Introduction
Cisplatin is widely used as an effective
chemotherapeutic drug in the clinical treatment of lung, head and
neck and bladder cancer (1).
However, side-effects, such as ototoxicity, nephrotoxicity and
neurotoxicity, limit its wide application in clinical practice
(2,3). As regards ototoxicity, cisplatin
induces the loss of cochlear hair cells, the degradation of the
stria vascularis and a reduction in the number of spiral ganglion
cells, which ultimately results in high frequency hearing loss in
both ears (4,5). The mechanisms underlying
cisplatin-induced ototoxicity have not been fully elucidate to
date. It has been demonstrated that the increased production of
pro-inflammatory factors, such as tumor necrosis factor-α (TNF-α),
interleukin (IL)-1β and IL-6 is involved in cisplatin-induced
ototoxicity in cochlear hair cells (6). The production of these
pro-inflammatory factors is regulated by the signal transducer and
activator of transcription (STAT) proteins (7).
STAT proteins are important transcription factors.
They promote signal conveying between cytokines and their receptors
to induce the transcription of downstream target genes (8). Activated STATs mediate a variety of
biological processes, such as cell development, proliferation,
differentiation, migration and apoptosis, as well as the response
to different stimuli in cells. Therefore, the STAT family is
closely associated with a variety of diseases (9,10).
STAT6, one of the crucial members of the STAT family, plays a key
role in cisplatin-induced ototoxicity. During the development of
ototoxicity induced by cisplatin, STAT6 is activated by the binding
of IL-4 to its receptor (11),
and then conveys signals to the nucleus to promote the
transcription of downstream genes which are associated with
inflammatory responses. The IL-4/STAT6 signaling pathway is
associated with cisplatin-induced ototoxicity (11).
Trichostatin A (TSA) is an anti-inflammatory agent
derived from the metabolites of streptavidin (12). It has been shown that TSA protects
cochlear hair cells from aminoglycoside antibiotics-induced cell
death, thereby exhibiting anti-ototoxic acitivity (13). Coincidentally, the ototoxicity
caused by cisplatin is similar to that induced by aminoglycoside
antibiotics; thus, it is conjectured that TSA may inhibit
cisplatin-induced ototoxicity as well (14). This conjecture was confirmed by a
study in which cisplatin-induced auditory cell toxicity was
successfully inhibited by treatment with 200 nM TSA (15). Although the protective mechanisms
of TSA against cisplatin-induced ototoxicity are largely unclear,
the implications of its anti-inflammatory responses have been
demonstrated in a number of previous studies (16,17). In the present study, we
hypothesized that TSA may protect cochlear hair cells from
cisplatin-induced damage by regulating the IL-4/STAT6 signaling
pathway which is associated with inflammatory responses, and we
succeeded in confirming this hypothesis.
Materials and methods
Animals
Wistar rats (newborn 3–4 days old) were provided by
the Animal Center of Jilin University [SCXK (JI) 2007-0003]. All
experimental procedures were approved by the Animal Ethics
Committee of Jilin University, Changchun, China.
Reagents
TSA, cisplatin and rhodamine phalloidin were
purchased from Sigma (St. Louis, MO, USA). Dulbecco’s modified
Eagle’s medium (DMEM) and fetal bovine serum (FBS) were provided by
Invitrogen (Carlsbad, CA, USA). The IL-1β, IL-4 and IL-6
enzyme-linked immunosorbent assay (ELISA) kits were obtained from
Wuhan Boster Biological Engineering Co., Ltd. (Wuhan, China).
Rabbit polyclonal antibody to STAT6 (sc-981) was obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse
monoclonal anti-phosphorylated (p-)STAT6 antibody (05-590) was
purchased from Millipore (Bedford, MA, USA).
Gene microarray and data analysis
To identify the differentially expressed genes
(DEGs), we used our previous microarray based on cochlear explants
treated for 6 h with cisplatin or cisplatin plus TSA compared with
cochlear explants without drug treatment (15). We calculated the DEGs between the
control and cisplatin-treated explants, as well as between the
cisplatin-and cisplatin plus TSA-treated explants. The fold change
values were calculated. The genes with a fold change in expression
of >2 or <0.5 were selected as the DEGs. Finally, to
implement subpathway enrichment, we matched the DEGs of the Wistar
rats with the corresponding orthologous human genes. A total of 467
differentially expressed orthologous human genes were obtained
between the control samples and those treated with cisplatin. A
total of 2,588 differentially expressed orthologous human genes
were obtained between the samples treated with cisplatin alone and
those treated with cisplatin plus TSA.
Identification of subpathways using
Subpathway Miner
To identify the perturbed subpathways following
treatment with cisplatin alone or with cisplatin plus TSA, we used
the Subpathway Miner software package which is a flexible
subpathway identification method (18). With this software, the pathway
structure data of KEGG are converted to R undirected graph objects.
The enzymes are then considered as nodes and 2 nodes are connected
by an edge if they are in the same reactions. This software package
can transform the entire pathway into subpathways using the
‘k-clique’ method, which is defined as a subgraph in which the
distance between any 2 nodes is no greater than a parameter k. In
this study, the value of this parameter was set as k=3.
We then imported 2 sets of DEGs (control vs.
cisplatin; cisplatin vs. cisplatin plus TSA) into Subpathway Miner
and this software identified 2 sets of significantly enriched
subpathways. A value of P <0.01 was selected as the cut-off
criterion for the statistically significant subpathways.
Culture of basilar membrane
The culture of the basilar membrane was performed as
previously described (19). The
rats were sacrificed by decapitation after being cleaned with 75%
ethanol, and the parietal bone was opened and the brain tissue was
removed. The otic vesicles were removed and placed into cold
D-Hank’s solution without Ca2+ and Mg2+. The
connective tissues around the spiral case was carefully removed
under an anatomical microscope, and the spiral case was opened and
the membranous labyrinth was completely exposed. The modiolus,
spiral ligament, vestibular membrane and tectorial membrane were
carefully dissected, leaving the whole basilar membrane with the
organ of Corti and spiral ganglion neurons. Briefly, the entire
basilar membrane was placed on a 35 mm culture dish with the convex
side up and incubated with 1 ml of DMEM supplemented with 10% FBS
in an incubator containing 5% CO2 at 37°C for 24 h. The
culture medium was then replaced with medium supplemented with TSA
(at final concentrations of 50, 100, 200 or 300 nM), cisplatin (150
µM), or a combination of TSA and cisplatin (150 µM
cisplatin plus TSA 200 nM), followed by a further 24 h of
incubation. The samples incubated with drug-free medium were
considered as the controls. Each experiment was repeated at least 3
times.
Staining of hair cells
At the end of the incubation period, the cultures
were fixed with 4% paraformaldehyde for 30 min and washed 3 times
with 0.1 mM phosphate-buffered saline (PBS). They were then
incubated in 0.25% Triton X-100 for 5 min. The cultures were then
incubated with rhodamine phalloidin in the dark at room temperature
for approximately 30 min. The cultures were observed under a laser
confocal microscope (FluoView FV1000, serial no. 08004045; Olympus,
Tokyo, Japan) and the images were captured using FV10-ASW 1.7
Viewer software (FluoView FV1000; Olympus). The number of hair
cells per 0.24 mm length of the cochlea was counted from the apical
back to the basal turn.
Scanning electron microscopy of hair
cells
The specimens were prepared according to methods
described by Segawa et al (20). The basilar membranes were washed
with PBS and fixed with 2.5% glutaraldehyde for 24 h and then
further fixed with 1% osmium tetroxide (OsO4) for 2 h.
The specimens were dehydrated in a graded series of ethanol (50,
70, 80, 90 and 100%, 10 min each) and then in tert-butyl alcohol
for 10 min. After drying with a carbon dioxide critical point
dryer, the specimens were attached to aluminum stubs and coated
with Au by an ion sputtering coating machine. Finally, a Hitachi
scanning electron microscope (S-3400N, serial no. 09008178; Hitachi
High Technologies, Tokyo, Japan) was employed to examine the
surface of the basilar membranes and capture images.
ELISA
The specimens were grouped according to the
treatment concentrations of TSA or cisplatin, 15 per group, as
follows: i) control group (not treated with drugs); ii) group
treated with 150 µM cisplatin; iii) group treated with 200
nM TSA; and iv) group treated with cisplatin 150 µM + TSA
200 nM. Following treatment for 24 h, the levels of cytokines in
the supernatant of the cultured basilar membranes were measured
using commercial two-site rat IL-4, IL-6 and IL-1β ELISA kits
according to the manufacturer’s instructions, respectively. The
absorbance was detected at 450 nm to evaluate the levels of IL-4,
IL-6 and IL-1β at different time points following treatment.
Western blot analysis
Western blot analysis was performed as previously
described (11). Total proteins
from the basilar membranes were extracted with RIPA-containing
phenylmethanesulfonylfluoride and protein phosphatase inhibitor.
The concentrations of proteins were determined using BCA protein
assay kits, after which proteins were separated by 8% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto polyvinylidene difluoride (PVDF) membranes. After
washing with 1X Tris-buffered saline/Tween-20 (TBST), the PVDF
membranes were stained in Ponceau S staining solution on a shaking
table at low velocity for 3–5 min, followed by blocking in 1X TBST
including 5% skim milk for 2 h at room temperature or overnight at
4°C. The PVDF membranes were probed with primary antibodies
(1:1,000) with gentle shaking at room temperature for 2 h and then
at 4°C overnight. Subsequently, the membranes was washed with 1X
TBST and incubated with corresponding horseradish
peroxidase-conjugated secondary antibodies (1:1,000) for 2 h at
room temperature. Finally, the protein bands were identified by
enhanced chemiluminescence kits. Image J 1.41 software (National
Institutes of Health, Bethesda, MD, USA) was used to analyze the
optical density of the bands. β-actin was used as a normalizing
protein.
Statistical analysis
The data were analyzed utilizing SPSS 17.0 software.
All results are expressed as the means ± SD. The analysis of hair
cell loss was performed by one-way analysis of variance (ANOVA).
The difference between any 2 groups was determined by the Q test. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
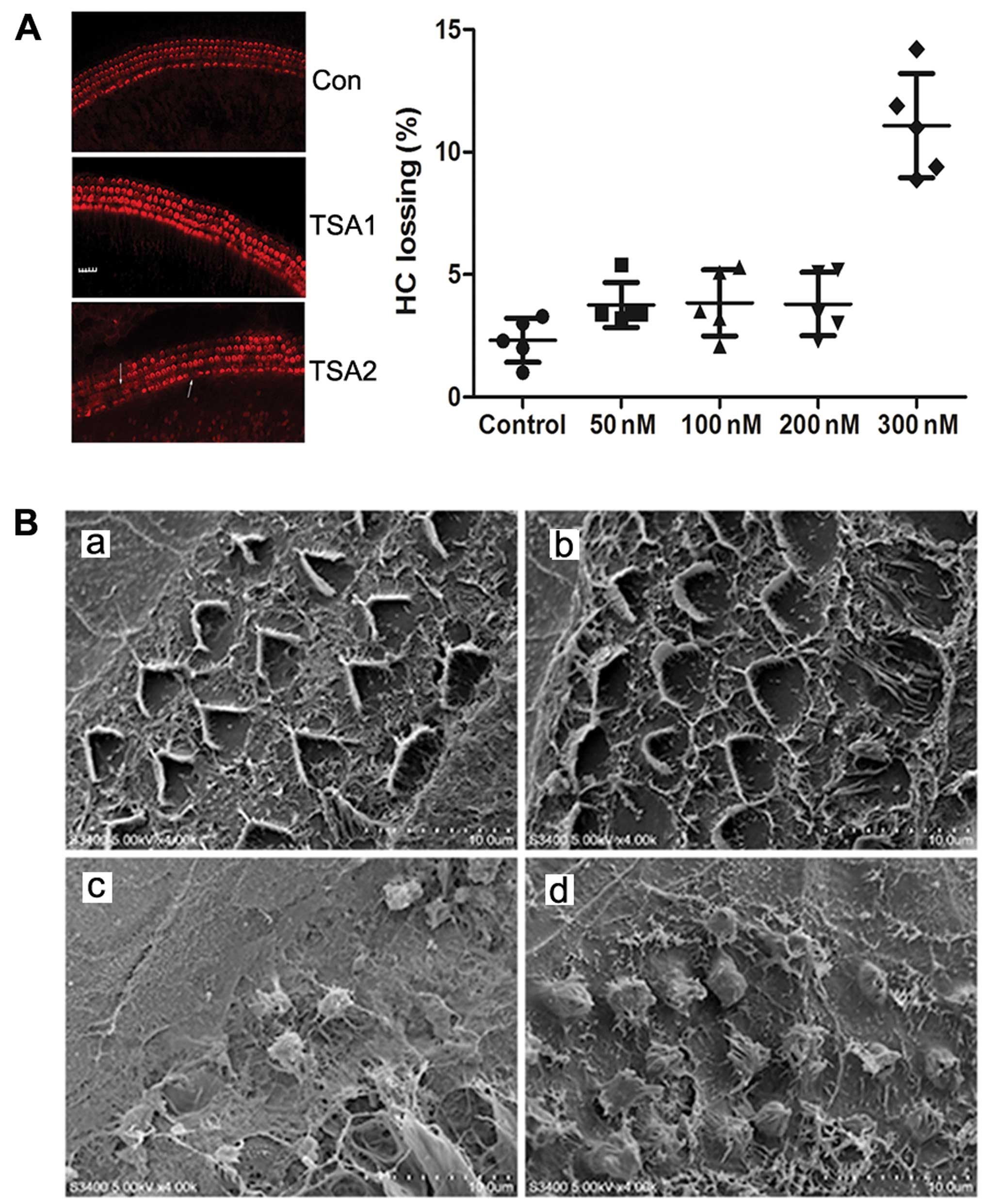
Hair cell survival in the basilar
membrane following treatment with TSA
Immunocytochemical staining revealed that the hair
cells of the basilar membrane grew well following culture. Three
lines of cochlear outer hair cells formed an orderly ‘V’ glyph with
a clearly visible outline and the cilia of the inner hair cells
aligned as a ‘-’ glyph. The loss of a few hair cells was observed
at the top back to the basal turn of the cochlea. Following
treatment with TSA (50, 100 or 200 nM) for 24 h, only a slight loss
of outer and inner hair cells was observed. However, there was a
significant increase in the loss of hair cells following treatment
with TSA 300 nM (Fig. 1A). Thus,
treatment with TSA at a low concentration had no obvious effect on
cochlear hair cells.
TSA reduces cisplatin-induced damage to
hair cells in the basilar membrane
Scanning electron microscopy revealed that the hair
cells had a normal morphology with an orderly arrangement
regardless of the absence or presence of 200 nM TSA in the medium.
However, following treatment with 150 µM cisplatin for 24 h,
the number of hair cells was markedly reduced and the cilia of the
hair cells became disorganized, fused and detached. Treatment with
a combination of 200 nM TSA and 150 µM cisplatin
significantly decreased hair loss compared to treatment with
cisplatin alone (Fig. 1B).
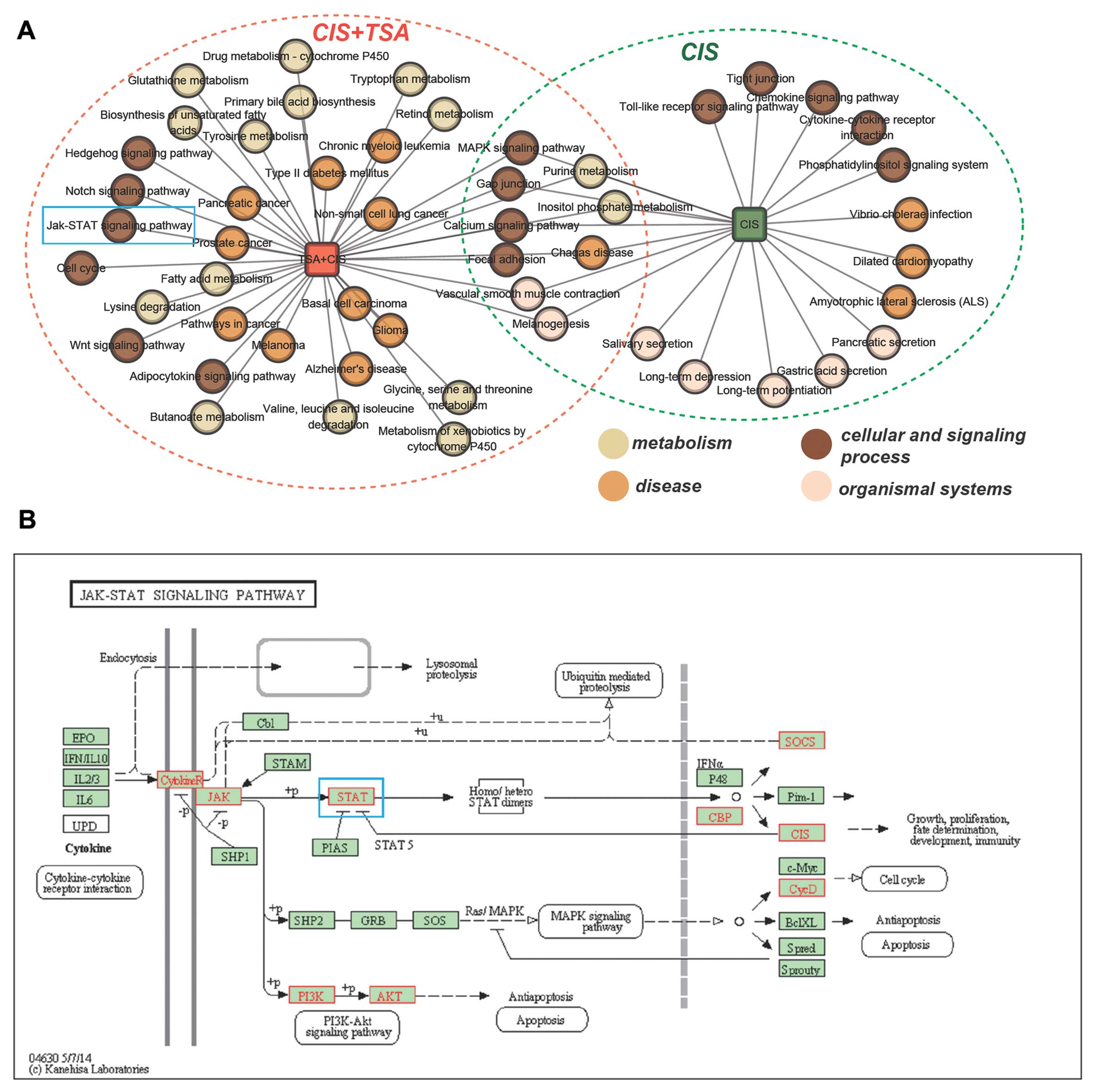
Identification of the mechanisms
repsonsible for the protective effects of TSA against
cisplatin-induced ototoxicity based on subpathway enrichment
analysis
To elucidate the mechanisms responsible for the
protective effects of TSA against cisplatin-induced ototoxicity, we
identified 2 sets of corresponding dysregulated subpathways using
Subpathway Miner (see Materials and methods; with a strict cut-off
value of P<0.01) One set corresponded to dysregulated
subpathways under conditions of treatment with cisplatin alone (62
subpathways corresponding to 21 entire pathways), and the other set
corresponded to dysregulated subpathways under conditions of
treatment with cisplatin plus TSA compared to treatment with
cisplatin alone (89 subpathways corresponding to 38 entire
pathways). We then compared the 2 sets of subpathways. Fig. 2A shows a network of entire
pathways under 2 treatment conditions (cisplatin alone or cisplatin
plus TSA). We found that there were 8 common pathways and that
cisplatin plus TSA treatment affected extra pathways compared to
treatment with cisplatin alone. These extra pathways were
considered to be associated with the potential molecular mechanisms
responsible for the TSA-mediated protective effects against
cisplatin ototoxicity (Fig. 2A).
We focused on the cell signaling process. Of these pathways, the
JAK-STAT signaling pathway (blue rectangle in Fig. 2A) was shown to play a pivotal role
in cisplatin-mediated pro-inflammatory cytokine production and
ototoxicity. In order to investigate the mechanisms responsible for
the protective effects of TSA against cisplatin ototoxicity, we
mapped the DEGs of cisplatin vs. cisplatin plus TSA into the
JAK-STAT signaling pathway using the KEGG mapping tool. We found
that some key proteins were annotated, including JAK and STAT (blue
rectangle in Fig. 2B).
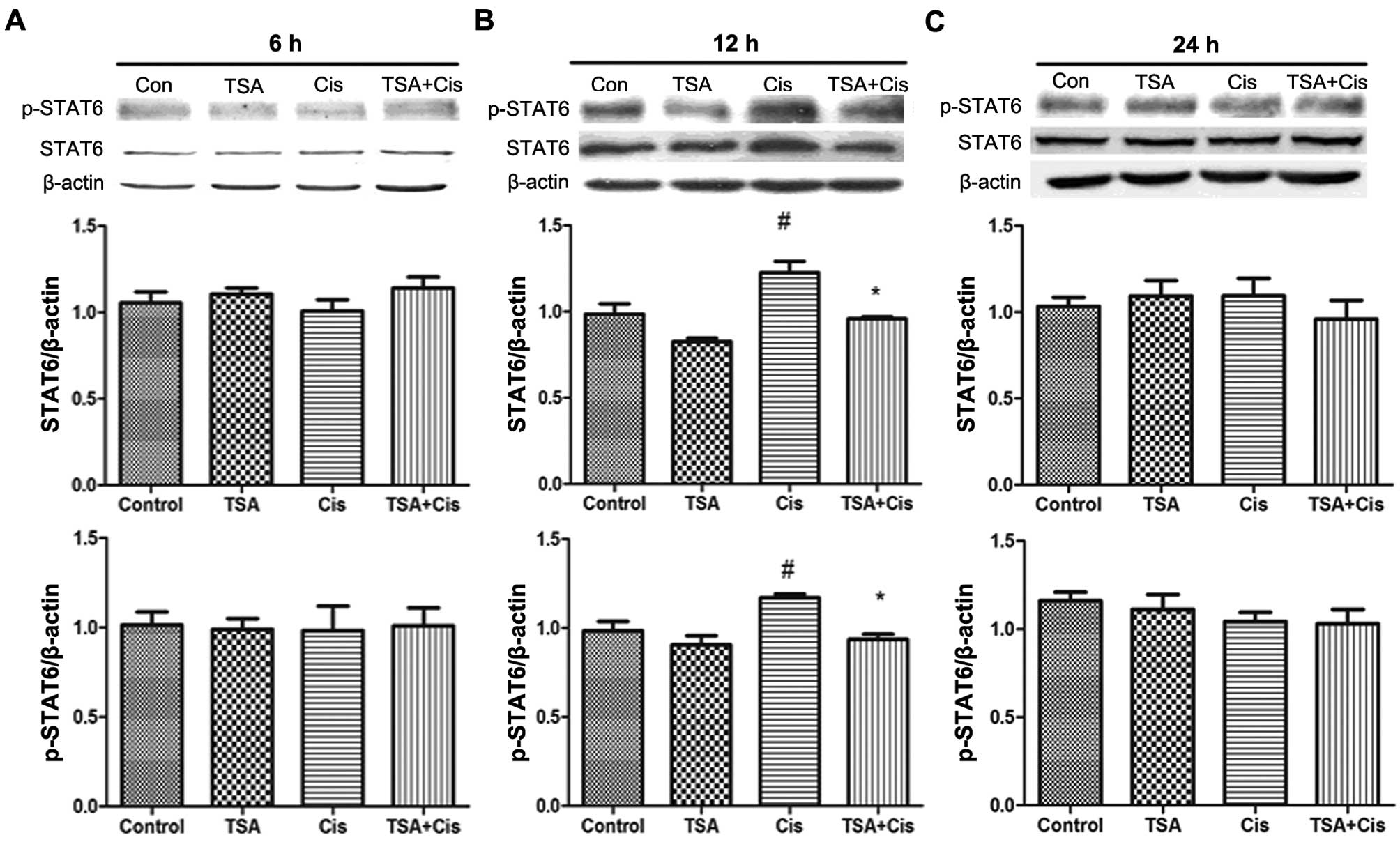
TSA increases the expression of STAT6 in
the basement membrane
The expression levels of STAT6 in the basement
membrane were measured by western blot analysis (Fig. 3). STAT6 protein and p-STAT6
expression levels were increased following treatment with cisplatin
for 12 h. By contrast, no changes were observed in the expression
of STAT6 and p-STAT6 following treatment with cisplatin for 6 and
24 h. Furthermore, the increase in the expression of STAT6 and
p-STAT6 induced by cisplatin was downregulated by treatment with
TSA. These findings indicated that TSA exerts its protective
effects against cisplatin-induced ototoxicity through the STAT6
pathway.
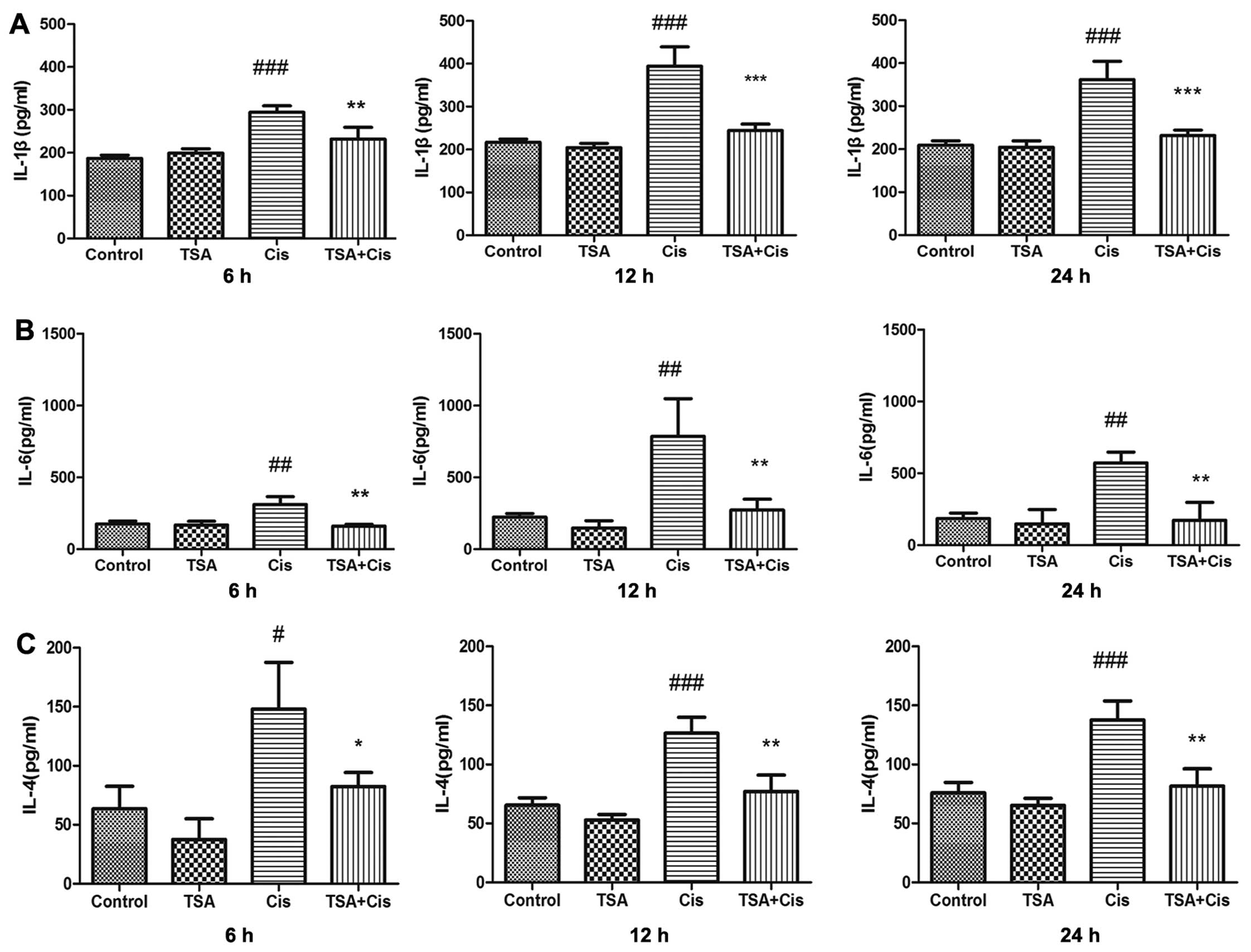
TSA decreases the expression inflammatory
cytokines in the cochlea following treatment with cisplatin
The levels of IL-4, IL-1β and IL-6 in the
supernatant of the cultured basilar membrane were measured by
ELISA. The concentrations of these cytokines in the treatment
groups were upregulated by cisplatin compared with the control
group. The expression levels of IL-1β and IL-6 reached maximum
levels at 12 h following treatment with cisplatin. The increase in
the expression levels of IL-1β and IL-6 induced by cisplatin was
inhibited by treatment with TSA (Fig.
4A and B). Subsequently, treatment with TSA inhibited the
increase in the expression of IL-4 induced by cisplatin (Fig. 4C). Thus, the secretion of
inflammatory cytokines related to STAT6 in the basilar membrane
induced by cisplatin was time-dependent and was inhibited by
TSA.
Discussion
The incidence of cisplatin-induced ototoxicity is
high in patients who have undergone cisplatin-based chemotherapy.
The damage caused by cisplatin mainly occurs in the outer hair
cells of the organ of Corti (21). In the present study, cisplatin
induced hair cell loss, as well as the collapse and disorder of
cultured basilar membranes. This effect was more prominent in the
outer hair cells than in the inner hair cells, further confirming
that cochlear hair cells are the main target of cisplatin
ototoxicity. Treatment with TSA alleviated the cisplatin-induced
loss of hair cells of the basilar membrane, which suggests that TSA
exerts a protective effect against cisplatin-induced
ototoxicity.
We identified 8 common pathways in the network of
entire pathways following treatment with cisplatin alone or
cisplatin plus TSA using Subpathway Miner. We focused on the role
of the JAK-STAT signaling pathway in the cisplatin-mediated
production of pro-inflammatory cytokines and ototoxicity. TSA has
been shown to suppress the growth of colorectal cancer cells
through the regulation of downstream targets of JAK2/STAT3
signaling (22). We found that
some key proteins were annotated, including JAK and STAT.
Recently, more attention has been paid to the
mechanisms responsible for cell damage induced by cisplatin, such
as inflammation. For example, in a previous study, the protective
effects of α-lipoic acid against cisplatin-induced ototoxicity were
shown to be mediated through the regulation of mitogen-activated
protein kinases (MAPKs) and pro-inflammatory cytokines in
cisplatin-treated HEI-OC1 cells (23). Reactive oxygen species (ROS) and
inflammation are the major contributors to cisplatin-induced
hearing loss. This suggests that controlling inflammation by the
inhibition of STAT1-dependent pathways in the cochlea may serve as
an effective approach for the treatment of cisplatin-induced
ototoxicity (24). The activation
of transient receptor potential vanilloid 1 (TRPV1)-mediated
temporary hearing loss has been shown to occur by initiating an
inflammatory process in the cochlea through the activation of NOX3
and STAT1 (25). Corticosteroid
therapy reduces inflammation and inhibits apoptosis while
activating pro-survival pathways in the organ of Corti following
exposure to noise, vibration, cisplatin, aminoglycoside,
ischemia/reperfusion injury, bacterial meningitis and electrode
insertion trauma (26).
Pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6 play a
critical role in cisplatin-induced cochlear injury (27). The cochlea, due to its unique
anatomical position and isolation, is a closed system, and is
therefore unable to flush out the accumulated toxins at the rapid
pace of their generation. The accumulated toxins induce the
overload of ROS and the dysfunction of the antioxidant system,
eventually leading to cell injury and apoptosis (3,28).
It has been demonstrated that cochlear and
vestibular functions, such as hearing and balance are related to
immune responses (29,30). Although the immune response plays
an important role in preventing infectious diseases, such as
labyrinthitis in the inner ear, it can also damage the inner ear
tissue, causing cochlear degradation and permanent hearing loss
(31,32). A previous study demonstrated that
cisplatin promoted the production of pro-inflammatory factors
involved in the development of ototoxicity in cochlear cells
(11). Inflammatory cells are
transported to the cochlea through 2 pathways. One involves the
infiltration of inflammatory cells into the cochlea from the spiral
modiolar vein. The other is that inflammatory cells generated in
the endolymphatic sac are directly transferred into the cochlea.
Once the inner ear is attacked by bacteria, endotoxins or other
inflammatory substances, inflammatory cells, such as monocytes and
macrophages migrate to the perilymph and secrete pro-inflammatory
cytokines, such as IL-1β and TNF-α during the early phase of
inflammation. These pro-inflammatory cytokines induce cochlear
spiral ganglionic fibrocytes to produce a variety of inflammatory
secretions and mediators that in turn activate inflammatory cells
to prolong the inflammatory response in the inner ear (33). So et al verified that the
secretion and expression of TNF-α, IL-1β and IL-6 were increased in
the cochleae of cisplatin-injected rats and in cultured cochleae
(6). Pro-inflammatory factors are
directly involved in the process of the cisplatin-induced apoptosis
of cochlear cells. The secretion of pro-inflammatory cytokines
stimulated by cisplatin in auditory cells in the early phase was
unrelated to the change in mRNA expression (6). These data suggest that cisplatin
promotes the release of pro-inflammatory factors stored in cells in
the early phase and then stimulates the de novo synthesis of
pro-inflammatory factors. Pro-inflammatory factor secretion
mediated by cisplatin may be the upstream signaling pathway of ROS
generation. Pro-inflammatory factor secretion may be responsible
for the ototoxicity induced by cisplatin (27). To further confirm this, we
measured the levels of IL-1β and IL-6 in the supernatant of
cultured basilar membrane and found that cisplatin increased the
expression levels of pro-inflammatory factors; the maximum levels
were observed at at 12 h following treatment. These results
confirmed that inflammatory responses are involved in
cisplatin-induced damage to the cultured basilar membrane.
The increase in the expression levels of STAT6 and
p-STAT6 following treatment with cisplatin for 12 h is in
accordance with gene chip analysis, indicating that the expression
of STAT6 was significantly enhanced in cochlear cultures following
treatment with cisplatin. The IL-1β and IL-6 expression reached
maximum levels at 12 h following treatment with cisplatin. The
effects of cisplatin on IL-4, IL-1β and IL-6 expression were
inhibited by TSA. Taken together, cisplatin time-dependently
promoted the secretion of inflammatory cytokines in the basilar
membrane, and this effect was inhibited by TSA. The level of IL-4
in the supernatant of the cultured basilar membrane reached a
maximum at 6 h following with cisplatin and then decreased rapidly.
Thus, IL-4 and the downstream STAT6 signaling cascade play a
crucial role in cisplatin-induced ototoxicity.
A previous in vivo study revealed that sodium
butyrate provided almost complete protection against cisplatin
induced ototoxicity (34). The
pathology of the ototoxicity induced by cisplatin is similar to
that caused by aminoglycoside antibiotics (35). The application of TSA may rescue
cochlear hair cells from aminoglycoside-induced death. TSA
suppresses the expression of pro-inflammatory cytokines and
inhibits the development of inflammatory diseases, such as
ulcerative colitis and rheumatoid arthritis (36). TSA exerts its anti-inflammatory
effects by inhibiting the activity of histone deacetylases that
aggravate cellular inflammatory responses (36). In the present study, the increase
in the expression of pro-inflammatory cytokines and STAT6 and
p-STAT6 mediated by cisplatin was significantly decreased by the
administration of TSA, demonstrating that TSA exerts a protective
effect against cisplatin-induced damage in cochlear hair cells by
inhibiting the expression of STAT6 and p-STAT6.
Our findings suggest that TSA protects cochlear hair
cells from cisplatin-induced damage by regulating the intracellular
levels of histone acetylation, inhibiting the expression of IL-4
and STAT6, and further attenuating excessive inflammatory
reactions. In addition, this study provides evidence supporting the
novel application of TSA in the pharmacotherapy of
cisplatin-induced ototoxicity.
Acknowledgments
This study was supported by a grant from the
National Natural Science Foundation of China (no. 30873131).
References
|
1
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barabas K, Milner R, Lurie D and Adin C:
Cisplatin: A review of toxicities and therapeutic applications. Vet
Comp Oncol. 6:1–18. 2008. View Article : Google Scholar
|
|
3
|
Rybak LP, Mukherjea D, Jajoo S and
Ramkumar V: Cisplatin ototoxicity and protection: Clinical and
experimental studies. Tohoku J Exp Med. 219:177–186. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakamoto M, Kaga K and Kamio T: Extended
high-frequency ototoxicity induced by the first administration of
cisplatin. Otolaryngol Head Neck Surg. 122:828–833. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rybak LP: Mechanisms of cisplatin
ototoxicity and progress in otoprotection. Curr Opin Otolaryngol
Head Neck Surg. 15:364–369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
So H, Kim H, Lee JH, Park C, Kim Y, Kim E,
Kim JK, Yun KJ, Lee KM, Lee HY, et al: Cisplatin cytotoxicity of
auditory cells requires secretions of proinflammatory cytokines via
activation of ERK and NF-kappaB. J Assoc Res Otolaryngol.
8:338–355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou BR, Zhang JA, Zhang Q, Permatasari F,
Xu Y, Wu D, Yin ZQ and Luo D: Palmitic acid induces production of
proinflammatory cytokines interleukin-6, interleukin-1β, and tumor
necrosis factor-α via a NF-κB-dependent mechanism in HaCaT
keratinocytes. Mediators Inflamm. 2013:5304292013. View Article : Google Scholar
|
|
8
|
Aittomäki S and Pesu M: Therapeutic
targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol.
114:18–23. 2014. View Article : Google Scholar
|
|
9
|
O’Shea JJ and Plenge R: JAK and STAT
signaling molecules in immunoregulation and immune-mediated
disease. Immunity. 36:542–550. 2012. View Article : Google Scholar
|
|
10
|
Kiu H and Nicholson SE: Biology and
significance of the JAK/STAT signalling pathways. Growth Factors.
30:88–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HJ, Oh GS, Lee JH, Lyu AR, Ji HM, Lee
SH, Song J, Park SJ, You YO, Sul JD, et al: Cisplatin ototoxicity
involves cytokines and STAT6 signaling network. Cell Res.
21:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grabiec AM, Tak PP and Reedquist KA:
Targeting histone deacetylase activity in rheumatoid arthritis and
asthma as prototypes of inflammatory disease: Should we keep our
HATs on? Arthritis Res Ther. 10:2262008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen FQ, Schacht J and Sha SH:
Aminoglycoside-induced histone deacetylation and hair cell death in
the mouse cochlea. J Neurochem. 108:1226–1236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cunningham LL and Brandon CS: Heat shock
inhibits both aminoglycoside- and cisplatin-induced sensory hair
cell death. J Assoc Res Otolaryngol. 7:299–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang P, Zhang P, Huang J, Li M and Chen X:
Trichostatin A protects against cisplatin-induced ototoxicity by
regulating expression of genes related to apoptosis and synaptic
function. Neurotoxicology. 37:51–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han SB and Lee JK: Anti-inflammatory
effect of Trichostatin-A on murine bone marrow-derived macrophages.
Arch Pharm Res. 32:613–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS
and Chuang DM: Histone deacetylase inhibitors exhibit
anti-inflammatory and neuroprotective effects in a rat permanent
ischemic model of stroke: multiple mechanisms of action. J
Pharmacol Exp Ther. 321:892–901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li C, Li X, Miao Y, Wang Q, Jiang W, Xu C,
Li J, Han J, Zhang F, Gong B and Xu L: SubpathwayMiner: A software
package for flexible identification of pathways. Nucleic Acids Res.
37:e1312009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zheng JL and Gao WQ: Differential damage
to auditory neurons and hair cells by ototoxins and neuroprotection
by specific neurotrophins in rat cochlear organotypic cultures. Eur
J Neurosci. 8:1897–1905. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Segawa A, Loffredo F, Puxeddu R, Yamashina
S, Testa Riva F and Riva A: Exocytosis in human salivary glands
visualized by high-resolution scanning electron microscopy. Cell
Tissue Res. 291:325–336. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamers FP, Wijbenga J, Wolters FL, Klis
SF, Sluyter S and Smoorenburg GF: Cisplatin ototoxicity involves
organ of Corti, stria vascularis and spiral ganglion: Modulation by
alphaMSH and ORG 2766. Audiol Neurootol. 8:305–315. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong H, Du W, Zhang YJ, Hong J, Su WY,
Tang JT, Wang YC, Lu R, Fang JY and Trichostatin A: Trichostatin A,
a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling
via inducing the promoter-associated histone acetylation of SOCS1
and SOCS3 in human colorectal cancer cells. Mol Carcinog.
51:174–184. 2012. View
Article : Google Scholar
|
|
23
|
Kim J, Cho HJ, Sagong B, Kim SJ, Lee JT,
So HS, Lee IK, Kim UK, Lee KY and Choo YS: Alpha-lipoic acid
protects against cisplatin-induced ototoxicity via the regulation
of MAPKs and proinflammatory cytokines. Biochem Biophys Res Commun.
449:183–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaur T, Mukherjea D, Sheehan K, Jajoo S,
Rybak LP and Ramkumar V: Short interfering RNA against STAT1
attenuates cisplatin-induced ototoxicity in the rat by suppressing
inflammation. Cell Death Dis. 2:e1802011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mukherjea D, Jajoo S, Sheehan K, Kaur T,
Sheth S, Bunch J, Perro C, Rybak LP and Ramkumar V: NOX3 NADPH
oxidase couples transient receptor potential vanilloid 1 to signal
transducer and activator of transcription 1-mediated inflammation
and hearing loss. Antioxid Redox Signal. 14:999–1010. 2011.
View Article : Google Scholar :
|
|
26
|
Abi-Hachem RN, Zine A and Van De Water TR:
The injured cochlea as a target for inflammatory processes,
initiation of cell death pathways and application of related
otoprotectives strategies. Recent Patents CNS Drug Discov.
5:147–163. 2010. View Article : Google Scholar
|
|
27
|
So H, Kim H, Kim Y, Kim E, Pae HO, Chung
HT, Kim HJ, Kwon KB, Lee KM, Lee HY, et al: Evidence that
cisplatin-induced auditory damage is attenuated by downregulation
of pro-inflammatory cytokines via Nrf2/HO-1. J Assoc Res
Otolaryngol. 9:290–306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kopke R, Allen KA, Henderson D, Hoffer M,
Frenz D and Van de Water T: A radical demise. Toxins and trauma
share common pathways in hair cell death. Ann NY Acad Sci.
884:171–191. 1999. View Article : Google Scholar
|
|
29
|
Ma C, Billings P, Harris JP and Keithley
EM: Characterization of an experimentally induced inner ear immune
response. Laryngoscope. 110:451–456. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rahman MU, Poe DS and Choi HK: Autoimmune
vestibulo-cochlear disorders. Curr Opin Rheumatol. 13:184–189.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ryan AF, Harris JP and Keithley EM:
Immune-mediated hearing loss: Basic mechanisms and options for
therapy. Acta Otolaryngol Suppl. 122:38–43. 2002. View Article : Google Scholar
|
|
32
|
Stone JH and Francis HW: Immune-mediated
inner ear disease. Curr Opin Rheumatol. 12:32–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fujioka M, Kanzaki S, Okano HJ, Masuda M,
Ogawa K and Okano H: Proinflammatory cytokines expression in
noise-induced damaged cochlea. J Neurosci Res. 83:575–583. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Drottar M, Liberman MC, Ratan RR and
Roberson DW: The histone deacetylase inhibitor sodium butyrate
protects against cisplatin-induced hearing loss in guinea pigs.
Laryngoscope. 116:292–296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schacht J, Talaska AE and Rybak LP:
Cisplatin and aminoglycoside antibiotics: Hearing loss and its
prevention. Anat Rec (Hoboken). 295:1837–1850. 2012. View Article : Google Scholar
|
|
36
|
Toussirot E, Abbas W, Khan KA, Tissot M,
Jeudy A, Baud L, Bertolini E, Wendling D and Herbein G: Imbalance
between HAT and HDAC activities in the PBMCs of patients with
anky-losing spondylitis orrheumatoid arthritis and influence of
HDAC inhibitors on TNF alpha production. PLoS One. 8:e709392013.
View Article : Google Scholar
|