Introduction
Glioblastoma multiforme (GBM) (World Health
Organization grade IV) is the most fatal form of malignant brain
cancer in humans and accounts for 12–15% of all intracranial tumors
and 50–60% of all primary brain tumors (1,2).
Treatment usually involves radiotherapy combined with temozolomide
(TMZ), a DNA-alkylating agent that mispairs nucleotide bases during
DNA replication (3). Although TMZ
has been shown to increase the two-year survival rate by
suppressing the proliferation of glioblastoma cells and
upregulation of apoptotic pathways, the overall prognosis of
patients with GBM remains poor (4). To date, >75% of patients treated
with TMZ succumb within 2 years due to recurrent GBM (5). The development of new combination
therapies that increase the sensitivity of TMZ may improve the
survival rate of patients with GBM.
Autophagy is a fundamental cellular process
responsible for the bulk degradation of cytoplasmic components
through an autophagosomal-lysosomal pathway (6). It is generally associated with cell
survival or a protective response to stress or inflammation;
however, sometimes it promotes cell death depending on specific
circumstances (7). Autophagic
cell death is regarded as an alternative tumor-suppressing
mechanism in chemotherapy-resistant cancers, such as glioblastoma
(8–10). The autophagic signaling pathway,
comprising PI3K, Akt and mammalian target of rapamycin (mTOR), is
an important signaling network involved in cell proliferation,
survival and tumorigenesis, and is becoming an important target for
treatment of several types of cancer (11). mTOR, a downstream effector of Akt,
has a well-known critical role in suppressing autophagy by
activating the downstream molecule p70S6 kinase (p70S6K). The role
of autophagy in oncogenesis and anticancer therapy is contradictory
(12). Although certain studies
suggest that autophagy, rather than apoptosis, is associated with
TMZ-induced chemoresistance in GBM (13), other studies have shown that TMZ
is an effective tumor suppressor by inducing autophagic cell death
in tumor cells, particularly in glioblastoma (14).
Carbazole alkaloids, obtained from natural sources,
such as the oleoresin of curry leaves (Murraya koenigii
Spreng.) (15), and their
derivatives from synthesized sources, are well known for their
various pharmacological activities, including anti-proliferation
(16), anti-angiogenesis and
anti-inflammation (17)
activities, their ability to inhibit DNA topoisomerase (18), and their ability to sensitize
cancer cells to anticancer drugs (19). Previous studies have demonstrated
the selective antitumor activity of carbazole in several human
cancer cell lines, including lung, colon, liver and leukemia
(20). However, the effects of
carbazole derivatives on glioblastoma cells remain poorly
understood. In our recent study, we proposed a mechanism by which
carbazole derivatives effectively trigger cell cycle arrest and
programmed cell death in breast cancer cells (20). Carbazole derivatives have also
been shown to increase autophagolysosomal membrane permeability,
which may resensitize drug-resistant cancer cells to
chemotherapeutic agents (21).
Therefore, the sensitizing effects of carbazole derivatives may be
mediated by the activation of the autophagy signaling pathway.
The aim of the present study was to determine the
anti-glioblastoma effects of synthetic carbazole derivatives. In
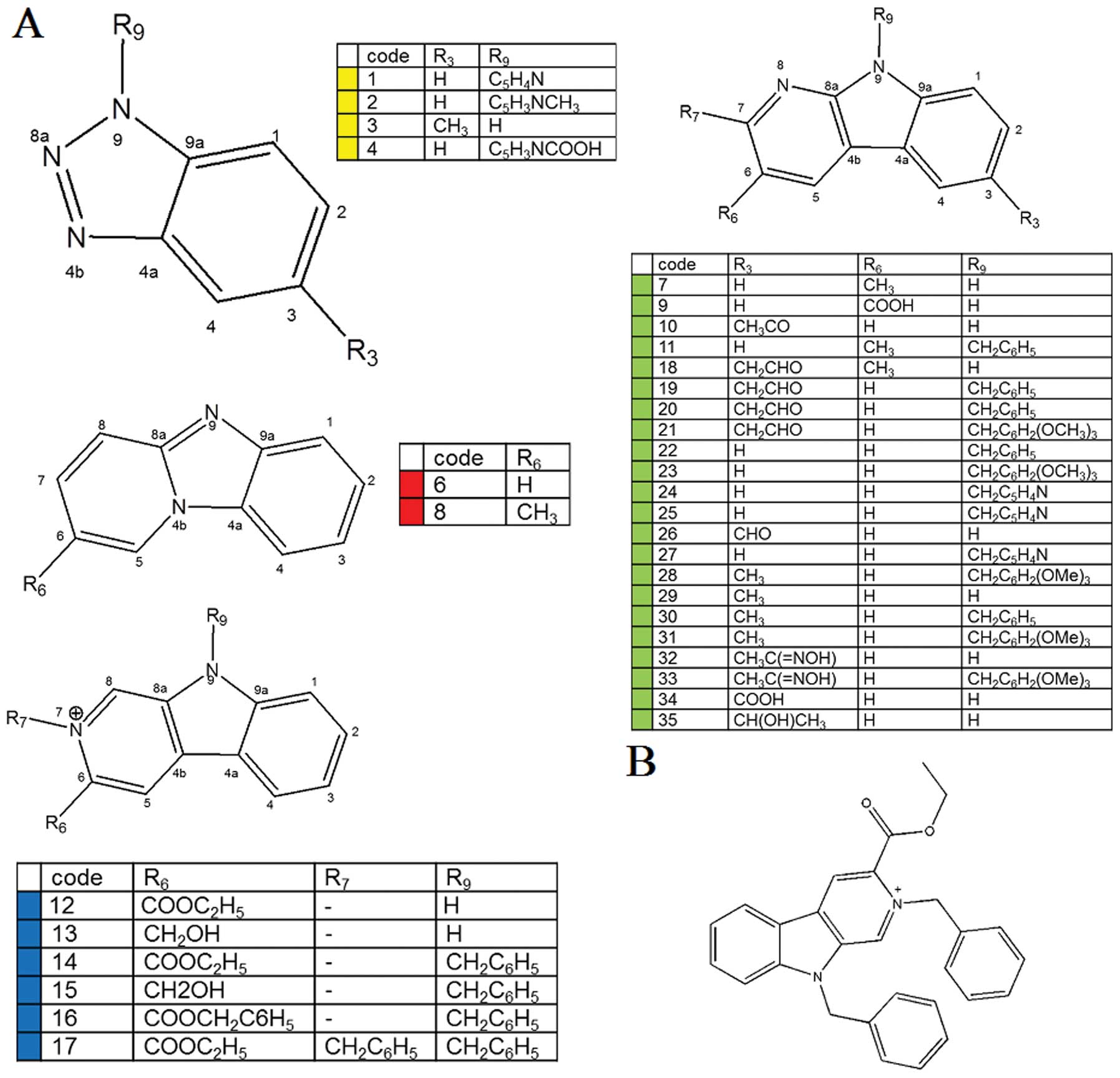
total, 35 different carbazole derivatives were synthesized and
their ability to inhibit the proliferation of GBM8401 and GBM8901
cells was measured. Of the derivatives analyzed,
bis(carbazole-2,9N-benzyl)-3-ethyl ethanoate (BC3EE2,9B) exhibited
the most promising anti-proliferation and autophagic-induced cell
death activities. Exposure of GBM8901 and GBM8401 cells to
BC3EE2,9B induced autophagy-mediated cell death through suppression
of the Akt-mTOR signaling pathway and the cells were
synergistically sensitized to TMZ cytotoxicity. The results of the
study provide a rationale for the mechanism of action through which
BC3EE2,9B, a carbazole derivative, sensitizes drug-resistant
glioblastoma cells to the chemotherapeutic agent TMZ.
Materials and methods
Chemical reagents
TMZ, 4′,6-diamidino-2-phenylindole (DAPI), propidium
iodide (PI) and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich (München, Germany). Antibodies to
β-actin (#sc-47778), anti-adenosine monophosphate-activated protein
kinase (AMPK; #sc-25792), anti-p-AMPK (#sc-33524), and anti-p-mTOR
(#sc-101738) were obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Antibodies to anti-p-Akt (#05-736) and anti-Akt
(#05-591) were obtained from Millipore (Bedford, MA, USA). Anti-LC3
antibodies (#NB100-2220) were purchased from Novus Biologicals
(Littleton, CO, USA). Primary antibodies were used at a dilution of
1:1,000 in 0.1% Tween-20 and secondary antibodies were used at
1:5,000 dilutions. Acridine orange was obtained from Life
Technologies (Rockville, MD, USA). Carbazole derivatives were
synthesized in the laboratory of the Graduate Institute of
Pharmaceutical Chemistry, College of Pharmacy, China Medical
University (Taichung, Taiwan), under the supervision of Dr L.J.
Huang. Carbazole derivatives were synthesized and obtained by the
laboratory of Dr L.J. Huang.
Cell cultures and cytotoxicity
assays
GBM8901 and GBM8401 cells were maintained in
RPMI-1640, and SK-N-MC and Detroit 551 cells were maintained in
minimum essential medium (MEM), and were routinely tested for
mycoplasma or any other bacterial contamination. All the cultures
were supplemented with 10% fetal bovine serum (FBS), penicillin (50
IU/ml) and streptomycin (50 µg/ml), and cells were grown at
37°C in a humidified 5% CO2 atmosphere. Following
treatment, cells were treated with MTT (1 mg/ml) and incubated for
2 h at 37°C. Cell viability was measured by the modified MTT assay.
Absorbance of the converted dye was measured at a wavelength of 550
nm with a 96-well microplate reader.
Flow cytometry assay
For the cell cycle analysis, cells were cultured in
10-cm culture dishes and seeded at 1×106 cells/dish.
Subsequently, cell pellets were fixed with methanol at −20°C
overnight. Following fixation, cell pellets were incubated at 37°C
for 30 min with 0.5% Triton X-100 in phosphate-buffered saline
(PBS) and 0.5 µg/ml RNase A. Cells were exposed to 1 ml of
PI solution (50 µg/ml) for 30 min on ice. The nuclei were
analyzed in an FACScan laser flow cytometer (Becton-Dickinson, San
Jose, CA, USA). Data were acquired and analyzed using WinMDI 2.8
software (Scripps Research Insititute, La Jolla, CA, USA).
Cell migration and invasion assay
The cell migration and invasion assays were
performed in a 24-well Boyden chamber with an 8-µm pore size
polycarbonate membrane (Corning, Inc., Corning, NY, USA). For the
migration assay, 1×105 cells in 200 µl of
serum-free medium were added to the upper compartment of the
chamber; the lower compartment was filled with 600 µl of
RPMI-1640 supplemented with 10% FBS. After incubation at 37°C for
24 h, cells remaining in the upper chamber were removed using
swabs. The cells on the lower surface of the membrane were fixed
with methanol and stained with 0.1% crystal violet. Images were
captured and the cells were counted using a light microscope. The
invasion assay was performed using the same procedure, except that
the membrane was coated with Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA) to form a matrix barrier and 3×104
GBM8901 cells were added to the upper compartment of the
chamber.
TUNEL assay
GBM8901 cells were seeded in 24-well plates, exposed
for 48 h to microglial-conditioned medium, incubated for 48 h with
either protein samples or a control solution, fixed in 4%
paraformaldehyde for 30 min at 37°C and subsequently treated with
DNase for 15 min. The TUNEL assay (Invitrogen, Carlsbad, CA, USA)
was performed according to the manufacturer's instructions.
Immunofluorescence and acridine-orange
staining assay
For the immunofluorescence assay, cells were
cultured on coated slides and treated for 24 h. Following
treatment, cells were fixed with 2% buffered paraformaldehyde,
permeabilized in 0.25% Triton X-100 for 5 min at 4°C, and gently
agitated in the presence of anti-LC3 at 4°C overnight.
Subsequently, the slides were incubated with an fluorescein
isothiocyanate-labeled secondary antibody, depending on the origin
of the primary antibody, and 4′,6-diamidino-2-phenylindole. For the
acridine-orange assay, cells were cultured on coated slides and
treated for 24 h. Following treatment, cells were exposed to
acridine orange solution (10 µg/ml) in PBS for 5 min and
green fluorescence was detected using a fluorescence microscope
(DP80/BX53; Olympus, Tokyo, Japan).
Western blot analysis
GBM8901 cells were harvested and homogenized in
lysis buffer [50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5%
Nonidet P-40, 0.5 mM phenylmethylsulfonyl fluoride and 0.5 mM
dithiothreitol] for 30 min at 4°C. Equal amounts of total cellular
proteins (50 µg) were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred
onto polyvinylidene difluoride membranes (Millipore) and probed
using primary antibodies, followed by horseradish
peroxidase-conjugated secondary antibodies. The immunocomplexes
were visualized using an enhanced chemiluminescence kit
(Millipore).
Plasmid transfection
DN-AKT and CE-AKT plasmids were obtained from Santa
Cruz Biotechnology, Inc. Cells were transfected with transfection
reagent Lipofectamine™ 2000 and incubated for 6 h. RPMI-1640 was
used instead of Opti-MEM.
Statistical analysis
Statistical comparisons of differences between
groups were conducted using the Student's t-test. P≤0.05 was
considered to indicate a statistically significant difference. All
the statistical analyses were performed with the statistical
package SPSS for Windows (version 16.0; SPSS, Inc., Chicago, IL,
USA).
Results
Anti-proliferative activity of carbazole
derivatives in GBM8901 and GBM8401 cells
To date, only a few carbazole derivatives have been
shown to have antitumor activity (23). In the present study, 35 carbazole
derivatives were designed and synthesized, and the
anti-proliferation activity of each derivative was tested in two
human glioblastoma multiforme cell lines, GBM8901 and GBM8401
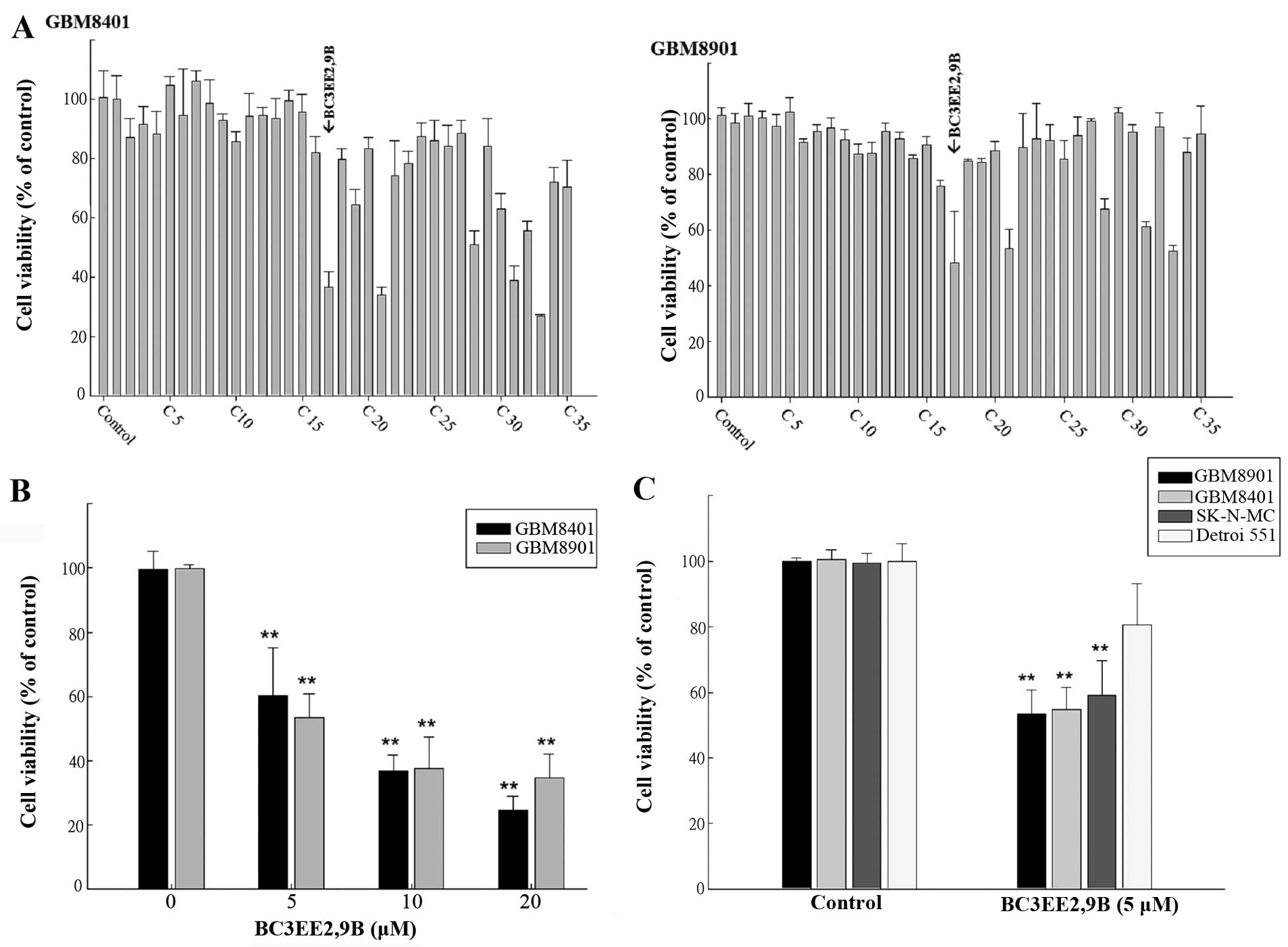
(Fig. 1A). Five of the synthetic
carbazole derivatives (nos. 17, 21, 28, 31 and 33) significantly
reduced the growth of glioblastoma cells at a concentration of 10
µM (Fig. 2A). Of those
derivatives, BC3EE2,9B (no. 17) (Fig.
1B) had the most significant anti-proliferative effect.
Viability, as a percent of control, of the GBM8901 cells was
48.13±18.6% and that of GBM8401 cells was 36.74±4.95% following
exposure to BC3EE2,9B (Fig. 2A).
Therefore, BC3EE2,9B was used as the primary carbazole derivative
in the following experiments. To further determine the inhibitory
effect of BC3EE2,9B on glioblastoma cells, a dose-range experiment
was performed by treating cells with various concentrations of
BC3EE2,9B for 24 h. The inhibitory effects of BC3EE2,9B were
significant at concentrations ranging from 5 to 20 µM in
GBM8401 (IC50=7 µM) and GBM8901
(IC50=5 µM) cells (Fig. 2B). To establish whether BC3EE2,9B
is toxic to malignant tumor cells, various cell lines, including
the human neuroblastoma cell line SK-N-MC and the normal skin cell
line Detroit 551, were treated with 5 µM BC3EE2,9B for 24 h.
BC3EE2,9B treatments significantly inhibited growth of GBM8901,
GBM8401 and SK-N-MC cells, however, Detroit 551 cells at 5
µM were not significantly inhibited (Fig. 2C). These results show that
BC3EE2,9B suppresses the proliferation of glioblastoma cells but
not healthy cells.
Combination of BC3EE2,9B and TMZ
synergistically increases GBM8901 cell death
Studies have shown that treatment of glioblastoma
cells with TMZ is associated with concentration-limiting toxicity.
In our previous experiment, BC3EE2,9B was relatively nontoxic to
normal cells. The present study aimed to improve the cytotoxic
efficacy of TMZ by reducing its concentration in the presence of
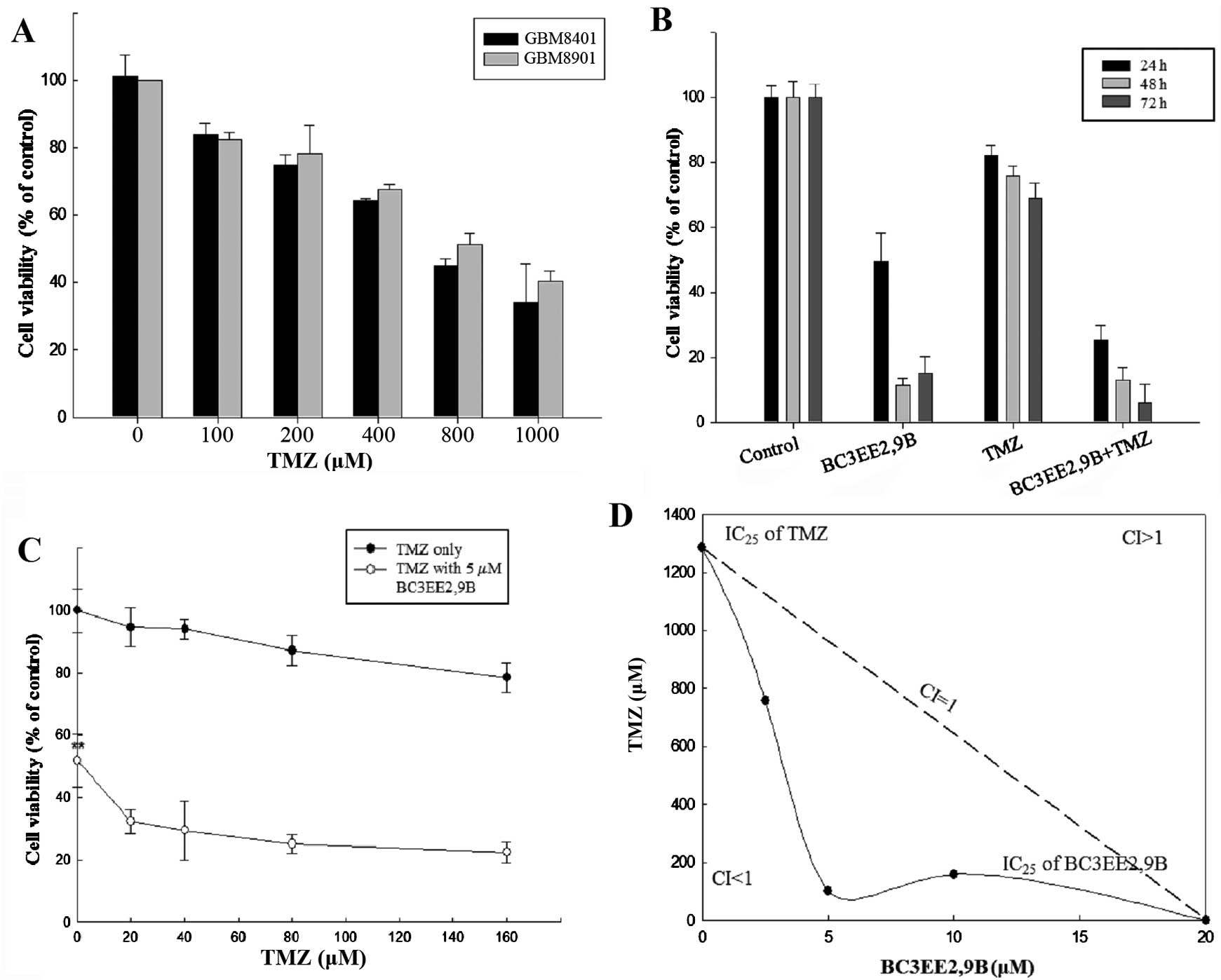
BC3EE2,9B. First, a dose-range experiment was performed by treating
glioblastoma cells with various concentrations of TMZ alone. A high
concentration of TMZ inhibited the proliferation of GBM8901
(IC50, ~800 µM) and GBM8401 (IC50,
~600 µM) cells after 24 h treatment (Fig. 3A). This indicates that TMZ at a
high concentration inhibited growth on glioblastoma cells,
particularly the GBM8901 cell line. Therefore, the GBM8901 cell
line was used in the following experiments. Our previous data
showed that BC3EE2,9B exhibits inhibitory activity against
glioblastoma cells; therefore, we hypothesized that combining this
compound with TMZ will increase the anti-proliferative effect. As
shown in Fig. 3B, BC3EE2,9B (5
µM) combined with TMZ (100 µM) significantly
inhibited GBM8901 cell growth for ≤72 h in a dose-dependent manner
(Fig. 3C). To further assess
whether the two compounds have synergistic benefits on growth
inhibition of glioblastoma cells, an isobologram curve was plotted
(Fig. 3D). The data demonstrated
a strong synergistic effect (confidence interval <1) among three
different ratios (TMZ:BC3EE2,9B, 750:2.5, 100:5 and 200:10,
respectively) at 5 µM BC3EE2,9B combined with 100 µM
TMZ. These results show that GBM8901 cells were more sensitive to
combined treatment with BC3EE2,9B and TMZ.
Effects of BC3EE2,9B on GBM8901 cell
morphology, cell cycle and migration/invasion
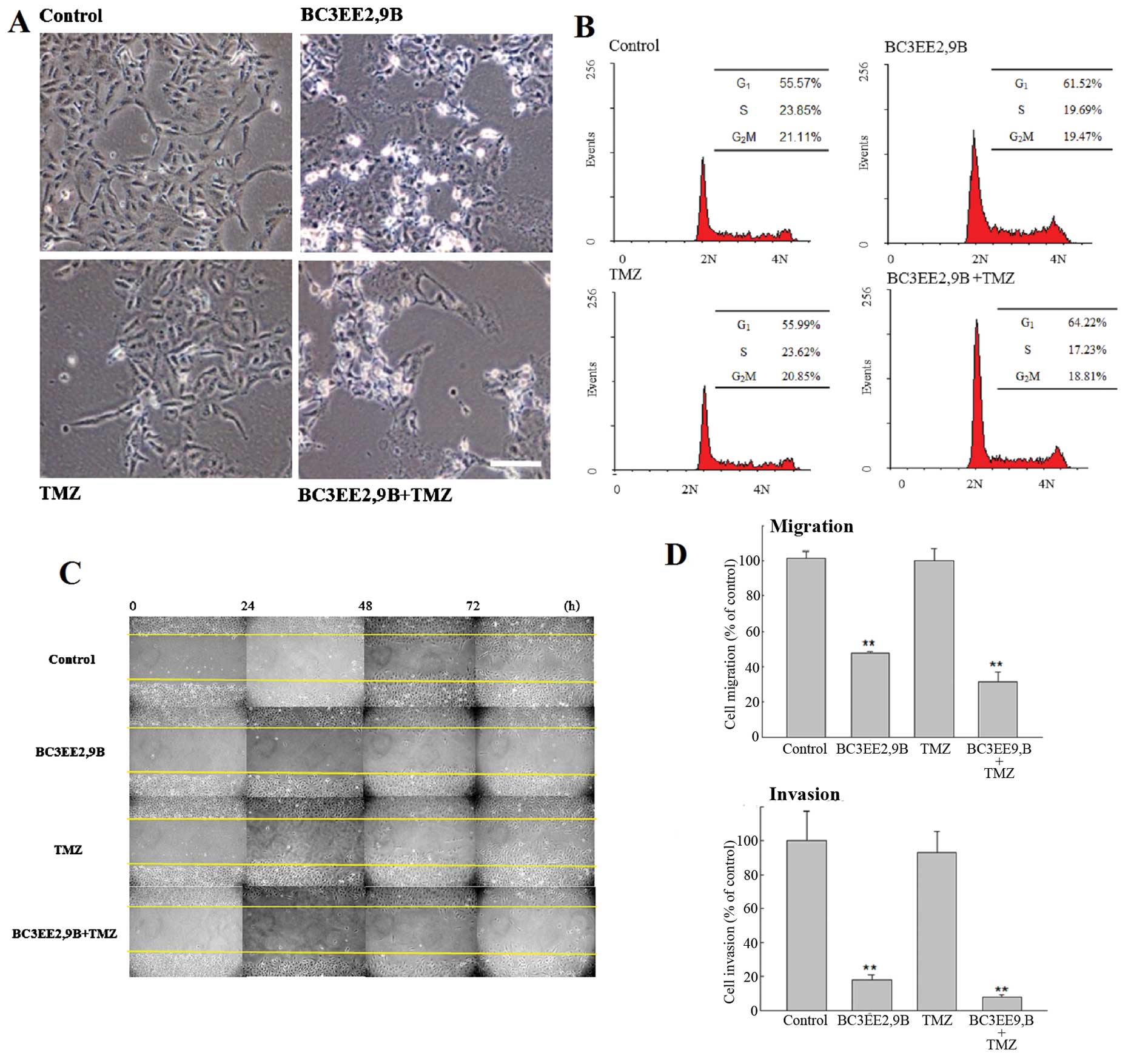
Microscopic analysis revealed that GBM8901 cells
exposed to 5 µM BC3EE2,9B for 24 h showed more evidence of
cell shrinkage and detachment from culture plates, as well as more
cytoplasmic vacuoles compared with the untreated cells (Fig. 4A). By contrast, no clear
morphological changes were observed in cells treated with 100
µM of TMZ. These results suggest that BC3EE2,9B induces cell
death in glioblastoma. To further analyze the nature of cell death,
flow cytometric analysis was performed in GBM8901 cells following
exposure to BC3EE2,9B with or without TMZ for 24 h (Fig. 4B). The results showed that 5
µM BC3EE2,9B combined with 100 µM TMZ slightly
induced G1 arrest (8.65%); however, no significant
sub-G1 hypoploid cell population was observed,
indicating that apoptosis may not be the mechanism governing cell
death. As motility and metastatic spread are well-known hallmarks
of all malignant tumors, particularly glioblastoma, wound healing
assays were performed to verify the effects of BC3EE2,9B on
migration and invasion of GBM8901 glioblastoma cells. Cells were
allowed to grow to confluence, and were scratched to create a
cleared area within the monolayer. Following treatments, the
movement of glioblastoma cells was imaged and quantified by
measuring the migration distance at different time points and
comparing it with the front area at time zero. Even a low
concentration of BC3EE2,9B (2.5 µM) combined with TMZ
treatment significantly inhibited cell migration at 72 h
(68.81±5.71%) (Fig. 4C).
Subsequently, a Transwell assay was employed to further measure the
effects of BC3EE2,9B on the migratory and invasive capacities of
glioblastoma cells. BC3EE2,9B (2.5 µM) alone and combined
with TMZ (100 µM) markedly suppressed the migration of
GBM8901 cells (Fig. 4D). As cell
migration is believed to be a fundamental step in tumor invasion,
the effect of chrysin on the invasive capacity of GBM8901 cells was
examined. Treatment of Matrigel-coated wells with BC3EE2,9B (2.5
µM) or combined with TMZ (100 µM) significantly
reduced cell invasion. In particular, the number of invasive
GBM8901 cells in cultures exposed to BC3EE2,9B and TMZ decreased by
91.98±1.07% relative to the number of cells treated with TMZ alone.
These findings suggest that low levels of BC3EE2,9B combined with
TMZ effectively stimulate cell death via a non-apoptotic
mechanism.
BC3EE2,9B combined with TMZ induces
autophagy, but not apoptosis, in GBM8901 cells
Our previous results indicated that treatment of
GBM8901 cells with BC3EE2,9B results in cell death via a
non-apoptotic mechanism. Therefore, the TUNEL assay was used to
confirm these finding (Fig. 5A).
No TUNEL-positive cells were detected after 24 h of BC3EE2,9B
treatment. Notably however, another carbazole derivative,
αc-6K-9TriMoB (no. 21), significantly induced apoptosis. These
findings confirmed that cell death due to BC3EE2,9B combined with
TMZ is not governed by an apoptotic pathway. A previous study
reported that TMZ can induce autophagic cell death in malignant
glioblastoma cells (14).
Therefore, whether BC3EE2,9B combined with TMZ treatment also
induces autophagic cell death was examined. Cells showed an
increase in orange fluorescence, indicating the accumulation of
acridine orange in the acidic compartments of TMZ-treated cells
after exposure to BC3EE2,9B for 24 h (Fig. 5B). However, treatment with
αc-6K-9TriMoB resulted in significantly less orange fluorescence.
Furthermore, cells treated with combination therapy showed the
presence of LC3-II aggregation, a specific marker associated with
autophagosome formation (Fig.
5C). These results indicate that the combination of BC3EE2,9B
with TMZ induces autophagy, but not apoptosis, in GBM8901
cells.
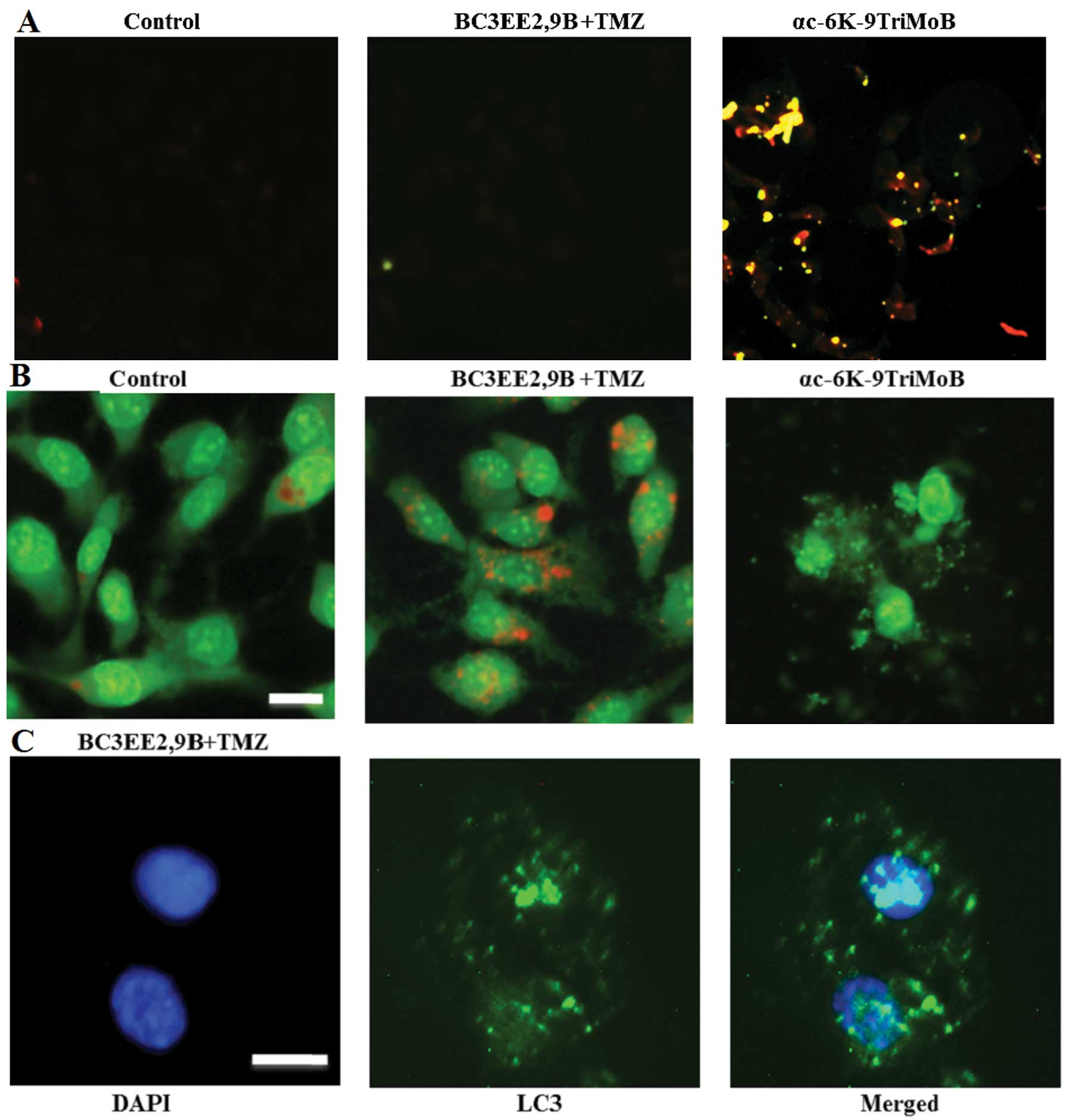 | Figure 5Combination of
bis(carbazole-2,9N-benzyl)-3-ethyl ethanoate (BC3EE2,9B) and
temozolomide (TMZ) induced autophagy, but not apoptosis, in
glioblastoma multiforme (GBM)8901 cells. (A) Cell apoptosis was
measured by the TUNEL assay and fluorescence microscopy. GBM8901
cells were treated with αc-6K-9TriMoB or TMZ combined with
BC3EE2,9B for 24 h. (B) Fluorescence microscopy analysis of GBM8901
cells treated with TMZ combined with BC3EE2,9B for 24 h, followed
by acridine orange staining. Formation of acridine
orange-accumulating autophagic vacuoles (orange-red fluorescence)
in cells treated with BC3EE2,9B and TMZ. (C) Cells stained with
4′,6-diamidino-2-phenylindole (DAPI) and green fluorescent
anti-LC3-II. The images show that cells treated with TMZ combined
with BC3EE2,9B have significant LC3-II aggregation. |
BC3EE2,9B combined with TMZ enhances
autophagy through inactivation of the Akt-mTOR pathway in GBM8901
cells
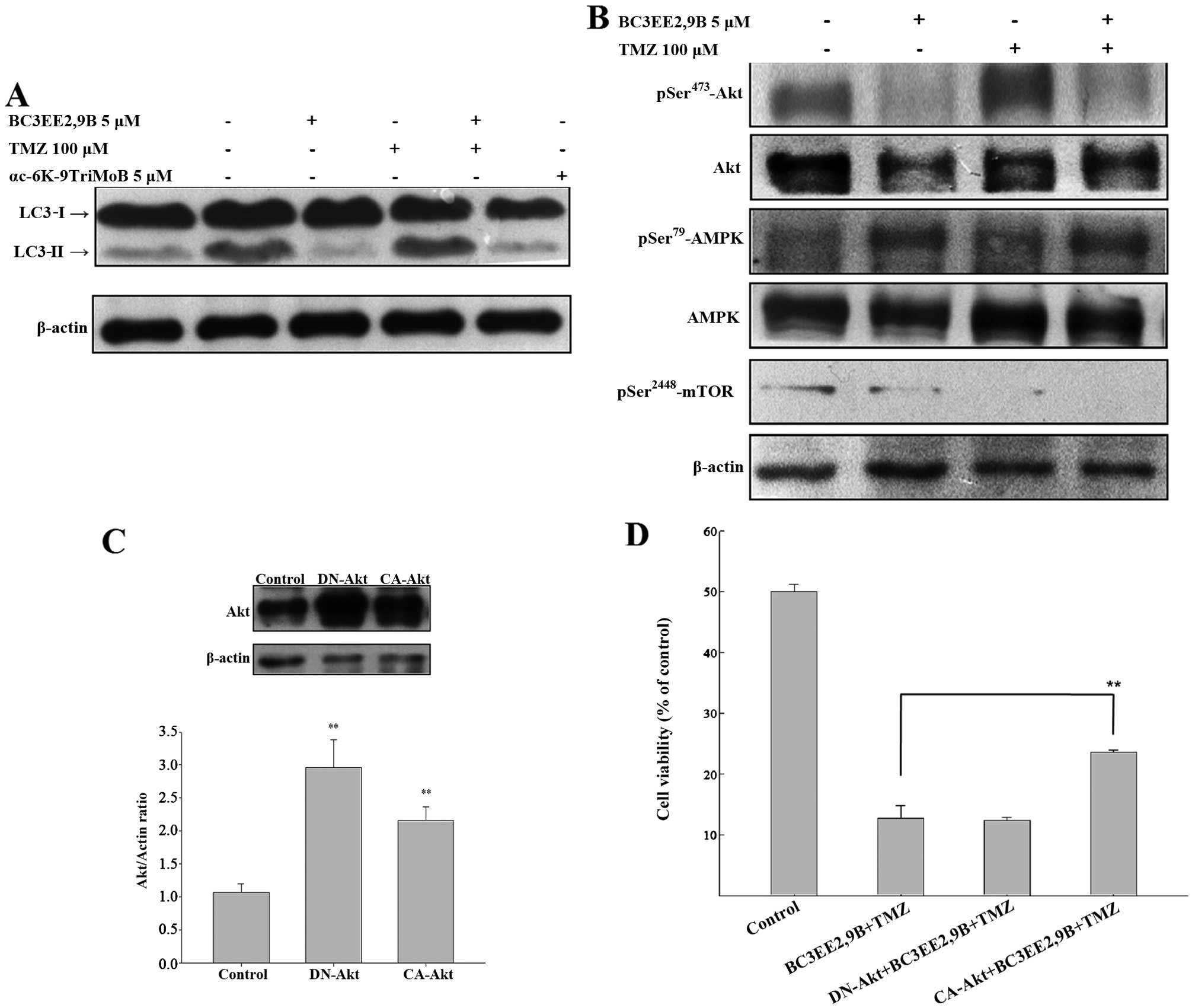
The detection of LC3-I to LC3-II conversion is a
useful and sensitive marker of autophagy (22). Western immunoblotting was
performed to probe for LC3. BC3EE2,9B combined with TMZ resulted in
a significant increase in the LC3-II level after 24 h (Fig. 6A); however, αc-6K-9TriMoB
treatment did not lead to an increase in LC3-II level. It is
well-known that inhibition of Akt and its downstream target mTOR
initiates autophagy. As autophagy is negatively regulated by the
PI3K-Akt-mTOR pathway, interference with this pathway promotes
autophagy. To elucidate whether BC3EE2,9B enhances autophagy
through inactivation of the Akt-mTOR pathway, western blot assays
using specific antibodies were performed. BC3EE2,9B combined with
TMZ resulted in a significant decrease in Akt and mTOR
phosphorylation, as well as a significant increase in AMPK
phosphorylation after 24 h (Fig.
6B), suggesting that BC3EE2,9B/TMZ-induced autophagy may
involve activation of AMPK and the attenuation of the Akt and mTOR
downstream signaling pathway. To further confirm whether the
inactivation of the Akt pathway is essential for combination
treatment-induced autophagy, constitutively active Akt (CA-Akt) or
dominant negative Akt (DN-Akt) plasmids were transfected into
GBM8901 cells. The two plasmids were successfully transfected and
expressed in glioblastoma cells (Fig.
6C). The cells were subsequently treated with BC3EE2,9B
combined with TMZ to observe the treatment effect on cell
viability. The results showed that the DN-Akt significantly
protected against cytotoxicity of BC3EE2,9B combined with TMZ
treatments (21.76%) (Fig. 6D).
These results indicate that BC3EE2,9B combined with TMZ induces
autophagic cell death via the AKT-mTOR pathway in GBM8901
cells.
Discussion
GBM is one of the most treated refractory tumors.
Standard therapy for GBM includes surgical resection, focal
radiotherapy and treatment with the alkylating agent TMZ. However,
this therapeutic approach results in only a modest increase in
survival of patients with the disease. Novel therapeutic approaches
aimed at improving the efficacy of TMZ are, therefore, urgently
required. The present study identified that treatment with the
carbazole derivative, BC3EE2,9B, resulted in a marked decrease in
cell viability, migration and invasion in GBM8901 glioblastoma
cells, but not in Detroit 551 normal cells. Furthermore,
combination treatment with BC3EE2,9B and TMZ inhibited glioblastoma
growth to a much higher extent compared with the treatment with TMZ
or BC3EE2,9B alone. In addition, co-administration of BC3EE2,9B and
TMZ significantly enhanced autophagic death, most likely through
inhibition of the Akt-mTOR signaling pathway. This supports the
idea that BC3EE2,9B synergistically sensitizes glioblastoma cells
to TMZ-induced cytotoxicity. Acquired drug resistance is associated
with TMZ treatment in GBM patients. The results indicate that
combination therapy, comprising a toxic chemotherapeutic agent
(TMZ) with a nontoxic naturally occurring compound that has
anticancer properties (BC3EE2,9B) could be a new strategy to
overcome this problem.
Glioblastoma cells frequently carry mutations in the
PTEN tumor-suppressor gene, a gene that inhibits the
continuous activation of Akt (3),
making them relatively resistant to TMZ treatment (4). As mutations in glioblastoma cells
often inactivate the apoptotic pathway, they are likely to be more
sensitive to autophagy as an alternative response to therapeutic
agents (23). Furthermore, agents
that induce autophagy may result in fewer side effects than those
that induce apoptosis, as apoptotic bodies are removed by
phagocytic cells, thus preventing an inflammatory response
(9,10). Carbazole derivatives have been
shown to act as inhibitors of DNA topoisomerase and exert their
cytotoxic effects in replicating cells by interfering with DNA
repair and inducing DNA strand breaks (24). O6-methylguanine-DNA
methyltransferase (MGMT), a DNA repair protein that removes
O6-methylguanine adducts from damaged DNA, is the most
important determinant of TMZ resistance in patients with GBM. As a
result, proficient DNA repair activities promote glioblastoma cell
survival, leading to TMZ resistance and poor clinical outcome.
These observations support the present findings that although
glioblastoma cells treated with TMZ alone failed to show
significant autophagy, TMZ combined with BC3EE2,9B treatment
inhibited Akt-mTOR signaling enhanced sensitivity of these cells by
increasing the extent of autophagy and appeared to synergistically
promote cell death. The autophagic effect of carbazole derivatives
provides insight into the anticancer activities of this compound,
particularly in TMZ-resistant and apoptosis-resistant glioblastoma
cells.
AMPK is a sensor of energy status. Although it is
best known for its effects on metabolism, AMPK is involved in
numerous important pathways that govern a variety of physiological
activities including cell growth, survival, migration and
cell-cycle regulation (25).
Recent studies have shown that the activation of the AMPK pathway
is associated with autophagy in cancer cells (26). In the present study, BC3EE2,9B
treatment decreased the phosphorylation of Akt and activated AMPK.
In fact, various active compounds isolated from natural products
are effective in inducing autophagic cell death in
apoptosis-resistance cells. For example, ursolic acid has been
shown to induce cell death and modulate autophagy in
apoptosis-resistant colorectal cancer cells (27). Additionally, coibamide A, a potent
anti-proliferative cyclic depsipeptide isolated from a marine
cyanobacterium, has been shown to induce autophagic cell death in
apoptosis-resistant glioblastoma cells (28). Therefore, a number of
autophagy-inducing carbazole compounds that can activate autophagic
cell death independent of the apoptotic process were synthesized in
the present study. These results point to a potential therapeutic
role of alkaloids in apoptosis-resistant cancers, such as
TMZ-treated glioblastoma cells.
In the present study, BC3EE2,9B, a carbazole
derivative, was demonstrated to inhibit glioblastoma cell
migration, invasion and growth. The data also indicate that
BC3EE2,9B induced autophagy-mediated cell death and synergistically
sensitized GBM cells to TMZ cytotoxicity. The possible mechanism
underlying BC3EE2,9B-induced autophagy may involve activation of
AMPK and the attenuation of the Akt and mTOR downstream signaling
pathway. Taken together, the data provide molecular evidence for
the mode of action governing the ability of BC3EE2,9B to sensitize
drug-resistant glioblastoma cells to the chemotherapeutic agent
TMZ.
Acknowledgments
The present study was supported by grants from the
Changhua Christian Hospital (no. 103-CCH-IRP-025) and from the
Ministry of Science and Technology (no. 101-2320-B-040-015-MY3).
Fluorescence microscopy and imaging analysis were performed at the
Instrument Center of the Chung Shan Medical University, which is
supported by the Ministry of Science and Technology, Ministry of
Education and the Chung Shan Medical University.
References
|
1
|
Hess KR, Broglio KR and Bondy ML: Adult
glioma incidence trends in the United States 1977–2000. Cancer.
101:2293–2299. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Adamson C, Kanu OO, Mehta AI, Di C, Lin N,
Mattox AK and Bigner DD: Glioblastoma multiforme: A review of where
we have been and where we are going. Expert Opin Investig Drugs.
18:1061–1083. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion
O6-methylguanine. Oncogene. 26:186–197. 2007. View Article : Google Scholar
|
|
5
|
Brandes AA, Tosoni A, Franceschi E, Sotti
G, Frezza G, Amistà P, Morandi L, Spagnolli F and Ermani M:
Recurrence pattern after temozolomide concomitant with and adjuvant
to radiotherapy in newly diagnosed patients with glioblastoma:
Correlation with MGMT promoter methylation status. J Clin Oncol.
27:1275–1279. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
7
|
Shen HM and Codogno P: Autophagic cell
death: Loch Ness monster or endangered species? Autophagy.
7:457–465. 2011. View Article : Google Scholar
|
|
8
|
Lefranc F, Facchini V and Kiss R:
Proautophagic drugs: A novel means to combat apoptosis-resistant
cancers, with a special emphasis on glioblastomas. Oncologist.
12:1395–1403. 2007. View Article : Google Scholar
|
|
9
|
Edinger AL and Thompson CB: Death by
design: Apoptosis, necrosis and autophagy. Curr Opin Cell Biol.
16:663–669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochim Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen C, Chen J and Zhao KN: Editorial:
Signalling pathways in anti-cancer drug resistance. Curr Med Chem.
21:3007–3008. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng X and Kinsella TJ: A novel role for
DNA mismatch repair and the autophagic processing of chemotherapy
drugs in human tumor cells. Autophagy. 3:368–370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Math M and Balasubramaniam P: Curry leaves
(Murraya Koenigii spreng) and halitosis. BMJ (South Asia ED).
19:2112003.
|
|
16
|
Tsao LT, Lee CY, Huang LJ, Kuo SC and Wang
JP: Inhibition of lipopolysaccharide-stimulated nitric oxide
production in RAW 264.7 macrophages by a synthetic carbazole,
LCY-2-CHO. Biochem Pharmacol. 63:1961–1968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arbiser JL, Govindarajan B, Battle TE,
Lynch R, Frank DA, Ushio-Fukai M, Perry BN, Stern DF, Bowden GT,
Liu A, et al: Carbazole is a naturally occurring inhibitor of
angiogenesis and inflammation isolated from antipsoriatic coal tar.
J Invest Dermatol. 126:1396–1402. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hajbi Y, Neagoie C, Biannic B, Chilloux A,
Vedrenne E, Baldeyrou B, Bailly C, Mérour JY, Rosca S, Routier S,
et al: Synthesis and biological activities of new
furo[3,4-b]carbazoles: Potential topoisomerase II inhibitors. Eur J
Med Chem. 45:5428–5437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoon S, Kim JH, Lee YJ, Ahn MY, Choi G,
Kim WK, Yang Z, Lee HJ, Moon HR and Kim HS: A novel carbazole
derivative, MHY407, sensitizes cancer cells to doxorubicin-,
etoposide-, and radiation treatment via DNA damage. Eur J
Pharmacol. 697:24–31. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu CH, Lin C, Tsai KJ, Chuang YC, Huang
YL, Lee TH, Huang LJ and Chan HC: Biological evaluation of
9-[(6-chloro-pyridin-4-yl)methyl]-9H-carbazole-3-carbinol as an
anticancer agent. Oncol Rep. 29:1501–1509. 2013.PubMed/NCBI
|
|
21
|
Kang CC, Huang WC, Kouh CW, et al:
Chemical principles for the design of a novel fluorescent probe
with high cancer-targeting selectivity and sensitivity. Integr
Biol. 5:1217–1228. 2013. View Article : Google Scholar
|
|
22
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zembower DE, Xie Y, Koohang A, Kuffel MJ,
Ames MM, Zhou Y, Mishra R, Mar AA, Flavin MT and Xu ZQ:
Methylenedioxy-and ethylenedioxy-fused indolocarbazoles: Potent
human topoisomerase I inhibitors and antitumor agents. Anticancer
Agents Med Chem. 12:1117–1131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee YK, Park SY, Kim YM, Kim DC, Lee WS,
Surh YJ and Park OJ: Suppression of mTOR via Akt-dependent and
-independent mechanisms in selenium-treated colon cancer cells:
Involvement of AMPKalpha1. Carcinogenesis. 31:1092–1099. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puissant A, Robert G, Fenouille N, Luciano
F, Cassuto JP, Raynaud S and Auberger P: Resveratrol promotes
autophagic cell death in chronic myelogenous leukemia cells via
JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res.
70:1042–1052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xavier CP, Lima CF, Pedro DF, Wilson JM,
Kristiansen K and Pereira-Wilson C: Ursolic acid induces cell death
and modulates autophagy through JNK pathway in apoptosis-resistant
colorectal cancer cells. J Nutr Biochem. 24:706–712. 2013.
View Article : Google Scholar
|
|
28
|
Hau AM, Greenwood JA, Löhr CV, Serrill JD,
Proteau PJ, Ganley IG, McPhail KL and Ishmael JE: Coibamide A
induces mTOR-independent autophagy and cell death in human
glioblastoma cells. PLoS One. 8:e652502013. View Article : Google Scholar : PubMed/NCBI
|