Introduction
Hepatocellular carcinoma (HCC) is one of the most
common and lethal malignancies worldwide (1,2).
An estimated 350,000 deaths from liver cancer occur worldwide each
year (3). The highest liver
cancer rates are to be found in East and Southeast Asia, and in
Central and Western Africa; chronic hepatitis B virus (HBV) and C
(HCV) infection are responsbile for approximately 75–80% of the HCC
cases worldwide, particularly in Asian and African populations
(4,5).
The fidelity of DNA replication is generally
considered an important characteristic of cancer progression and
during the cell cycle. Dysfunctional DNA damage repair and
checkpoints during the cell cycle process contribute to genomic
instability. Replication factor C (RFC) is a heteropentameric
primer-recognition protein complex involved in DNA replication, DNA
damage repair and checkpoint control during cell cycle progression
(6–10). The RFC complex functions to load
proliferating cell nuclear antigen (PCNA), a ring-shaped
homotrimer, onto DNA in an ATP-dependent manner in order to provide
a sliding clamp for various proteins involved in DNA replication
processes (11).
RFC is comprised of one large subunit [replication
factor C, subunit 1 (RFC1)] and four small subunits [replication
factor C, subunits 2–5 (RFC2-5)]. Of these subunits, replication
factor C, subunit 3 (RFC3), a 38-kDa subunit, has been reported to
be overexpressed in esophageal adenocarcinomas and ovarian
carcinomas (12,13). Moreover, RFC3 knockdown has been
shown to result in the inhibition of cancer cell proliferation and
growth (12,14). The disruption of the RFC3-PCNA
complex induced by 9-cis retinoic acid-activated retinoid X
receptor α (RXRα) has been shown to inhibit the growth of cancer
and embryonic cells and to arrest S phase entry (15). These findings suggest that RFC3
may be one of the most important cancer antigens. However, its role
in the development of HCC remains unclear.
In this study, we found that RFC3 was overexpressed
in HCC tissues and cells. Further investigations revealed that RFC3
is a critical factor in promoting the development of HCC, as the
silencing of RFC3 by shRNA led to cell cycle arrest. Our data
provide new insight into the role of RFC3 in the development of
HCC.
Materials and methods
Tissue samples
Liver tumor tissue samples were obtained from 24
patients (age: mean 55, rage 40–68, gender: female 5, male 19) who
were diagnosed with HCC at the Third Affiliated Hospital, Sun
Yat-Sen University, Guangzhou, China in 2012. A total of 24 human
HCC tissues and 12 adjacent non-tumor tissue samples were examined
in this study. For each case, tumor samples with matched adjacent
non-tumor tissue samples were collected during surgical resection
and frozen in liquid nitrogen and stored at −80°C. This study was
approved by the Ethics Committee of Sun Yat-Sen University and all
patients provided written informed consent prior to obtaining the
samples.
Cell lines and culture
In this study, we used 1 human hepatocyte cell line
(L02) and 5 HCC cell lines (HepG2, BEL-7402, Hep3B, SMMC-7721 and
LM3), obtained from Shanghai Cell Bank (Chinese Academy of
Science), to detect RFC3 expression. The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Paisley,
Scotland, UK) supplemented with 10% fetal calf serum, 100 IU/ml
penicillin, 100 µg/ml streptomycin, and 2% L-glutamine (all
from Biological Industries Israel Beit-Haemek Ltd. Kibbutz
Beit-Haemek, Israel) at 37°C in an atmosphere with 5%
CO2.
RNA isolation and RT-qPCR
Total RNA was extracted from the tissues and cells
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to
the instructions provided by the manufacturer. Reverse
transcription was performed using a reverse transcription kit
(Takara Bio, Dalian, China), and primers were designed as follows:
RFC3 forward, 5′-GCC TGCAGAGTGCAACAATA-3′ and reverse,
5′-TCAAGGAGCCTTTGTGGAGT-3′; and GAPDH forward,
5′-GAGTCAACGGATTTGGTCGT-3′ and reverse, 5′-GACAAGCTT
CCCGTTCTCAG-3′. Amplification reactions were performed in a 20
µl volume of SYBR-Green PCR Master mix (from Takara Bio).
All the reactions were performed in triplicate in a LightCycler
Real-Time PCR system. The RFC3 mRNA expression levels were
standardized to the GAPDH mRNA levels using the comparative Ct
method. All experiments were performed at least 3 times.
Immunohistochemistry (IHC)
IHC was performed as previously described (16). Briefly, the tumor sections were
deparaffinized using xylene and rehydrated with graded ethyl
alcohol, and a solution of 3% (v/v) H2O2 was
then added to halt the peroxidase activity. Antigen retrieval was
performed by heating the tumor sections in 10 mM sodium citrate
buffer (pH 6.0) at 95–100°C for 20 min. After being washed 3 times
with phosphate-buffered saline (PBS; Sigma-Aldrich, St. Louis, MO,
USA), the sections were blocked with 3% bovine serum albumin (BSA;
Sigma-Aldrich) at room temperature for 1 h, and this was followed
by overnight incubation at 4°C with RFC3 antibody (sc-390293; 1:100
dilution; Santa Cruz Biotechnology, Inc., CA, USA). After being
washed 3 times with PBS, the sections were incubated at 37°C for 2
h with secondary antibodies. Finally, the sections were
counterstained with hematoxylin.
Western blot analysis
The cells were harvested and then lysed in
radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris-HCl (pH
7.4), 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, 1 mM EDTA and 1
mM NaF], containing protease inhibitor cocktail (Sigma-Aldrich).
The cell lysates were boiled for 5 min and refrigerated on ice, and
this was followed by centrifugation at 10,000 × g for 30 sec.
Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and electrotransferred onto
polyvinylidene fluoride (PVDF) membranes. The membranes were
blocked in 5% non-fat dry milk and then probed with the primary
antibodies against RFC3 (sc-390293; 1:500 dilution; Santa Cruz
Biotechnology, Inc.) and p53 (ab31333; 1:500 dilution), p21
(ab7960; 1:200 dilution), p57 (ab75974; 1:500 dilution), cyclin A
(ab137769; 1:1,000 dilution) and cyclin B1 (ab32053; 1:3,000
dilution) (all from Abcam, Cambridge, MA, USA). Subsequently, the
membranes were washed twice with TBST and incubated with
horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG
(H+L) (115-035-003; 1:5,000 dilution) or goat anti-rabbit IgG (H+L)
(111-035-003; 1:5,000 dilution; both from Jackson ImmunoResearch,
West Grove, PA, USA) secondary antibodies at room temperature for 1
h. The membranes were washed another 3 times and then visualized
using an ECL kit (Forevergen Biosciences Co., Ltd., Guangzhou,
China).
Construction of shRFC3 lentivirus and
gene silencing
The lentiviral vector, LV-008 (Forevergen
Biosciences Co., Ltd.), expressing short hairpin RNA (shRNA) and
containing the green fluorescent protein (GFP) gene was used as a
reporter. The recombinant lentiviruses were designed to generate
shRNA targeting the sequence of the RFC3 gene
(5′-AAGTAACTACCACCTTGAAGTTA-3′) and negative control (NC)
(5′-TGGTTTACATGTCGACTAA-3′). The LV-008-shRFC3 plasmids were
transfected into 293T cells (Shanghai Cell Bank, Chinese Academy of
Science), together with the lentiviral packaging vectors, to
generate the respective lentiviruses. Infection lentiviruses were
collected at 72 h post-transfection, and the lentiviruses were
concentrated by ultracentrifugation for 1.5 h at 25,000 rpm in an
SW28 rotor (Bekcman Instruments Inc., Fullerton, CA, USA). For
lentiviral infection, the SMMC-7721 cells were seeded in a 6-well
plate at a density of 50,000 cells/well and infected with the
lentiviruses in the presence of 5–10 µg/ml of polybrene. The
cells in which RFC3 was knocked down were screened out with 2
µg/ml puromycin for 10–15 days. The knockdown efficiency was
validated by RT-qPCR and western blot analysis on day 5
post-infection. Each experiment was performed in triplicate.
Colony formation assay
The cells were digested at the logarithmic growth
phase and seeded into 6-well plates at density gradients of 50, 100
and 200 cells/well. Following 2 weeks of culture, the cells were
washed and fixed with 4% paraformaldehyde for 30 min at room
temperature, and then stained with crystal violet. The number of
colonies was counted under a fluorescence microscope (BX-50;
Olympus, Tokyo, Japan). Each experiment was performed in
triplicate.
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) viability assay
The viability of the cells was determiend by MTS
assay (Sigma-Aldrich, St. Louis, MO, USA). Cells in the logarithmic
growth phase were collected and seeded at a density of
1×103 cells/well in 96-well plates, in triplicate. On
days 1, 2, 3 and 4, MTS reagent was added to the cells at a ratio
of 1:10 followed by incubation at 37°C for 4 h. The solution was
removed, and the cells were dissolved with dimethyl sulfoxide
(Sigma-Aldrich). The absorbance of each well was measured using an
LW R96 ELISA microplate reader (Diatek, West Bengal, India) at a
wavelength of 490 nm. Each experiment was performed in
triplicate.
Cell growth curves
The cells were digested, and the number of living
cells was counted using the method described by Freshney (17). The cells were then seeded in 3
wells of a 12-well plate at approximately 1×105
cells/well. The living cells were digested and counted on days 1, 2
and 3. The experiments were repeated 3 times, and averages were
used to plot the cell growth curves.
Flow cytometric analysis
The cells were harvested and washed in PBS, and
fixed in ice-cold 70% ethanol for 1 h. Following treatment with
RNase A (50 µg/ml; Sigma-Aldrich), the cells were stained
with propidium iodide (PI; Sigma-Aldrich) for 30 min at room
temperature and then analyzed and recorded using a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA). Cell cycle analysis
was performed using FlowJo software (TreeStar Inc., Ashland, OR,
USA).
Statistical analysis
SPSS 18.0 statistical software was used for
statistical analysis. Data are presented as the means ± SD, and all
experiments were performed in triplicate (n=3). Statistical
analysis was performed using analysis of variance (ANOVA). A
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
RFC3 is overexpressed in human liver
tumor tissue
It has previously been reported that RFC3 has
oncogenic activity and is overexpressed in epithelial carcinomas
(12,13). In this study, in order to
determine whether the overexpression of RFC3 is associated with the
development of HCC, liver tumor tissue samples from 24 patients
were examined by RT-qPCR using RFC3-specific primers. Paired
adjacent normal tissue samples were used as the controls. As shown
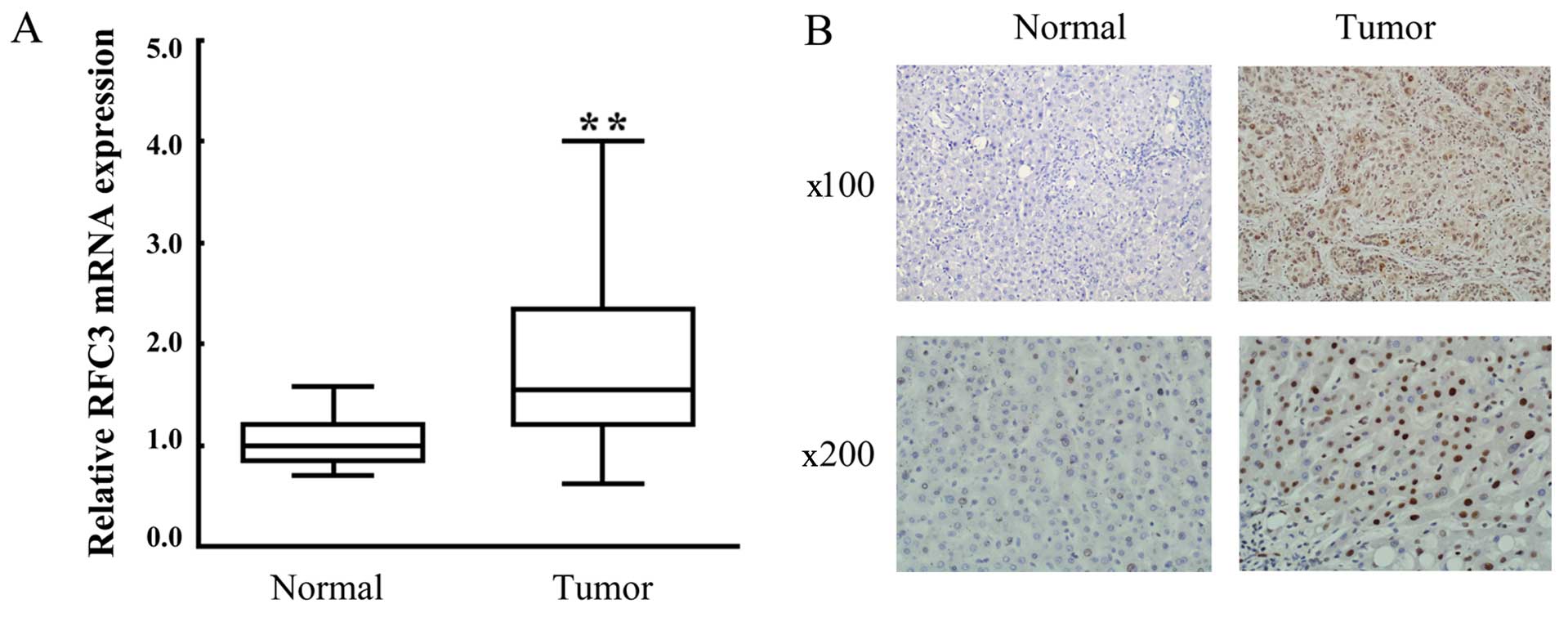
in Fig. 1A, the mRNA expression
level of RFC3 in tumor tissue samples was markedly upregulated
compared with the adjacent non-tumor tissues. Moreover, IHC
analysis revealed that strong positive staining in the liver tumor
tissues, indicating the overexpression of RFC3 protein (Fig. 1B; compare 'Tumor' to 'Normal').
Taken together, these results indicated that RFC3 was upregulated
in the liver tumor tissues.
RFC3 is overexpressed in HCC cell
lines
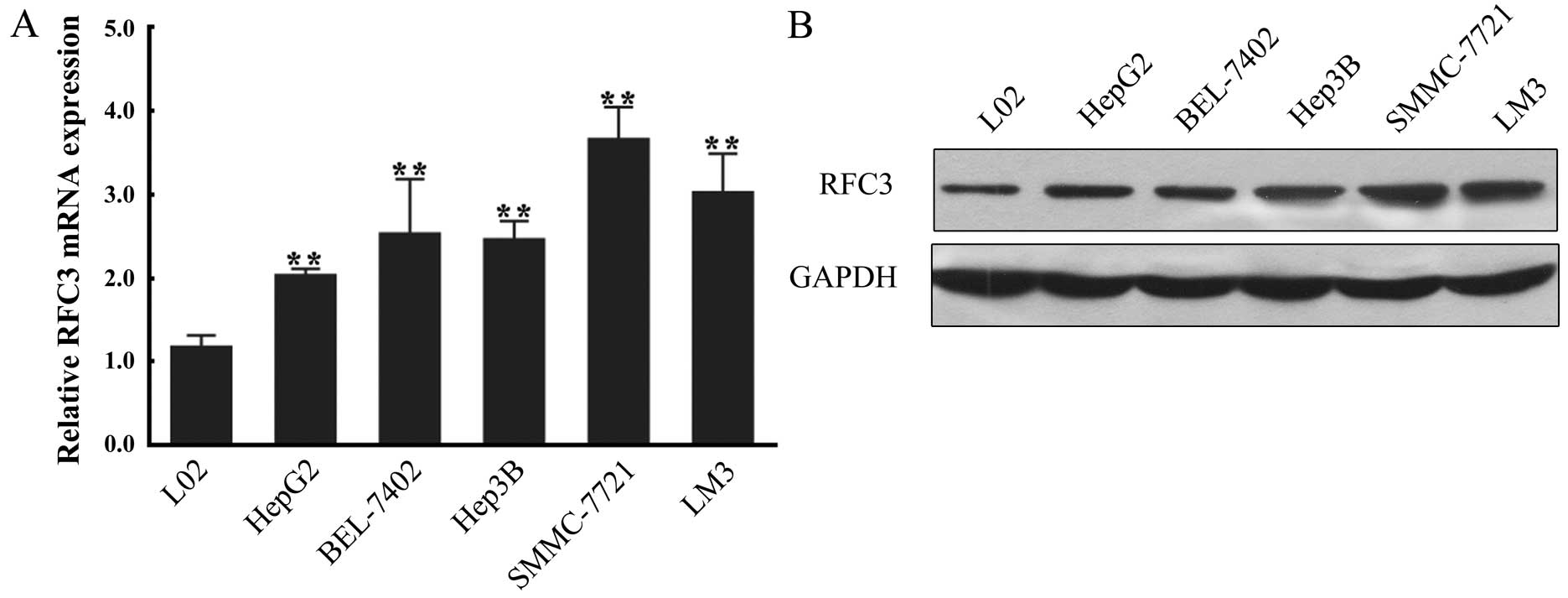
To further confirm the stimulatory effect of RFC3 on
HCC, we sought to identify an RFC3-sensitive cell line. For this
purpose, 5 HCC cell lines (HepG2, BEL-7402, Hep3B, SMMC-7721 and
LM3) were used to measure the mRNA and protein expression of RFC3
by RT-qPCR and western blot analysis, respectively. A normal
hepatocyte cell line (L02) was used as the negative control. In
brief, we found that both the mRNA and protein levels of RFC3 were
increased in all HCC cell lines compared to the hepatocyte cell
line, further confirming that RFC3 overexpression is associated
with HCC (Fig. 2). Of the HCC
cell lines, the SMMC-7721 cells exhibited the highest mRNA and
protein expression of RFC3 and were thus used in subsequent
experiments.
Downregulation of RFC3 through
lentivirus-mediated shRNA in the SMMC-7721 cell line
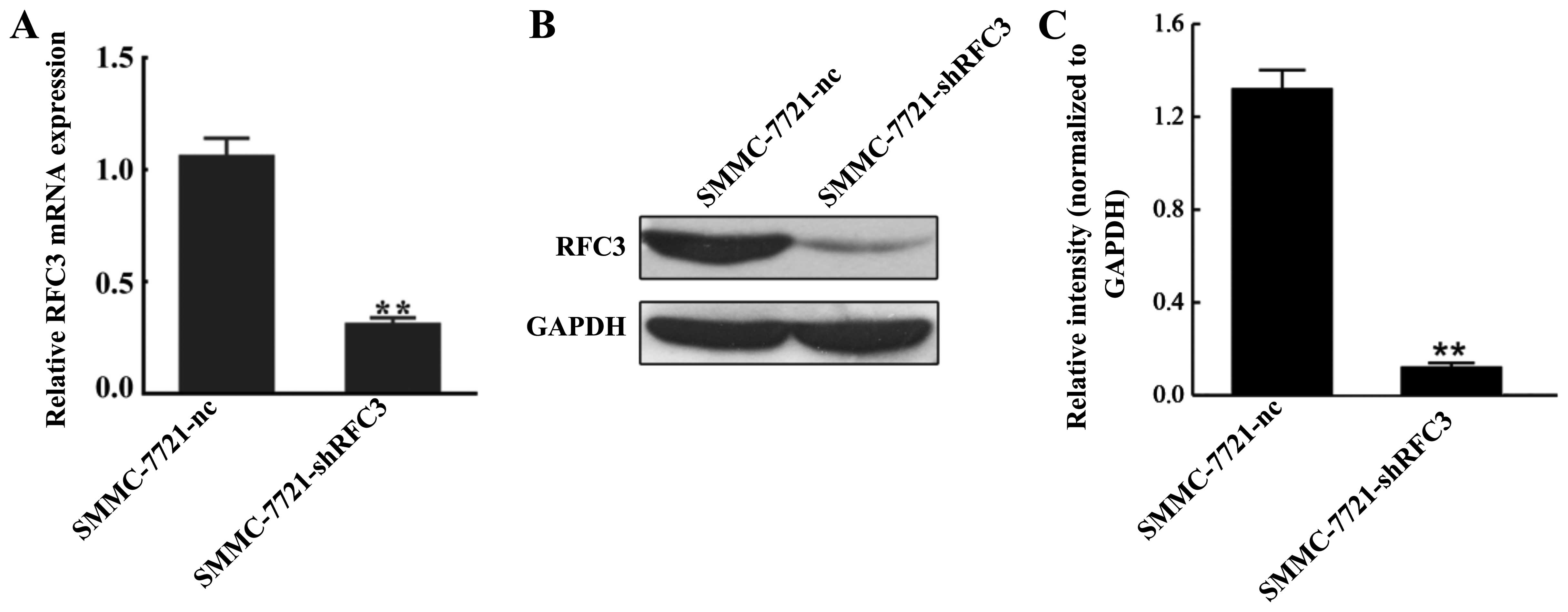
To examine the role RFC3 plays in HCC, a stable HCC
cell line in which RFC3 was knocked down was established using
lentivirus-mediated RNA interference (RNAi) technology. The
SMMC-7721 cell line was selected to establish the HCC cell line in
which RFC3 would be knocked down. The knockdown effect was
evaluated by RT-qPCR and western blot analysis. As shown in
Fig. 3A, RFC3 mRNA expression was
reduced by approximately 70% in the SMMC-7721-shRFC3 cells compared
to the NC cells (P<0.01). Western blot analysis further
confirmed that almost 90% of RFC3 expression was markedly
suppressed in the SMMC-7721-shRFC3 cells (Fig. 3B and C).
Knockdown of RFC3 inhibits HCC cell
proliferation and viability
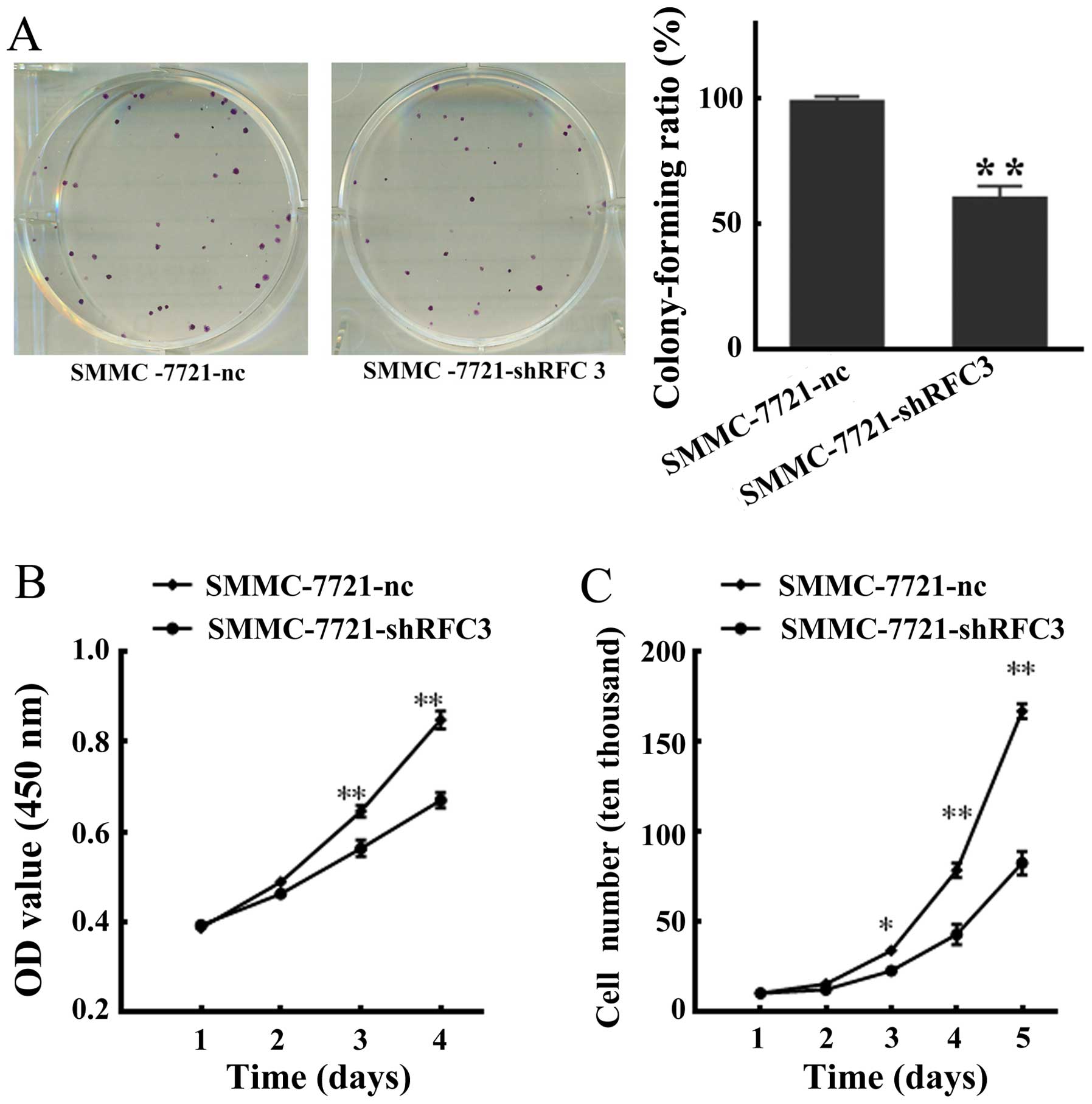
We sought to examine the effects of RFC3 knock-down
on HCC cells. To this end, we examined the proliferation and
viability of SMMC-7721-shRFC3 cells using a cell colony formation
assay, MTS viability assay and cell growth curve assay,
respectively. As shown in Fig.
4A, statistical analysis indicated that the colony-forming
ability of the SMMC-7721-shRFC3 cells decreased to 62% (P<0.01)
compared with that of the NC cells which was 97%. Moreover, a
marked decrease in cellular viability was observed in the cells in
which RFC3 was knocked down (SMMC-7721-shRFC3 cells). Furthermore,
the cell growth curve assay revealed that the population of
SMMC-7721-shRFC3 living cells was considerably lower compared with
the NC cells (Fig. 4C).
Collectively, these data indicated that the knockdown of RFC3
inhibited HCC cell proliferation and viability.
Knockdown of RFC3 induces HCC cell cycle
arrest at the S phase
As abnormal cell proliferation is closely associated
with the dysregulation of the cell cycle (18), we examined whether the knockdown
of RFC3 affects the HCC cell cycle using flow cytometric analysis.
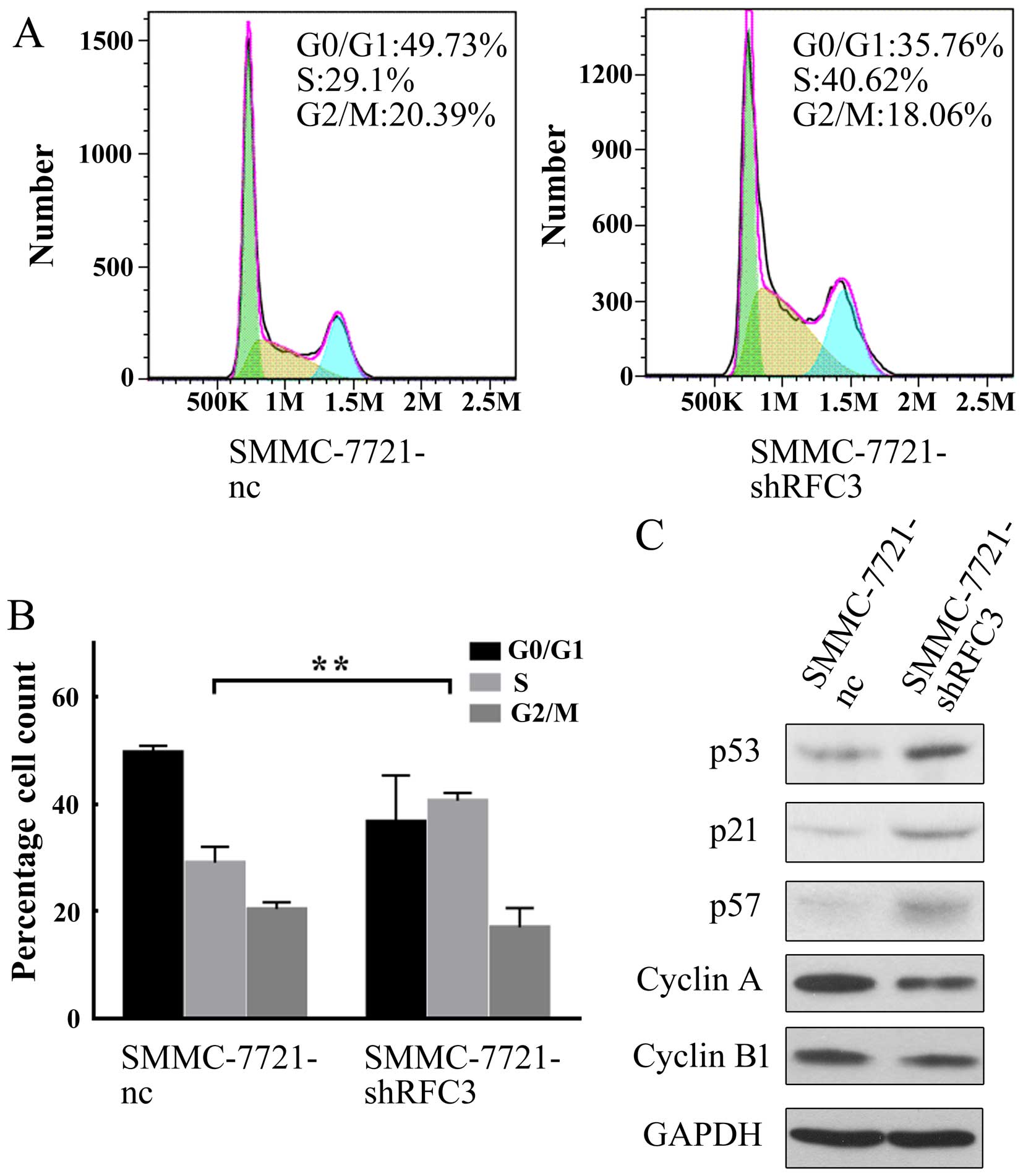
As shown in Fig. 5A and B, the
knockdown of RFC3 significantly increased the percentage of cells
in the S phase, but decreased that in the G0/G1 phase; moreover,
the downregulation of RFC3 did not significantly alter the
percentage of cells in the G2/M phase, indicating that the cell
cycle was arrested at the S phase, when RFC3 was knocked down.
To further elucidate the mechanisms behind the cell
cycle arrest at the S phase following the knockdown of RFC3, we
measured the expression levels of cell cycle-related proteins by
western blot analysis. As shown in Fig. 5C, in the HCC cells in which RFC3
was knocked down, the tumor suppressor genes, p53, p21 and p57 were
all upregulated. In the cell cycle, p21 functions as a negative
regulator that inhibits DNA synthesis and arrests the cell cycle at
the G1/S phase by binding to and inhibiting the activity of
cyclin-dependent kinase (CDK)2, CDK1, CDK4 and CDK6 complexes
(19). p53 upregulates the
expression of p21 (20). p57 is
also a tight-binding inhibitor of CDK2, CDK4 and CDK6 complexes and
a negative regulator of cell proliferation (21). Based on this information, our data
indicated that RFC3 knockdown upregulated p53 expression,
subsequently inducing the upregulation of p21, and eventually
inhibiting the cell cycle. Similarly, the knockdown of RFC3
upregulated p57 and directly resulted in the blocking of CDK
complex activity. Indeed, the expression of cyclin A, a known cell
cycle protein which is associated with the CDK2 complex required
for G1-S phase transition (22),
was downregu-lated (Fig. 5C). Of
note, the expression of cyclin B1, a protein required for G2-M
phase transition (23), was not
markedly affected (Fig. 5C),
suggesting that the knockdown of RFC3 specifically regulates G1-S
phase transition. Taken together, these findings demonstrate that
the knockdown of RFC3 induces HCC cell cycle arrest at the S phase
by regulating tumor suppressor genes involved in G1-S phase
transition.
Discussion
HCC is one of the most common and lethal
malignancies in the world (1,2).
The identification of novel therapeutic targets that will
contribute to the development and progression of HCC is obviously
desirable if we are to combat this lethal disease. RFC3 is clearly
one of the most important cancer antigens since it plays an
indispensable role in DNA replication (7,8,24,25). Previous studies have shown that
the overexpression of RFC3 is closely related to esophageal
adenocarcinomas and ovarian carcinomas, implying that this gene
plays a role in tumor development (12,13); however, its role in the
development of HCC remains unclear. In the present study, we aimed
to investigate the expression and biological functions of RFC3 in
HCC tissues and cells.
The RFC complex has been identified as an important
component of the cell cycle (10). The overexpression of the RFC
complex was has been found to be responsible for DNA replication,
DNA damage repair, checkpoints and inducing tumor formation
(7,10,12,25). Previous studies have demon-strated
that RFC subunits are upregulated in different types of
malignancies: RFC2, RFC3, RFC4 and RFC5 are upregulated in
nasopharyngeal (13), ovarian
(12), HCC (26) and human papillomavirus-positive
squamous cell carcinomas (27),
respectively. In agreement with these findings, we found that the
expression of RFC3 was significantly upregulated in human HCC
tissues and cell lines. Owing to the importance of RFC3 in the
formation of DNA replication complex, it has been suggested that
the overexpression of RFC3 is responsible for inducing tumor
formation (13). Based on this
hypothesis, RFC3 can be identified as one of the most important
cancer antigens.
After noting that RCF3 was associated with HCC, we
focused on whether the downregulation of RFC3 affects HCC cells.
Lentivirus-mediated RNAi methods provide an attractive approach to
efficiently suppress gene expression. The RNAi knockdown assays
revealed that the suppression of RFC3 expression in HCC cells led
to a considerable suppression of HCC cell viability and
proliferation. These results are consistent with those of a
previous study which demonstrated that RFC3 was overexpressed in
esophageal adenocarcinoma and that RFC3 knockdown had an
anti-proliferative effect (13).
This suppression may be partly due to the induction of cancer cell
cycle arrest at the S phase, the checkpoint of which is activated
upon the formation and function of DNA replication complexes
(28,29). Given that RFC3 is one of the key
components of DNA replication complexes, the downregulation of RFC3
is likely to result in the blockade of DNA replication complex
formation and eventually suppress DNA replication.
The knockdown of RFC3 increased the levels of S
phase-associated proteins, such as p21, p53 and p57, but reduced
the expression of cyclin A. In the cell cycle, the CDK2/cyclin A
complex leads to progression through the G1-S phase transition, a
step that is strictly regulated in the process of cell
proliferation. p21 and p51, CDK inhibitors, bind to CDK2 and
inhibit its activity. The overexpression of p21 and/or p51 results
in cells remaining in the G1/S phase and the arrest of cell cycle
progression. p53 is known to be an activator of p21 expression
(20). It was proposed herein
that the knockdown of RFC3 upregulates p53 expression, and
subsequently induces p21 and/or p51 upregulation, and eventually
inhibits G1-S phase transition. Further studies are required
however, to focus on the detailed mechanisms behind the RFC3
regulation of cell cycle-related proteins.
In conclusion, the present study demonstrated that
RFC3 was notably upregulated in HCC tissues and cell lines. The
downregulation of RFC3 suppressed HCC cell viability and
proliferation. Further experiments demonstrated that the knockdown
of RFC3 induced HCC cell cycle arrest at the S phase. Tumor
suppression was likely accomplished partially by inducing S phase
arrest and regulating cell cycle-related proteins. These results
indicate that RFC3 plays an important role in the development of
HCC. Therefore, we suggest that a specific enzymatic inhibitor to
RFC3 may have therapeutic significance in the treatment of HCC.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (No. 8157111144) and the
Science and Technology Planning Project of Guangzhou, Guangdong
Province, China (No. 1563000226).
References
|
1
|
Slotta JE, Kollmar O, Ellenrieder V,
Ghadimi BM and Homayounfar K: Hepatocellular carcinoma: Surgeon's
view on latest findings and future perspectives. World J Hepatol.
7:1168–1183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang CY, Lin CS, Tai WT, Hsieh CY, Shiau
CW, Cheng AL and Chen KF: Sorafenib enhances radiation-induced
apoptosis in hepatocellular carcinoma by inhibiting STAT3. Int J
Radiat Oncol Biol Phys. 86:456–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGlynn KA, Petrick JL and London WT:
Global epidemiology of hepatocellular carcinoma: an emphasis on
demographic and regional variability. Clin Liver Dis. 19:223–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang HI, Lee MH, Liu J and Chen CJ: Risk
calculators for hepatocellular carcinoma in patients affected with
chronic hepatitis B in Asia. World J Gastroenterol. 20:6244–6251.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bahri O, Ezzikouri S, Alaya-Bouafif NB,
Iguer F, Feydi AE, Mestiri H, Benazzouz M, Khalfallah T, Afifi R,
Elkihal L, et al: First multicenter study for risk factors for
hepatocellular carcinoma development in North Africa. World J
Hepatol. 3:24–30. 2011.PubMed/NCBI
|
|
6
|
Culligan KM and Hays JB: DNA mismatch
repair in plants. An Arabidopsis thaliana gene that predicts a
protein belonging to the MSH2 subfamily of eukaryotic MutS
homologs. Plant Physiol. 115:833–839. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pascucci B, Stucki M, Jónsson ZO,
Dogliotti E and Hübscher U: Long patch base excision repair with
purified human proteins. DNA ligase I as patch size mediator for
DNA polymerases delta and epsilon. J Biol Chem. 274:33696–33702.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimada M, Okuzaki D, Tanaka S, Tougan T,
Tamai KK, Shimoda C and Nojima H: Replication factor C3 of
Schizosaccharomyces pombe, a small subunit of replication factor C
complex, plays a role in both replication and damage checkpoints.
Mol Biol Cell. 10:3991–4003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia ST, Xiao LT, Bi DL and Zhu ZH:
Arabidopsis replication factor C subunit 1 plays an important role
in embryogenesis. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao.
33:179–187. 2007.PubMed/NCBI
|
|
10
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mossi R and Hübscher U: Clamping down on
clamps and clamp loaders - the eukaryotic replication factor C. Eur
J Biochem. 254:209–216. 1998.PubMed/NCBI
|
|
12
|
Shen H, Cai M, Zhao S, Wang H, Li M, Yao S
and Jiang N: Overexpression of RFC3 is correlated with ovarian
tumor development and poor prognosis. Tumour Biol. 35:10259–10266.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiong S, Wang Q, Zheng L, Gao F and Li J:
Identification of candidate molecular markers of nasopharyngeal
carcinoma by tissue microarray and in situ hybridization. Med
Oncol. 28(Suppl 1): S341–S348. 2011. View Article : Google Scholar
|
|
14
|
Xia S, Xiao L, Gannon P and Li X: RFC3
regulates cell proliferation and pathogen resistance in
Arabidopsis. Plant Signal Behav. 5:168–170. 2010. View Article : Google Scholar :
|
|
15
|
Maeng S, Kim GJ, Choi EJ, Yang HO, Lee DS
and Sohn YC: 9-Cis-retinoic acid induces growth inhibition in
retinoid-sensitive breast cancer and sea urchin embryonic cells via
retinoid X receptor α and replication factor C3. Mol Endocrinol.
26:1821–1835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu K, Wang J, Yao Z, Liu B, Lin Y, Liu L
and Xu L: Expression of cytoskeleton regulatory protein Mena in
human hepatocellular carcinoma and its prognostic significance. Med
Oncol. 31:9392014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Freshney RI, Sherry A, Hassanzadah M,
Freshney M, Crilly P and Morgan D: Control of cell proliferation in
human glioma by glucocorticoids. Br J Cancer. 41:857–866. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Gong L, Zhu SJ, Zhu Q, Yao L, Han
XJ, Zhang JR, Li YH and Zhang W: The human homolog of Drosophila
headcase acts as a tumor suppressor through its blocking effect on
the cell cycle in hepatocellular carcinoma. PLoS One.
10:e1375792015.
|
|
19
|
Gartel AL and Radhakrishnan SK: Lost in
transcription: p21 repression, mechanisms, and consequences. Cancer
Res. 65:3980–3985. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee MH, Reynisdóttir I and Massagué J:
Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique
domain structure and tissue distribution. Genes Dev. 9:639–649.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guadagno TM and Newport JW: Cdk2 kinase is
required for entry into mitosis as a positive regulator of
Cdc2-cyclin B kinase activity. Cell. 84:73–82. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Souza CP, Ellem KA and Gabrielli BG:
Centrosomal and cytoplasmic Cdc2/cyclin B1 activation precedes
nuclear mitotic events. Exp Cell Res. 257:11–21. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnson A, Yao NY, Bowman GD, Kuriyan J
and O'Donnell M: The replication factor C clamp loader requires
arginine finger sensors to drive DNA binding and proliferating cell
nuclear antigen loading. J Biol Chem. 281:35531–35543. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X and Burgers PM: Molecular cloning and
expression of the Saccharomyces cerevisiae RFC3 gene, an essential
component of replication factor C. Proc Natl Acad Sci USA.
91:868–872. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arai M, Kondoh N, Imazeki N, Hada A,
Hatsuse K, Matsubara O and Yamamoto M: The knockdown of endogenous
replication factor C4 decreases the growth and enhances the
chemosensitivity of hepatocellular carcinoma cells. Liver Int.
29:55–62. 2009. View Article : Google Scholar
|
|
27
|
Martinez I, Wang J, Hobson KF, Ferris RL
and Khan SA: Identification of differentially expressed genes in
HPV-positive and HPV-negative oropharyngeal squamous cell
carcinomas. Eur J Cancer. 43:415–432. 2007. View Article : Google Scholar :
|
|
28
|
Koch HB, Zhang R, Verdoodt B, Bailey A,
Zhang CD, Yates JR III, Menssen A and Hermeking H: Large-scale
identification of c-MYC-associated proteins using a combined
TAP/MudPIT approach. Cell Cycle. 6:205–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green CM, Erdjument-Bromage H, Tempst P
and Lowndes NF: A novel Rad24 checkpoint protein complex closely
related to replication factor C. Curr Biol. 10:39–42. 2000.
View Article : Google Scholar : PubMed/NCBI
|