Introduction
Cervical cancer is the third most commonly diagnosed
cancer after breast and colorectal cancers and one of the most
common causes of cancer-related mortality in women in developing
countries (1). Infection with
human papillomavirus (HPV) is an important contributing factor in
the development of cervical cancer. Mechanistically, the insertion
of viral DNA into chromosomal DNA results in either the activation
of proto-oncogenes or the inactivation of tumor suppressor genes,
which leads to uncontrolled cell proliferation and cervical
intraepithelial neoplasia (CIN) (2). Eventually, approximately 30% of all
CIN cases will develop into cervical cancer (3). Previous studies using different
cancer cell models, including HeLa cells have demonstrated that
interferon (IFN)-α is capable of inhibiting cancer cell
proliferation and inducing apoptosis (4). In addition, IFNs has also been
reported to be used in cervical cancer treatment (5). However, the efficacy of IFN in
cervical cancer therapy has proven to be inconsistent and sometimes
controversial (6). Moreover,
resistance to IFNs has also been observed in the treatment of
certain types of HPV-associated cervical cancers (7). Understanding the mechanisms
responsible for the effects of IFN-α on cancer cells may aid in the
clinical application of IFN-α in cervical cancer therapies.
Type I interferons (IFN-α and IFN-β), a family of
secreted proteins, were originally identified as cytokines with
important antiviral properties (8), and therefore have been widely used
as important anti-viral drugs since their discovery in 1957
(9). Mechanistically, they bind
their specific receptors (IFNAR1 and IFNAR2) presenting on the cell
surface and activate Janus-activated kinase (JAK)/signal transducer
and activator of transcription (STAT) signaling pathways,
activating different cellular cascades, including antiviral defense
and immune modulation (10). At
the molecular level, IFNs initiate a genetic program that
orchestrates the regulation of hundreds of genes known as
IFN-stimulated genes (ISGs). Previous studies have suggested that
IFNs may also possess anticancer functions by inhibiting cancer
cell growth and inducing apoptosis (11,12).
IFN-stimulated gene 15 (ISG15), an ubiquitin-like
protein, is highly upregulated by IFNs (13). It is initially translated as a 165
amino acids precursor and is subsequently processed to expose its
functional C-terminal sequence, LRLRGG. ISG15 contains two
ubiquitin-like domains with the N- and C-terminal domain bearing 33
and 32% identity to that of ubiquitin, respectively (13,14). Also like ubiquitin, ISG15 exists
either as a free moiety or is conjugated to a myriad of proteins
(15). Since ISG15 was originally
found to cross-react with certain ubiquitin antibodies, it has been
also known as ubiquitin cross-reactive protein (16). The conjugation of ISG15 to target
proteins is processed through three consecutive steps, including
activation, conjugation and ligation. This process is widely known
as ISGylation, involving at least three classes of enzymes: the E1
activating enzyme UBE1L, the E2 conjugating enzymes UBCH8 and
UBCH6, and the E3 ligases EFP and HERC5, which are all regulated by
IFNs (8,17). In addition, the identification of
the deconjugating enzyme ISG15-specific protease USP18 (UBP43)
provides an explanation for the dynamic ISGylation status of a
particular protein. Functionally, ISGylation can either activate or
inhibit the activity of a specific target protein (18). Nevertheless, unconjugated free
ISG15 itself also possesses antiviral activities (19).
Significant efforts have been made to identify the
substrates of ISG15 conjugation and their roles in pathogen
infection and tumorigenesis (20,21). In this study, HeLa cells were used
as a model system to examine the roles of ISG15 in cervical cancer.
ISG15 expression was found to be increased in HeLa cells treated
with IFN-α or transiently transfected with ISG15 overexpression
plasmid. Both p53 and p21 were also upregulated when the HeLa cells
were either treated with IFN-α or transiently transfected with
ISG15 overexpression plasmid. Our study reveals the existence of
the IFN-α/ISG15/p53 axis in cervical cancer cells, and suggest that
manipulating ISG15 expression may prove to be effective in the
treatment of cervical cancer.
Materials and methods
Cell culture and IFN-α treatment
HeLa cells (obtained from Department of Molecular
Biology, Central South University, Changsha, China) were grown in
DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (Biological Industries, Beit Haemek, Israel), 100 U/ml
of penicillin and 100 µg/ml of streptomycin, at 37°C in an
atmosphere with 5% CO2. The HeLa cells were treated with
IFN-α (Sigma-Aldrich, St. Louis, MO, USA) at various concentrations
(0, 500, 1,000 and 2,000 U/ml) for 48 h in 6-well plates.
Plasmid transfection
The PCDNA3.1 and PCDNA3.1-ISG15 plasmids were
previously constructed in our laboratory (22). The cells were seeded in 6-well
plates (8×105 cells/well) and transfected with
Lipofectamine 2000 transfection reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) in accordance with the
manufacturer's instructions. The transfected cells were then
cultured in DMEM supplemented with 10% fetal bovine serum for 48 h
for use in further experiments as described below.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The cells (104 cells/well) were plated in
96-well plates. MTT assays were conducted following the different
treatments. The cells were incubated with MTT for 4 h at 37°C. The
dye produced by viable cells was dissolved in DMSO and the
absorbance values were measured at 490 nm using an EnSpire
Multilabel Reader (Perkin Elmer Singapore Pte. Ltd., Singapore).
All assays were performed at least 3 times.
RNA interference
Specific small interfering RNA (siRNA) targeting
ISG15 (AY168648; ISG15-siRNA) (5′-UGAGC ACCGUGUUCAUGAATT-3′,
5′-UUCAUGAACACGGUGCUCATT-3′), p53 (AB082923)
(5′-CCACCAUCCACUACAACUATT-3′, 5′-UAGUUGUAGUGGAUGGUGGTT-3′), and
scrambled siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′,
5′-ACGUGACACGUUCGGAGAATT-3′) were obtained from GenePharm
(Shanghai, China). The cells (5×105/well) were
transfected with 100 pmol siRNA using Lipofectamine 2000
transfection reagent in accordance with the manufacturer's
instructions. Following culture for 48 h post-transfection, the
cells were collected for use in further experiments.
Western blot analysis
The cells were harvested and lysed with cold RIPA
buffer (Beyotime Institute of Biotechnology, Inc., Shanghai, China)
supplemented with 1 mM proteinase inhibitor PMSF (Solarbio,
Beijing, China) for 30 min on ice, and then centrifuged at 12,000
rpm for 16 min at 4°C. The protein concentration was measured using
a BCA protein assay kit (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Proteins were first separated by SDS-PAGE and then
transferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA) by electro-blotting and blocked in
TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 0.2% Tween-20)
containing 5% non-fat milk for 4 h at 4°C. The blots were incubated
with primary antibodies (1:1,000) against the control protein
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000; Cat. no.
10494-1-AP), rabbit anti-p53 (1:1,000; Cat. no. 10442-1-AP), rabbit
anti-p21 (1:1,000; Cat. no. 10355-1-AP), rabbit anti-UBE1L (1:500;
Cat no. 15818-1-AP), rabbit anti-UBCH8 (1:500; Cat. no. 11844-1-AP)
and rabbit anti-HERC5 (1:500; Cat. no. 22692-1-AP) (all from
Proteintech Group, Inc., Chicago, IL, USA), rabbit anti-ISG15
(1:1,000; Cat. no. 2743; Cell Signaling Technology, Inc., Boston,
MA, USA) at 4°C overnight and washed 3 times with TBST. The blots
were then incubated with the secondary antibody (1:1,000) at room
temperature for 1 h. The horseradish peroxidase (HRP)-conjugated
secondary antibody was purchased from Beyotime Institute of
Biotechnology, Inc.
Flow cytometry and caspase-3 activity
detection
After being washed with PBS (pH 7.4), the cells were
suspended in binding buffer and Annexin V-FITC and DNA dye
propidium iodide (PI) were added followed by incubation at 37°C for
30 min in a dark room. The cells were analyzed by Well
Biotechnology, Co., Ltd. (China) using Cytomics FC500 (Beckman
Coulter, Brea, CA, USA). Caspase-3 activity was determined using
the caspase-3 activity assay kit (Beyotime Insititute of
Biotechnology, Inc.). Following treatment, the 96-well plate was
incubated for 1 h at 37°C. The absorbance values were measured at
405 nm using an EnSpire Multilabel Reader (Perkin Elmer Singapore
Pte. Ltd.).
Statistical analysis
Data were analyzed by one-way analysis of variance
(ANOVA) and LSD using SPSS 16.0 software (SPSS, Inc., Chicago, IL,
USA). Densitometric analysis of the western blot bands was carried
out using ImageJ software. The results were presented as the means
± standard deviation (SD). Differences were considered significant
if the P-value was ≤0.05.
Results
Expression and induction of ISG15 by
IFN-α
The cells were treated with various concentrations
of IFN-α for 48 h, and ISG15 expression was detected by western
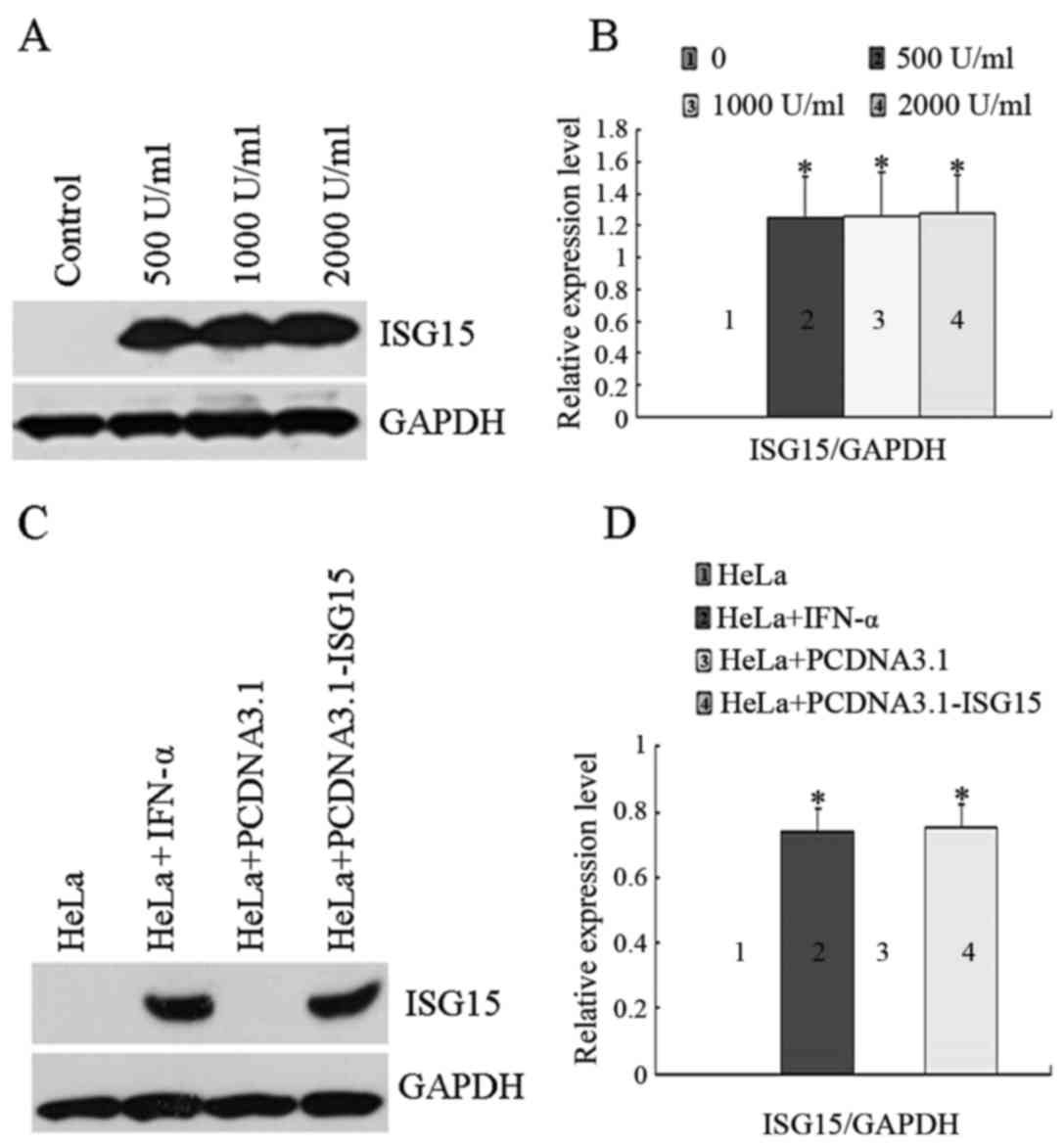
blot analysis. As shown in Fig. 1A
and B, ISG15 was undetectable in the untreated control cells,
and its expression level was increased in response to IFN-α
treatment, although a statistically significant
concentration-dependent effect (500 U/ml; 1,000 U/ml; 2,000 U/ml)
of IFN-α was not observed. The concentration of 1,000 U/ml IFN-α
was selected for use in the subsequent experiments based on the
commonly used dosage in clinical practice. As shown in Fig. 1C and D, the transient transfection
of the cells with the empty plasmid vector, PCDNA3.1, had no effect
on ISG15 expression. Compared with the control group, the
expression levels of ISG15 were increased in response to both IFN-α
treatment and transient transfection with PCDNA3.1-ISG15
plasmid.
Effect of ISG15 on cell viability
Cell viability was examined by MTT assay after the
cells were treated with IFN-α or transiently transfected with the
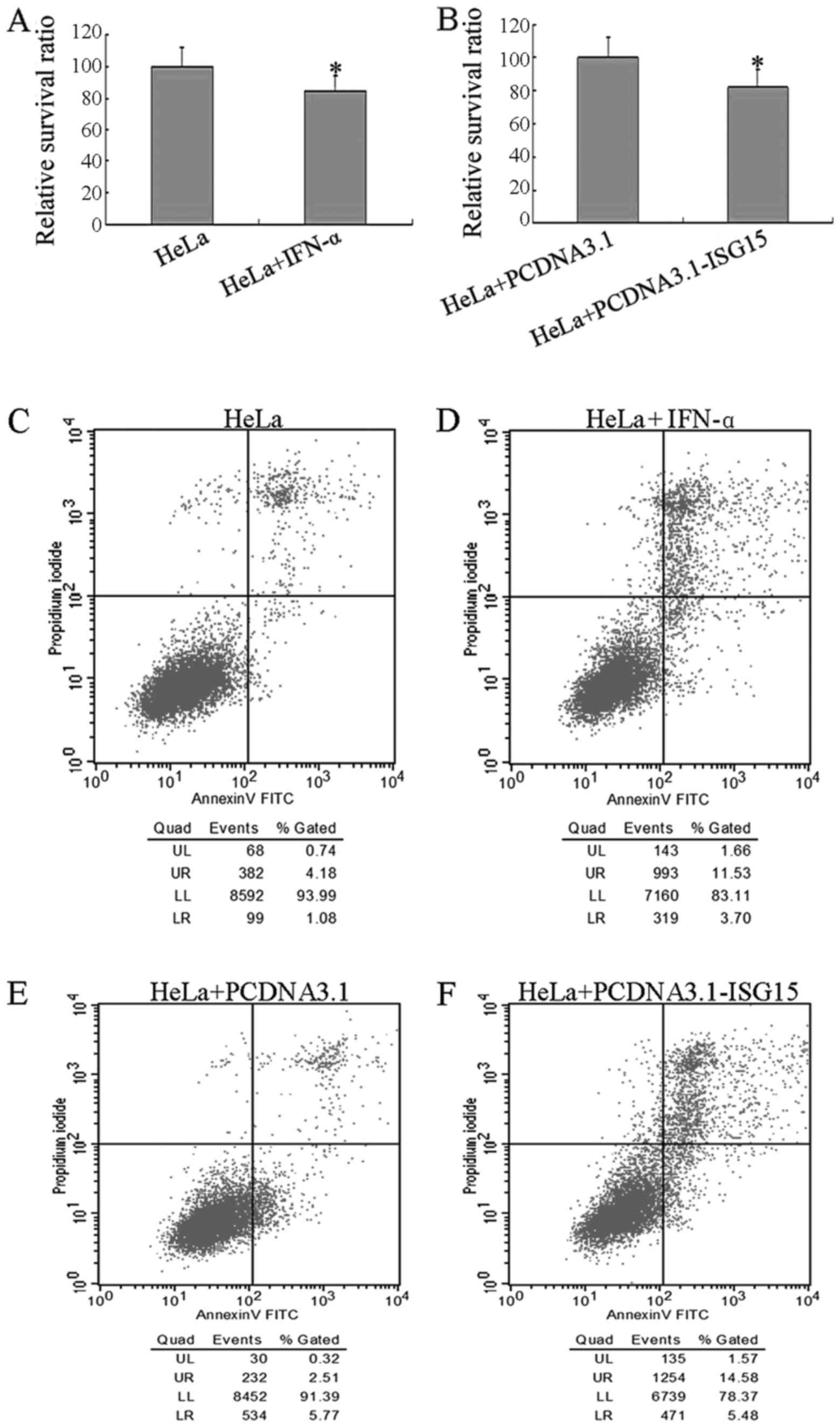
PCDNA3.1-ISG15 plasmid. As shown in Fig. 2A and B, the viability of the cells
was significantly decreased (100 vs. 84.93%) when they were exposed
to 1,000 U/ml of IFN-α, and the viability of the cells decreased
from 100% (the PCDNA3.1 plasmid) to 82.02% when the cells were
transfected with the PCDNA3.1-ISG15 plasmid. These data indicated
that IFN-α treatment and ISG15 overexpression have similar effects
on HeLa cell viability. The cell apoptotic status was further
analyzed by flow cytometry under different conditions. As shown in
Fig. 2C–F, the proportion of
apoptotic cells was increased by IFN-α treatment or transient
transfection with the PCDNA3.1-ISG15 plasmid. The detailed
apoptotic data (Table I) revealed
that the number of apoptotic cells in both the early and late
stages of apoptosis was significantly elevated by IFN-α treatment
or ISG15 over-expression. In addition, the caspase-3 enzymatic
activity in the HeLa cells either treated with IFN-α or transiently
transfected with the PCDNA3.1-ISG15 plasmid was measured. As shown
in Table II, compared with the
basal levels in the control HeLa cells, either treatment with IFN-α
or transfection with the PCDNA3.1-ISG15 plasmid increased caspase-3
enzymatic activity. These results suggest that the overexpression
of ISG15 or treatment with IFN-α play the same important roles in
HeLa cell growth inhibition and the induction of apoptosis.
 | Table IEffect of IFN-α or ISG15 on HeLa cell
apoptosis. |
Table I
Effect of IFN-α or ISG15 on HeLa cell
apoptosis.
| Groups | Early
apoptosis
(%) | Late
apoptosis
(%) |
|---|
| HeLa | 1.08 | 4.18 |
| HeLa + IFN-α | 3.70 | 11.53 |
| HeLa +
PCDNA3.1 | 5.77 | 2.51 |
| HeLa +
PCDNA3.1-ISG15 | 5.48 | 14.58 |
| Table IIEffect of IFN-α or ISG15 on caspase-3
activity in HeLa cells (n=3). |
Table II
Effect of IFN-α or ISG15 on caspase-3
activity in HeLa cells (n=3).
| Groups | Caspase-3 enzymatic
activity (U/µg) |
|---|
| HeLa | 3.13±1.45 |
| HeLa + IFN-α | 7.73±2.08a |
| HeLa +
PCDNA3.1 | 3.39±1.96 |
| HeLa +
PCDNA3.1-ISG15 | 8.37±2.25a |
Effects of ISG15 on p53, p21, UBE1L,
UBCH8 and HERC5 expression
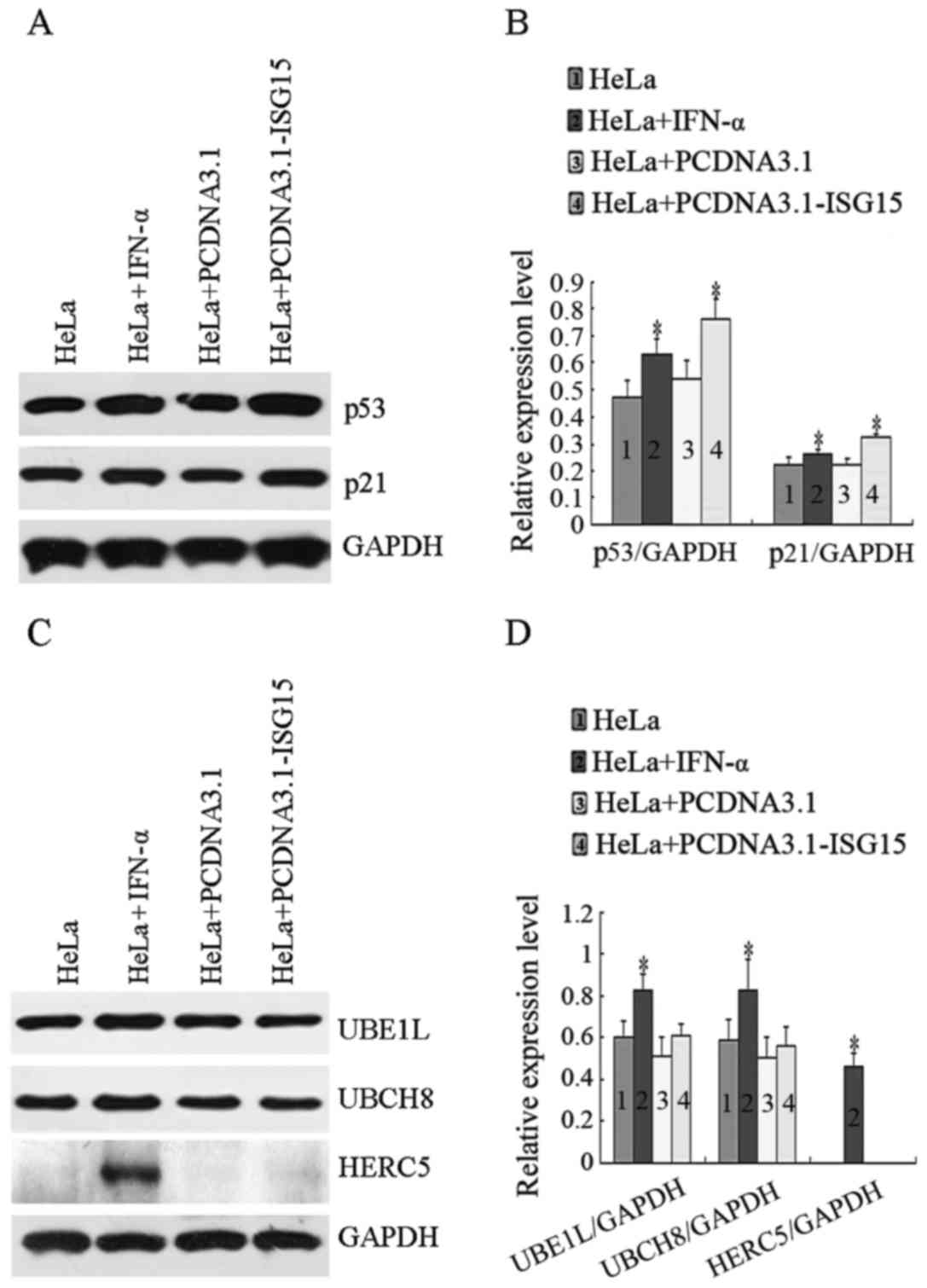
Since p53 is one of the key tumor suppressors in the
regulation of tumor cell survival and apoptosis, the effects of
IFN-α treatment or ISG15 overexpression on the expression of p53
and its downstream target, p21, were investigated. The protein
levels of p53 and p21 were examined by western blot analysis using
specific antibodies. As shown in Fig.
3A, compared to the controls, the levels of p53 and p21 were
markedly elevated in the cells treated with IFN-α or transfected
with PCDNA3.1-ISG15 plasmid. As shown in Fig. 3C, the expression levels of UBE1L,
UBCH8 and HERC5 were upregulated in the cells treated with IFN-α.
However, the overexpression of ISG15 did not affect the levels of
UBE1L, UBCH8 and HERC5 expression. The gray scale protein band
intensity from 3 experiments was quantified by ImageJ software
(Fig. 3B and D). These results
indicate that free ISG15 is related to inhibition of HeLa cell
proliferation.
Knockdown and expression of ISG15 and
p53
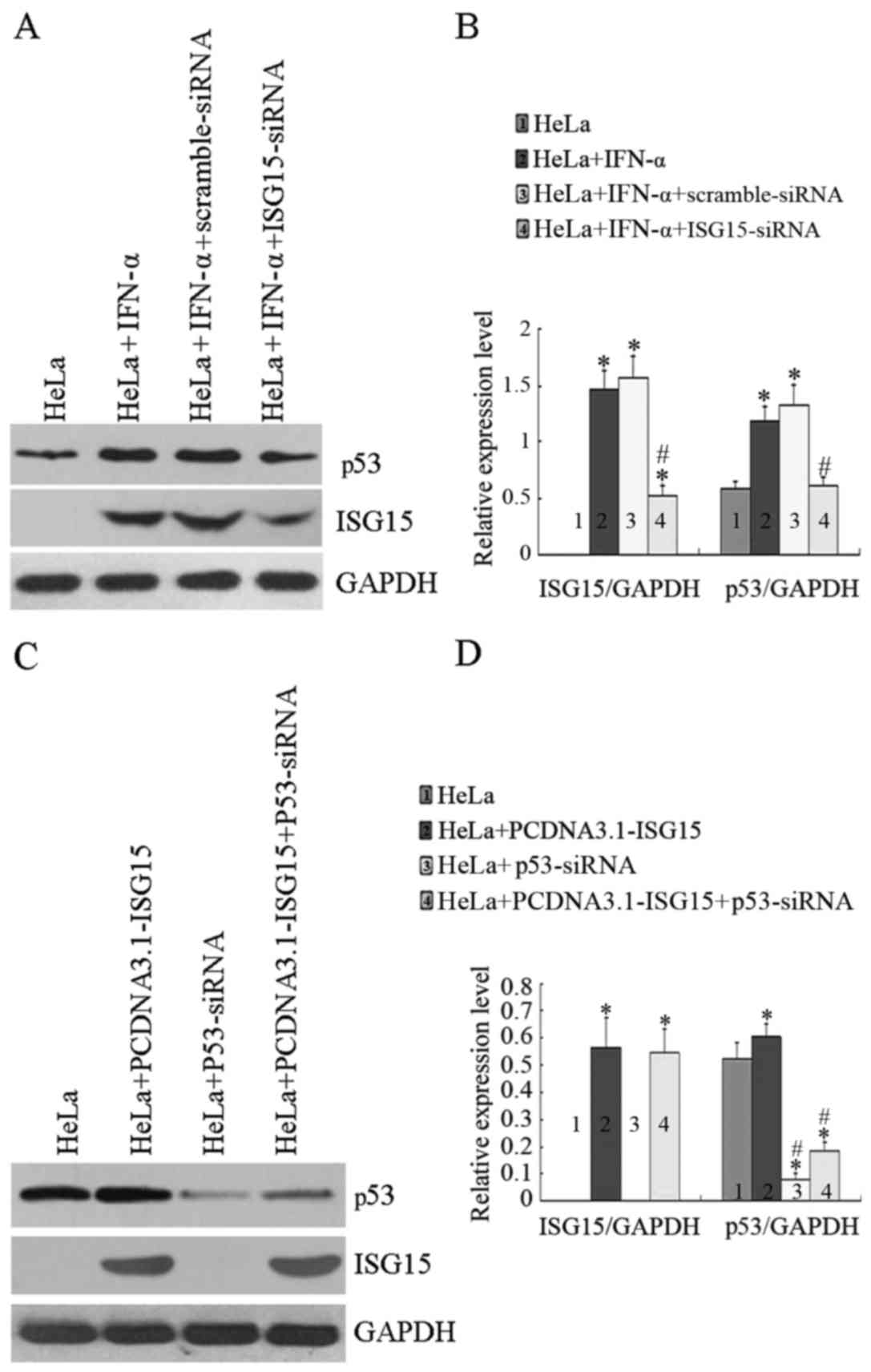
In order to elucidate the role of ISG15 in the
regulation of p53 expression, ISG15 was knocked down by siRNA and
the level of p53 was analyzed under different conditions. As shown
in Fig. 4A and B, transfection of
the cells with siRNA against ISG15 downregulated ISG15 by >50%;
however, transfection with scrambled siRNA did not affect the
IFN-α-induced ISG15 expression. More importantly, IFN-α-induced p53
expression was also significantly decreased when ISG15 was knocked
down; however, transfection with scrambled siRNA had no effect on
the IFN-α-induced p53 expression. As shown in Fig. 4C, compared with the untreated
controls, the expression of p53 was increased in the cells
transfected with the PCDNA3.1-ISG15 plasmid. Transfection with
siRNA against p53 was able to knock down both the endogenous, as
well as the ISG15-induced p53 expression. Moreover, cell viability
was measured by MTT assay when ISG15 and/or p53 were knocked down.
As shown in Table III, cell
viability was reduced to a similar degree when the cells were
either treated with IFN-α (87.5%) or transfected with the ISG15
overexpression plasmid (87%). However, the effect of IFN-α on cell
viability was completely diminished when siRNA against ISG15, but
not scrambled siRNA, was transfected into the cells, indicating
that ISG15 is essential in the IFN-α-mediated reduction of HeLa
cell viability. Furthermore, the effect of the overexpression of
ISG15 on cell viability was not observed when siRNA against p53,
but not scrambled siRNA, was transfected into the cells, suggesting
that the above-observed effects of ISG15 are p53-dependent. Taken
together, these data demonstrate that p53 plays an essential role
in the IFN-α/ISG15-mediated apoptosis of HeLa cells.
| Table IIIThe relative survival ratio of HeLa
cells (n=3). |
Table III
The relative survival ratio of HeLa
cells (n=3).
| Groups | Relative survival
ratio (%) |
|---|
| HeLa | 100.0±8.4 |
| HeLa + IFN-α | 87.5±8.0a |
| HeLa +
IFN-α+scramble-siRNA | 88.3±7.7a |
| HeLa +
IFN-α+ISG15-siRNA | 98.8±8.0 |
| HeLa +
PCDNA3.1-ISG15 | 87.0±8.7a |
| HeLa +
p53-siRNA | 100.0±7.9 |
| HeLa +
PCDNA3.1-ISG15 + p53-siRNA | 99.5±7.7 |
Discussion
Although type I interferons, particularly IFN-α,
have been used in the treatment of various types of cancer, the
precise molecular mechanisms of action of IFNs in cancer treatment
are far from being elucidated (23,24), and this severely hinders the
further application of IFNs in cancer therapy. One of the most
well-studied IFN-α signaling mechanisms is the JAK/STAT-mediated
rapid induction of ISGs (25,26). Among the ISGs, ISG15 is the most
important one and it exerts its effects on cancer cells through
protein ISGylation via its C-terminal LRLRGG sequence or in the
form of a free moiety (15,27). However, the effect of ISGylation
on a specific protein could be inhibitive or simulative (18). Since high levels of ISG15 and
upregulated ISGylation have been observed in certain types of
cancer, it has also been postulated that ISG15 may directly or
indirectly be involved in tumor development (28). In this study, the human cervical
cancer cell line, HeLa, was used as the model system to demonstrate
that IFN-induced ISG15 inhibits cell growth and induces apoptosis
in a p53-dependent manner; we therefore revealed the existence of
the IFN-α/ISG15/p53 signaling axis in cervical cancer cells.
In addition to the upregulation of ISG15, IFN-α also
induces the expression of the E1 activating enzyme UBE1L, the E2
conjugating enzyme UBCH8, and the major E3 ligase HERC5. Therefore,
one of the immediate effects of IFN-α is to increase ISGylation
through the sequential actions of these three enzymes (29). Although the E1 activating enzyme
UBE1L is 45% homologous to the ubiquitin E1 enzyme (UBE1), it does
not affect the ubiquitin level and function, and therefore UBE1L is
specific for ISGylation (30).
However, the E2 conjugating enzyme UBCH8 and the E3 ligase HERC5
can acquire either ISG15 or ubiquitin as substrates. Therefore, the
effect of IFN-α on the E1 enzyme UBE1L may be specific for
ISGylation, but its effect on E2 enzyme UBCH8 and E3 ligase HERC5
can affect both ISGylation and ubiquitination. This study
demonstrated that the level of ISG15 in HeLa cells can be either
induced by IFN-α treatment or can be artificially overexpressed by
transfection with a ISG15 overexpression plasmid. The ISG15-induced
inhibition of HeLa cell proliferation is associated with free ISG15
regardless of ISGylation. Additionally, the anti- and pro-tumor
effects of ISG15 and ISGylation have been reported, and it has also
been suggested that increased ISGylation and decreased free ISG15
may lead to cancer (31–36). Moreover, it has been indicated
that ISG15 exerts an anti-apoptotic effect on pancreatic beta MIN6
cells (37). Thus, free ISG15 is
a suppressor of tumorigenesis.
It has been well accepted that p53 is a key factor
in tumor suppression and the main activating pathway is DNA damage
(38). Activated-p53 in turn
induces cell cycle arrest and/or apoptosis. IFN-treated cells are
more susceptible to p53-dependent apoptosis and HeLa cells undergo
apoptosis in response to DNA damage in a p53-dependent manner
(39,40). We explored the expression of p53
and its downstream protein p21 in HeLa cells treated with IFN-α or
in cells overexpressing ISG15. Consistent with what we have
observed previously in HepG2 cells (22), the levels of p53 and p21 were
significantly upregulated in HeLa cells when they were either
treated with IFN-α or transfected with ISG15 overexpression vector.
However, the knockdown of ISG15 significantly attenuated the
IFN-α-induced increase in p53 expression, suggesting that ISG15
plays an essential role in IFN-α-induced p53 expression, as well as
apoptosis. The role of ISG15 in this process was also corroborated
by the fact that p53 and p21 expression were upregulated by the
overexpression of ISG15. Furthermore, the overexpression of ISG15
not only upregulated p53 and p21, but also promoted HeLa cell
apoptosis. However, ISG15-induced apoptosis was markedly abrogated
when the cells were transfected with siRNA against p53. This
demonstrated that ISG15-induced HeLa cell apoptosis is
p53-dependent. However, the ISGylation-mediated p53 degradation
revealed another pathway regulating p53 stability (41–43). Taken together, these data
demonstrate the existence of the IFN-α/ISG15/p53 axis in HeLa
cells, and demonstrate that is plays pivotal roles in IFN-α-induced
cancer cell growth inhibition, as well as apoptotic cell death.
In conclusion, in this study, using HeLa cells as a
cancer model system, we explored the roles of ISG15 in
IFN-α-induced cancer cell growth inhibition and apoptotic cell
death. By treating the cells with IFN-α and manipulating the
expression of ISG15, we identified the IFN-α/ISG15/p53 axis in HeLa
cells. Therefore, any strategies manipulating the expression of
ISG15 without IFNs may prove to be effective in the treatment of
cervical cancer. Furthermore, given the fact that resistance to
IFN-α has often been observed in clinical trails, ISG15 may serve
as a novel therapeutic target in the treatment of cervical and
possibly, other types of cancer.
Acknowledgments
This study was supported by the Open-End Fund for
the Valuable and Precision Instruments of Central South University
and National Basic Research Program of China (grant no.
2011CB910700-704).
References
|
1
|
Duenas-Gonzalez A, Serrano-Olvera A,
Cetina L and Coronel J: New molecular targets against cervical
cancer. Int J Womens Health. 6:1023–1031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dasari S, Wudayagiri R and Valluru L:
Cervical cancer: biomarkers for diagnosis and treatment. Clin Chim
Acta. 445:7–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiffman MH: Recent progress in defining
the epidemiology of human papillomavirus infection and cervical
neoplasia. J Natl Cancer Inst. 84:394–398. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Massad LS, Turyk ME, Bitterman P and
Wilbanks GD: Interferon-alpha and all-trans-retinoic acid
reversibly inhibit the in vitro proliferation of cell lines derived
from cervical cancers. Gynecol Oncol. 60:428–434. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim KY, Blatt L and Taylor MW: The effects
of interferon on the expression of human papillomavirus oncogenes.
J Gen Virol. 81:695–700. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koromilas AE, Li S and Matlashewski G:
Control of interferon signaling in human papillomavirus infection.
Cytokine Growth Factor Rev. 12:157–170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barnard P, Payne E and McMillan NA: The
human papillomavirus E7 protein is able to inhibit the antiviral
and anti-growth functions of interferon-α. Virology. 277:411–419.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lenschow DJ, Lai C, Frias-Staheli N,
Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE,
García-Sastre A, et al: IFN-stimulated gene 15 functions as a
critical antiviral molecule against influenza, herpes, and Sindbis
viruses. Proc Natl Acad Sci USA. 104:1371–1376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedman RM: Clinical uses of interferons.
Br J Clin Pharmacol. 65:158–162. 2008. View Article : Google Scholar :
|
|
10
|
Sadler AJ and Williams BR:
Interferon-inducible antiviral effectors. Nat Rev Immunol.
8:559–568. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang T, Sun HC, Zhou HY, Luo JT, Zhang
BL, Wang P, Wang L, Qin LX, Ren N, Ye SL, et al: Interferon alpha
inhibits hepatocellular carcinoma growth through inducing apoptosis
and interfering with adhesion of tumor endothelial cells. Cancer
Lett. 290:204–210. 2010. View Article : Google Scholar
|
|
12
|
Shi WY, Cao C and Liu L: Interferon α
induces the apoptosis of cervical cancer HeLa cells by activating
both the intrinsic mitochondrial pathway and endoplasmic reticulum
stress-induced pathway. Int J Mol Sci. 17:pii: E1832. 2016.
View Article : Google Scholar
|
|
13
|
Kim KI, Yan M, Malakhova O, Luo JK, Shen
MF, Zou W, de la Torre JC and Zhang DE: Ube1L and protein
ISGylation are not essential for alpha/beta interferon signaling.
Mol Cell Biol. 26:472–479. 2006. View Article : Google Scholar :
|
|
14
|
Hishiki T, Han Q, Arimoto K, Shimotohno K,
Igarashi T, Vasudevan SG, Suzuki Y and Yamamoto N:
Interferon-mediated ISG15 conjugation restricts dengue virus 2
replication. Biochem Biophys Res Commun. 448:95–100. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loeb KR and Haas AL: The
interferon-inducible 15-kDa ubiquitin homolog conjugates to
intracellular proteins. J Biol Chem. 267:7806–7813. 1992.PubMed/NCBI
|
|
16
|
Haas AL, Ahrens P, Bright PM and Ankel H:
Interferon induces a 15-kilodalton protein exhibiting marked
homology to ubiquitin. J Biol Chem. 262:11315–11323.
1987.PubMed/NCBI
|
|
17
|
Darb-Esfahani S, Sinn BV, Rudl M, Sehouli
J, Braicu I, Dietel M and Denkert C: Interferon-stimulated gene, 15
kDa (ISG15) in ovarian high-grade serous carcinoma: prognostic
impact and link to NF-κB pathway. Int J Gynecol Pathol. 33:16–22.
2014. View Article : Google Scholar
|
|
18
|
Jeon YJ, Yoo HM and Chung CH: ISG15 and
immune diseases. Biochim Biophys Acta. 1802:485–496. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burks J, Reed RE and Desai SD: Free ISG15
triggers an antitumor immune response against breast cancer: a new
perspective. Oncotarget. 6:7221–7231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao C, Hsiang TY, Kuo RL and Krug RM:
ISG15 conjugation system targets the viral NS1 protein in influenza
A virus-infected cells. Proc Natl Acad Sci USA. 107:2253–2258.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeon YJ, Jo MG, Yoo HM, Hong SH, Park JM,
Ka SH, Oh KH, Seol JH, Jung YK and Chung CH: Chemosensitivity is
controlled by p63 modification with ubiquitin-like protein ISG15. J
Clin Invest. 122:2622–2636. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wan XX, Chen HC, Khan MA, Xu AH, Yang FL,
Zhang YY and Zhang DZ: ISG15 inhibits IFN-α-resistant liver cancer
cell growth. BioMed Res Int. 2013:5709092013. View Article : Google Scholar
|
|
23
|
Strander H and Einhorn S: Interferons and
the tumor cell. Biotherapy. 8:213–218. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pfeffer LM, Dinarello CA, Herberman RB,
Williams BR, Borden EC, Bordens R, Walter MR, Nagabhushan TL,
Trotta PP and Pestka S: Biological properties of recombinant
alpha-interferons: 40th anniversary of the discovery of
interferons. Cancer Res. 58:2489–2499. 1998.PubMed/NCBI
|
|
25
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
O'Shea JJ, Gadina M and Schreiber RD:
Cytokine signaling in 2002: new surprises in the Jak/Stat pathway.
Cell. 109(Suppl): S121–S131. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sainz B Jr, Martín B, Tatari M, Heeschen C
and Guerra S: ISG15 is a critical microenvironmental factor for
pancreatic cancer stem cells. Cancer Res. 74:7309–7320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takaoka A, Hayakawa S, Yanai H, Stoiber D,
Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K and
Taniguchi T: Inte gration of interferon-α/β signalling to p53
responses in tumour suppression and antiviral defence. Nature.
424:516–523. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malakhov MP, Malakhova OA, Kim KI, Ritchie
KJ and Zhang DE: UBP43 (USP18) specifically removes ISG15 from
conjugated proteins. J Biol Chem. 277:9976–9981. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan W and Krug RM: Influenza B virus NS1
protein inhibits conjugation of the interferon (IFN)-induced
ubiquitin-like ISG15 protein. EMBO J. 20:362–371. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
D'Cunha J, Knight E Jr, Haas AL, Truitt RL
and Borden EC: Immunoregulatory properties of ISG15, an
interferon-induced cytokine. Proc Natl Acad Sci USA. 93:211–215.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Owhashi M, Taoka Y, Ishii K, Nakazawa S,
Uemura H and Kambara H: Identification of a ubiquitin family
protein as a novel neutrophil chemotactic factor. Biochem Biophys
Res Commun. 309:533–539. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Campbell JA and Lenschow DJ: Emerging
roles for immunomodulatory functions of free ISG15. J Interferon
Cytokine Res. 33:728–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Desai SD, Reed RE, Burks J, Wood LM,
Pullikuth AK, Haas AL, Liu LF, Breslin JW, Meiners S and Sankar S:
ISG15 disrupts cytoskeletal architecture and promotes motility in
human breast cancer cells. Exp Biol Med (Maywood). 237:38–49. 2012.
View Article : Google Scholar
|
|
35
|
Burks J, Reed RE and Desai SD: ISGylation
governs the oncogenic function of Ki-Ras in breast cancer.
Oncogene. 33:794–803. 2014. View Article : Google Scholar
|
|
36
|
Kiessling A, Hogrefe C, Erb S, Bobach C,
Fuessel S, Wessjohann L and Seliger B: Expression, regulation and
function of the ISGylation system in prostate cancer. Oncogene.
28:2606–2620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yoshikawa A, Imagawa A, Nakata S, Fukui K,
Kuroda Y, Miyata Y, Sato Y, Hanafusa T, Matsuoka TA, Kaneto H, et
al: Interferon stimulated gene 15 has an anti-apoptotic effect on
MIN6 cells. Endocr J. 61:883–890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Butz K, Shahabeddin L, Geisen C,
Spitkovsky D, Ullmann A and Hoppe-Seyler F: Functional p53 protein
in human papillomavirus-positive cancer cells. Oncogene.
10:927–936. 1995.PubMed/NCBI
|
|
39
|
Mendonca MS, Howard KL, Farrington DL,
Desmond LA, Temples TM, Mayhugh BM, Pink JJ and Boothman DA:
Delayed apoptotic responses associated with radiation-induced
neoplastic transformation of human hybrid cells. Cancer Res.
59:3972–3979. 1999.PubMed/NCBI
|
|
40
|
Yang J, Dai LX, Chen M, Li B, Ding N, Li
G, Liu YQ, Li MY, Wang BN, Shi XL and Tan HB: Inhibition of
antiviral drug cidofovir on proliferation of human
papillomavirus-infected cervical cancer cells. Exp Ther Med.
12:2965–2973. 2016.PubMed/NCBI
|
|
41
|
Huang YF, Wee S, Gunaratne J, Lane DP and
Bulavin DV: Isg15 controls p53 stability and functions. Cell Cycle.
13:2200–2210. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang YF and Bulavin DV: Oncogene-mediated
regulation of p53 ISGylation and functions. Oncotarget.
5:5808–5818. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Menendez D and Anderson CW: p53 vs. ISG15:
stop, you're killing me. Cell Cycle. 13:2160–2161. 2014. View Article : Google Scholar : PubMed/NCBI
|