT and Cx26-c.257C>G proteins revealed distinct changes at
residue 86 (Fig. 2A). Anolea mean
force potential energy analysis revealed one region with marked
changes (Fig. 2B). The
significant increase of atomic potential energy in this region may
change the Cx26 structure from a stable low-energy state to an
unstable higher-energy state. Overlapping analysis of the Cx26
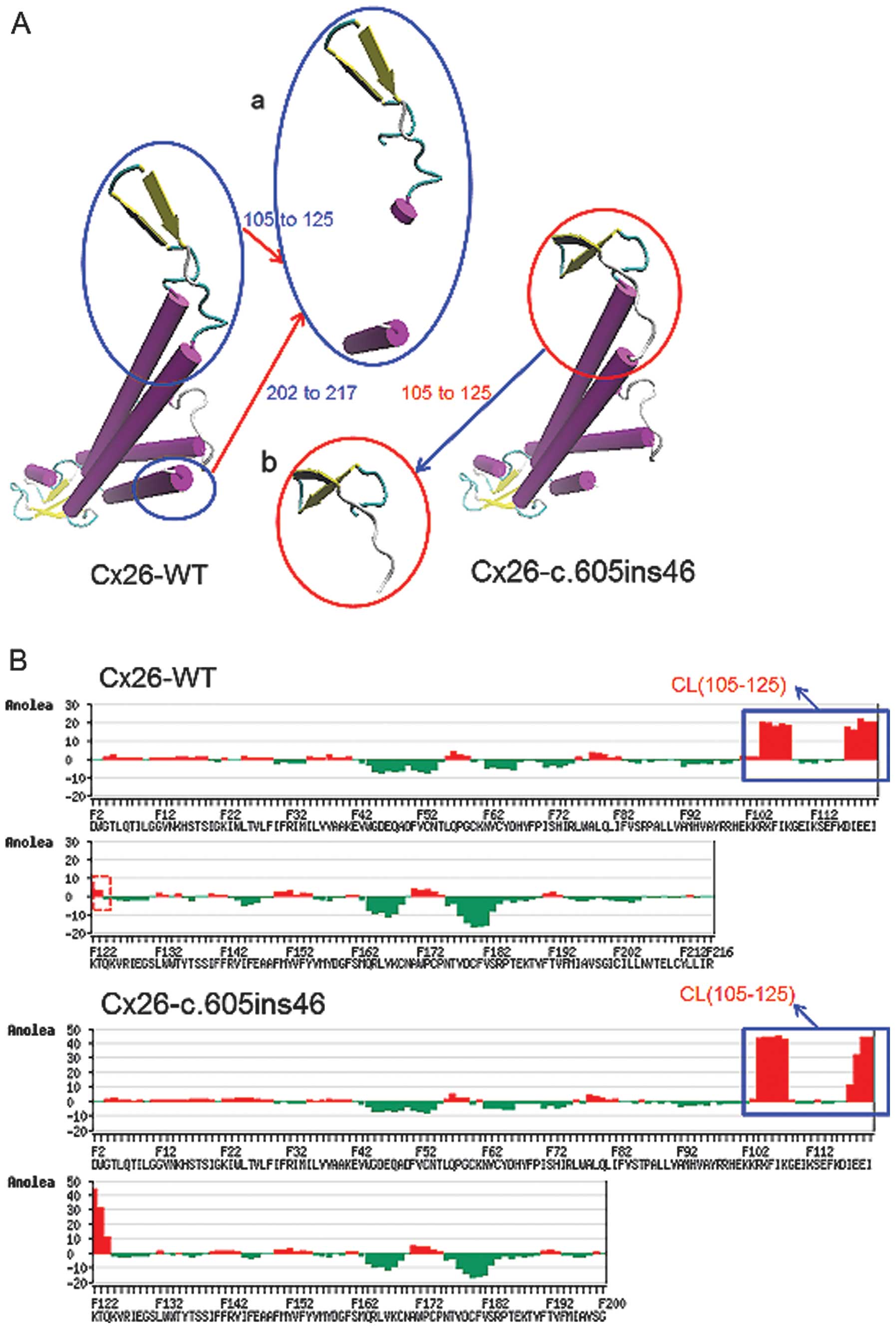
structure with the c.605ins46 mutant (Fig. 3A) also showed distinct changes of
spatial structure at residues 105–125 and 202–215, located in the
cytoplastic loop (CL) and TM4, respectively. Anolea mean force
potential energy analysis revealed two regions with marked changes
(Fig. 3B). As a result, the
c.257C>G and c.605ins46 mutations are capable of altering the
spatial structures of some amino acid residues in the transmembrane
domain, thereby preventing the effective functioning of Cx26.
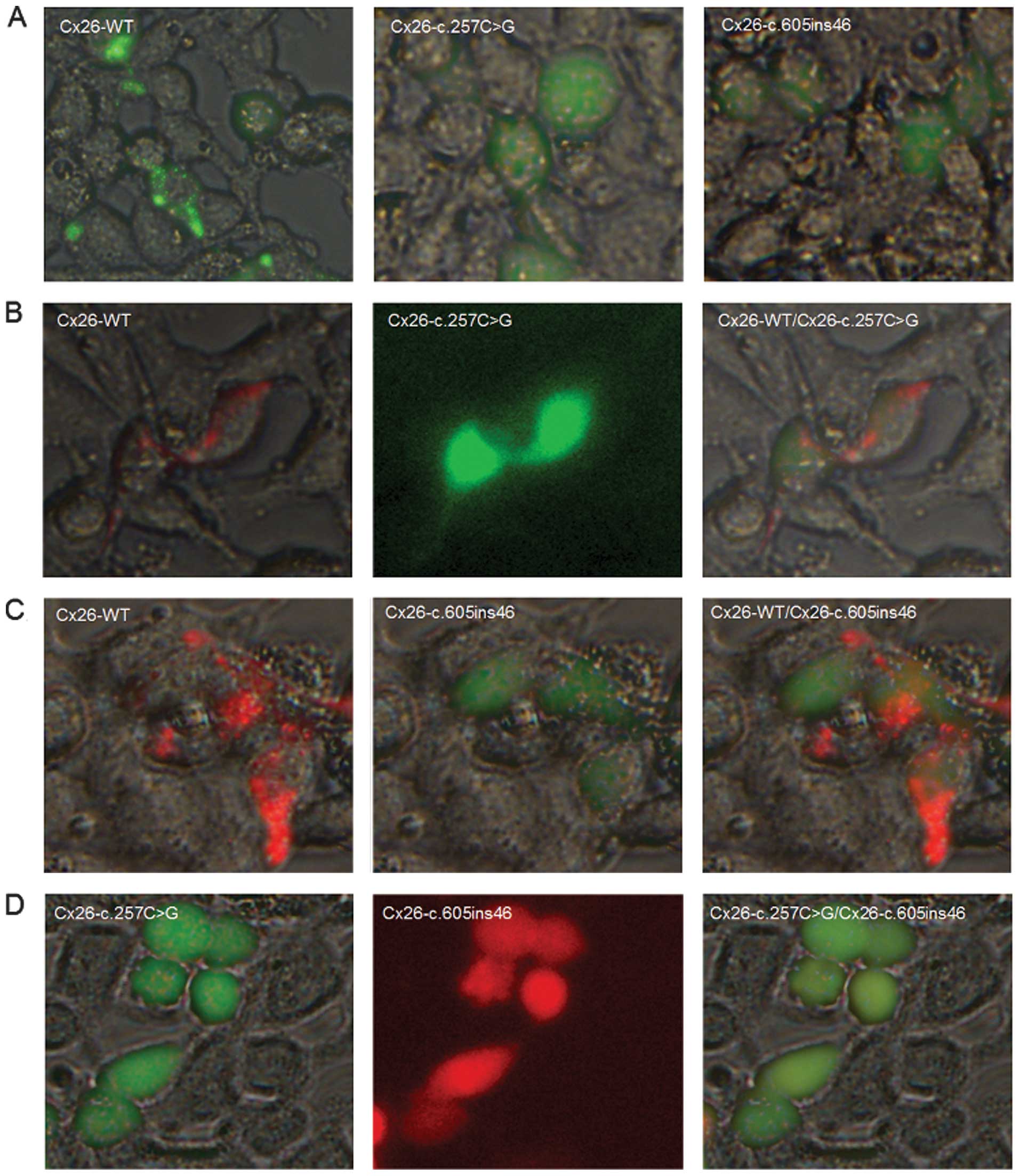
Cx26 protein expression and subcellular
localization in HEK293 cells
To examine the effects of the c.257C>G and
c.605ins46 mutations on the cellular localization of Cx26, we
transfected expression plasmids encoding WT or mutant Cx26 into
HEK293 cells. The proteins were fluorescently tagged to track their
intracellular locations. Cx26-WT localized at the cell membrane and
formed gap junctions, as indicated by the characteristic plaques
between two adjacent cells (Fig.
4A). By contrast, the Cx26-c.257C>G and Cx26-c.605ins46
mutants were not expressed at the cell membrane and lacked the
ability to form gap junctions. When the mCherry-tagged Cx26-WT
protein was co-transfected with the Cx26-c.257C>G mutant, the
mutant protein did not interact with Cx26-WT to co-assemble into
the same gap junction plaques, however, gap junction plaques were
still evident at the cell-cell contact areas (Fig. 4B). The Cx26-c.605ins46 mutant
behaved in a similar manner (Fig.
4C). However, the Cx26-c.257C>G protein was unable to
interact with the Cx26-c.605ins46 mutant to co-assemble into the
same gap junction plaques (Fig.
4D). Therefore, we suggest that the mechanism of hearing loss
caused by the c.257C>G/Cx26-c.605ins46 mutation is due to the
inability of the mutant Cx26 proteins to be transported to the cell
membrane.
Discussion
In the current study, we present a novel compound
heterozygous mutation in the GJB2 gene, the
c.257C>G/c.605ins46 mutation, which resulted in non-syndromic
sensorineural hearing loss in a Chinese family. Both of the
affected siblings suffered from prelingual hearing loss, while
their father and mother (who were each heterozygous for one of the
individual mutations) exhibited normal hearing function, suggesting
an autosomal recessive pattern of inheritance in this family. This
information may advance our understanding of the pathogenic
mechanism of GJB2 mutations associated with hearing
loss.
c.257C>G and c.605ins46 are rare GJB2
mutations that have previously been reported to segregate with
hearing loss exclusively in East Asian populations, either
homozygously (8,14–16) or as part of compound heterozygous
mutations with other more prevalent mutations such as c.235delC and
c.299delAT (8). It is
theoretically a very improbable event for these two rare mutations
to occur in one patient. To the best of our knowledge, this is the
first international study to demonstrate that the compound
heterozygous mutations c.257C>G and c.605ins46 are associated
with non-syndromic recessive hearing loss in a family. Results of
this study demonstrate the limitations of screening as only the
most prevalent mutations of the GJB2 gene were used to
identify causative factors in hearing loss patients. Therefore, it
is preferable to sequence the entire coding sequence of this gene
when a genetic cause cannot be excluded.
GJB2 mutations have been shown to cause
variable hearing loss phenotypes even among family members
(17,18). However, the c.257C>G/c.605ins46
compound heterozygous state led to profound deafness in the two
affected siblings in the studied family. The c.257C>G mutation,
which is located in the second transmembrane domain of Cx26,
converts an uncharged amino acid (threonine) at codon 86 to a
positively charged amino acid (arginine) and produces a
functionally null protein (14).
It is believed that the hearing loss caused by this mutation stems
from its inability to localize to the cell membrane (16). c.605ins46, a frame-shift mutation,
is sited in the fourth transmembrane domain of Cx26 and has a
tandem repeat of 46 nucleotides at position 605. A stop codon (TGA)
is introduced at the 202nd amino acid, leading to premature
termination of polypeptide synthesis. To clarify the function of
these proteins and the pathological mechanisms of the mutations, we
constructed a 3D characterization of protein structures (13,19). Based on 3D modeling and Anolea
mean force potential energy analysis (10–13,20), we investigated the molecular
mechanisms of the c.257C>G/c.605ins46 compound heterozygous
mutation in the GJB2 gene by analyzing the protein structure
of Cx26. The c.257C>G/c.605ins46 mutation caused changes in the
spatial structure of neighboring amino acids, preventing Cx26 from
functioning efficiently (21–23). In the current study, protein
localization and gap junction function were investigated by
transfecting fluorescently tagged WT or mutant Cx26 into HEK293
cells, which allowed visual confirmation of homozygous or
heterozygous mutant gap junctions. When the Cx26-c.605ins46 or the
Cx26-c.257C>G mutant was transfected together with the Cx26-WT
protein, gap junction plaques were observed at cell-cell contact
areas, although the mutant proteins were unable to interact with
Cx26-WT to co-assemble into the same gap junction plaques. However,
the Cx26-c.257C>G mutant was unable to interact with the
Cx26-c.605ins46 mutant to co-assemble into gap junction plaques. A
similar biological effect would be expected if the above event
occurred in the hair cells of the inner ear, in other words, that
the individual’s normal hearing function would be affected.
In conclusion, to the best of our knowledge, this is
the first description of the compound c.257C>G/c.605ins46
mutation of GJB2 in a Chinese family with non-syndromic
hearing loss. The c.605ins46 mutation causes a frame-shift and
premature termination at amino acid 202, while c.257C>G is a
missense mutation converting codon 86 from threonine (T) to
arginine (R). Further bioinformatic structural analysis and
cell-based functional assays indicated the pathogenic mechanism of
this compound heterozygous mutation in the GJB2 gene
associated with hearing loss. This information is valuable for
understanding the pathogenic roles of Cx26 mutations associated
with hearing loss and for determining their genetic diagnosis.
Acknowledgements
This study was supported by a grant from the Jiangsu
Health Administration (LJ201120), by a research grant award from
the National Natural Science Foundation of China (no. 31171217),
and by a grant funded by the Priority Academic Program Development
of Jiangsu Higher Education Institutions.
References
|
1
|
Morton CC: Genetics, genomics and gene
discovery in the auditory system. Hum Mol Genet. 11:1229–1240.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hilgert N, Smith RJ and Campa G: Forty-six
genes causing nonsyndromic hearing impairment: which ones should be
analyzed in DNA diagnostics. Mutat Res. 681:189–196. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kenneson A, Van Naarden Braun K and Boyle
C: GJB2 (connexin 26) variants and nonsyndromic sensorineural
hearing loss: a HuGE review. Genet Med. 4:258–274. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Estivill X, Fortina P, Surrey S, et al:
Connexin-26 mutations in sporadic and inherited sensorineural
deafness. Lancet. 351:394–398. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morell RJ, Kim HJ, Hood LJ, et al:
Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with
nonsyndromic recessive deafness. N Engl J Med. 339:1500–1505. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abe S, Usami S, Shinkawa H, Kelley PM and
Kimberling WJ: Prevalent connexin 26 gene (GJB2) mutations in
Japanese. J Med Genet. 37:41–43. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sobe T, Vreugde S, Shahin H, et al: The
prevalence and expression of inherited connexin 26 mutations
associated with nonsyndromic hearing loss in the Israeli
population. Hum Genet. 106:50–57. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dai P, Yu F, Han B, et al: GJB2 mutation
spectrum in 2,063 Chinese patients with nonsyndromic hearing
impairment. J Transl Med. 7:262009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li CX, Pan Q, Guo YG, et al: Construction
of a multiplex allele-specific PCR-based universal array (ASPUA)
and its application to hearing loss screening. Hum Mutat.
29:306–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL workspace: a web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwede T, Kopp J, Guex N and Peitsch MC:
SWISS-MODEL: An automated protein homology-modeling server. Nucleic
Acids Res. 31:3381–3385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guex N and Peitsch MC: SWISS-MODEL and the
Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis. 18:2714–2723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maeda S, Nakagawa S, Suga M, Yamashita E,
Oshima A, Fujiyoshi Y and Tsukihara T: Structure of the connexin 26
gap junction channel at 3.5 A resolution. Nature. 458:597–602.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee KY, Choi SY, Bae JW, et al: Molecular
analysis of the GJB2, GJB6 and SLC26A4 genes in Korean deafness
patients. Int J Pediatr Otorhinolaryngol. 72:1301–1309. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi SY, Park HJ, Lee KY, et al: Different
functional consequences of two missense mutations in the GJB2 gene
associated with non-syndromic hearing loss. Hum Mutat.
30:E716–E727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuge I, Ohtsuka A, Matsunaga T and Usami
S: Identification of 605ins46, a novel GJB2 mutation in a Japanese
family. Auris Nasus Larynx. 29:379–382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murgia A, Orzan E, Polli R, et al: Cx26
deafness: mutation analysis and clinical variability. J Med Genet.
36:829–832. 1999.PubMed/NCBI
|
|
18
|
Cohn ES, Kelley PM, Fowler TW, et al:
Clinical studies of families with hearing loss attributable to
mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics.
103:546–550. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang WH, Liu YF, Su CC, Su MC, Li SY and
Yang JJ: A novel missense mutation in the connexin30 causes
nonsyndromic hearing loss. PLoS One. 6:e214732011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anfinsen CB: Principles that govern the
folding of protein chains. Science. 181:223–230. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han SH, Park HJ, Kang EJ, Ryu JS, Lee A,
Yang YH and Lee KR: Carrier frequency of GJB2 (connexin-26)
mutations causing inherited deafness in the Korean population. J
Hum Genet. 53:1022–1028. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mani RS, Ganapathy A, Jalvi R, et al:
Functional consequences of novel connexin 26 mutations associated
with hereditary hearing loss. Eur J Hum Genet. 17:502–509. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pollak A, Skórka A, Mueller-Malesińska M,
et al: M34T and V37I mutations in GJB2 associated hearing
impairment: evidence for pathogenicity and reduced penetrance. Am J
Med Genet. 143A:2534–2543. 2007. View Article : Google Scholar : PubMed/NCBI
|