Introduction
Dental agenesis is one of the most common congenital
anomaly of human dentition, with a prevalence (excluding third
molars) ranging from 2.6 to 11.3% (1). Relative to the number of missing
teeth, dental agenesis can be classified as hypodontia (when 1 to 5
teeth are missing), oligodontia (when 6 or more teeth are missing),
or anodontia (when the complete failure of dentition development
occurs). The most frequently missing teeth are the mandibular
second premolars, followed by the maxillary lateral incisors and
maxillary second premolars (2).
It is generally accepted that both genetic and environmental
factors play a significant role in the pathogenesis of this disease
(3). Tooth agenesis may occur
either in association with genetic syndromes characterized by other
inherited anomalies as an isolated non-syndromic familial trait, or
as sporadic cases. Familial tooth agenesis is characterized by
moderate genetic heterogeneity and has been reported to have an
autosomal-dominant, autosomal-recessive or X-linked mode of
inheritance (4,5).
Tooth development requires a complex network of
molecular interactions in which the activation of the nuclear
factor-κB (NF-κB) and WNT signaling pathways indeed play a central
role. The transcription factor, NF-κB, controls several cellular
functions and it is activated by members of the tumor necrosis
factor receptor (TNFR) superfamily, including EDA receptor (EDAR).
Ectodysplasin (EDA) is a member of the tumor necrosis factor
(TNF)-related ligand family and binds to its membrane receptor,
EDAR, that in turn binds to its adaptor, EDAR-associated death
domain (EDARADD). The EDA-EDAR-EDARADD complex activates the
NEMO-IKK signaling cascade, allowing the nuclear translocation of
NF-κB and thus the transcriptional control of genes that are
crucial for tooth development (6).
WNT signaling molecules play important roles in the
differentiation of tissues and organs during embryonic development.
The secretion of WNTs, including WNT4, WNT6, and WNT10 from the
dental epithelium is essential for tooth development. WNT10A is
expressed in the dental epithelium and mesenchyme. Similar to other
WNT proteins, it binds to the Frizzled (Fz) transmembrane receptors
and to lipoprotein receptor-related protein 5/6 (LRP5/6)
coreceptors, leading to the activation of the β-catenin pathway
(7).
One of the direct downstream targets of
WNT/β-catenin signaling during craniofacial development is Msh
homeobox 1 (MSX1). MSX1 is a homeobox gene encoding a transcription
factor that regulates the expression of bone morphogenetic protein
4 (BMP4), a member of the transforming growth factor-β (TGF-β)
superfamily, during the bud and cap stages of the tooth
development. In these phases, MSX1 interacts with paired box 9
(PAX9) both at the gene and protein levels, with MSX1 and PAX9
being intimately involved in the genetic networks regulating tooth
development (8). PAX9 is a
transcriptional factor that plays a key role during embryogenesis
by modifying the transcriptional activity of downstream genes. In
particular, by physically associating with MSX1, it enhances both
MSX1 and BMP4 gene expression during tooth development (9,10).
On the other hand, Axin 2 (AXIN2), also known as conductin/axil, is
an inhibitor of the WNT signaling pathway (11).
Previously, dominant loss-of-function mutations in
MSX1, PAX9 and AXIN2 have been described in familial forms of
non-syndromic tooth agenesis (12). Recently, the WNT10A variant has
been described in up to 50% of analyzed patients with dental
agenesis and also in various ectodermal dysplasia syndromes
(13,14). Mutations in EDA, EDAR and EDARADD
have also been shown to be associated with both isolated tooth
agenesis and syndromic tooth agenesis, such as X-linked
hypohidrotic ectodermal dysplasia (XLHED), a rare anomaly involving
sparse hair, dental abnormalities, skin lesions and hypoplasia of
sweat glands (15).
The initial aim of this study was to evaluate the
MSX1 and PAX9 mutation rate in a cohort of patients and
subsequently to identify the causative gene mutations associated
with auto-somal tooth agenesis in some families by employing whole
exome sequencing (WES).
Subjects and methods
Study subjects
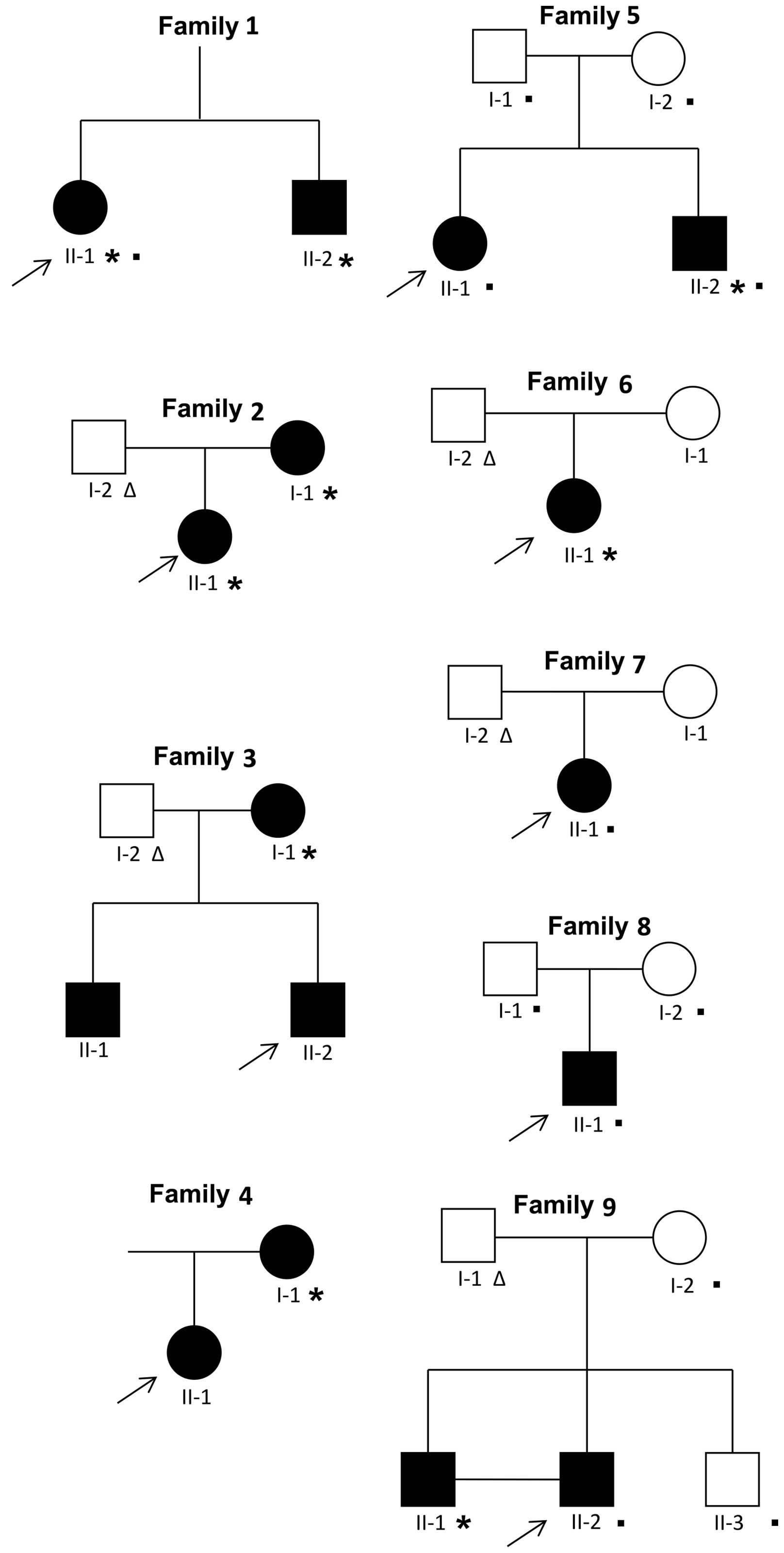
The probands (n=9; Fig. 1) were patients who were 10 to 20
years of age enrolled at the Dental Clinic of the University of
Brescia, Italy, from 2008 to 2014, with a confirmed diagnosis of
dental agenesis, but without systemic or syndromic diseases. When
available, parents and siblings either affected or not, were also
enrolled in the study. All individuals enrolled were informed of
the purpose of this study and signed an informed consent. All
clinical and genetic studies were approved by the Ethics Committee
of the Spedali Civili of Brescia (AOBS-GENI-2011; NP 1119) and were
conducted according to the principles expressed in the Declaration
of Helsinki. In addition to panoramic radiography, a clinical
examination was conducted on a dental chair, under artificial
light, with a probe and a dental mirror by two trained
dentists.
Mutation analysis by Sanger
sequencing
Genomic DNA was isolated from the peripheral blood
of affected individuals and available affected/non-affected
relatives using the Purgene Blood Core kit (Qiagen, Valencia, CA,
USA) following the instructions of the manufacturer. DNA from
peripheral blood of a control (CTRL) unaffected subject was also
isolated. The PAX9 and MSX1 coding sequences were amplified by PCR
using the primers and the thermal cycler protocols previously
described (16–18). The amplified products were
sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit and
analyzed using the ABI PRISM 310xl Genetic Analyzer (both from
Applied Biosystems, Foster City, CA, USA). The results were
compared with the reference sequences for each (MSX1, NM_002448.3;
PAX9, NM_006194.3).
The exons and approximately 30 bp of the flanking
introns of the WNT10A and EDARADD genes were amplified with the
primers previously described (19). The results were compared with the
reference sequences for each gene (WNT10A, NM_025216.2; EDARADD,
NM_080738.3).
Whole exome sequencing (WES)
WES was carried out in selected affected individuals
and their relatives. In total, 12 cases were examined by WES and
the details of the subjects analyzed are reported in Fig. 1. The exomes of the probands of
families 1, 5, 7 and 8 were sequenced at Personal Genomics
(University of Verona, Italy) using the TruSeq Exome Enrichment kit
(Illumina, San Diego, CA, USA) for whole exome capture. The members
of family 5 (I-1, I-2, II-2), family 8 (I-1, I-2) and family 9
(I-2, II-2, II-3) had their exomes sequenced at Erasmus MC,
University Medical Center Rotterdam using the Nimblegen SeqCap EZ
Exome version 2.0 exome capture kit. In both protocols, paired-end
libraries were sequenced on the Illumina HiSeq instrument.
Sequencing reads were aligned to the human genome reference
sequence (hg19) using BWA-mem (20) and then processed according to GATK
3.4–46 best practices (21) for
variant discovery. Briefly, aligned reads were processed for
duplicate removal using Picard and the base quality score
recalibration was performed using GATK. The processed BAM files of
single probands or the proband and relatives were then analyzed
using GATK HaplotypeCaller for the identification of both single
nucleotide polymorphisms (SNPs) and indel variants. Reported
variants in VCF file format were then filtered removing those with
a read depth <6 and a genotype quality (GQ) <20.
Subsequently, the filtered variants were annotated using ANNOVAR
(22) and selected according to
the following criteria: i) exonic, non-synonymous variants; ii)
reported allele frequency in 1000G phase 3 data and ExAc 0.3<1%;
and iii) not located in segmental duplicated region. These variants
were searched for mutations in known genes related to dental
agenesis. Where no mutations emerged, the data of the family
pedigree were analyzed to select variants segregating according to
the disease hereditary model. These variants were further annotated
with known phenotypes from the ClinVar database (23) and the RVIS score (24) that provides an estimate of the
tolerance to functional variations for each gene. Candidate genes
variants were then prioritized based on PolyPhen2 (25) and FATHMM (26) deleteriousness predictions and the
RVIS score of the associated gene.
Results
Study subjects
A total of 9 females and 7 males ranging in age from
8 to 48 years affected by tooth agenesis from 9 different families
were enrolled in this study (Fig.
1). In particular, 5 individuals were affected by oligodontia
(number of missing teeth ≥6) and 11 individuals by hypodontia
(number of missing teeth ≤5). The most common missing teeth were
the mandibular second premolars (35 in 10/16 affected; 45 in 9/16
affected), maxillary lateral incisors (12 in 5/16 affected; 22 in
5/16 affected) and maxillary second premolars (15 in 4/16 affected;
25 in 4/16 affected) (Tables I
and II).
 | Table ICharacteristics of the probands and
the affected relatives. |
Table I
Characteristics of the probands and
the affected relatives.
| Characteristic | N | % |
|---|
| Males | 7 | 44 |
| Females | 9 | 56 |
| Type of tooth
agenesis (total number of cases) | 16 | |
| Hypodontia (1–5
permanent teeth missing) | 11 | 69 |
| Oligodontia (>6
permanent teeth missing) | 5 | 31 |
| Type of permanent
teeth missing (total teeth missing) | 81 | |
| Central incisor (11,
21, 31, 41) | 8 | 9.9 |
| Lateral incisor
(12, 22, 32, 42) | 16 | 19.7 |
| Canine (13, 23,
33, 43) | 4 | 4.9 |
| First premolar
(14, 24, 34, 44) | 9 | 11.1 |
| Second premolar
(15, 25, 35, 45) | 27 | 33.3 |
| First molar (16,
26, 36, 46) | 1 | 1.2 |
| Second molar (17,
27, 37, 47) | 6 | 7.4 |
| Third molar (18,
28, 38, 48) | 10 | 12.3 |
| No. of missing
incisors | 35 | 43.2 |
| No. of missing
canines | 4 | 4.9 |
| No. of missing
premolars | 25 | 30.8 |
| No. of missing
molars | 17 | 21 |
| Table IIDescription of oligodontia and
hypodontia phenotypes. |
Table II
Description of oligodontia and
hypodontia phenotypes.
Mutation analysis of PAX9 and MSX1
genes
To identify causative mutations in subjects with
tooth agenesis, we performed direct sequencing of the PAX9 and MSX1
genes in 9 affected subjects (Fig.
1, individuals marked with an asterisk). Ultimately, the
mutation analysis of the PAX9 and MSX1 genes did not highlight the
presence of any causative mutation in the 9 affected
individuals.
WES
Since the genetic heterogeneity of tooth agenesis
has become apparent and has expanded since our first screening, to
include 17 different genes, we decided to apply WES to identify
novel and/or previously described causative mutations, as well as
new candidate genes. Overall, we performed WES on 12 subjects from
5 distinct families (Fig. 1,
individuals marked with a filled square; Table II, individuals highlighted in
grey) with a mean coverage across target region of 75X-171X and
22,482-37,874 variants identified per subject in exonic regions
(Table III).
| Table IIISummary of whole exome sequencing
results. |
Table III
Summary of whole exome sequencing
results.
| Sample | Family | Mean coverage in
target region | % bases covered at
least 20X | No. Of exonic
variants |
|---|
| II-1 | 5 | 100X | 93.6 | 23,297 |
| II-1 | 1 | 171X | 95.5 | 22,801 |
| II-1 | 7 | 77X | 91.8 | 22,482 |
| II-1 | 8 | 82X | 92.2 | 23,564 |
| I-1 | 5 | 75X | 83.9 | 22,603 |
| II-2 | 5 | 82X | 85.3 | 22,822 |
| I-2 | 5 | 82X | 85.3 | 22,706 |
| I-1 | 8 | 83X | 85.8 | 22,825 |
| I-2 | 8 | 80X | 84.9 | 23,129 |
| I-2 | 9 | 82X | 91.6 | 37,874 |
| II-2 | 9 | 100X | 92.7 | 37,822 |
| II-3 | 9 | 88X | 91.1 | 37,556 |
For 2 families, it was possible to identify known
causative mutations, thus providing a molecular diagnosis. The
already described p.F228I mutation (rs121908120) in the homozygous
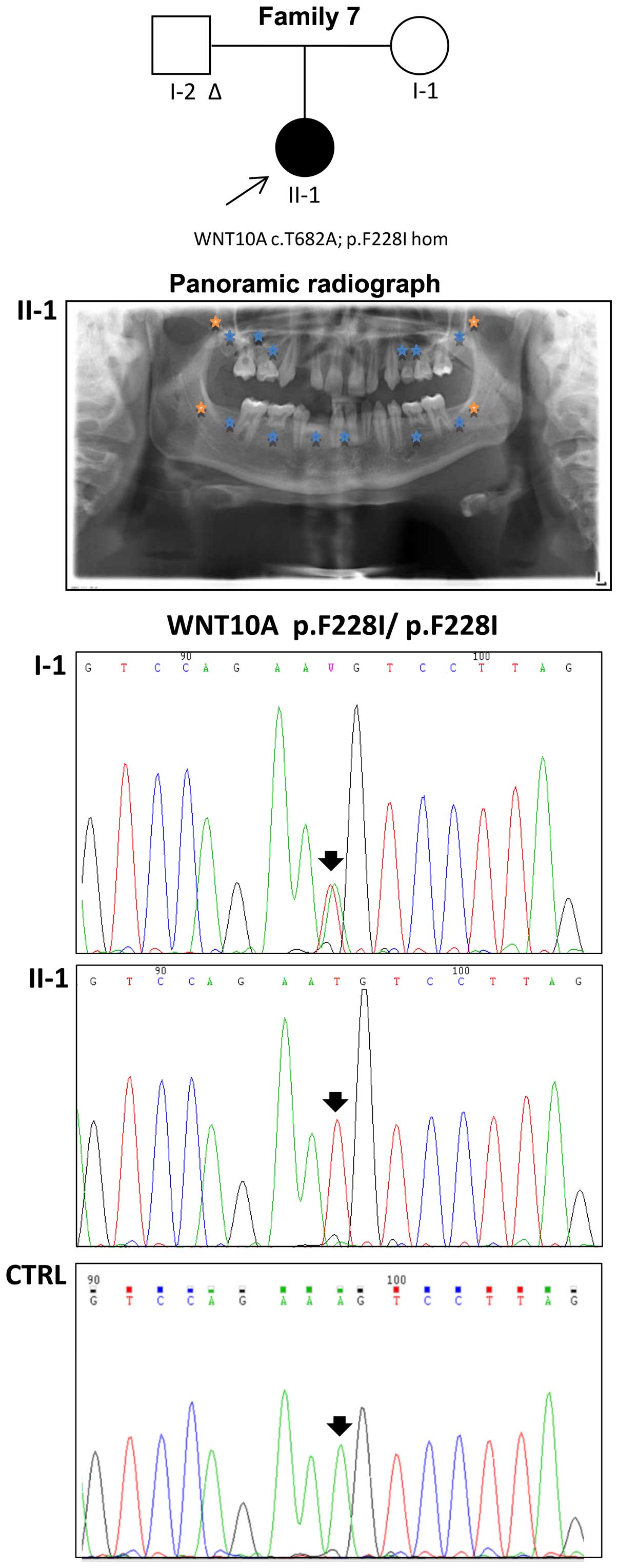
state in the WNT10A gene was detected in the proband of family 7
and confirmed by Sanger sequencing; her unaffected mother displayed
the same variant in the heterozygous state (Fig. 2). The same mutation was also
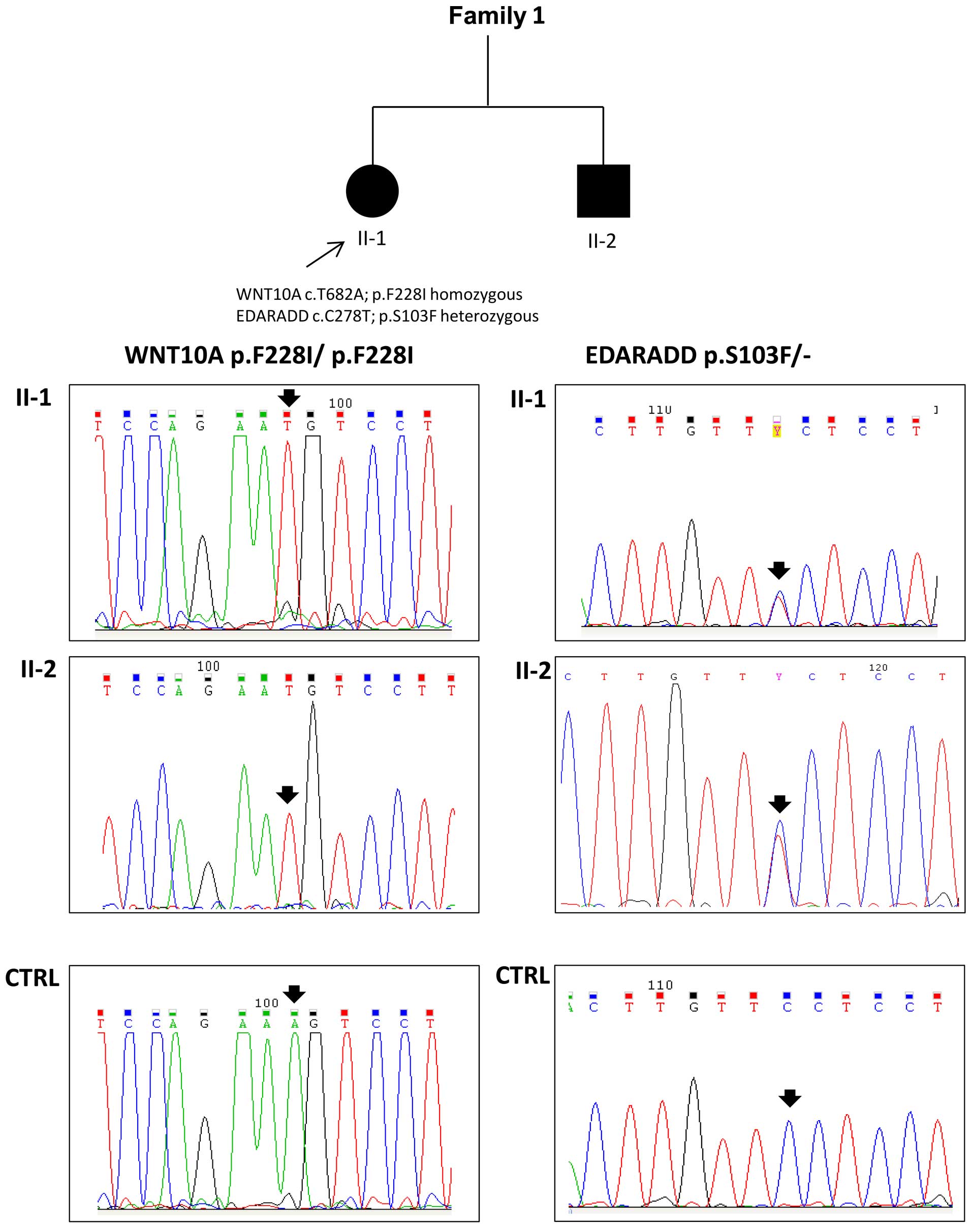
detected in both the affected siblings from family 1 (Fig. 3). Moreover, the proband (II-1,
oligodontia) and her brother (II-2, more severe phenotype) also
carried a p.S103F mutation (rs114632254) in the EDARADD gene in the
heterozygous state (Fig. 3). Both
mutations were confirmed by Sanger sequencing. In 3 other families,
our analysis led to the identification of novel candidate variants
in genes with a potential role in tooth agenesis.
In family 5, an autosomal recessive form of
inheritance seemed to be most probable (Fig. 1). In both the affected siblings,
we found compound heterozygous mutations in the Mucin 16 (MUC16)
and Titin (TTN) genes and a homozygous genetic variation
c.-387delC/G in the 5′UTR portion of the paired-like homeodomain 2
(PITX2) gene. Furthermore, in the proband (II-1) and in her
brother, we detected an SNP (c. T455C:p.V152A; rs17563) in the
homozygous state in the BMP4 gene previously associated with tooth
agenesis (Table IV).
| Table IVSelected candidate genes and variants
in family 5. |
Table IV
Selected candidate genes and variants
in family 5.
| Gene.refGene | ExonicFunc.
refGene | Chr | Ref | Alt | Func.refGene |
AAChange.refGene |
|---|
| Compound | | | | | | |
| MUC16 | Non-synonymous
SNV | chr19 | G | T | Exonic |
MUC16:NM_024690:exon3:c.C12953A:p.A4318D |
| MUC16 | Non-synonymous
SNV | chr19 | T | C | Exonic |
MUC16:NM_024690:exon3:c.A23897G:p.E7966G |
| MUC16 | Non-synonymous
SNV | chr19 | C | T | Exonic |
MUC16:NM_024690:exon12:c.G36581A:p.R12194Q |
| TTN | Non-synonymous
SNV | chr2 | C | G | Exonic |
TTN:NM_003319:exon186:c.G75952C:p.
E25318Q,
TTN:NM_133432:exon187:c.G76327C:p.E25443Q,
TTN:NM_133437:exon187:c.G76528C:p.E25510Q,
TTN:NM_133378:exon307:c.G95443C:p.E31815Q,
TTN:NM_001256850:exon308:c.G98224C:p.E32742Q,
TTN:NM_001267550:exon358:c.G103147C:p.E34383Q |
| TTN | Non-synonymous
SNV | chr2 | C | T | Exonic |
TTN:NM_003319:exon73:c.G18133A:p.
D6045N,
TTN:NM_133432:exon74:c.G18508A:p.D6170N,
TTN:NM_133437:exon74:c.G18709A:p.D6237N,
TTN:NM_133378:exon194:c.G37624A:p.D12542N,
TTN:NM_001256850:exon195:c.G40405A:p.D13469N,
TTN:NM_001267550:exon245:c.G45328A:p.D15110N |
| TTN | Non-synonymous
SNV | chr2 | T | C | Exonic |
TTN:NM_133378:exon90:c.A23131G:p.I7711V,
TTN:NM_001256850:exon91:c.A25912G:p.I8638V,
TTN:NM_001267550:exon93:c.A26863G:p.I8955V |
| TTN | Non-synonymous
SNV | chr2 | G | A | Exonic |
TTN:NM_133378:exon77:c.C19445T:p.S6482L,
TTN:NM_001256850:exon78:c.C22226T:p.S7409L,
TTN:NM_001267550:exon80:c.C23177T:p.S7726L |
| Known | | | | | | |
| PITX2 | UTR5 | chr4 | C | – | NM_000325:
c.-387delG | |
| BMP4 | Exonic | chr14 | T | C | NM_001202: c.T455C:
p.V152A | |
Family 8 was also likely to present a recessive form
of inheritance (Fig. 1). The
proband displayed homozygous non-synonymous single nucleotide
variants (SNVs) in the aryl-sulfatase family member H (ARSH),
proline rich 32 (PRR32) and apurinic/apyrimidinic
endodeoxyribonuclease 2 (APEX2) genes, a nucleotide change in
homozygosis in transglutaminase 4 (TGM4) resulting in a premature
stop codon and a 40 bp frameshift insertion in homozygosis in the
placenta specific 4 (PLAC4) gene. Putative de novo
heterozygous genetic variants were identified in 14 genes, 4 of
whom displayed compound heterozygous gene alterations. Furthermore,
the homozygous genetic variant in the 5′UTR portion of the PITX2
gene identified in family 6 was also present in the proband of
family 8 (Table V).
| Table VSelected candidate genes and variants
in family 8. |
Table V
Selected candidate genes and variants
in family 8.
| Chr | Ref | Alt | Func.refGene | Gene.refGene |
GeneDetail.refGene |
ExonicFunc.refGene |
|---|
| Recessive | | | | | | |
| chrX | G | A | Exonic | ARSH | NA | Non-synonymous
SNV |
| chrX | G | A | Exonic | PRR32 | NA | Non-synonymous
SNV |
| chr3 | C | T | Exonic | TGM4 | NA | Stopgain |
| chr21 | – | 40 bp | Exonic | PLAC4 | NA | Frameshift
insertion |
| chrX | G | A | Exonic | APEX2 | NA | Non-synonymous
SNV |
| De novo | | | | | | |
| chr17 | 21 bp | – | Splicing | MYO19 | NA | NA |
| chr16 | T | G | Exonic | IFT140 | NA | Non-synonymous
SNV |
| chr1 | A | C | Splicing | NFIA |
NM_001145511:exon9:c.12312A>C,
NM_001145512:exon10:c.1390-2A>C,
NM_005595:exon9:c.12552A>C,
NM_001134673:exon9:c.1255-2A>C | NA |
| chr11 | – | T | Exonic | MUC6 | NA | Frameshift
insertion |
| chr6 | G | A | Exonic | GPANK1 | NA | Non-synonymous
SNV |
| chr1 | G | C | Splicing | NFIA |
NM_001145511:exon9:c.1231-1G>C,
NM_001145512:exon10:c.1390-1G>C,
NM_005595:exon9:c.12551G>C,
NM_001134673:exon9:c.1255-1G>C | NA |
| chr6 | A | G | Exonic | HSPA1L | NA | Non-synonymous
SNV |
| chr11 | G | A | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr19 | – | 18 bp | Exonic | RSPH6A | NA | Non-frameshift
insertion |
| chr9 | C | G | Exonic | TRIM14 | NA | Non-synonymous
SNV |
| chr19 | – | AGC | Exonic | CHST8 | NA | Non-frameshift
insertion |
| chr6 | CACC
ACCA
CCAT | – | Exonic | SYNGAP1 | NA | Non-frameshift
insertion |
| chrX | C | T | Exonic | RBMX | NA | Non-synonymous
SNV |
| chr11 | G | T | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr11 | A | G | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr11 | G | – | Exonic | MUC6 | NA | Frameshift
deletion |
| chr1 | G | – | Splicing | SEC22B |
NM_004892:exon6:c.1066-1G>- | NA |
| chr3 | C | G | Exonic | IL17RE | NA | Non-synonymous
SNV |
| Compound | | | | | | |
| chr2 | – | CTGC | Exonic | DNAH7 | NA | Frameshift
insertion |
| chr2 | A | G | Exonic | DNAH7 | NA | Non-synonymous
SNV |
| chr2 | C | T | Exonic | DNAH7 | NA | Non-synonymous
SNV |
| chr11 | – | T | Exonic | MUC6 | NA | Frameshift
insertion |
| chr11 | G | A | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr11 | – | TA | Exonic | MUC6 | NA | Frameshift
insertion |
| chr11 | G | T | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr11 | A | G | Exonic | MUC6 | NA | Non-synonymous
SNV |
| chr11 | G | – | Exonic | MUC6 | NA | Frameshift
deletion |
| chr1 | A | C | Splicing | NFIA |
NM_001145511:exon9:c.1231-2A>C,
NM_001145512:exon10:c.1390-2A>C,
NM_005595:exon9:c.1255-2A>C,
NM_001134673:exon9:c.1255-2A>C | NA |
| chr1 | G | C | Splicing | NFIA |
NM_001145511:exon9:c.1231-1G>C,
NM_001145512:exon10:c.1390-1G>C,
NM_005595:exon9:c.1255-1G>C,
NM_001134673:exon9:c.1255-1G>C | NA |
| chr10 | A | T | Splicing | NRAP |
NM_006175:exon38:c.4431+2T>A,
NM_198060:exon39:c.4536+2T>A,
NM_001261463:exon39:c.4536+2T>A | NA |
| chr10 | T | G | Exonic | NRAP | NA | Nonsynonymous
SNV |
| Known | | | | | | |
| PITX2 | UTR5 | chr4 | C | – |
NM_000325:c.-387delG | |
Finally, an autosomal recessive form of inheritance
was likely also for family 9. The father could not be included in
the study and the analysis was conducted on the twin proband (II-2)
and on his unaffected mother and brother (Fig. 1). The proband evidenced a
non-synonymous SNP in the heterozygous state in the ankyrin repeat
and EF-hand domain containing 1 (ANKEF1), COMM domain-containing
protein 7 (COMMD7), DDHD domain containing 1 (DDHD1), early B-cell
factor 2 (EBF2), envoplakin (EVPL), SPT6 homolog, histone chaperone
(SUPT6H) genes and a compound heterozygous mutation in the
succinate-CoA ligase GDP-forming beta subunit (SUCLG2) gene
(Table VI).
| Table VISelected candidate genes and variants
in family 9. |
Table VI
Selected candidate genes and variants
in family 9.
| Chr | Ref | Alt | Func. refGene | Gene. refGene | GeneDetail.
refGene | ExonicFunc.
refGene |
AAChange.refGene |
|---|
| Compound | | | | | | | |
| chr20 | A | G | Exonic | ANKEF1 | NA | Non-synonymous
SNV |
ANKEF1:NM_198798:exon2:c.A332G:p.D111G,
ANKEF1:NM_022096:exon3:c.A332G:p.D111G |
| chr20 | C | T | Exonic | COMMD7 | NA | Non-synonymous
SNV |
COMMD7:NM_001099339:exon1:c.G5A:p.G2D,
COMMD7:NM_053041:exon1:c.G5A:p.G2D |
| chr14 | T | C | Exonic | DDHD1 | NA | Non-synonymous
SNV |
DDHD1:NM_001160148:exon6:c.A1411G:p.I471V,
DDHD1:NM_030637:exon6:c.A1411G:p.I471V,
DDHD1:NM_001160147:exon7:c.A1432G:p.I478V |
| chr8 | A | G | Exonic | EBF2 | NA | Non-synonymous
SNV |
EBF2:NM_022659:exon7:c.T560C:p.L187S |
| chr17 | G | T | Exonic | EVPL | NA | Non-synonymous
SNV |
EVPL:NM_001988:exon11:c.C1213A:p.L405M |
| chr3 | C | T | Exonic | SUCLG2 | NA | Non-synonymous
SNV |
SUCLG2:NM_001177599:exon10:c.G1124A:p.G375E,
SUCLG2:NM_003848:exon10:c.G1124A:p.G375E |
| chr3 | C | T | Exonic | SUCLG2 | NA | Non-synonymous
SNV |
SUCLG2:NM_001177599:exon10:c.G1123A:p.G375R,
SUCLG2:NM_003848:exon10:c.G1123A:p.G375R |
| chr17 | T | C | Exonic | SUPT6H | NA | Non-synonymous
SNV |
SUPT6H:NM_003170:exon32:c.T4393C:p.C1465R |
| De novo | | | | | | | |
| chr3 | C | T | Exonic | SUCLG2 | NA | Non-synonymous
SNV |
SUCLG2:NM_001177599:exon10:c.G1124A:p.G375E,
SUCLG2:NM_003848:exon10:c.G1124A:p.G375E |
| chr3 | C | T | Exonic | SUCLG2 | NA | Non-synonymous
SNV |
SUCLG2:NM_001177599:exon10:c.G1123A:p.G375R,
SUCLG2:NM_003848:exon10:c.G1123A:p.G375R |
Discussion
The main objective of this study was to identify the
causative genetic defects in subjects affected by non-syndromic
tooth agenesis. We began with Sanger sequencing of the PAX9 and
MSX1 genes, since they were considered in the literature the most
probable candidate genes (27).
This analysis performed on 9 individuals affected by agenesis did
not evidence any mutation, thus suggesting that other genes may be
involved in tooth agenesis, in agreement with other published data
(28) Mutations in the WNT10A
gene emerged as frequently involved (13,14) We decided to use the WES technology
to identify causative mutations. We identified pathogenic mutations
in the proband of family 1 and the same alterations in her brother.
The subjects displayed a known homozygous disease mutation in the
WNT10A gene (p.F228I, rs121908120) and a heterozygous mutation in
the EDARADD gene (p.S103F, rs114632254). The frequent p.F228I
aminoacid substitution is associated with the autosomal dominant or
autosomal recessive form of isolated hypodontia and ectodermal
dysplasia (29). The EDARADD
mutation p.S103F was previously predicted to be harmful for protein
function by bioinformatics analysis (19). This particular combination of
genetic variants involving two genes related to teeth development
has never been previously described, at least to the best of our
knowledge. Both the affected individuals had oligodontia, but
individual II-1 had 17 missing teeth and subject II-2 had 6 missing
teeth. By WES, we identified the pathogenic mutation in the proband
of family 8 that also displays the p.F228I mutation in WNT10A in
the homozygous state.
To extend the WES analysis, we focused on families
5, 8 and 9, as genomic DNA was available from all the members with
the exception of the unaffected father (I-1) of family 9. For
family 5, an autosomal recessive form of inheritance seemed most
probable. Our analysis identified the PITX2, TTN and MUC16 genes as
affected by candidate mutations. The TTN and MUC16 genes were not
considered good candidate genes for agenesis prediction, given
their high RVIS score, confirming the propensity of these genes to
accumulate rare functional mutations with neutral effect (30). Of note, we found a homozygous
c.-387delC/G variation in the 5′UTR of the PITX2 gene in the
siblings that could be associated with tooth agenesis as we
mentioned above. PITX2 is a transcription factor that initiates
tooth development, activating amelogenin expression, whose protein
product is necessary for enamel formation. Since the mutation is
localized in the promoter region of the gene, it may affect its
transcriptional activity (31). A
more in depth analysis of genetic variants in this family also
identified an interesting variant in the BMP4 gene, a
non-synonymous SNP in the homozygous state (rs17563;
c.T455C:p.V152A) in the affected siblings II-1 and II-2. In a
Brazilian study conducted on 46 individuals with tooth agenesis and
88 control cases, the CC genotype of BMP4 was more frequent in
individuals with 3 or more missing teeth than in the control group
(p<0.0001), leading the authors to the conclusion that this
variant was associated with tooth agenesis (32). The same SNP was previously found
in two Mexican families with oligodontia (33). Capasso et al (34) observed that the c455T>C
substitution altered the BMP4 mRNA secondary structure and that the
BMP4 mRNA and protein expression levels were higher for the T
allele in a population in southern Italy with cutaneous melanoma.
PAX9 and MSX1 synergistically activate BMP4, and BMP4 is a gene
crucial to tooth development as it regulates the passage from bud
to cap stages. Since the unaffected parents displayed the same
genetic variations of PITX2 and BMP4 in the homozygous state, it
was deemed that these variations alone were unlikely to be the
direct cause of the observed tooth hypodontia; however, they may
still play a role as risk factors or modulators of the
phenotype.
Family 8 (a trio with unaffected parents) displayed
a probable autosomic recessive form of inheritance. Unfortunately,
we were unable to identify a strong candidate gene with a clear
role in the physiopathology of tooth formation and development.
Based on RVIS score prioritization criteria, 6 genes emerged
affected by functional mutation in this family and not expected to
accumulate functional mutation by chance, namely PRR32, PLAC4,
nuclear factor 1A (NFIA), tripartite motif containing 14 (TRIM14),
carbohydrate sulfotransferase 8 (CHST8) and synaptic Ras GTPase
activating protein 1 (SYNGAP1). Of these, 3 genes were unlikely to
be involved in tooth agenesis: PLAC4 is mainly involved in placenta
tissue formation (35); CHST8 has
been associated with autosomal recessive peeling skin syndrome
(36); SYNGAP1 is responsible for
non-syndromic mental retardation autosomal dominant form 5 (OMIM
612621) (37), a phenotype not
reported in this family. The PRR32 gene is poorly characterized and
difficult to evaluate. NFIA and TRIM14 are two transcription factor
genes and they may represent good candidates for further studies
aimed at assessing their involvement in the regulatory network
driving teeth morphogenesis. NFIA may be of particular interest,
since mutations in this gene cause craniofacial abnormalities in
mice and craniosynostosis in humans (38). However, homozygous null mutations
in the gene have been associated with kidney, nervous and fertility
phenotypes that were not observed in this family. Of note, this
family was also a carrier of the genetic variant c.387delC/G in the
5′UTR of PITX2 (found in the homozygous state in the proband and in
his unaffected brother and in the heterozygous state in their
unaffected mother in family 5; discussed above), suggesting that a
complex interplay of genetic risk factors may be involved in this
disease.
Finally, family 9 included a pair of monozygotic
twins with hypodontia and the recessive inheritance model was
likely. Unfortunately, DNA for the father was not available for the
study. Among the genetic variants found, only the one in COMMD7 may
have a link with the disease. COMMD7 is in fact a NEMO interacting
protein involved in the termination of NF-κB signaling that is
activated by the EDA-EDAR-EDARADD complex (39). This mutation is predicted to be
damaging with a score of 1 by PolyPhen-2. Further studies are
necessary to establish whether this heterozygous mutation in the
proband twin may be involved in the pathological phenotype.
In conclusion, in this study, using WES analysis, we
identified the causative mutations in cases with a more severe
phenotype (families 1 and 7, where 17 and 16 teeth were missing,
respectively). We did not immediately identify variants that may be
disease-causing for families with a light phenotype, such as
families 5 and 9 (maximum 4 missing teeth) and with a mild
oligodontia, such as family 8 (6 missing teeth). However, our
analysis of these families highlighted some interesting candidate
genes that may be considered targets of future functional studies.
However, we cannot exclude that the manifestation of the phenotypic
traits may also depend or be modulated by other mechanisms,
including long and small non-coding RNAs (i.e., miRNAs), epigenetic
modifications, such as DNA methylation and histone modifications or
regulatory DNA variants. Finally, since a complex regulatory
network contributes to tooth formation with multitudes of genes
potentially involved, we consider that WES may be an effective
strategy with which to detect the genetic defects related to tooth
agenesis, particularly in severe cases of oligodontia in sporadic
and familial cases. The determination of the genetic causes of
tooth agenesis is important for genetic counseling and for
anticipating the problems related to the clinical management of
dental anomalies, in order to ensure a correct occlusion,
particularly during developing dentition in children.
Acknowledgments
We warmly thank Dr M. Crosatti (University of
Leicester, UK) for the linguistic revision of the manuscript. This
study was supported by Ministero dell'Istruzione, dell'Università e
della Ricerca (MIUR) local funds of the University of Brescia.
Publication costs were covered by Grant 'New Opportunities and Ways
towards ERC' (NOW ERC, Project: 2014-2256) from Fondazione Cariplo
and Regione Lombardia.
References
|
1
|
Bozga A, Stanciu RP and Mănuc D: A study
of prevalence and distribution of tooth agenesis. J Med Life.
7:551–554. 2014.
|
|
2
|
Brook AH, Jernvall J, Smith RN, Hughes TE
and Townsend GC: The dentition: The outcomes of morphogenesis
leading to variations of tooth number, size and shape. Aust Dent J.
59(Suppl 1): 131–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brook AH: Multilevel complex interactions
between genetic, epigenetic and environmental factors in the
aetiology of anomalies of dental development. Arch Oral Biol.
54(Suppl 1): S3–S17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cobourne MT: Familial human hypodontia -
is it all in the genes? Br Dent J. 203:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin W and Bian Z: The Gene Network
Underlying Hypodontia. J Dent Res. 94:878–885. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang YD, Chen Z, Song YQ, Liu C and Chen
YP: Making a tooth: Growth factors, transcription factors, and stem
cells. Cell Res. 15:301–316. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng C, Xu Z, Li Z, Zhang D, Liu Q and Lu
L: Down-regulation of Wnt10a by RNA interference inhibits
proliferation and promotes apoptosis in mouse embryonic palatal
mesenchymal cells through Wnt/β-catenin signaling pathway. J
Physiol Biochem. 69:855–863. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nassif A, Senussi I, Meary F, Loiodice S,
Hotton D, Robert B, Bensidhoum M, Berdal A and Babajko S: Msx1 role
in cranio-facial bone morphogenesis. Bone. 66:96–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogawa T, Kapadia H, Feng JQ, Raghow R,
Peters H and D'Souza RN: Functional consequences of interactions
between Pax9 and Msx1 genes in normal and abnormal tooth
development. J Biol Chem. 281:18363–18369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suryadeva S and Khan MB: Role of homeobox
genes in tooth morphogenesis: A review. J Clin Diagn Res.
9:ZE09–ZE12. 2015.PubMed/NCBI
|
|
11
|
Lammi L, Arte S, Somer M, Jarvinen H,
Lahermo P, Thesleff I, Pirinen S and Nieminen P: Mutations in AXIN2
cause familial tooth agenesis and predispose to colorectal cancer.
Am J Hum Genet. 74:1043–1050. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galluccio G, Castellano M and La Monaca C:
Genetic basis of non-syndromic anomalies of human tooth number.
Arch Oral Biol. 57:918–930. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song S, Zhao R, He H, Zhang J, Feng H and
Lin L: WNT10A variants are associated with non-syndromic tooth
agenesis in the general population. Hum Genet. 133:117–124. 2014.
View Article : Google Scholar
|
|
14
|
Arzoo PS, Klar J, Bergendal B, Norderyd J
and Dahl N: WNT10A mutations account for ¼ of population-based
isolated oligodontia and show phenotypic correlations. Am J Med
Genet A. 164A:353–359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nikopensius T, Annilo T, Jagomägi T,
Gilissen C, Kals M, Krjutškov K, Mägi R, Eelmets M, Gerst-Talas U,
Remm M, et al: Non-syndromic tooth agenesis associated with a
nonsense mutation in ectodysplasin-A (EDA). J Dent Res. 92:507–511.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klein ML, Nieminen P, Lammi L, Niebuhr E
and Kreiborg S: Novel mutation of the initiation codon of PAX9
causes oligodontia. J Dent Res. 84:43–47. 2005. View Article : Google Scholar
|
|
17
|
De Muynck S, Schollen E, Matthijs G,
Verdonck A, Devriendt K and Carels C: A novel MSX1 mutation in
hypodontia. Am J Med Genet A. 128A:401–403. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JW, Simmer JP, Lin BP and Hu JC: Novel
MSX1 frameshift causes autosomal-dominant oligodontia. J Dent Res.
85:267–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arte S, Parmanen S, Pirinen S, Alaluusua S
and Nieminen P: Candidate gene analysis of tooth agenesis
identifies novel mutations in six genes and suggests significant
role for WNT and EDA signaling and allele combinations. PLoS One.
8:e737052013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: the Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:111011–33. 2013.
|
|
22
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Landrum MJ, Lee JM, Benson M, Brown G,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al:
ClinVar: Public archive of interpretations of clinically relevant
variants. Nucleic Acids Res. 44:D862–D868. 2016. View Article : Google Scholar :
|
|
24
|
Petrovski S, Wang Q, Heinzen EL, Allen AS
and Goldstein DB: Genic intolerance to functional variation and the
interpretation of personal genomes. PLoS Genet. 9:e10037092013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shihab HA, Gough J, Cooper DN, Stenson PD,
Barker GL, Edwards KJ, Day IN and Gaunt TR: Predicting the
functional, molecular, and phenotypic consequences of amino acid
substitutions using hidden Markov models. Hum Mutat. 34:57–65.
2013. View Article : Google Scholar :
|
|
27
|
Vieira AR, Meira R, Modesto A and Murray
JC: MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J
Dent Res. 83:723–727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tallón-Walton V, Manzanares-Céspedes MC,
Carvalho-Lobato P, Valdivia-Gandur I, Arte S and Nieminen P:
Exclusion of PAX9 and MSX1 mutation in six families affected by
tooth agenesis. A genetic study and literature review. Med Oral
Patol Oral Cir Bucal. 19:e248–e254. 2014. View Article : Google Scholar :
|
|
29
|
van den Boogaard MJ, Créton M, Bronkhorst
Y, van der Hout A, Hennekam E, Lindhout D, Cune M and Ploos van
Amstel HK: Mutations in WNT10A are present in more than half of
isolated hypodontia cases. J Med Genet. 49:327–331. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
MacArthur DG, Manolio TA, Dimmock DP, Rehm
HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE,
Ashley EA, et al: Guidelines for investigating causality of
sequence variants in human disease. Nature. 508:469–476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lubitz SA, Sinner MF, Lunetta KL, Makino
S, Pfeufer A, Rahman R, Veltman CE, Barnard J, Bis JC, Danik SP, et
al: Independent susceptibility markers for atrial fibrillation on
chromosome 4q25. Circulation. 122:976–984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Antunes LS, Küchler EC, Tannure PN, Lotsch
PF, Costa MC, Gouvêa CV, Olej B and Granjeiro JM: TGFB3 and BMP4
polymorphism are associated with isolated tooth agenesis. Acta
Odontol Scand. 70:202–206. 2012. View Article : Google Scholar
|
|
33
|
Mu Y, Xu Z, Contreras CI, McDaniel JS,
Donly KJ and Chen S: Phenotype characterization and sequence
analysis of BMP2 and BMP4 variants in two Mexican families with
oligodontia. Genet Mol Res. 11:4110–4120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Capasso M, Ayala F, Russo R, Avvisati RA,
Asci R and Iolascon A: A predicted functional single-nucleotide
polymorphism of bone morphogenetic protein-4 gene affects mRNA
expression and shows a significant association with cutaneous
melanoma in Southern Italian population. J Cancer Res Clin Oncol.
135:1799–1807. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang L, Sun HY, Chen DZ, Lu MD, Tang Y and
Xiao JP: Explore the dynamic alternation of gene PLAC4 mRNA
expression levels in maternal plasma in second trimester for
nonivasive detection of trisomy 21. Obstet Gynecol Sci. 58:261–267.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cabral RM, Kurban M, Wajid M, Shimomura Y,
Petukhova L and Christiano AM: Whole-exome sequencing in a single
proband reveals a mutation in the CHST8 gene in autosomal recessive
peeling skin syndrome. Genomics. 99:202–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Parker MJ, Fryer AE, Shears DJ, Lachlan
KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park SM, Benoit
V, et al: De novo, heterozygous, loss-of-function mutations in
SYNGAP1 cause a syndromic form of intellectual disability. Am J Med
Genet A. 167A:2231–2237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nyboe D, Kreiborg S, Kirchhoff M and Hove
HB: Familial craniosynostosis associated with a microdeletion
involving the NFIA gene. Clin Dysmorphol. 24:109–112. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Esposito E, Napolitano G, Pescatore A,
Calculli G, Incoronato MR, Leonardi A and Ursini MV: COMMD7 as a
novel NEMO interacting protein involved in the termination of NF-κB
signaling. J Cell Physiol. 231:152–161. 2016. View Article : Google Scholar
|