Introduction
Inflammatory ocular responses are associated with
the pathophysiology of several retinal degenerative diseases,
including uveitis (1), diabetic
retinopathy (2), and age-related
macular degeneration (AMD) (3).
In the eye, the retinal pigment epithelium (RPE), which is a
pigmented layer of the neural retina, provides metabolic support to
the photoreceptors that provide visual functionality (4). This epithelium is in the unique
position to sense the circulating immune system status and has both
macrophage and microglia-like activities in the retina (5,6).
The RPE plays a critical role in innate immunity through the
expression of pattern recognition receptors (PRRs), such as
toll-like receptors (TLR), to detect the unique molecular patterns
associated with microbial pathogens prior to evoking RPE cell
inflammatory responses via the production of inflammatory mediators
(7). If this response is
prolonged, then the subsequent atrophy of RPE and photoreceptors
may occur, which is the leading cause of legal blindness.
Activation of an inflammatory response upon
encountering a pathogen cause the release of pro-inflammatory
mediators. One such pro-inflammatory mediator is the phospholipid
platelet-activating factor (PAF,
1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine), which acts via its
G protein-coupled receptor to stimulate numerous complex signaling
pathways, producing diverse biological actions (8). Bacterial pathogen
lipopolysaccharides (LPS) are known inducers of numerous
pro-inflammatory events, including the production of PAF (9). PAF is a lipid molecule involved in
inflammatory processes and cell-to-cell communication. PAF is
produced by a variety of cells that may be involved in the
development of the inflammatory reaction, such as
monocytes/macrophages, polymorphonuclear neutrophils (PMN),
eosinophils, basophils, and platelets (10–12). PAF is rapidly hydrolyzed into
lyso-PAF, which is an inactive phospholipid, by specific enzymes
with PAF-AH (13). PAF-AH
hydrolyzes the oxidized phospholipids that are structurally similar
to PAF and therefore exerts anti-inflammatory activity (14).
Peroxisome proliferator-activated receptors (PPAR)
are members of the nuclear receptor superfamily with at least three
identified subtypes (PPARα, PPARδ and PPARγ) (15). PPARγ are important in a variety of
biological processess, including adipogenesis, glucose metabolism,
and inflammation (16). Of the
various PPARγ ligands,
15-deoxy-δ12,14-prostaglandin J2
(15d-PGJ2) has exhibited a potent immuno-modulatory
effect on several cell types, including monocytes/macrophages,
microglia, astrocytes, neutrophils and lymphocytes (17–19). RPE cells express all three forms
of PPARs, although PPARβ is the dominant isoform (20). In particular, PPARγ may be
important for processing the lipids generated during the
phagocytosis of the outer segments of photoreceptors in RPE cells
(20). In earlier studies,
15d-PGJ2 attenuated the degree of inflammation via
modulation of the production of cytokines, chemokines, and adhesion
molecules (21,22). Therefore, 15d-PGJ2 may
be a therapeutic candidate for ocular inflammatory diseases. Human
RPE is clinically involved in many ocular inflammatory diseases. In
this study, we investigated whether 15d-PGJ2 was capable
of attenuating ocular inflammatory responses and elucidate its
regulatory molecular mechanisms for the RPE cells that are induced
by LPS.
Materials and methods
Cell culture and reagents
Human ARPE19 retinal pigment epithelial cells (RPE)
were obtained from the American Type Culture Collection (Manassas,
VA, USA). The cells were grown to confluency in a standard
incubator in Dulbecco’s MEM/Nut MIX F-12 medium (Gibco, Paisley,
UK) including 10% fetal bovine serum, penicillin/streptomycin
(Gibco/BRL, Gaithersburg, MD, USA), and 2 mM glutamine (Life
Technologies, UK). LPS was purchased from Sigma-Aldrich (St. Louis,
MO, USA). The reverse transcription polymerase chain reaction
(RT-PCR) reagents were purchased from Promega (Madison, WI, USA).
The light shift chemiluminescent electrophoretic mobility shift
assay reagents, nuclear and cytoplasmic extraction reagents, and
the enhanced chemiluminescence (ECL)-detecting reagent were
purchased from Pierce (Rockford, IL, USA). Rabbit anti-mouse p65
and IκBα antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Antibodies against ERK, phosphor
(p)-ERK 1/2, p38, p-p38, JNK/SAPK, p-JNK/SAPK, and p-IκBα were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Determination of cell viability
The cell viability of the ARPE19 cells was
determined using the Cell Counting Kit-8 (CCK-8) according to the
manufacturer’s instructions (Dojindo Laboratories, Kumamoto,
Japan). Briefly, the cells were plated onto 96-well plates at a
density of 1xl04 cells/well. Subsequently, 10 μl of
CCK-8 reagent was added to each well followed by incubation for 2
h. The amount of CCK-8 reagent that was reduced to yield formazan
via cellular dehydrogenase indicated whether the cell was viable.
The results were measured by reading the absorbance at 450 nm in a
96-well plate reader (Model EL800, BioTek Instruments, Inc.,
Winooski, VT, USA). The absorbance reading was subtracted from the
background control and reported as the mean of three
measurements.
RT-PCR
The ARPE19 cells were plated overnight in 6-well
culture plates at a density of 2×105 cells/well, and the
cells were incubated in a serum-free medium for at least 4 h prior
to treatment. Total RNAs were isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s
instructions. The cDNA was generated using ImProm-II Reverse
Transcription System (Promega) before being amplified via PCR with
specific primers for interleukin-6 (IL-6) (forward, 5′-GAT GGC TGA
AAA AGA TGG ATG C-3′; reverse, 5′-TGG TTG GGT CAG GGG TGG TT-3′),
monocyte chemoattractant protein-1 (MCP-1) (forward, 5′-AAT GCC CCA
GTC ACC TGC TGT TAT-3′; reverse, 5′-GCA ATT TCC CCA AGT CTC TGT
ATC-3′), and intercellular adhesion molecule-1 (ICAM-1) (forward,
5′-ACT TTC CCA CTG CCC ATC GG-3′; reverse, 5′-GTG GCT TGT GTG TTC
GGT TTC A-3′). After the amplification process, sections of the PCR
reactions were subjected to agarose gel electrophoresis.
Enzyme-linked immunosorbent assay
(ELISA)
Cytokine levels were determined by ELISA. ELISA kits
purchased from BioLegend (San Diego, CA, USA) were used to measure
IL-6 and MCP-1 levels, and a kit obtained from R&D Systems
(Minneapolis, MN, USA) was used to measure ICAM-1 levels. The
absorbance at 450 nm was determined using a microplate reader
(Model EL800, BioTek Instruments, Inc.).
PAF-acetylhydrolase (PAH-AH) assay
Cytosolic PAF-AH levels were determined with PAF-AH
assay kits purchased from Cayman Chemical (Ann Arbor, MI, USA).
Briefly, cells (1×105 cells/ml, in 60-mm culture dishes)
were collected by centrifugation for 10 min at 4°C followed by
sonication of the cell pellet in 1 ml of cold buffer (0.1 M
Tris-HCl, pH 7.2). The mixture was centrifuged at 10,000 × g for 15
min at 4°C. The supernatant was removed and 10 μl of DTNB was added
to it, followed by addition of 10 μl of cell lysate, and 5 μl of
assay buffer (0.1 M Tris-HCl, pH 7.2) to the wells. The wells were
incubated for 30 min at room temperature and 200 μl of substrate
solution was added to all of the wells. The 96-well plate was
agitated for 30 sec. The absorbance at 412 nm was determined using
a microplate reader (SpetraMax M2, Molecular Devices, Sunnyvale,
CA, USA).
Western blot analysis
The ARPE19 cells were plated overnight in a 100-mm
culture dish at a density of 1×106 cells/dish.
Subsequently, the cells were incubated in serum-free medium for at
least 4 h before treatment. The cells were washed three times with
PBS buffer before being lysed in a lysis buffer (1% Triton X-100,
1% deoxycholate, 0.1% NaN3) containing protease
inhibitor cocktail tablets (Roche Diagnostics, Mannheim, Germany).
Equal amounts of protein were separated on 10% SDS polyacrylamide
mini-gels and transferred to a nitrocellulose transfer membrane
(Whatman, Florham Park, NJ, USA). Following incubation with the
appropriate primary antibodies, the membranes were incubated for 1
h at room temperature with a secondary antibody conjugated to
horseradish peroxidase. After three washes in TBST, the
immunoreactive bands were visualized using the ECL detection system
(Pierce). In a parallel experiment, a nuclear protein was prepared
using nuclear extraction reagents (Pierce) according to the
manufacturer’s instructions.
Preparation of nuclear extracts and
EMSA
Nuclear extracts were prepared with the NE-PER
nuclear extraction reagent (Pierce). An oligonucleotide containing
the immunoglobulin κ-chain binding site (κB, 5′-CCGGTT
AACAGAGGGGGCTTTCCGAG-3′) was synthesized as a probe for the gel
retardation assay, and the probe was labeled with biotin (Pierce).
The binding reactions contained 5 μg of nuclear extract protein,
buffer (10 mM Tris, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM
dithiothreitol, 0.05% Nonidet P-40, and 2.5% glycerol), 50 ng of
poly-(dI-dC), and a 20 fM solution of biotin-labeled DNA. The
reactions were incubated for 20 min at room temperature at a final
volume of 20 μl. During the competition reactions, the nuclear
extracts were incubated for 15 min at room temperature with a
competing cold oligonucleotide (100-fold excess) before the
addition of a labeled probe. The reaction mixture was
electrophoretically analyzed in a 5% polyacrylamide gel with 0.5X
Tris-borate EDTA buffer. The reactions were transferred to nylon
membranes and the biotinylated DNA was detected using a LightShift
chemiluminescent EMSA kit (Pierce).
Statistical analysis
Statistical analyses were conducted using the
Student’s t-test. The results are presented as the means ± SD of at
least three separate experiments. P<0.05 was considered to be
statistically significant.
Results
Effects of 15d-PGJ2 on IL-6,
MCP-1, and ICAM-1 production in LPS-stimulated ARPE19
Initially, to exclude the possibility that the
inhibition of the production of inflammatory mediators was caused
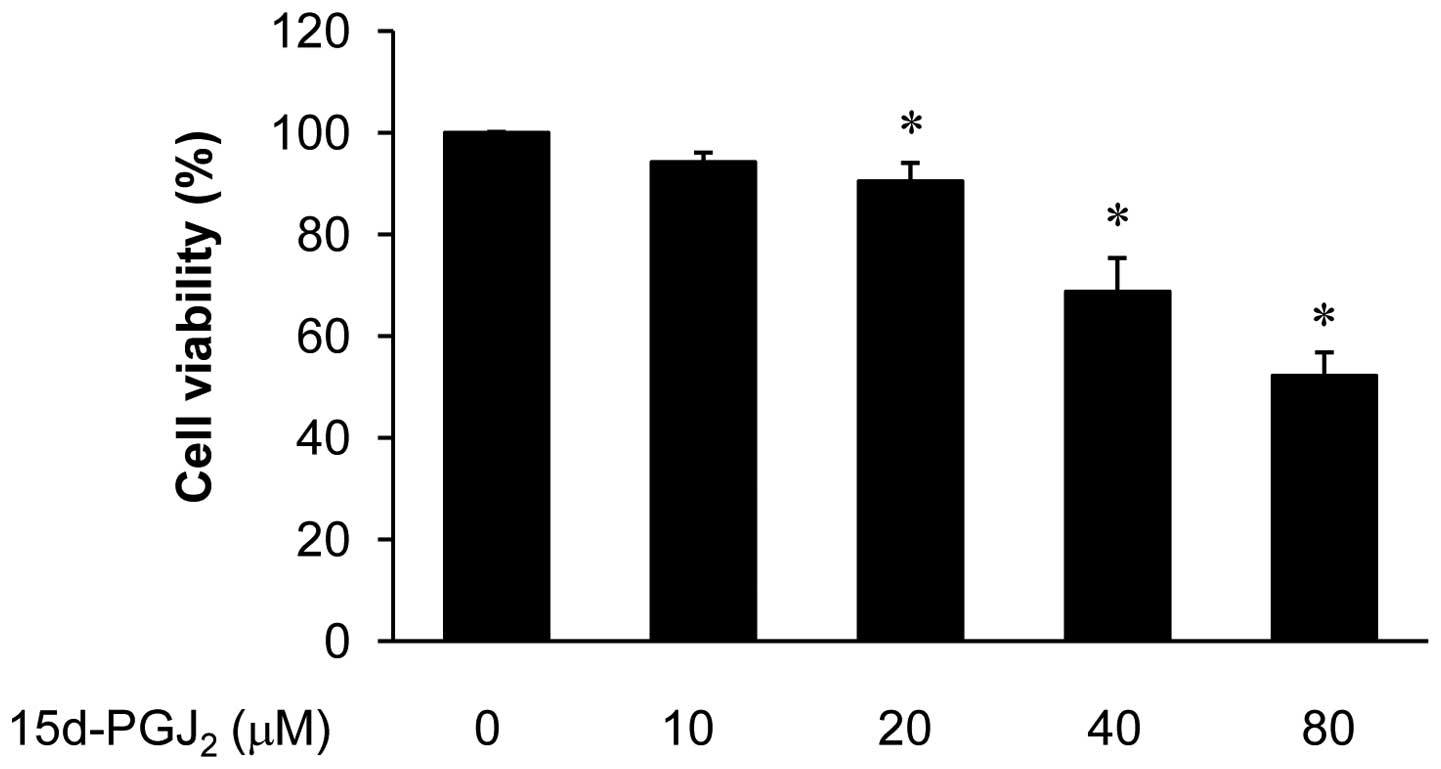
by the cytotoxity of 15d-PGJ2, we performed CCK-8 assays
in ARPE19 cells treated with 15d-PGJ2 (10–80 μM)
(Fig. 1). A decrease in cell
survival was detected at concentrations from 40 to 80 μM. The
concentrations (10–20 μM) of 15d-PGJ2 alone did not
affect cell viability in this investigation. Therefore,
concentrations of 10–20 μM 15d-PGJ2 were used in
subsequent experiments.
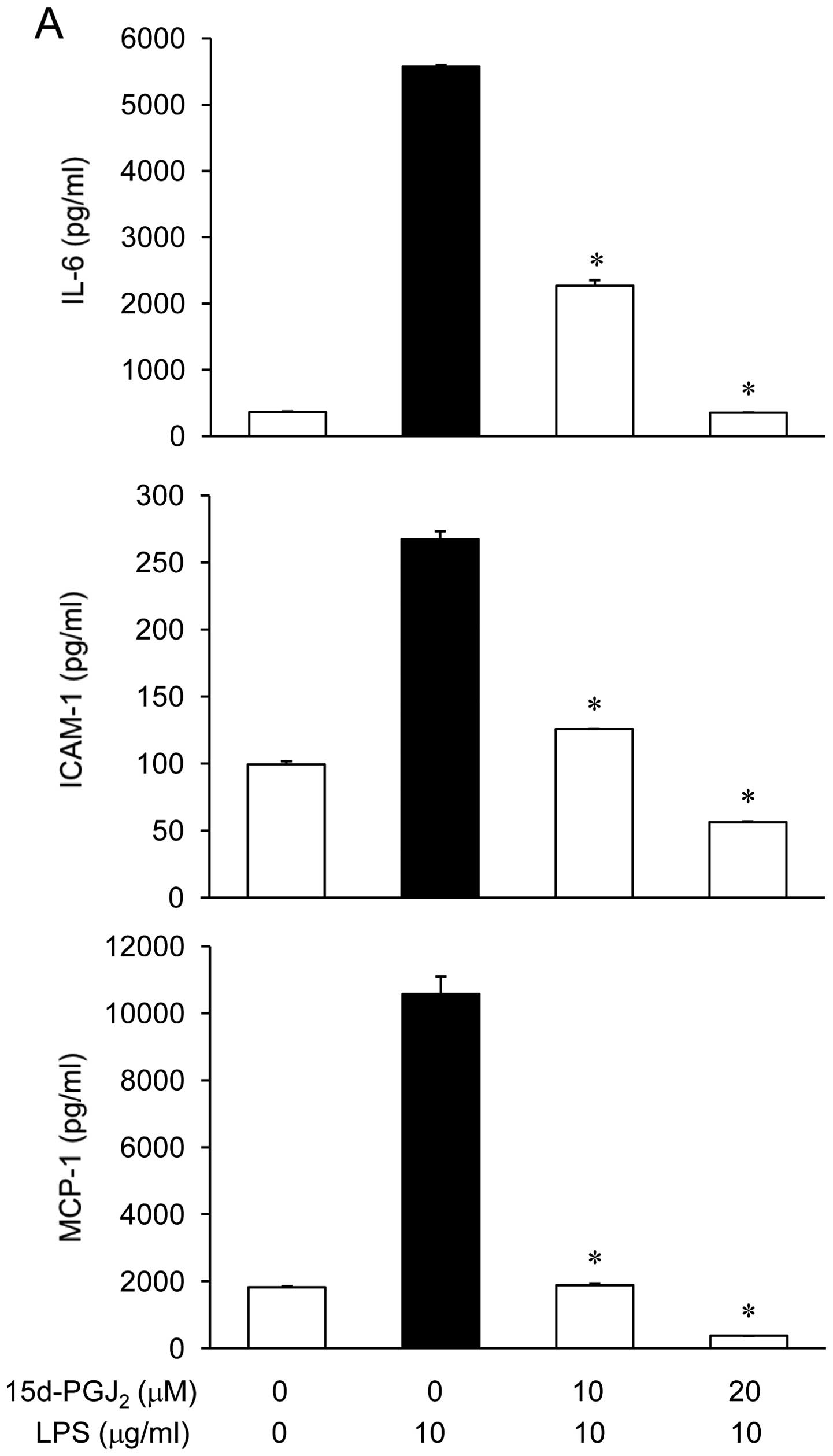
The effect of 15d-PGJ2 on IL-6, MCP-1,
and ICAM-1 production in LPS-stimulated ARPE19 cells was examined.
The ARPE19 cells were incubated with 15d-PGJ2 (10 or 20
μM) in the presence of LPS (10 μg/ml) for 24 h, and the mediator
levels in the culture media were measured via ELISA. As shown in
Fig. 2A, the IL-6, MCP-1, and
ICAM-1 levels were increased in the culture media from the
LPS-stimulated ARPE19. These increases were significantly decreased
in a concentration-dependent manner by treatment with
15d-PGJ2. In a parallel experiment, RT-PCR was performed
to determine whether 15d-PGJ2 inhibits the expression of
these mediators at the transcriptional level. As shown in Fig. 2B, the treatment of ARPE19 cells
with different concentrations of 15d-PGJ2 2 h prior to
LPS treatment caused a dose-dependent decrease in the IL-6, MCP-1,
and ICAM-1 mRNA. These results suggested that 15d-PGJ2
acts primarily by preventing the expression of IL-6, MCP-1, and
ICAM-1 at the transcriptional level. Therefore, the results
indicated that 15d-PGJ2 inhibited the expression of
these mediators, which are involved in the inflammatory process. We
further explored the mechanism of inhibitory action for
15d-PGJ2.
Relationship between 15d-PGJ2
levels and PAF on IL-6, MCP-1, and ICAM-1 expression
The effect of CV-3988 on IL-6, MCP-1, and ICAM-1
production in LPS-stimulated ARPE19 cells was examined. The PAF
released in response to LPS is a major contributor to the
pathological events associated with numerous pro-inflammatory
events. PAF may be a major mediator of retinal inflammation.
Rosenbaum et al reported that intravitreal injection of PAF
induces retinitis in experimental animals (23). To validate the above information,
we examined the relationship between 15d-PGJ2 levels and
the repression of PAF. In order to exclude the possibility that the
inhibition of inflammatory mediators production was caused by the
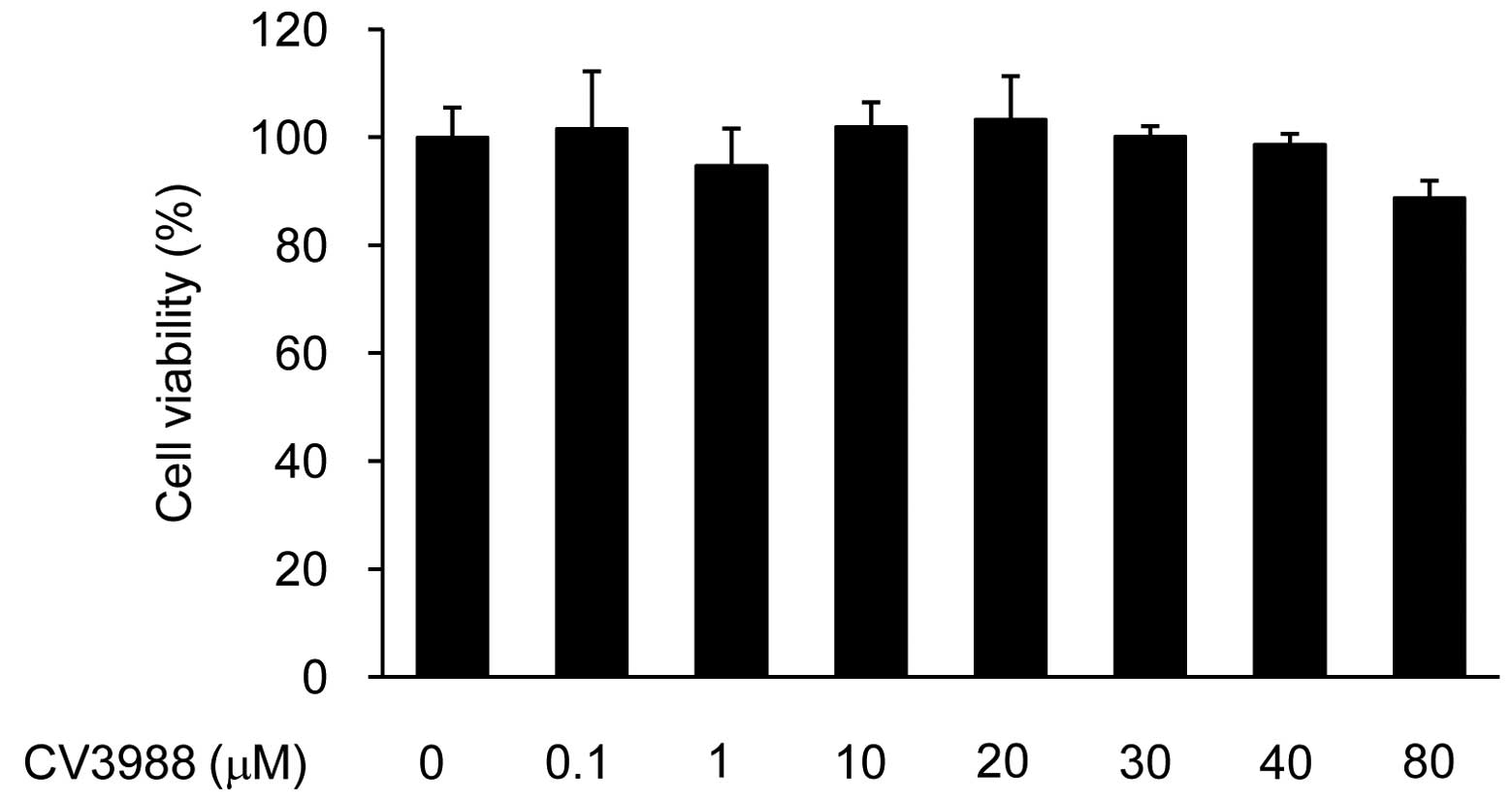
cytotoxity of CV-3988, we initially performed CCK-8 assays in
ARPE19 cells treated with CV-3988 (0.1–80 μM) (Fig. 3). A decrease in cell survival was
detected at 80 μM. The concentrations (10–40 μM) CV-3988 alone did
not affect cell viability in this investigation. Therefore,
concentrations of 10–40 μM CV-3988 were used in the subsequent
experiments.
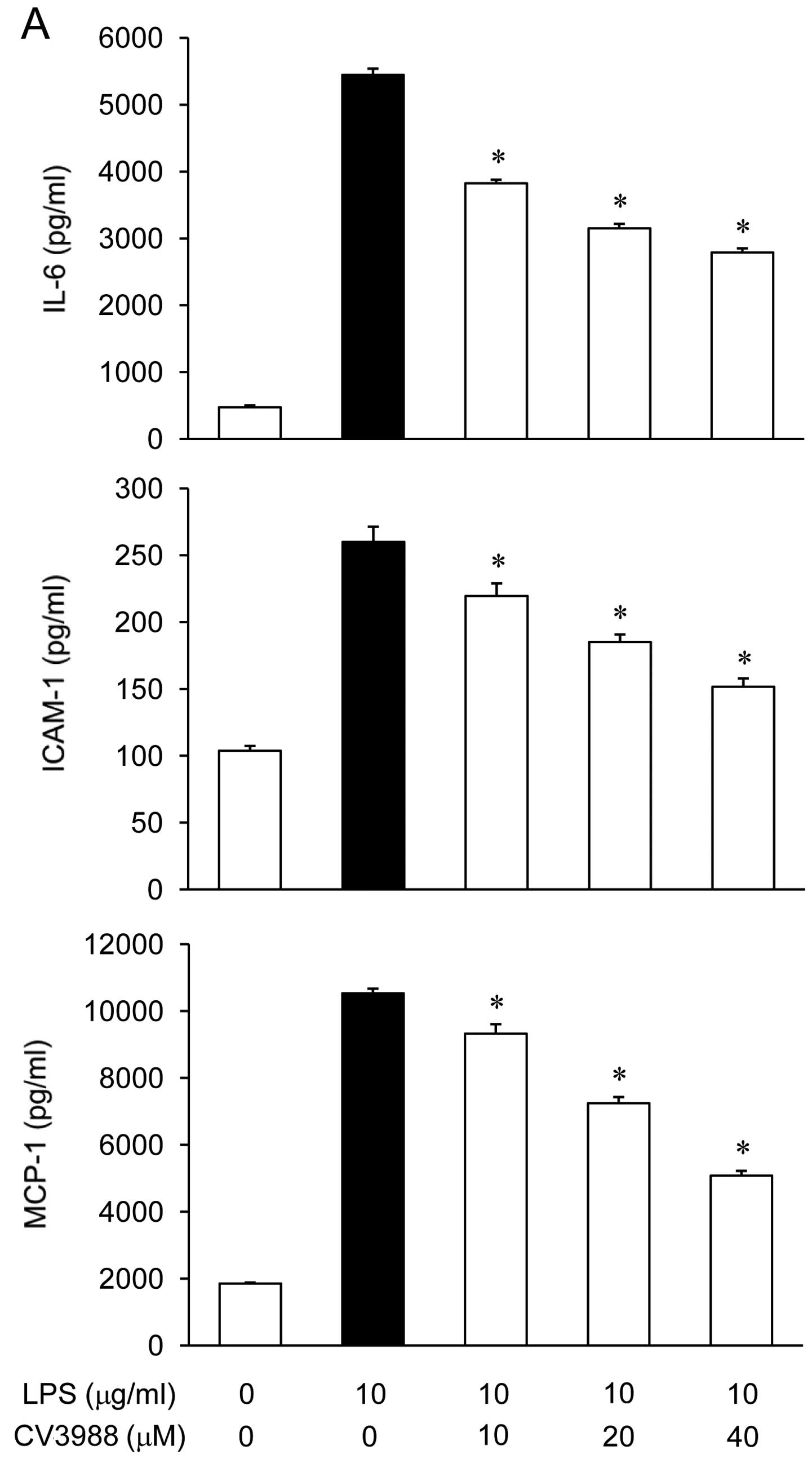
ARPE-19 cells were incubated with CV-3988 (10, 20 or
40 μM) in the presence of LPS (10 μg/ml) for 24 h, and the mediator
levels in the culture media were measured via ELISA. As shown in
Fig. 4A, the IL-6, MCP-1, and
ICAM-1 levels were increased in the culture media of LPS-stimulated
ARPE19 cells, and these increases were significantly decreased in a
concentration-dependent manner by treatment with CV-3988.
Bulger et al demonstrated that treatment of
alveolar macrophages with PAF-acetylhydrolase (PAF-AH) in
vitro caused significant inhibition of the cytokine response to
the endotoxin (24). To determine
whether 15d-PGJ2 and CV-3988 were capable of
upregulating PAF-AH activity in ARPE19 cells, the cells were
treated with 15d-PGJ2 (20 μM) and CV-3988 (40 μM) in the
presence of LPS (10 μg/ml) for 24 h, and then PAF-AH activity in
the cytosols was measured. As shown in Fig. 4B, the PAF-AH activity decreased in
response to LPS treatment in the cultured ARPE19 cells, and these
decreases were reversed by the administration of
15d-PGJ2 and CV-3988. The results suggested that the
anti-inflammatory effects of 15d-PGJ2 are generated via
enhancement of PAH-AH activity.
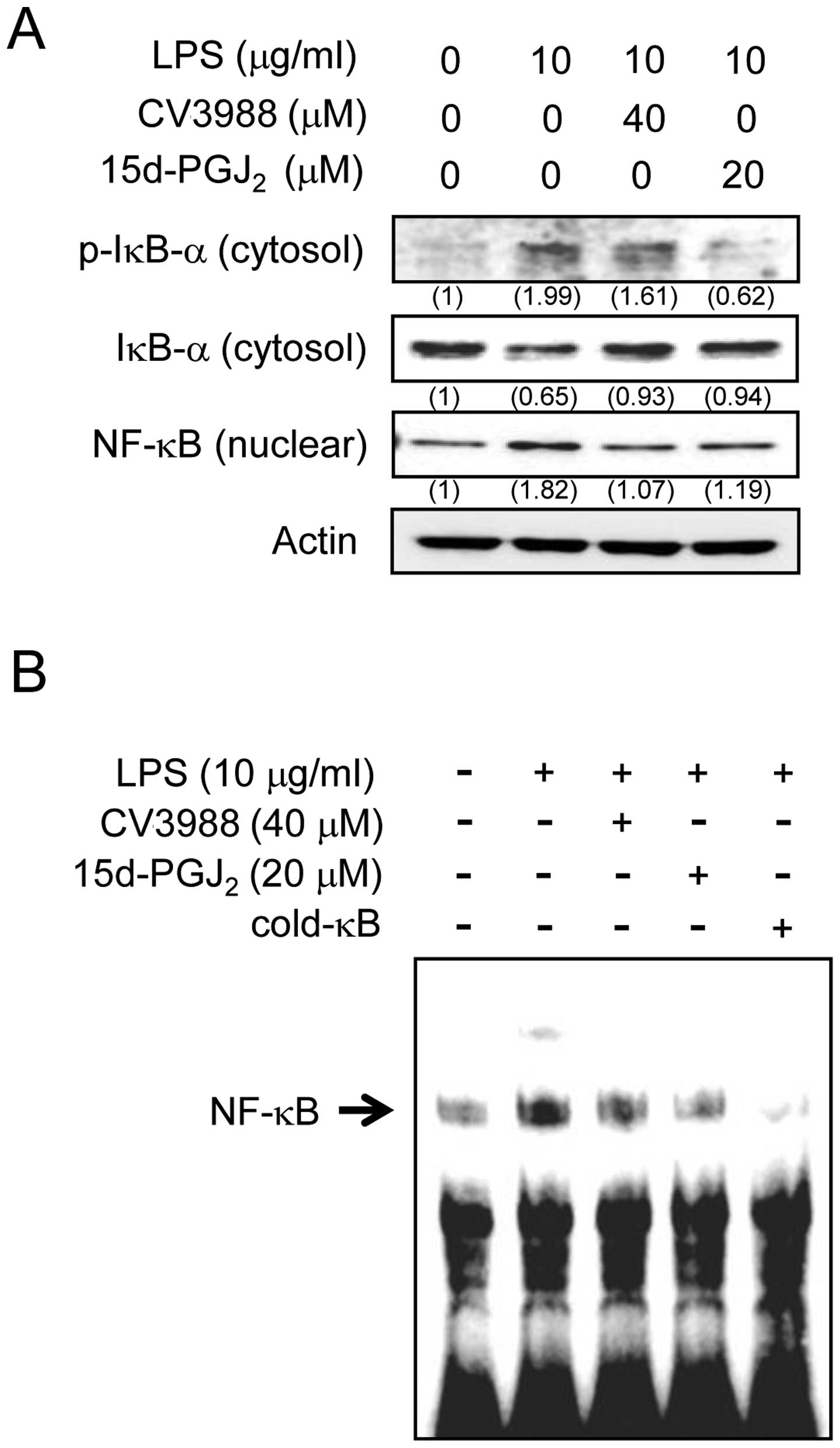
Effects of 15d-PGJ2 on the
activation of NF-κB in LPS-stimulated ARPE19 cells
Since 15d-PGJ2 inhibited the expression
of the inflammatory mediators in ARPE19 cells, we examined the
mechanism of inhibitory action for 15d-PGJ2. Activation
of nuclear transcription factor-κB (NF-κB) is necessary to induce
the IL-6, MCP-1, and ICAM-1 genes. Therefore, using western blot
and EMSA, we investigated whether 15d-PGJ2 acts on NF-κB
activity. The effect of 15d-PGJ2 on LPS-induced IκBα
phosphorylation and degradation was also examined. The
imunoblotting results shown in Fig.
5A reveal that the LPS-induced IκBα phosphorylation/degradation
were inhibited after 1 h of exposure to 15d-PGJ2.
Additionally, 15d-PGJ2 inhibited the LPS
exposure-induced translocation of the NF-κB p65 subunit from the
cytosol to the nucleus. To further characterize the mechanism, the
effect of 15d-PGJ2 on the DNA-binding activity of NF-κB
was determined by EMSA (Fig. 5B).
LPS treatment caused a significant increase in the DNA-binding
activity of NF-κB. By contrast, treatment with 15d-PGJ2
markedly reduced the LPS-induced DNA-binding activity of NF-κB.
Additionally, pretreatment with a PAF antagonist significantly
abrogated the LPS-induced NF-κB activation. When combined, these
results suggested that 15d-PGJ2 may inhibit NF-κB
activation in ARPE19 cells by suppressing the IκBα
phosphorylation/degradation, as well as the binding of NF-κB.
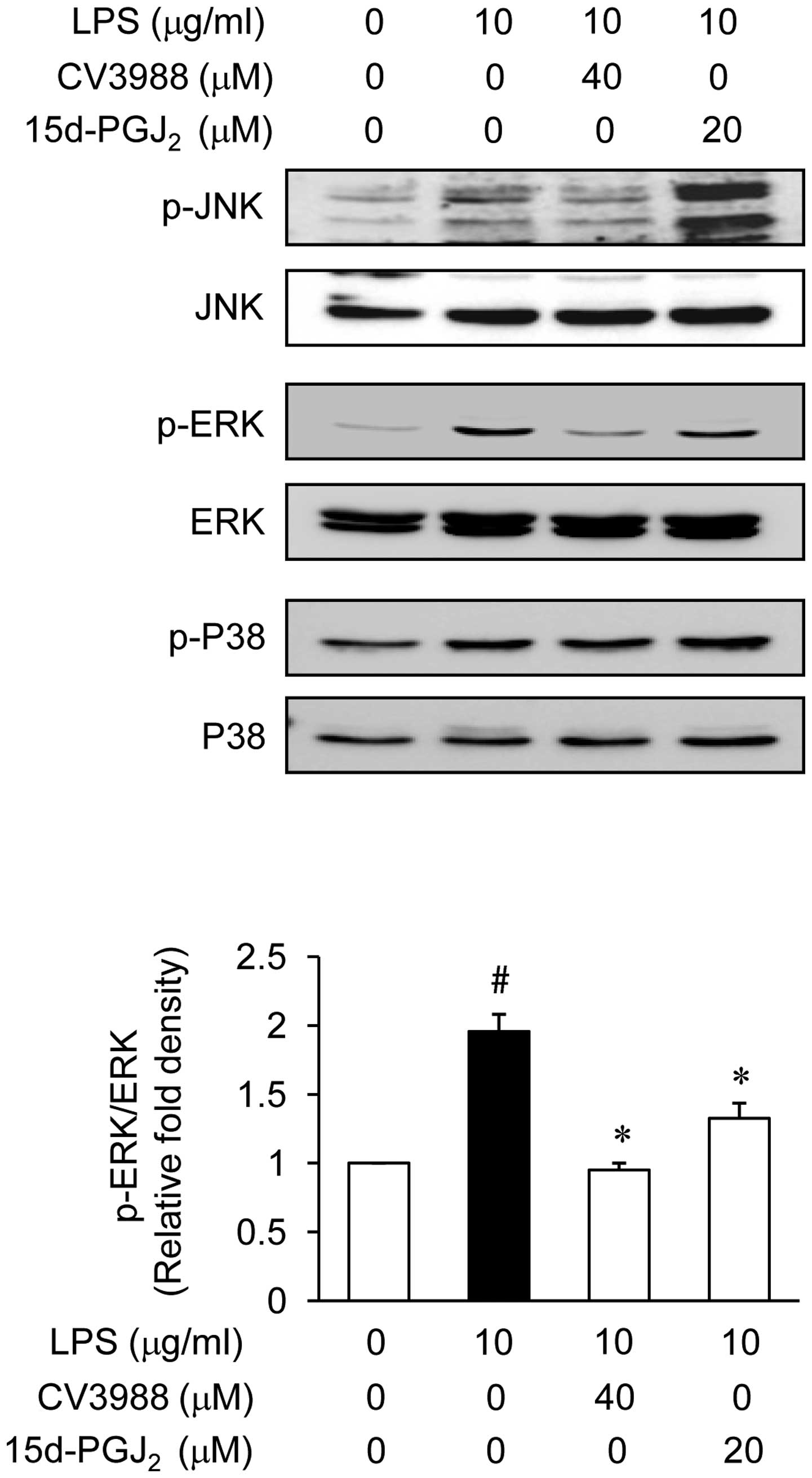
Effect of 15d-PGJ2 on the
phosphorylation of MAP kinases in LPS-stimulated ARPE19
Experiments were designed to elucidate the signaling
cascades that regulate the expression of the inflammatory mediators
in ARPE19 cells in responding to LPS stimulation. MAP kinases are
important for the expression of IL-6, MCP-1 and ICAM-1. MAP kinases
therefore act as a specific target for inflammatory responses. To
investigate whether the inhibition of inflammation by
15d-PGJ2 is regulated by the MAP kinase pathway, we
examined the effect of 15d-PGJ2 on LPS-induced
phosphorylation of ERK, JNK, and p38 kinase in ARPE19 cells using
western blot analysis. We first demonstrated that ERK, JNK, and p38
kinase were phosphorylated following ARPE19 cell stimulation with
LPS, and then examined the effect of 15d-PGJ2 on the
LPS-induced activation of MAP kinases. As shown in Fig. 6, 15d-PGJ2 (20 μM)
markedly inhibited ERK activation, while the phosphorylation of JNK
and the p38 kinase were not affected. The total amount of ERK was
not affected by treatment with LPS or 15d-PGJ2
treatment. These results suggested that the ERK pathways are
relevant during the LPS-mediated expression of IL-6, MCP-1, and
ICAM-1.
15d-PGJ2 inhibition of the
LPS-stimulated IL-6, MCP-1, and ICAM-1 production via
PPARγ-independent pathways
To verify whether the action of 15d-PGJ2
was PPARγ-dependent or -independent, we examined the effects of
GW9662, which is a potent, irreversible and selective PPARγ
antagonist, on the expression of IL-6, MCP-1, and ICAM-1. Fig. 7 shows that, in the presence of
LPS, GW9662 did not reverse the inhibitory effect of
15d-PGJ2 on the expression of IL-6, MCP-1, and ICAM-1.
Additionally, we compared the inhibitory effects of different PPARγ
agonists on the LPS-stimulated production of IL-6, MCP-1, and
ICAM-1. 15d-PGJ2 inhibited the LPS-stimulated production
of IL-6, MCP-1, and ICAM-1, but other PPARγ agonists, such as
troglitazone and rosiglitazone, did not inhibit the LPS-stimulated
production of IL-6, MCP-1, and ICAM-1 in ARPE19 cells. These
results supported the hypothesis that 15d-PGJ2 regulates
the LPS-stimulated production of IL-6, MCP-1, and ICAM-1 via
PPARγ-independent mechanisms.
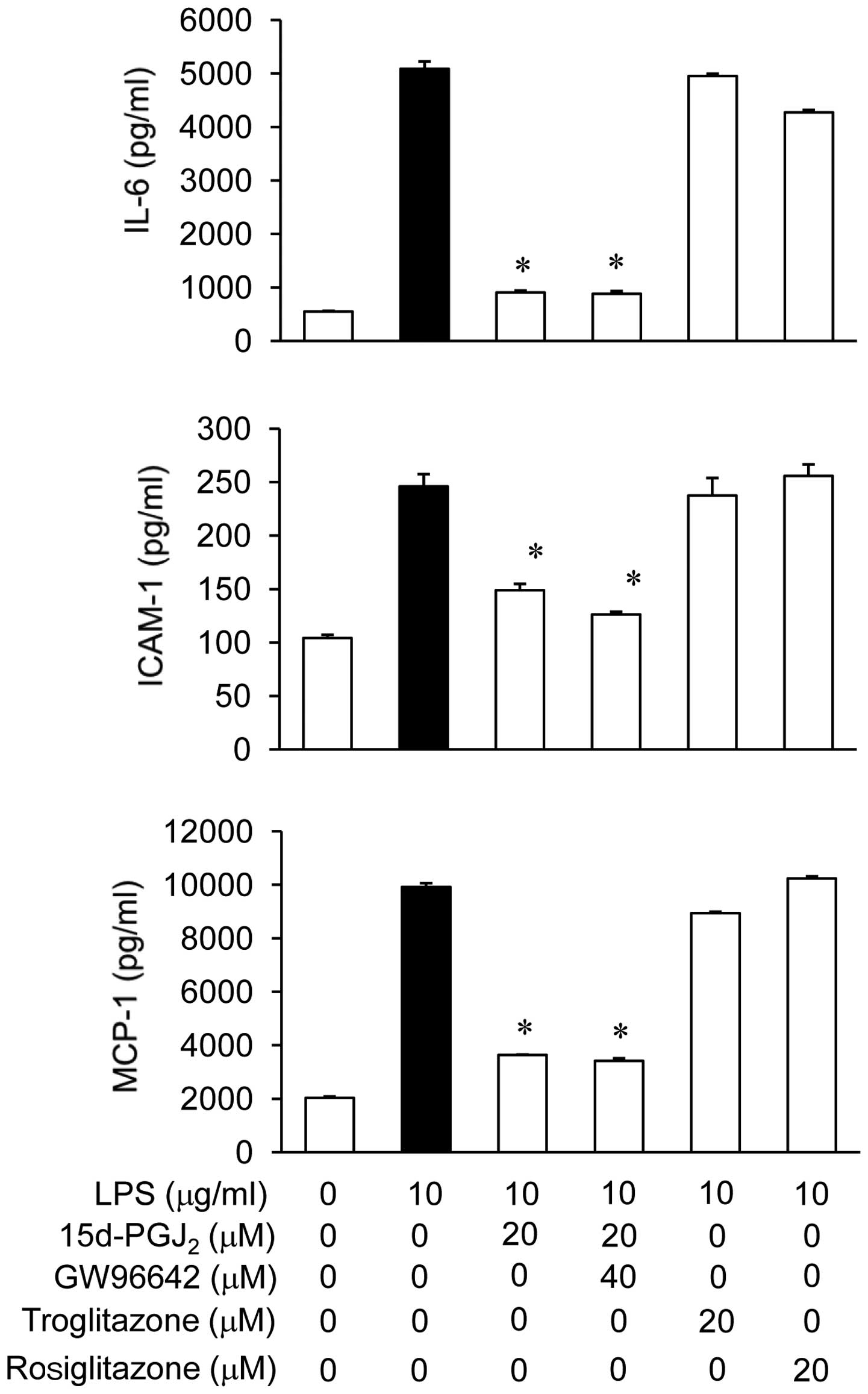 | Figure 7Effects of
15-deoxy-δ12,14-prostaglandin J2
(15d-PGJ2) on lipopolysaccharide (LPS)-stimulated
interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1),
and intercellular adhesion molecule-1 (ICAM-1) production in ARPE19
cells via PPARγ-independent mechanisms. The ARPE19 cells were
pretreated with PPARγ agonists, such as troglitazone,
rosiglitazone, or 15d-PGJ2, at the indicated
concentrations for 2 h, followed by stimulation with
lipopolysaccharide (LPS) (10 μg/ml) for 24 h. In addition, the
cells were pretreated with a specific PPARγ antagonist, GW9662, at
40 μM for 0.5 h followed by treatment with 15d-PGJ2 for
2 h; subsequently, they were stimulated with LPS at the indicated
concentrations. Twenty-four hours later, the cultured supernatants
were isolated and analyzed for IL-6, MCP-1, and ICAM-1 levels using
commercial ELISA kits. *P<0.05 indicates a
significant difference from the value obtained for cells treated
with only LPS. The experiments are reported as the mean
representative of three independent experiments. |
Discussion
The present study was undertaken to elucidate the
anti-inflammatory effects and mechanisms of 15d-PGJ2 on
the production of inflammatory mediators in RPE cells stimulated
with LPS. In particular, we investigated whether the
anti-inflammatory effects of 15d-PGJ2 were associated
with PAF activation. The bacterial LPS activates the RPE cells to
produce and release potent inflammatory mediators, including IL-6,
MCP-1, and ICAM-1. The results suggest that 15d-PGJ2 is
an effective inhibitor of the LPS-induced inflammatory mediators
through the blockade of the NF-κB and mitogen-activated protein
kinase (MAPK) pathways via PAF release in the retinal pigment
epithelium cell line, ARPE19. The inhibitory effect of
15d-PGJ2 on the inflammatory mediator expression
suggests efficacy, which is responsible for its anti-inflammatory
action, as well as its potential use as a therapeutic agent for
treating LPS-stimulated ocular diseases.
Bacterial LPS can elicit acute ocular inflammation
in animals and lead to uveitis, various degrees of degeneration of
the retina, and loss of vision (1,25).
The RPE is a major component of the blood retinal barrier and
controls the nutrient flow to the photoreceptors. It has been
reported that the bacterial endotoxin activates the RPE to enhance
the expression of various inflammatory mediators, such as IL-6,
MCP-1, and ICAM-1 (26,27). IL-6 is an important mediator of
the acute-phase response and possesses biological activities that
support host immune reactions. The pro-inflammatory cytokine IL-6
is an important mediator of inflammation and has chemotactic
activity for neutrophils and macrophages, in addition to activating
T lymphocytes, stimulating the secretion of immunoglobulin, and
triggering the release of acute phase proteins (28,29). The local production of IL-6 by
resident cells and infiltrating inflammatory cells has been
detected during a variety of inflammatory ocular conditions
(30,31). MCP-1 is overexpressed in human
eyes during acute anterior uveitis and is known to have strong
chemotactic activity for monocytes/macrophages (32). Retinal detachment-induced
photoreceptor apoptosis has been associated with MCP-1 (33). ICAM-1 expression is upregulated in
the iris and ciliary bodies following LPS application (34). Leukocyte adhesion to the vessel
walls is an important process during inflammation. When leukocytes
are recruited to inflammatory sites, the adhesion molecules play
essential roles during the first step of inflammation. Therefore,
the downregulation of IL-6-, MCP-1-, and ICAM-1-mediated migration
and adhesion of leukocytes may lead to the suppression of ocular
inflammation. In the present study, the application of
15d-PGJ2 reduced the retinal expression of IL-6, ICAM-1,
and MCP-1 (Fig. 2), suggesting
that the anti-inflammatory effects of 15d-PGJ2 on the
eye resulted from inhibition of the inflammation-related
molecules.
In a previous report, when LPS was injected
intracamerally, PAF was detected in the aqueous humor and found to
enhance the intraocular inflammation (35). In this study, we found that LPS
induces the mRNA expression of the PAF receptor in ARPE19 cells. In
addition, the LPS-induced expression of the PAF receptor mRNA was
completely inhibited by the PAF receptor antagonist, CV3988.
Therefore, we hypothesized that the anti-inflammatory effects of
15d-PGJ2 may be mediated through inhibition of the PAF
secretion. To prove this hypothesis, we investigated the
relationship of 15d-PGJ2 and PAF with the expression of
inflammatory mediators (IL-6, MCP-1, and ICAM-1) in LPS-stimulated
ARPE19 cells. We initially examined whether CV3988 reduced the
expression of these inflammatory mediators in the presence of LPS
in the ARPE19 cells. As demonstrated in Fig. 4A, the expression levels of the
inflammatory LPS-induced mediators were significantly suppressed
when the cells were pretreated with 20 and 40 μM CV3988 for 2 h
prior to LPS stimulation for 24 h. To elucidate whether
15d-PGJ2 is related to the PAF response, we examined
PAF-acetylhydrolase (PAH-AH) activity in the presence of LPS. In a
previous study, the PAF-AH protein was upregulated by
15d-PGJ2 (13). This
previous finding, in accordance with our data, indicated that the
decreased activities of PAH-AH, which were stimulated by LPS, were
significantly enhanced when the cells were pretreated with 20 μM
15d-PGJ2 and 40 μM CV3988 for 2 h prior to LPS
stimulation for 24 h (Fig. 4).
This suggest that 15d-PGJ2 induces PAF-AH, leading to
the prevention of inflammatory reactions in RPE cells, considering
that PAF-AH rapidly hydrolyzed PAF to form lyso-PAF.
Bacterial LPS stimulate the transcription of several
genes involved in inflammatory and immune responses, including the
NF-κB and MAPK pathways. Previously, NF-κB and MAPK signaling
pathways were demonstrated to be involved in the anti-inflammatory
responses of various primary human ocular cells (36). To determine the mechanisms by
which 15d-PGJ2 inhibited the production of inflammatory
mediators, we examined its effects on LPS-induced NF-κB activation
(Fig. 5). NF-κB is a pleiotropic
regulator of various genes involved in cell responses to infection
and participates in inflammatory responses, leading to organ
dysfunction and death in patients with sepsis (37). In addition, PAF is a potent
inducer of NF-κB activity. IκBα is one of the inhibitor proteins
that binds to NF-κB. Degradation of IκBα after its phosphorylation
is required to translocate NF-κB to the nucleus.
15d-PGJ2 represses the NF-κB transcriptional activity
(38). Our findings show that 20
μM 15d-PGJ2 significantly inhibits the LPS-stimulated
IκBα phosphorylation/degradation and nuclear translocation of p65,
as well as the DNA binding activity of NF-κB in ARPE19 cells.
Therefore, inhibiting the NF-κB signaling pathways in RPE cells
with 15d-PGJ2 may cause the downregulation of
inflammatory mediators, generating an anti-inflammatory effect. The
involvement of various intracellular signaling pathways, such as
MAPKs, is needed to induce and maintain the inflammatory process.
Activation of these kinases leads to the nuclear translocation of
the NF-κB. Previous studies have revealed that MAPKs have a
significant role in the regulation of IL-6, MCP-1, and ICAM-1
production in LPS-stimulated ocular cells (36). Therefore, experiments were
performed to determine whether the 15d-PGJ2 tightly
regulates the expression of MAPKs to induce anti-inflammatory
effects in LPS-stimulated ARPE19 cells. In the present study,
15d-PGJ2 inhibits the activation of ERK, which is
induced by LPS stimulation in ARPE19 cells (Fig. 6). Therefore, the inhibition of ERK
and the NF-κB signaling pathways in ARPE19 cells by
15d-PGJ2 may cause the downregulation of inflammatory
mediators, resulting in an anti-inflammatory effect.
Previous studies have indicated that
15d-PGJ2 exerts its anti-inflammatory action via a
PPARγ-dependent (39) or
-independent (40) mechanism. To
verify the PPARγ-dependence or -independence of
15d-PGJ2, we examined the effects of PPARγ antagonists
and agonist on the production of IL-6, MCP-1, and ICAM-1 (Fig. 7). In this study, we investigated
the mechanism of action for 15d-PGJ2 with respect to the
LPS response. GW9662, which is a selective PPAR antagonist, was not
able to efficiently reduce the LPS response. The inhibition of the
expression of LPS-stimulated inflammatory mediators by
15d-PGJ2 was not reversed by GW9662. Additionally,
troglitazone and rosiglitazone, which are synthetic PPARγ agonists,
were administered at concentrations that induced PPARγ activity,
but did not affect the inhibitory effect on the expression of
inflammatory mediators. Taken together, these results suggest that
the higher potency and efficiency of 15d-PGJ2 for
attenuating the expression of inflammation-related molecules (IL-6,
MCP-1, and ICAM-1) triggered by LPS are mainly achieved through
PPARγ-independent mechanisms.
In conclusion, the results of the present study have
demonstrated that 15d-PGJ2 significantly suppresses the
LPS-induced expression of pro-inflammatory mediators. Specifically,
15d-PGJ2 significantly inhibited the release of IL-6,
MCP-1, and ICAM-1 by inhibiting NF-κB and ERK activation, as well
as through a PPARγ-independent pathway in the RPE cells.
Additionally, the anti-inflammatory properties of
15d-PGJ2 are mediated by the enhancement of PAF-AH
activity. Therefore, our results suggest that 15d-PGJ2
is considered for use as a treatment for ocular inflammatory
disorders. Future studies are to address this aspect in ocular
inflammation models performed in vivo.
Acknowledgements
This study was supported by a grant of the Korea
Healthcare Technology R&D Project, Ministry of Health &
Welfare Affairs, Republic of Korea (A120006).
Abbreviations:
|
15d-PGJ2
|
15-deoxy-δ12,14-prostaglandin
J2
|
|
PAF
|
platelet activating factor, PAF-AH,
PAF-acetylhydrolase
|
|
hRPE
|
human retinal pigment epithelial
|
|
IL-6
|
interleukin-6, MCP-1, monocyte
chemoattractant protein-1
|
|
ICAM-1
|
intercellular adhesion molecule-1
|
|
NF-κB
|
nuclear transcription factor-κB
|
|
MAPKs
|
mitogen-activated protein kinases
|
References
|
1
|
Shen DF, Chang MA, Matteson DM, Buggage R,
Kozhich AT and Chan CC: Biphasic ocular inflammatory response to
endotoxin-induced uveitis in mouse. Arch Ophthalmol. 118:521–527.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joussen AM, Poulaki V, Le M, et al: A
central role for inflammation in the pathogenesis of diabetic
retinopathy. FASEB J. 18:1450–1452. 2004.PubMed/NCBI
|
|
3
|
Rodrigues EB: Inflammation in dry
age-related macular degeneration. Ophthalmologica. 221:143–152.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strauss O: The retinal pigment epithelium
in visual function. Physiol Rev. 85:845–881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kumaki N, Anderson DM, Cosman D and Kumaki
S: Expression of interleukin-15 and its receptor by human fetal
retinal pigment epithelial cells. Curr Eye Res. 15:876–882. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McLaren MJ: Kinetics of rod outer segment
phagocytosis by cultured retinal pigment epithelial cells.
Relationship to cell morphology. Invest Ophthalmol Vis Sci.
37:1213–1224. 1996.PubMed/NCBI
|
|
7
|
Yu FS and Hazlett LD: Toll-like receptors
and the eye. Invest Ophthalmol Vis Sci. 47:1255–1263. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ishii S and Shimizu T: Platelet-activating
factor (PAF) receptor and genetically engineered PAF receptor
mutant mice. Prog Lipid Res. 39:41–82. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han SJ, Choi JH, Ko HM, et al:
Glucocorticoids prevent NF-kappaB activation by inhibiting the
early release of platelet-activating factor in response to
lipopolysaccharide. Eur J Immunol. 29:1334–1341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Camussi G, Tetta C and Baglioni C: The
role of platelet activating factor in inflammation. Clin Immun
Immunopathol. 57:331–338. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lotner GZ, Lynch JM, Betz SJ and Henson
PM: Human neutrophil-derived platelet activating factor. J Immunol.
124:676–684. 1980.PubMed/NCBI
|
|
12
|
Triggiani M, Schleimer RP, Warner JA and
Chilton FH: Differential synthesis of
1-acyl-2-acetyl-sn-glycero-3-phosphocholine and platelet-activating
factor by human inflammatory cells. J Immunol. 147:660–666.
1991.PubMed/NCBI
|
|
13
|
Sumita C, Maeda M, Fujio Y, et al:
Pioglitazone induces plasma platelet activating
factor-acetylhydrolase and inhibits platelet activating
factor-mediated cytoskeletal reorganization in macrophage. Biochim
Biophys Acta. 1673:115–121. 2004. View Article : Google Scholar
|
|
14
|
Tjoelker LW, Wilder C, Eberhardt C, et al:
Anti-inflammatory properties of a platelet-activating factor
acetylhydrolase. Nature. 374:549–553. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tontonoz P, Nagy L, Alvarez JG, Thomazy VA
and Evans RM: PPARγ promotes monocyte/macrophage differentiation
and uptake of oxidized LDL. Cell. 93:241–252. 1998.
|
|
16
|
Mueller E, Sarraf P, Tontonoz P, et al:
Terminal differentiation of human breast cancer through PPARγ. Mol
Cell. 1:465–470. 1998.
|
|
17
|
Buckingham RE: Thiazolidinediones:
pleiotropic drugs with potent anti-inflammatory properties for
tissue protection. Hepatol Res. 33:167–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Consoli A and Devangelio E:
Thiazolidinediones and inflammation. Lupus. 14:794–797. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Drew PD and Chavis JA: The cyclopentone
prostaglandin 15-deoxy-δ12,14 prostaglandin
J2 represses nitric oxide, TNF-, and IL-12 production by
microglial cells. J Neuroimmunol. 115:28–35. 2001.
|
|
20
|
Garg TK and Chang JY: Oxidative stress
causes ERK phosphorylation and cell death in cultured retinal
pigment epithelium: prevention of cell death by AG126 and
15-deoxy-delta12,14-PGJ2. BMC Ophthalmol.
3:52003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Surh YJ, Na HK, Park JM, et al:
15-Deoxy-Δ12,14-prostaglandin J2, an
electrophilic lipid mediator of anti-inflammatory and pro-resolving
signaling. Biochem Pharmacol. 82:1335–1351. 2011.
|
|
22
|
Abdelrahman M, Sivarajah A and Thiemermann
C: Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion
injury, inflammation and shock. Cardiovasc Res. 65:772–781. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosenbaum JT, Angell E, Wilson D, Broquet
C, Boney RS and Braquet P: Intravitreally injected platelet
activating factor induces retinitis in experimental animals. Curr
Eye Res. 18:342–348. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bulger EM, Arbabi S, Garcia I and Maier
RV: The macrophage response to endotoxin requires platelet
activating factor. Shock. 17:173–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cousin SW, Guss RB, Howes EL Jr and
Rosenbaum JT: Endotoxin-induced uveitis in the rat: observations on
alters vascular permeability, clinical findings and histology. Exp
Eye Res. 35:665–676. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suzuki M, Noda K, Kubota S, et al:
Eicosapentaenoic acid suppresses ocular inflammation in
endotoxin-induced uveitis. Mol Vis. 16:1382–1388. 2010.PubMed/NCBI
|
|
27
|
Leung KW, Barnstable CJ and Tombran-Tink
J: Bacterial endotoxin activates retinal pigment epithelial cells
and induces their degeneration through IL-6 and IL-8 autocrine
signaling. Mol Immunol. 46:1374–1386. 2009. View Article : Google Scholar
|
|
28
|
Planck SR, Dang TT, Graves D, Tara D,
Ansel JC and Rosenbaum JT: Retinal pigment epithelial cells secrete
interleukin-6 in response to interleukin-1. Invest Ophthalmol Vis
Sci. 33:78–82. 1992.PubMed/NCBI
|
|
29
|
Feghali CA and Wright TM: Cytokines in
acute and chronic inflammation. Front Biosci. 2:d12–d26.
1997.PubMed/NCBI
|
|
30
|
Biswas PS, Banerjee K, Kinchington PR and
Rouse BT: Involvement of IL-6 in the paracrine production of VEGF
in ocular HSV-1 infection. Exp Eye Res. 82:46–54. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
De Vos AF, Hoekzema R and Kijlstra A:
Cytokines and uveitis, a review. Curr Eye Res. 11:581–597.
1992.PubMed/NCBI
|
|
32
|
Verma MJ, Lloyd A, Rager H, et al:
Chemokines in acute anterior uveitis. Curr Eye Res. 16:1202–1208.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakazawa T, Hisatomi T, Nakazawa C, et al:
Monocyte chemoattractant protein 1 mediates retinal
detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA.
104:2425–2430. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kanagawa T, Matsuda S, Mikawa Y, et al:
Role of ICAM-1 and LFA-1 in endotoxin-induced uveitis in mice. Jpn
J Ophthalmol. 40:174–180. 1996.PubMed/NCBI
|
|
35
|
Tsuji F and Shirasawa E: The role of
platelet-activating factor in cell infiltration in
endotoxin-induced uveitis in guinea pigs. Curr Eye Res. 17:501–505.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang JZ, Cavet ME, VanderMeid KR,
Salvador-Silva M, López FJ and Ward KW: BOL-303242-X, a novel
selective glucocorticoid receptor agonist, with full
anti-inflammatory properties in human ocular cells. Mol Vis.
15:2606–2616. 2009.PubMed/NCBI
|
|
37
|
Rodrigues CE, Sanches TR, Volpini RA, et
al: Effects of continuous erythropoietin receptor activator in
sepsis-induced acute kidney injury and multi-organ dysfunction.
PLoS One. 7:e298932012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Straus DS, Pascual G, Li M, et al:
15-deoxy-delta12,14-prostaglandin J2 inhibits
multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad
Sci USA. 97:4844–4849. 2000.PubMed/NCBI
|
|
39
|
Inoue H, Tanabe T and Umeson K: Feedback
control of cyclooxygenase-2 expression through PPARgamma. J Biol
Chem. 275:28028–28032. 2000.PubMed/NCBI
|
|
40
|
Thieringer R, Fenyk-Melody JE, Le Grand
CB, et al: Activation of peroxisome proliferator-activated receptor
gamma does not inhibit IL-6 or TNF-alpha responses of macrophages
to lipopolysaccharide in vitro or in vivo. J Immunol.
164:1046–1054. 2000. View Article : Google Scholar : PubMed/NCBI
|