Introduction
Myocardial infarction (MI) is the leading cause of
morbidity and mortality worldwide, and extended ischaemia leads to
irreversible myocardial damage (1,2),
which results in pathological ventricular remodelling (3). Therefore, strategies with which to
reduce the myocardial injury have been the focus of studies on MI
(4,5).
The transforming growth factor-β (TGF-β)-inducible
early gene-1 (TIEG1), also known as Kruppel-like factor 10, was
first discovered in foetal osteoblasts using a polymerase chain
reaction (PCR) assay (6). TIEG1
expression is induced by TGF-β, bone morphogenetic proteins (BMPs)
and other cytokines (7). It is
located in multiple cell types and tissues, such as myocytes,
tumour tissues and osteoblasts (8,9).
The TIEG1 gene encodes a 72 kDa protein, which regulates cell
growth and apoptosis (10,11).
There has been significant progress regarding the role of TIEG1 in
various diseases. In the field of cancer studies, Yao et al
found that TIEG1 blocked the proliferation and induced the
apoptosis of leukaemic cell lines, including HL-60, U937, Raji and
K562 in both a time- and dose-dependent manner (12). Jiang et al proved that the
transactivation of TIEG1 inhibited the growth of hepatocellular
carcinoma cells in a dose-dependent manner (13). Moreover, Jin et al
demonstrated that TIEG1 inhibited the invasion and metastasis of
breast cancer by suppressing epidermal growth factor receptor
(EGFR) transcription and the EGFR-related signalling pathway
(14). However, the biological
function of TIEG1 in the cardiovascular field remains unclear. In
2011, Miyake et al discovered that TIEG1 was a feedback
regulator of myostatin in myoblasts (9), and Li et al illustrated that
TIEG1 played a novel role as a blocker in angiotensin II (Ang
II)-induced hypertrophy via the GATA binding protein 4 (GATA4)
signalling pathway (15).
In this study, we investigated both the functional
changes and underlying pathway variations caused by TIEG1
deficiency in ischaemic heart disease, in both in vitro and
in vivo experiments in an aim to fully determine the role of
TIEG1 in heart disease.
Materials and methods
Experimental animals
All animal experiments were approved by the Animal
Care and Use Committee of Zhejiang Chinese Medical University,
Hangzhou, China and met the guidelines for the Care and Use of
Laboratory Animals published by the National Institutes of Health.
Wild-type (WT) C57BL/6 and 10 TIEG1 knockout (KO) mice (aged 6–8
weeks; C57BL/6 background) were purchased from Nanjing University,
Nanjing, China for research purposes only. The mice were housed in
cages in a controlled environment (12 h light/dark) with an ambient
humidity of 50–80% and a temperature of 21±4°C. Food and water were
available ad libitum.
Isolation of neonatal ventricular
cardiomyocytes (CMs)
Neonatal ventricular CMs were prepared as described
previously (16,17). Briefly, the hearts from 10 other
24-h-old TIEG1 KO and WT mice were removed and dissociated with
0.1% trypsin (Gibco, Grand Island, NY, USA). The dispersed cells
were cultured with 10% fetal bovine serum (FBS)-supplemented high
glucose Dulbecco's modified Eagle's medium (DMEM) (glucose
concentration, 4 g/l) at 37°C with 5% CO2 for 90 min.
Unadherent cells were harvested and seeded on the bottom of 24-well
plates (2.5×105 cells/well) or 6-well plates
(1×106 cells/well).
Isolation of endothelial cells
Murine endothelial cells were isolated from 10 WT
and TIEG KO adult mice as previously described (18) with some modifications. The mice
were sacrificed by an overdose of isoflurane, the lungs were
separated and minced into small pieces, washed in Hank's buffer and
digested with dispase for 1 h. The homogenate was passed through a
filter and centrifuged at 300 × g for 5 min. The resulting cells
were suspended and purified with anti-mouse VE-cadherin
antibody-coated magnetic beads (BD Pharmingen, Heidelberg,
Germany). The collected cells were cultured in DMEM (Invitrogen,
Darmstadt, Germany) supplemented with 20% fetal calf serum,
endothelial cell growth factor (Sigma-Aldrich, St. Louis, MO, USA)
penicillin (50 U/ml) and streptomycin (50 µg/ml). The
endothelial cells from the first two passages were >95%
Dil-Ac-LDL positive for purification. The cells at passage 3–6 were
then prepared for use in the in vitro experiments.
In vitro apoptosis assay
The neonatal CMs were seeded at 1×104
cells/well in 24-well plates and apoptosis was induced under
hypoxic conditions (0.1% O2, 5% CO2) in
FBS-free medium for 48 h. The apoptotic cells were detected by
terminal deoxynucleotidyltransferase-mediated dUTP nick-end
labelling (TUNEL) staining (In Situ Cell Death kit, TMR red; Roche
Applied Science, Indianapolis, IN, USA). Briefly, the cells were
fixed in 4% paraformaldehyde, permeabilised in 0.2% Triton X-100,
blocked with 5% BSA and incubated with primary antibodies [troponin
I (TnI; ab47003); Abcam, Cambridge, MA, USA] overnight followed by
the respective fluorescent conjugated secondary antibodies [goat
anti-rabbit IgG H&L (DyLight 488; ab96883); and goat
anti-rabbit IgG H&L (DyLight 550; ab96884); both from Abcam].
Following 3 washes with 0.1% Tween-20 phosphate-buffered saline
(PBS), the CMs were incubated with the reaction mixture provided
with the TUNEL kit following the manufacturer's instructions. Cell
nucleuses were stained with 4′,6-diamidino-2-phenylindole,
dihydrochloride (DAPI) (Thermo Fisher Scientific, Waltham, MA,
USA). Fluorescence images were acquired using a Leica fluorescence
microscope (Leica, Wetzlar, Germany).
In vitro proliferation assay
Cell proliferation was determined by Ki67
immunostaining. Neonatal CMs were seeded at 1×104
cells/well in 24-well plates and cultured under normoxic conditions
(37°C, 5% CO2, 5% O2) for 7 days. The
proliferating cells were detected by double staining with Ki67 and
TnI. Briefly, the cells were fixed in 4% paraformaldehyde, blocked
with 5% BSA after 10 min of permeabilization, and incubated with
primary antibodies [Ki67 (ab16667), TnI (ab30807) both from Abcam]
overnight followed by the respective fluorescent conjugated
secondary antibodies [donkey anti-rabbit IgG H&L (DyLight 488;
ab96891); and donkey anti-goat IgG H&L (DyLight 550; ab96932);
both from Abcam]. After 3 washes with PBST, the cell nuclei were
stained with Hoechst 33258 pentahydrate 1 µg/ml
(Invitrogen). Fluorescence images were then acquired using a Leica
fluorescence microscope.
Tube formation assay
To investigate the angiogenic potential of
endothelial cells in vitro, the cells were seeded at a
concentration of 2×104 cells onto each growth
factor-reduced Matrigel (BD Pharmingen, San Diego, CA, USA)-coated
well of a 96-well plate. Following 4–6 h of incubation, the
capillary network structures of the endothelial cells were
photographed using a phase-contrast microscope (Leica, Wetzlar,
Germany), and the total number of inter-branches was evaluated
using Image-Pro software.
Mouse model of MI
All animal research protocols met with the National
Institutes of Health (NIH) Publication no. 85-23, (revised in 1996)
and were approved by the Animal Care and Use Committee of Zhejiang
Province Medical Institute. The WT and TIEG1 KO mice were subjected
to MI as previously described (19,20). Briefly, the mice were
anaesthetised intraperitoneally with pentobarbital sodium (60
mg/kg) and ventilated with a rodent ventilator via tracheal
intubation. The hearts were exposed through a lateral thoracotomy,
and the left anterior descending artery (LAD) was permanently
ligated with a 7-0 silk suture. Paleness around and below the
ligation point indicated a successful operation. The chest was
closed, and the mice were placed back into cages following
surgery.
Assessment of cardiac function by
echocardiography and haemodynamics
Echocardiography was performed at 3 and 28 days
post-MI, whereas haemodynamics evaluation was performed 28 days
post-MI. Briefly, the mice were anaesthetised via the inhalation of
isoflurane at a concentration of 2%, and a transthoracic
parasternal M-mode echocardiogram was performed using a Vevo 2100
system. The left ventricular end-diastolic diameter (LVEDD and
end-systolic diameter (LVESD) were obtained from at least 3
separate cardiac cycles. The left ventricular end-systolic volume
(LVESV) and left ventricular end-diastolic volume (LVEDV) were
calculated as follows: 7.0 × LVESD3/(2.4 + LVESD), and
7.0× LVEDD3/(2.4 + LVEDD), respectively. The ejection
fraction (EF, %) and fractional shortening (FS, %) were calculated
as follows: [(LVEDV − LVESV)/LVEDV] ×100, and [(LVEDD −
LVESD)/LVEDD] ×100, respectively.
Cardiac catheterization was performed with a
catheter conducer (Millar Instrument, Houston, TX, USA) for
haemodynamic assessment. A 1.4F pressure catheter, SPR-671, was
inserted into the aorta and left ventricle through the right
carotid artery. The transducer was connected to measure the left
ventricular systolic pressure (LVSP), left ventricular end
diastolic pressure (LVEDP), and left ventricular maximum
±dp/dt.
Histological analysis
The animals were sacrificed, and the excised hearts
were immediately placed in a 10% KCl solution to induce cardiac
arrest during the diastolic phase. The left and right atria of the
heart were removed, leaving the left ventricle, and dehydrated in a
30% sucrose solution. The samples were embedded in Tissue-Tek OCT
(Sakura Finetek, Torrance, CA, USA) compound and snapfrozen in
liquid nitrogen. Frozen sections of the left ventricle were cut
into a 7-µm-thick sections.
After preparing 3-day-post-MI samples, TUNEL assay
was used to measure the ratio of CM apoptosis in the border zone
according to manufacturer's instructions. The slices were fixed in
4% paraformaldehyde, permeabilised with 0.2% Triton X-100, blocked
with 5% BSA and incubated with primary antibodies (TnI; Abcam)
overnight followed by the respective fluorescent conjugated
secondary antibodies. After 3 washes with 0.1% Tween-20 PBS, the
sections were incubated in equilibration buffer accroding to the
manufacturer's instructions. Nucleotide and TdT enzyme mixture were
added to the samples at 37°C for 1 h. Cell nuclei were stained with
Hoechst 33258 pentahydrate 1 µg/ml (Invitrogen).
Fluorescence images were obtained using a Leica fluorescence
microscope. The ratio of TUNEL-positive CMs was analysed using the
quantitative software Image-Pro Plus.
For BrdU incorporation assay, the 7 day-post-MI
heart sections were fixed in 4% paraformaldehyde for 10 min,
permeabilised in 0.2% Triton X-100, and blocked with 5% BSA in PBS
for 1 h. The samples were incubated with primary antibody reacted
with BrdU (ab1893, Abcam), which was injected into the heart on the
day of the surgery and primary antibody to TnI (ab47003, Abcam)
diluted 1:200 at 4°C overnight, followed by incubation with the
respective secondary fluorescent antibodies [donkey anti-sheep IgG
H&L (DyLight 488; ab96939); and donkey anti-rabbit IgG H&L
(DyLight 550; ab96892); both from Abcam]. Nuclei were stained with
DAPI (Thermo Fisher Scientific), and imaging was observed using a
fluorescence microscope (Leica, Wetzlar, Germany). The proportion
of BrdU-positive CMs was analysed using the quantitative soft-ware
Image-Pro Plus.
For α-smooth muscle actin (α-SMA)/CD31
immunostaining, the 7-day-post-MI heart tissue sections were fixed
in 4% paraformaldehyde, permeabilised with 0.2% Triton X-100 and
blocked with PBST containing 5% BSA. The samples were reacted with
anti-α-SMA antibody (ab21027) and anti-CD31 antibody (ab7388) (both
from Abcam) at 4°C overnight. After several washes with PBST, the
samples were reacted with fluorescence-conjugated secondary
antibody for 1 h at room temperature. Cell nuclei were stained with
Hoechst 33258 pentahydrate 1 µg/ml. The fluorescence
photographs were obtained at 4–5 randomly selected high visual
fields in the border zone of ischaemic myocardium/sample and
calculated average number of microvessels/visual field with a Leica
fluorescence microscope.
The scar size of the 28-day-post-MI samples was
measured using Masson's trichrome staining. Briefly, the frozen
tissue sections of heart tissues from different groups were fixed
in 4% paraformaldehyde and stained with the Masson's trichrome kit
(HT15, Sigma-Aldrich). The fibrosis and total left ventricular area
of each image were measured using Image-Pro Plus software. The
infarct area was calculated as the percentage (%) of the infarcted
area divided by the entire left ventricle.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the CMs using TRIzol
reagent (Invitrogen) according to the manufacturer's instructions.
cDNA was synthesized from 2 µg of RNA using Moloney murine
leukaemia virus (M-MLV) reverse transcriptase and an
oligo(dT)18 primer (Takara, Otsu, Japan). Quantitative
PCR (qPCR) was performed using the SYBR-Green Reaction Mix (Takara)
following the manufacturer's instructions. The PCR conditions were
95°C for 10 min and 40 cycles of 95°C for 30 sec, 60°C for 30 sec
and 40 cycles of 72°C for 1 min. The PCR primers were designed
using Primer3 Input online software and are listed in Table I. Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) was used as a control, and the relative
expression of the target genes were evaluated by a comparative CT
method and normalised to the control.
 | Table IName and sequence of primer sets for
RT-qPCR. |
Table I
Name and sequence of primer sets for
RT-qPCR.
| Gene name | Primer
sequence | Gene ID | Product size
(bp) |
|---|
| Mus-TIEG1 | F:
CATCCGTCACACAGCTGATG | NM 013692.3 | 250 |
| Mus-TIEG1 | R:
TGTCTCTGAGGAAGGCACAG | | |
| Mus-Pten | F:
GAAAGGGACGGACTGGTGTA | NM 008960.2 | 213 |
| Mus-Pten | R:
TCTTGTGAAACAGCAGTGCC | | |
| Mus-Bcl-2 | F:
TTGTAATTCATCTGCCGCCG | NM 009741.5 | 179 |
| Mus-Bcl-2 | R:
AATGAATCGGGAGTTGGGGT | | |
| Mus-Bax | F:
TCATGAAGACAGGGGCCTTT | NM 007527.3 | 197 |
| Mus-Bax | R:
GTCCACGTCAGCAATCATCC | | |
| Mus-Casp-3 | F:
CAGCCAACCTCAGAGAGACA | NM 009810.3 | 190 |
| Mus-Casp-3 | R:
ACAGGCCCATTTGTCCCATA | | |
| Mus-GAPDH | F:
CTGCGACTTCAACAGCAACT | NM 008084.3 | 330 |
| Mus-GAPDH | R:
GAGTTGGGATAGGGCCTCTC | | |
Western blot analysis
After a 48-h incubation under hypoxic conditions and
FBS-free stimulation, the CMs were rinsed with PBS, lysed in 2.5X
sodium dodecyl sulfate (SDS) gel loading buffer [30 mM Tris-HCl (pH
6.8), 1% SDS, 0.05% bromophenol blue, 12.5% glycerol and 2.5%
mercaptoethanol] and boiled for 30 min. We also rinsed the
endothelial cells with PBS, lysed them in 2.5X SDS gel loading
buffer [ mM Tris-HCl (pH 6.8), 1% SDS, 0.05% bromophenol blue,
12.5% glycerol and 2.5% mercaptoethanol] followed by boiling for 30
min. Equal amounts of the cell lysates were loaded, and the
proteins were separated on 8–12% SDS polyacrylamide gels and
electrotransferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Boston, MA, USA). After the non-specific binding sites
were blocked with 5% non-fat milk, the membranes were incubated
with the primary antibodies, TIEG1 (1:500; A302-015A, Bethyl,
Laboratories Inc., Montgomery, TX, USA), Pten (1:500; 9559), p-Akt
(1:500; 13038), t-Akt (1:500; 14702), Bcl-2 (1:500; 3498), Bax
(1:500; 2772), caspase-3 (1:500; 9664) (all from Cell Signaling
Technology, Beverly, MA, USA), VEGF (1:500; Abcam) and actin
(1:3,000; Kangchen, Shanghai, China), followed by horse radish
peroxidase (HRP)-conjugated secondary antibody (sc-2357; Santa Cruz
Biotechnology, Dallas, TX, USA) incubated at room temperature.
After washing with PBST, the protein bands were visualised using
the Gel Doc EZ Imaging System and analysed using Image Lab software
(Bio-Rad, Hercules, CA, USA).
Statistical analysis
All the data are presented as the means ± standard
deviation (SD). Statistical analyses for the measurement of
significant differences between the TIEG1 KO and WT groups were
performed using the Student's t-test. Probability values of
P<0.05 were considered to indicate statistically significant
differences.
Results
Suppression of TIEG1 results in decreased
apoptosis in vitro
To examine the biological role of TIEG1 in CM
apoptosis, CMs isolated from TIEG1 KO and WT mice underwent 48 h of
exposure to hypoxic conditions. We evaluated TUNEL-positive cells
and found that TIEG1 deficiency led to an approximately 15%
decrease in the apoptosis of myocytes in the border areas compared
to the WT group (P<0.05; Fig. 1A
and D).
Deficiency of TIEG1 results in increased
CM proliferation in vitro
To determine the functional role of TIEG1 in CM
proliferation, Ki67 immunostaining was performed. We calculated the
number of Ki67-positive cells and found that TIEG1 deficiency led
to a 20% greater number of proliferative CMs than the WT mice
(P<0.05; Fig. 1B and E).
Absence of TIEG1 results in the promotion
of the proliferation of endothelial cells in vitro
To determine the functional role of TIEG1 in
endothelial cell proliferation, tube formation was performed. We
calculated the length of capillary structure in the TIEG1 KO and WT
group, and found that the endothelial cells from the TIEG1 KO mice
had a greater powerful ability of microvessel formation than those
from the WT mice (P<0.05; Fig. 1C
and F).
Deletion of TIEG1 ameliorates cardiac
function and decreases infarct size in vivo
To determine whether TIEG1 plays a key role in
cardiac function, we evaluated the myocardial contractile
parameters at 3 and 28 days post-MI in mice. The echocardiographic
results indicated that the absence of TIEG1 led to an improvement
in heart function compared to the WT group at 28 days post-MI
(Fig. 2A), including a better EF
(TIEG1 KO, 41.496±2.131%; WT, 27.616±2.503%), and FS (TIEG1 KO,
23.522±0.870%; WT, 17.536±1.585%) (P<0.05; Fig. 2B and C). Furthermore, the LVEDD
and LVESD were shorter in the TIEG1 KO mouse hearts than in the WT
group at 28 days post-MI (LVEDD: TIEG1 KO, 3.841±0.122 mm; WT,
4.165±0.108 mm; LVESD: TIEG1 KO, 2.950±0.133 mm; WT, 3.420±0.112
mm) (P<0.05; Fig. 2D and E).
Moreover, the haemodynamic results revealed a similar effect.
Compared to the WT group, the TIEG1 KO mice had a significantly
higher LVSP, a lower LVEDP and a higher ±dp/dt (TIEG1 KO: LVSP,
125±12 mmHg; LVEDP, 2.4±0.8 mmHg; dp/dt, 11500±1050; −dp/dt,
−9700±790 mm; WT: LVSP, 95±7 mmHg; LVEDP, 5.9±1.1 mmHg; dp/dt,
7800±1100; −dp/dt, −5400±850) (P<0.05; Fig. 2F–I).
To evaluate the effect of TIEG1 on infarct size
variation, Masson's trichrome staining was used. The quantitative
assessment of myocardial fibrosis indicated that the
TIEG1-deficient mice had reduced 10% scar area compared to the WT
group (P<0.05; Fig. 4A and
B).
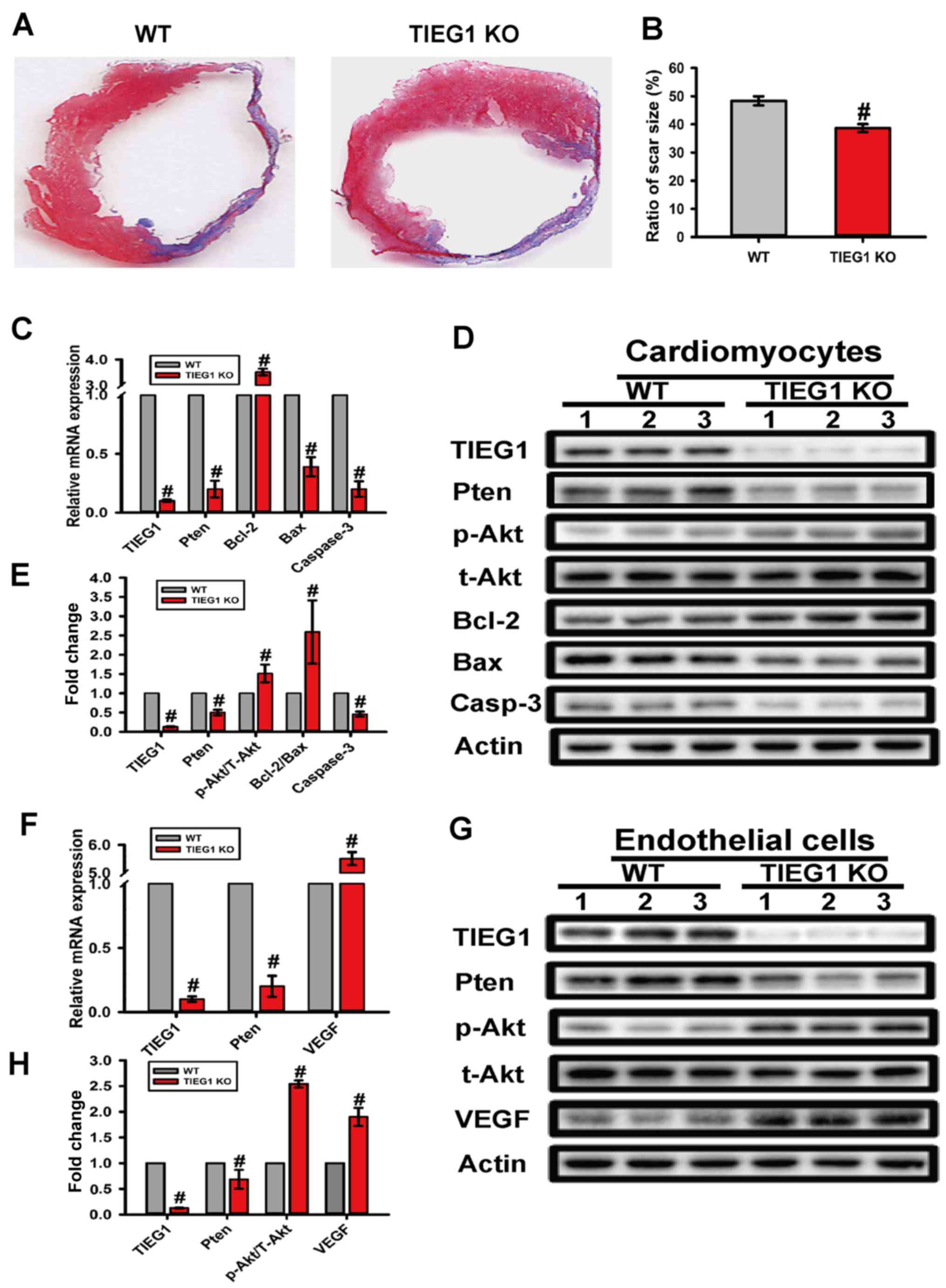 | Figure 4Deficiency of transforming growth
factor-β-inducible early gene-1 (TIEG1) decreases infarct size, and
alters the expression of molecules in the Pten/Akt and Bcl-2/Bax
signalling pathways in myocytes and endothelial cells at the mRNA
and protein level. (A) Representative Masson's trichrome staining
of the heart to show the infarct zone 28 days post-myocardial
infarction (MI). (B) Quantification of infarct zone in heart
tissue. #P<0.01 vs. the WT group. (C) Quantitative
analysis of mRNA expression of TIEG1, Pten, Akt, Bcl-2, Bax,
caspase-3 in cardiomyocytes (CMs). #P<0.01 vs. the WT
group. (D) Western blot analysis of the altered expression of
Pten/Akt and Bcl-2/Bax signalling pathways in CMs. (E)
Quantification of TIEG1, Pten, Akt, Bcl-2, Bax and caspase-3
expession in CMs, n=3. #P<0.01 vs. the WT group. (F)
Quantitative analysis of mRNA expression of TIEG1, Pten, Akt,
Bcl-2, Bax, caspase-3 in endothelial cells. #P<0.01
vs. the WT group. (G) Western blot analysis of the altered
expression of Pten/Akt and Bcl-2/Bax signalling pathways in
endothelial cells. (H) Quantification of TIEG1, Pten, Akt, Bcl-2,
Bax and caspase-3 expession in endothelial cells, n=3.
#P<0.01 vs. the WT group. |
Lack of TIEG1 reduces apoptosis and
enhances proliferation in vivo
We validated the role of TIEG1 in apoptosis in
vivo using TUNEL staining in 3-day samples. Our findings
revealed that the defect of TIEG1 markedly decreased the number of
apoptotic nuclei in the border zone compared to the WT group
(P<0.05; Fig. 3A and B).
The proliferation ratio of myocytes in the border
area of 7-day-samples was measured by immunostaining with BrdU
antibody. We observed more double-positive staining of BrdU and TnI
(P<0.05; Fig. 3C and D) in the
CMs in the border zones from the TIEG1 KO mice than those from the
WT mice.
The absence of TIEG1 promotes
angiogenesis in vivo
We validated the role of TIEG1 in angiogenesis in
vivo using immunofluorescence staining in 28-day-samples. Our
findings suggested that the defect of TIEG1 markedly increased
microvessel formation in the border zone compared to the WT group
(P<0.05; Fig. 3E–H).
Deficiency of TIEG1 influences the
Pten/Akt and Bcl-2/Bax signalling pathways at both the mRNA and
protein level in myocytes
In order to explore the underlying mechanisms
responsible for the anti-apoptotic effects observed with the loss
of TIEG1 in CMs, we analysed the relative expression levels of
Pten/Akt and Bcl-2/Bax signalling pathway molecules by RT-qPCR and
western blot analysis. Our results revealed that a deficiency in
TIEG1 led to the differential expression of the Pten/Akt and
Bcl-2/Bax signalling pathway molecules at the mRNA and protein
level. Specifically, the deficiency of TIEG1 decreased the
expression of Pten, Bax and caspase-3, and increased the
p-Akt/t-Akt ratio and the Bcl-2/Bax ratio (P<0.05; Fig. 4C–E).
Deficiency of TIEG1 influences the
Pten/Akt and Bcl-2/Bax signalling pathways at both the mRNA and
protein level in endothelial cells
We also examined the expression of related molecules
in the Pten/Akt and Bcl-2/Bax signalling pathways in endothelial
cells. Our results revealed the deficiency of TIEG1 also decreased
Pten expression in endothelial cells, as in the CMs, and increased
VEGF expression (P<0.05; Fig.
4F–H).
Discussion
Our study first assessed the role of TIEG1 in
reducing myocardial damage and promoting microvessel generation in
the infarcted heart. The major findings of our study are as
follows: i) the lack of TIEG1 improves heart function and
haemodynamics; ii) the absence of TIEG1 results in less myocardial
apoptosis and greater myocyte proliferation, as shown by both in
vitro and in vivo experiments; and iii) TIEG1 deficiency
causes the variable expression of the Pten/Akt and Bcl-2/Bax
signalling pathways; thus, these are the mechanisms through which
TIEG1 deficiency has cardioprotective effects.
A number of studies have revealed the biological
functions of TIEG1 in different cells (12,15,21). Of these properties, apoptosis
induction and proliferation inhibition by TIEG1 have garnered
attention from researchers. In the field of oncology, higher levels
of TIEG1 protein have been shown to be expressed in epithelial
cells compared to breast tumour cells, whereas it was absent in
infiltrative tumour tissues, demonstrating its inhibitory role in
cell growth (22). The results of
haematological tumour analyses have also demonstrated the effects
of TIEG1 (23,24). Jin et al found that TIEG1
is a key regulator that promotes the apoptosis of K562 leukaemic
cells (25). Furthermore, TIEG1
has been shown to promote the apoptosis of different leukaemia cell
lines, such as HL-60, U937, Raji and K562, in a dose- and
time-dependent manner (12). For
cardiovascular diseases, TIEG1 was first reported to be involved in
cardiac hypertrophy in 2007 (26), and this result was confirmed in a
later clinic study (27).
Currently, TIEG1 prevents CMs from Ang II-mediated hypertrophy,
which provides a novel strategy for the treatment of cardiac
hypertrophy (15). Although the
effect of TIEG1 on hypertrophy has been studied, the functional
role of TIEG1 in myocardial infarction remains unknown. In this
study, we examined cell apoptosis and proliferation in the
myocardium both in vitro and in vivo, in a
TIEG1-deficient mouse model of MI compared to a WT mouse model of
MI. Moreover, we characterised the heart function and haemodynamics
of the TIEG1 KO mice and investigated the underlying pathway
responsible for these phenotypic changes.
For our in vitro experiments, CMs from TIEG1
KO mice were significantly more resistant to apoptosis according to
the results of TUNEL assay. By contrast, Ki67 immunostaining
demonstrated that the lack of TIEG1 was also beneficial to myocyte
proliferation. These results suggest the acceleration of apoptosis
and the suppression of proliferation by TIEG1 in CMs, which have
not been previously reported, at least to the best of our
knowledge. Notably, our findings using the animal model of MI also
revealed that the TIEG1 KO mice had better cardiac functional
recovery than WT mice following coronary artery ligation surgery.
The results of histological analysis confirmed greater myocardium
preservation and a smaller scar area in the TIEG1 KO infarcted
mouse heart than in the WT mouse hearts. Our results provide strong
evidence for the apoptosis-promoting role of TIEG1 in CMs both
in vitro and in vivo.
Apoptosis can be initiated by the
mitochondria-mediated intrinsic pathway. This pathway is controlled
by the Bcl-2 protein family, which is critical for regulating
apoptosis and cell survival. Bcl-2 inhibits apoptosis by forming
heterodimers with Bax, a pro-apoptotic protein, to stop its release
(28). Bcl-2 and Bax are relevant
to apoptosis in CMs, and they are apoptosis involved factors in
myocytes under ischaemic conditions (29,30). A correlation between TIEG1 and
Bcl/Bax in tumour cells has been confirmed (12); however, this remains unclear in
cardiovascular cells.
The tumour suppressor gene, Pten, inactivates the
signalling pathway by promoting protein phosphatase to reduce
tumour cell growth and invasion (31,32). Similarly, Pten inhibition results
in cardioprotective effects against ischaemia reperfusion injury of
the myocardium by stimulating apoptotic survival signals (33). Furthermore, the inactivation of
the Pten gene is regulated by transcriptional modification
(34).
Pten and Bcl-2/Bax are associated with apoptosis,
and they are critical apoptosis factors in CMs. In this study, we
found an increased Bcl-2 expression, and decreased Bax and
downregulated expression of Pten caused by TIEG1 deficiency at both
the mRNA and protein level in CMs, which is key to protecting CMs
against apoptosis. However, the details of this process require
further investigation.
In conclusion, our data demonstrate that the
deletion of TIEG1 results in a significant anti-apoptotic effect
for myocardium salvage following ischaemic injury. The
cardioprotective effects caused by TIEG1 defects are associated
with variations in the expression of Pten, Akt and Bcl-2/Bax. These
data suggest that TIEG1 may be a novel target for the effective
therapy of MI.
References
|
1
|
Alpert JS, Thygesen K, Antman E and
Bassand JP: Myocardial infarction redefined - a consensus document
of the Joint European Society of Cardiology/American College of
Cardiology Committee for the redefinition of myocardial infarction.
J Am Coll Cardiol. 36:959–969. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kitsis RN, Peng CF and Cuervo AM: Eat your
heart out. Nat Med. 13:539–541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White HD, Norris RM, Brown MA, Brandt PW,
Whitlock RM and Wild CJ: Left ventricular end-systolic volume as
the major determinant of survival after recovery from myocardial
infarction. Circulation. 76:44–51. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanelidis A, Premer C, Lopez JG, Balkan W
and Hare JM: Route of delivery modulates the efficacy of
mesenchymal stem cell therapy for myocardial infarction: A
meta-analysis of preclinical studies and clinical trials. Circ Res
Dec 28: pii. CIRCRESAHA. 116:3098192016.
|
|
5
|
Shiba Y, Gomibuchi T, Seto T, Wada Y,
Ichimura H, Tanaka Y, Ogasawara T, Okada K, Shiba N, Sakamoto K, et
al: Allogeneic transplantation of iPS cell-derived cardiomyocytes
regenerates primate hearts. Nature. 538:388–391. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Subramaniam M, Harris SA, Oursler MJ,
Rasmussen K, Riggs BL and Spelsberg TCL: Identification of a novel
TGF-beta-regulated gene encoding a putative zinc finger protein in
human osteoblasts. Nucleic Acids Res. 23:4907–4912. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haldar SM, Ibrahim OA and Jain MK:
Kruppel-like factors (KLFs) in muscle biology. J Mol Cell Cardiol.
43:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spittau B and Krieglstein K: Klf10 and
Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue
Res. 347:65–72. 2012. View Article : Google Scholar
|
|
9
|
Miyake M, Hayashi S, Iwasaki S, Uchida T,
Watanabe K, Ohwada S, Aso H and Yamaguchi T: TIEG1 negatively
controls the myoblast pool indispensable for fusion during myogenic
differentiation of C2C12 cells. J Cell Physiol. 226:1128–1136.
2011. View Article : Google Scholar
|
|
10
|
Reinholz MM, An MW, Johnsen SA,
Subramaniam M, Suman VJ, Ingle JN, Roche PC and Spelsberg TC:
Differential gene expression of TGF beta inducible early gene
(TIEG), Smad7, Smad2 and Bard1 in normal and malignant breast
tissue. Breast Cancer Res Treat. 86:75–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JJ, Park K, Shin MH, Yang WJ, Song MJ,
Park JH, Yong TS, Kim EK and Kim HP: Accessible chromatin structure
permits factors Sp1 and Sp3 to regulate human TGFBI gene
expression. Biochem Biophys Res Commun. 409:222–228. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao K, Xing HC, Wu B, Li Y, Liao AJ, Yang
W and Liu ZG: Effect of TIEG1 on apoptosis and expression of
Bcl-2/Bax and Pten in leukemic cell lines. Genet Mol Res.
14:1968–1974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang L, Lai YK, Zhang JF, Chan CY, Lu G,
Lin MC, He ML, Li JC and Kung HF: Transactivation of the TIEG1
confers growth inhibition of transforming growth
factor-β-susceptible hepatocellular carcinoma cells. World J
Gastroenterol. 18:2035–2042. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin W, Chen BB, Li JY, Zhu H, Huang M, Gu
SM, Wang QQ, Chen JY, Yu S, Wu J and Shao ZM: TIEG1 inhibits breast
cancer invasion and metastasis by inhibition of epidermal growth
factor receptor (EGFR) transcription and the EGFR signaling
pathway. Mol Cell Biol. 32:50–63. 2012. View Article : Google Scholar :
|
|
15
|
Li Q, Shen P, Zeng S and Liu P: TIEG1
inhibits angiotensin II-induced cardiomyocyte hypertrophy by
inhibiting transcription factor GATA4. J Cardiovasc Pharmacol.
66:196–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ehler E, Moore-Morris T and Lange S:
Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp.
Sep 6;(79)2013. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Louch WE, Sheehan KA and Wolska BM:
Methods in cardiomyocyte isolation, culture, and gene transfer. J
Mol Cell Cardiol. 51:288–298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marelli-Berg FM, Peek E, Lidington EA,
Stauss HJ and Lechler RI: Isolation of endothelial cells from
murine tissue. J Immunol Methods. 244:205–215. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao J, Moon M, Yan L, Nian M, Zhang Y,
Liu C, Lu J, Guan H, Chen M, Jiang D, et al: Cellular
FLICE-inhibitory protein protects against cardiac remodelling after
myocardial infarction. Basic Res Cardiol. 107:2392012. View Article : Google Scholar
|
|
20
|
Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X,
Wang Y, Su H, Wang X, Gao E, et al: C1q/tumor necrosis
factor-related protein-9, a novel adipocyte-derived cytokine,
attenuates adverse remodeling in the ischemic mouse heart via
protein kinase A activation. Circulation. 128(Suppl 1): S113–S120.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Subramaniam M, Pitel KS, Withers SG,
Drissi H and Hawse JR: TIEG1 enhances Osterix expression and
mediates its induction by TGFbeta and BMP2 in osteoblasts. Biochem
Biophys Res Commun. 470:528–533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Subramaniam M, Hefferan TE, Tau K, Peus D,
Pittelkow M, Jalal S, Riggs BL, Roche P and Spelsberg TC: Tissue,
cell type, and breast cancer stage-specific expression of a
TGF-beta inducible early transcription factor gene. J Cell Biochem.
68:226–236. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Noti JD, Johnson AK and Dillon JD: The
leukocyte integrin gene CD11d is repressed by gut-enriched
Kruppel-like factor 4 in myeloid cells. J Biol Chem. 280:3449–3457.
2005. View Article : Google Scholar
|
|
24
|
Noti JD, Johnson AK and Dillon JD: The
zinc finger transcription factor transforming growth factor
beta-inducible early gene-1 confers myeloid-specific activation of
the leukocyte integrin CD11d promoter. J Biol Chem.
279:26948–26958. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin W, Di G, Li J, Chen Y, Li W, Wu J,
Cheng T, Yao M and Shao Z: TIEG1 induces apoptosis through
mitochondrial apoptotic pathway and promotes apoptosis induced by
homoharringtonine and velcade. FEBS Lett. 581:3826–3832. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rajamannan NM, Subramaniam M, Abraham TP,
Vasile VC, Ackerman MJ, Monroe DG, Chew TL and Spelsberg TC:
TGFbeta inducible early gene-1 (TIEG1) and cardiac hypertrophy:
Discovery and characterization of a novel signaling pathway. J Cell
Biochem. 100:315–325. 2007. View Article : Google Scholar
|
|
27
|
Bos JM, Subramaniam M, Hawse JR,
Christiaans I, Rajamannan NM, Maleszewski JJ, Edwards WD, Wilde AA,
Spelsberg TC and Ackerman MJ: TGFβ-inducible early gene-1 (TIEG1)
mutations in hypertrophic cardiomyopathy. J Cell Biochem.
113:1896–1903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cook SA, Sugden PH and Clerk A: Regulation
of bcl-2 family proteins during development and in response to
oxidative stress in cardiac myocytes: Association with changes in
mitochondrial membrane potential. Circ Res. 85:940–949. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Misao J, Hayakawa Y, Ohno M, Kato S,
Fujiwara T and Fujiwara H: Expression of bcl-2 protein, an
inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in
ventricular myocytes of human hearts with myocardial infarction.
Circulation. 94:1506–1512. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Myers MP, Stolarov JP, Eng C, Li J, Wang
SI, Wigler MH, Parsons R and Tonks NK: P-TEN, the tumor suppressor
from human chromosome 10q23, is a dual-specificity phosphatase.
Proc Natl Acad Sci USA. 94:9052–9057. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ruan H, Li J, Ren S, Gao J, Li G, Kim R,
Wu H and Wang Y: Inducible and cardiac specific PTEN inactivation
protects ischemia/reperfusion injury. J Mol Cell Cardiol.
46:193–200. 2009. View Article : Google Scholar
|
|
34
|
Liu Y, Nie H, Zhang K, Ma D, Yang G, Zheng
Z, Liu K, Yu B, Zhai C and Yang S: A feedback regulatory loop
between HIF-1α and miR-21 in response to hypoxia in cardiomyocytes.
FEBS Lett. 588:3137–3146. 2014. View Article : Google Scholar : PubMed/NCBI
|

















