Introduction
Mercury exposure is linked to a shift in the redox
status toward oxidative stress. It may enhance lipid peroxidation
in all tissues and may have deleterious effects on an organism
(1). As MeHg easily crosses the
blood-brain barrier, it is highly neurotoxic in exposed human
populations (2). Therefore, its
cytotoxic effect on neurons is stronger when compared to inorganic
HgCl2, even at low levels (3). Eventually, MeHg administration
reduces non-enzymatic and enzymatic antioxidants (6).
Mercury has been shown to affect several aspects of
glutamatergic signaling (4). In
this context, MeHg markedly increases the glutamate concentration
at the synaptic cleft by enhancing spontaneous glutamate release
from neurons (5). Eventual
excitotoxic activity of glutamate resulting from MeHg exposure
contributes to neuronal injury. N-methyl-D-aspartate (NMDA)
receptor-binding memantine attenuates MeHg-induced neurotoxicity
(6). It has also been shown that
the HgCl2-induced reduction of cell viability is
substantially attenuated by the application of a non-competitive
antagonist of NMDA receptors (7).
Although mercury-induced neuronal degeneration is suggested to
invoke glutamate-mediated excitotoxicity, the underlying mechanisms
remain poorly understood.
Caffeine is the most widely consumed psychoactive
substance and acts as an antagonist of adenosine A1 and A2A
receptors at non-toxic doses (8).
Although A1 receptors are located pre-synaptically on dopaminergic,
glutamatergic and cholinergic inputs to neurons, Brown et al
could not detect any evidence regarding the effect of caffeine on
mercury-induced toxicity (9).
On the other hand, mercury-exposed rats have been
shown to exhibit enhanced interferon-γ (IFN-γ) serum levels as
compared to the controls (10).
Furthermore, it is claimed that vascular endothelial growth factor
and interleukin-6 (IL-6) are released from human mast cells via the
stimulation of mercury and disrupt the blood-brain-barrier and
permit brain inflammation (11).
In neurodegenerative diseases, brain inflammation and the
facilitated entrance of immune cells through the blood-brain
barrier can potentially cause neuronal damage and cognitive
dysfunction (12,13). Thus, the disruption of the
blood-brain barrier allows the infiltration of immune cells to the
brain and enhances the responsiveness of neurons to IFN-γ (14). T-cell traffic across the
blood-brain barrier considerably increases, thereby exposing
neuronal cells to the potent effects of IFN-γ (15). Eventually, IFN-γ acts directly on
neural cells (16,17), and causes neurodegenerative
alterations in the central nervous system (CNS) (18). Nevertheless, the precise role of
IFN-γ during neuroinflammation remains unclear (19). Mizuno et al suggested that
IFN-γ synergistically enhances glutamate neurotoxicity mediated by
α-amino-3-hy droxy-5-methyl-4-isoxazolepropionic acid (AMPA)
receptors, but not NMDA receptors (20). By contrast, Lee et al
previously indicated that IFN-γ-mediated neuroprotection is
associated with an enhanced recovery of intracellular
Ca2+ concentrations following exposure to glutamate
(21). In this manner,
conflicting results have been presented regarding the effect of
IFN-γ on glutamate-induced signaling. Furthermore, there is less
information available on the association between mercury-induced
cytotoxicity and caffeine or IFN-γ during the presence or absence
of glutamine.
Thus, the aim of the present study was primarily to
investigate whether mercury-induced neuronal damage is associated
with glutamatergic excitotoxicity, and secondly, to determine
whether glutamate signal transmission participates in the
alteration of mercury-induced neurotoxicity through caffeine and
IFN-γ.
Materials and methods
Cell culture
The human neuroblastoma cell line, SH-SY5Y, was
cultured in EMEM:F12 (1:1) (Biochrom GmbH, Berlin, Germany)
supplemented with 15% fetal bovine serum (FBS; Biochrom GmbH) at
37°C, 5% CO2. The cells were divided into 2 groups and
cultured in either 292 mg/l L-glutamine containing or L-glutamine
free-medium. All the experiments were run in both cell groups. The
solutions of 1, 2 and 5 μM MeHgCl2 and
HgCl2 (Merck KGaA, Darmstadt, Germany), 10 and 20
μM caffeine (Sigma-Aldrich, St. Louis, MO, USA) were
prepared in L-glutamine-supplemented or glutamine-free medium
(Biochrom GmbH) and sterilized using a 0.2 μm syringe filter
(Fuxing Pharmaceutical Co., Ltd., Shanghai, China). Experiments
were repeated under either 1 ng/ml human IFN-γ (hIFN-γ)-containing
or hIFN-γ-free conditions.
Production of hIFN-γ
Active hIFN-γ was produced by using a bacterial
protein expression system. The pET28abased expression plasmid was
constructed using the SLICE cloning procedure, as previously
described (22). Briefly, codon
optimized synthetic gene that encodes hIFN-g mature peptide
(Uniprot accession P01579, amino acids between 24 and 161) was
purchased from Macrogen, Inc. (Seoul, Korea). Escherichia
coli BL21 cells were used as the expression host (Novagen Inc.,
Madison, WI, USA). pET expression system and expression host
bacterium E. coli BL21 cells were obtained from Novagen Inc.
(23). The cells were grown in
100 ml of terriffic broth until a turbidity of 0.5 absorbance was
reached at OD600. Subsequently, culture was induced by using 1 mM
IPTG (24). Following overnight
expression, the cells were harvested and lysed using BPER reagent
(Thermo Fisher Scientific, Waltham, MA, USA). IFN-γ from cleared
lysate was purified using immobilized nickel affinity
chromatography (GE Healthcare, Piscataway, NJ, USA). Imidazole
removal and a polishing step were performed using sephadex G25 (GE
Healthcare) gel filtration chromatography, as previously described
(25).
Experimental design
The SH-SY5Y human neuroblastoma cells
(104 cells/well) were seeded in 96-well plates. Twenty
four hours after seeding (one cell cycle), the cells were exposed
to various concentrations of HgCl2 and
MeHgCl2 in medium with or without 292 mg/l L-glutamine
for either 24 or 48 h. All the assays were performed in triplicates
in 3 sets of experiments.
In this study, in order to clarify the mechanisms
responsible for mercury-induced neuronal toxicity, we used two
different substances in addition to the various concentrations of
mercury compounds in SH-SY5Y cell cultures, caffeine and IFN-γ. The
concentrations of mercury compounds and caffeine that were used in
the experiments, were selected by the evaluation of possible
exposure doses (26–30). The exposure duration was
determined as one and two cell cycles. For further experiments,
104 cells were seeded in 96-well plates in medium with
or without 292 mg/l L-glutamine; each set was individually
pre-incubated for 30 min with 10 or 20 μM caffeine and after
this period, 1, 2 or 5 μM of either MeHgCl2 or
HgCl2 were added and the cells were incubated for 24 and
48 h in FBS-containing medium. Each set of experiments was repeated
with cells pre-incubated with hIFN-γ. In all samples
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was performed as described below. The cells were also counted
using trypan blue dye (Sigma-Aldrich) for each time point and for
each concentration in each assay condition.
Mitochondrial metabolic activity
Mitochondrial metabolic activity was assessed by MTT
assay according to a modified method of Mosmann (31). Briefly, the cells were exposed to
the compounds and MTT dye (Serva, Heidelberg, Germany) (0.5 mg/ml
in phosphate-buffered saline; Merck KGaA) was added to each well 4
h after the completion of the incubation period. Thereafter, the
produced formazan crystals were solubilized by the addition of 10%
SDS (Merck KGaA) in 1 N HCl solution (Merck KGaA). The resultant
absorbance was measured spectrophotometrically (VersaMax ELISA
Microplate Reader; Molecular Devices, Sunnyvale, CA, USA) at 550 nm
with the reference wavelength of 690 nm.
Total nitrite and nitrate (Merck KGaA)
(NO3+NO2; NOx) levels were measured using the
Griess method, as previously described (32). The oxidative stress intensity
coefficient (Q) was calculated by dividing the NOx produced per
cell (cell count; Cc) to the total mitochondrial metabolic activity
per cell (alteration in cell viability - MTT/Cc); Q =
[(NOx/Cc)/MTT/Cc] (33).
Statistical analysis
The significance of the differences between the
control and compound-treated cell groups were analyzed by a
Student's t-test and a value of P<0.05 was considered to
indicate a statistically significant difference. The calculations
were performed using the statistical package SPSS, version 13.0
(SPSS, Inc., Chicago, IL, USA).
Results
The effects of L-glutamine in caffeine-supplemented
medium on the total mitochondrial metabolic activity and oxidative
stress in mercury-exposed SH-SY5Y cells are shown in Tables I–IV. Only 5 μM MeHg led to a
significantly higher oxidative stress intensity score and lower
cell viability when compared with the controls in L-glutamine-free
medium (P<0.05) at the first 24-h incubation period (Table I). However, at the end of the 48-h
incubation period in glutamine-free medium, caffeine
supplementation generated more marked oxidative stress and caused a
significant decrease in cell viability, particularly in the 5
μM MeHg-exposed SH-SY5Y cells (Table II). Moreover, in glutamine-free
medium, caffeine supplementation enhanced mercury-induced oxidative
stress by approximately 10–28% at the end of 48-h incubation period
(Tables I and II). Following the addition of
L-glutamine to the medium, 5 μM MeHg increased oxidative
stress by 88.6% and decreased the total mitochondrial metabolic
activity/cell viability by 48.7% at the first 24-h in comparison to
glutamine-free medium (P<0.05; Table III), whereas the oxidative
stress intensity score increased by 118%, cell viability decreased
by 26% in the 5 μM MeHg-exposed SH-SY5Y cells after 48-h of
incubation in glutamine-containing medium when compared to the
cells cultued in glutamine-free medium (Table IV). In the glutamine-containing
medium, exposure to HgCl2 resulted in a smaller increase
in oxidative stress compared to exposure to MeHg. Cell viability
was higher when the cells were exposed to HgCl2 compared
to MeHg (Tables III and
IV). These results indicated
that MeHg was more toxic than HgCl2 to the SH-SY5Y
cells.
 | Table IOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-free medium. |
Table I
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-free medium.
| Without caffeine
| 10 μM
caffeine
| 20 μM
caffeine
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.4±0.16 | 0.18±0.13 | 114.7±1.33 | 0.17±0.03 | 100.3±0.15 | 0.16±0.01 |
| MeHg 1
μM | 91.9±0.18a | 0.23±0.07 | 98.5±0.58 | 0.23±0.04 | 92.1±0.54 | 0.19±0.02 |
| MeHg 2
μM | 94.4±0.51 | 0.24±0.02 | 94.1±0.42 | 0.24±0.03 | 94.8±0.92 | 0.18±0.04 |
| MeHg 5
μM | 56.0±1.02a | 0.35±0.04a | 57.8±1.87a | 0.26±0.01a | 44.4±1.14a | 0.43±0.22a |
| HgCl2 1
μM | 104.3±0.47 | 0.18±0.05 | 99.1±0.21 | 0.18±0.08 | 114.2±2.11 | 0.14±0.02 |
| HgCl2 2
μM | 104.8±0.01a | 0.23±0.06 | 114.1±13.4 | 0.16±0.04 | 95.6±0.54 | 0.15±0.00 |
| HgCl2 5
μM | 104.4±0.86 | 0.19±0.04 | 94.8±0.89 | 0.19±0.02 | 95.4±1.35 | 0.22±0.04 |
| Table IVOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-containing medium. |
Table IV
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-containing medium.
| Without caffeine
| 10 μM
caffeine
| 20 μM
caffeine
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 100.0±0.38 | 0.36±0.15 | 93.8±1.27 | 0.24±0.05 | 108.5±0.20a | 0.15±0.02a |
| MeHg 1
μM | 82.42±0.79a | 0.33±0.02 | 111.7±1.26 | 0.24±0.04 | 104.5±0.87 | 0.22±0.01 |
| MeHg 2
μM | 53.09±0.04a | 0.63±0.03a | 79.5±0.23a | 0.32±0.05 | 74.1±1.50a | 0.18±0.02 |
| MeHg 5
μM | 29.60±1.74a | 0.85±0.16a | 57.0±1.57a | 0.26±0.02a | 71.7±0.02a | 0.27±0.21a |
| HgCl2 1
μM | 89.16±0.13a | 0.25±0.11a | 103.8±1.27 | 0.15±0.03a | 117.1±0.09a | 0.14±0.02a |
| HgCl2 2
μM | 76.44±0.14 | 0.29±0.14 | 111.8±0.94 | 0.16±0.03a | 107.0±0.18a | 0.23±0.06 |
| HgCl2 5
μM | 61.12±0.40a | 0.35±0.11 | 98.3±0.82 | 0.18±0.00 | 86.8±1.09 | 0.28±0.09 |
| Table IIOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-free medium. |
Table II
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-free medium.
| Without caffeine
| 10 μM
caffeine
| 20 μM
caffeine
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.8±0.22 | 0.23±0.16 | 111.1±0.77 | 0.15±0.05 | 111.2±0.55a | 0.14±0.03 |
| MeHg 1
μM | 100.2±0.35 | 0.19±0.00 | 91.0±0.04a | 0.32±0.07 | 107.2±0.76 | 0.14±0.03 |
| MeHg 2
μM | 58.2±0.26a | 0.27±0.01 | 44.9±0.17a | 0.40±0.09a | 89.2±1.43 | 0.18±0.02 |
| MeHg 5
μM | 40.1±0.74a | 0.39±0.06a | 34.6±0.27a | 0.43±0.00a | 29.4±0.44a | 0.50±0.08a |
| HgCl2 1
μM | 85.0±0.31a | 0.19±0.01 | 104.0±0.99 | 0.17±0.01 | 122.3±0.52a | 0.12±0.04 |
| HgCl2 2
μM | 88.3±0.09a | 0.18±0.02 | 107.6±0.88 | 0.18±0.07 | 127.1±1.94 | 0.12±0.01a |
| HgCl2 5
μM | 89.2±0.91 | 0.19±0.03 | 116.1±0.23a | 0.18±0.03 | 113.1±2.00 | 0.17±0.00 |
| Table IIIOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-containing medium. |
Table III
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-containing medium.
| Without caffeine
| 10 μM
caffeine
| 20 μM
caffeine
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.5±0.33 | 0.19±0.07 | 102.40±1.12 | 0.21±0.07 | 101.1±0.33 | 0.18±0.03 |
| MeHg 1
μM | 82.2±0.74a | 0.27±0.02a | 103.20±0.57 | 0.24±0.02a | 84.80±0.67a | 0.24±0.05a |
| MeHg 2
μM | 73.79±0.63a | 0.36±0.03a | 94.50±0.38 | 0.21±0.03 | 84.70±1.00 | 0.20±0.05 |
| MeHg 5
μM | 28.7±0.56a | 0.66±0.02a | 67.30±0.54a | 0.28±0.01a | 73.1±0.25a | 0.26±0.04a |
| HgCl2 1
μM | 91.32±0.17a | 0.17±0.05a | 102.5±1.27 | 0.16±0.04 | 103.5±2.59 | 0.22±0.04 |
| HgCl2 2
μM | 80.81±0.37a | 0.36±0.07a | 92.7±1.52 | 0.25±0.07 | 113.8±1.02 | 0.23±0.02 |
| HgCl2 5
μM | 73.65±0.02a | 0.36±0.04a | 105.9±0.55 | 0.24±0.04a | 105.2±0.35 | 0.27±0.01a |
Of note, the addition of L-glutamine to the
incubation medium increased oxidative stress by 133 and 118% in the
cells exposed to 2 and 5 μM MeHg, respectively, at the end
of 48-h incubation period (Table
IV). Furthermore, in the presence of L-glutamine, caffeine
supplementation to 5 μM MeHg-containing medium decreased the
oxidative stress scores by 69 and 68% for 10 and 20 μM
caffeine, respectively. At the 48 h, the addition of 10 μM
caffeine to the culture medium containing L-glutamine augmented
cell viability by 93%, while 20 μM caffeine increased
viability by 142%, in comparison to the SH-SY5Y cells exposed only
to 5 μM MeHg. Moreover, incubation with 10 or 20 μM
caffeine attenuated MeHg-induced toxicity in the presence of
L-glutamine (P<0.05) (Table
IV). Overall, caffeine ameliorated the cytotoxic effects of
mercury at all concentrations. In these cases, following exposure
to mercury, alterations in mitochondrial metabolic activity/cell
viability inversely correlated with oxidative stress intensity
scores. Furthermore, the increase in the NOx generation-related
oxidative stress and the decrease in cell viability were also
inversely proportional in a dose-dependent manner, particularly in
MeHg-exposed cells in the presence of L-glutamine.
On the other hand, the addition of caffeine to the
glutamine-free medium had no significant effect on
HgCl2-related oxidative stress and mitochondrial
metabolic activity/cell viability. By contrast, caffeine
supplementation to the glutamine-containing medium significantly
attenuated MeHg- and HgCl2-related toxicity at the
matched doses and for all concentrations during the first and
second 24-h incubation periods. These results were interpreted as
the consumption of antioxidant capacity due to mercury-induced
toxicity. Thus, the most striking toxicity was observed with 5
μM MeHg. It should be noted that a significant amount of
extracellular glutathione is directly derived from glutamine.
Culture in glutamine-free medium reduces cell proliferation and
viability and abolishes glutathione excretion (34).
The effects of L-glutamine on IFN-γ- and
caffeine-supplemented medium on the total mitochondrial metabolic
activity and oxidative stress in mercury-exposed SH-SY5Y cells are
shown in Tables V–VIII. Stimulation of the SH-SY5Y cells
with IFN-γ in glutamine-free medium irregularly affected the
caffeine-controlled mercury-induced toxicity when compared to cells
exposed to mercury only. Moreover, 10 μM caffeine augmented
5 μM MeHg-induced oxidative stress by 125.6% in
glutamine-free medium, when the medium was supplemented with IFN-γ
at the end of the 48-h incubation period. When glutamine was added
to the IFN-γ-containing medium, the SH-SY5Y cells were 42% less
protected by 10 μM caffeine in comparison to only
glutamine-containing medium. In glutamine-containing medium, 10
μM caffeine decreased 5 μM MeHg-induced oxidative
stress by 58 and 69% at the first 24-h and second 24-h incubation
periods, respectively. However, under co-stimulation with glutamine
and IFN-γ, 10 μM caffeine reduced 5 μM MeHg-induced
oxidative stress in the SH-SY5Y cells by 44 and 56% at the first
24-h and second 24-h incubation periods, respectively. This
suggests that the IFN-γ-stimulated SH-SY5Y cells in glutamine-free
medium almost remained unresponsive to mercury-induced toxicity
despite caffeine supplementation (Tables V and VI). Eventually, at the second 24 h
incubation period, the addition of IFN-γ and caffeine to
glutamine-free medium significantly enhanced the toxicity of MeHg
(p<0.05).
| Table VOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-free and IFN-γ-containing medium. |
Table V
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine-free and IFN-γ-containing medium.
| Without caffeine
| 10 μM
caffeine + IFN-γ
| 20 μM
caffeine + IFN-γ
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.4±0.16 | 0.18±0.13 | 79.6±1.02a | 0.23±0.05a | 82.9±0.87 | 0.25±0.05 |
| MeHg 1
μM | 91.9±0.18a | 0.23±0.07 | 98.6±1.68 | 0.27±0.03 | 109.2±0.74 | 0.19±0.09a |
| MeHg 2
μM | 94.4±0.51 | 0.24±0.02a | 102.6±0.41 | 0.19±0.00a | 100.4±0.54 | 0.19±0.08a |
| MeHg 5
μM | 56.0±1.02a | 0.35±0.04a | 65.5±0.51a | 0.28±0.02 | 60.1±0.23a | 0.33±0.07a |
| HgCl2 1
μM | 104.3±0.47 | 0.18±0.05 | 96.2±1.22 | 0.19±0.01a | 96.6±1.80 | 0.18±0.00a |
| HgCl2 2
μM | 104.8±0.01a | 0.23±0.06 | 97.3±0.93 | 0.19±0.04a | 93.4±0.87 | 0.21±0.00a |
| HgCl2 5
μM | 104.4±0.86 | 0.19±0.04 | 101.8±0.79 | 0.17±0.02a | 91.9±0.35a | 0.23±0.03a |
| Table VIIIOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine- and IFN-γ-containing medium. |
Table VIII
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine- and IFN-γ-containing medium.
| Without caffeine
| 10 μM
caffeine + IFN-γ
| 20 μM
caffeine + IFN-γ
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 100.0±0.38 | 0.36±0.15 | 102.4±0.52 | 0.24±0.14 | 98.7±1.82 | 0.19±0.00a |
| MeHg 1
μM | 82.42±0.79a | 0.33±0.02 | 98.5±0.32 | 0.34±0.16 | 114.0±1.34 | 0.15±0.00a |
| MeHg 2
μM | 53.09±0.04a | 0.63±0.03a | 107.1±0.67 | 0.27±0.07 | 108.3±1.07 | 0.16±0.05 |
| MeHg 5
μM | 29.60±1.74a | 0.85±0.16a | 46.7±0.50a | 0.37±0.02a | 58.8±0.73a | 0.36±0.00a |
| HgCl2 1
μM | 89.16±0.13a | 0.25±0.11a | 104.1±0.03a | 0.17±0.05a | 116.6±0.40 | 0.20±0.03a |
| HgCl2 2
μM | 76.44±0.14 | 0.29±0.14 | 92.2±2.49 | 0.27±0.05 | 101.0±0.10 | 0.25±0.05a |
| HgCl2 5
μM | 61.12±0.40a | 0.35±0.11 | 91.0±1.20 | 0.39±0.02 | 85.2±1.28 | 0.23±0.08a |
| Table VIOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-free and IFN-γ-containing medium. |
Table VI
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the second 24-h incubation period with or
without caffeine in glutamine-free and IFN-γ-containing medium.
| Without caffeine
| 10 μM
caffeine + IFN-γ
| 20 μM
caffeine + IFN-γ
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.8±0.22 | 0.23±0.16 | 92.6±1.51 | 0.30±0.03 | 121.0±1.36a | 0.22±0.02a |
| MeHg 1
μM | 100.2±0.35 | 0.19±0.00 | 79.7±0.28a | 0.40±0.02a | 88.4±0.45a | 0.21±0.02a |
| MeHg 2
μM | 58.2±0.26a | 0.27±0.01 | 63.8±0.70a | 0.33±0.02 | 83.2±0.52a | 0.27±0.09 |
| MeHg 5
μM | 40.1±0.74a | 0.39±0.06a | 28.9±0.49a | 0.97±0.11a | 36.5±0.56a | 0.46±0.09a |
| HgCl2 1
μM | 85.0±0.31a | 0.19±0.01 | 93.2±1.79 | 0.18±0.01a | 93.4±0.78 | 0.19±0.03a |
| HgCl2 2
μM | 88.3±0.09a | 0.18±0.02 | 94.4±0.77 | 0.19±0.07a | 95.4±0.89 | 0.19±0.00a |
| HgCl2 5
μM | 89.2±0.91 | 0.19±0.03 | 94.4±0.93 | 0.21±0.12a | 85.4±0.11a | 0.25±0.02a |
In the L-glutamine-containing medium, MeHg treatment
decreased the average cell viability of IFN-γ-stimulated neuronal
cells following caffeine supplementation in comparison to the
controls (Tables VII and
VIII). Following the
stimulation of neuronal cells with IFN-γ, caffeine supplementation
provided a partial improvement in MeHg toxicity in comparison to
the unstimulated counterparts. When taking into account the
mitochondrial metabolic activities and oxidative stress scores,
IFN-γ and caffeine were more effective against
HgCl2-induced toxicity than MeHg. On the one hand,
L-glutamine increased mercury-induced toxicity, but on the other
hand, it was required for improving the effects of caffeine against
mercury-induced toxicity in IFN-γ-stimulated SH-SY5Y cells.
| Table VIIOxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine- and IFN-γ-containing medium. |
Table VII
Oxidative stress intensity
coefficient and total mitochondrial metabolic activity/viability of
MeHg- and HgCl2-exposed SH-SY5Y human neuroblastoma
cells at the end of the first 24-h incubation period with or
without caffeine in glutamine- and IFN-γ-containing medium.
| Without caffeine
| 10 μM
caffeine + IFN-γ
| 20 μM
caffeine + IFN-γ
|
|---|
| MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) | MTT (%) | Q
(μM/%) |
|---|
| Control | 99.5±0.33 | 0.19±0.07 | 92.1±1.91 | 0.17±0.01 | 107.4±1.44 | 0.23±0.06 |
| MeHg 1
μM | 82.2±0.74a | 0.27±0.02a | 82.60±1.21a | 0.32±0.06 | 104.9±0.37 | 0.20±0.16 |
| MeHg 2
μM | 73.79±0.63a | 0.36±0.03a | 87.00±0.15a | 0.20±0.07a | 89.8±0.32 | 0.20±0.02a |
| MeHg 5
μM | 28.7±0.56a | 0.66±0.02a | 59.50±0.72a | 0.37±0.03a | 54.3±1.03a | 0.32±0.04a |
| HgCl2
1μM2 | 91.32±0.17a | 0.17±0.05a | 99.1±0.81 | 0.20±0.01a | 129.6±2.37 | 0.13±0.00a |
| HgCl2 2
μM2 | 80.81±0.37a | 0.36±0.07a | 104.0±0.27 | 0.22±0.05a | 99.5±0.48 | 0.23±0.02a |
| HgCl2 5
μM | 73.65±0.02a | 0.36±0.04a | 103.4±0.85 | 0.18±0.02 | 95.3±0.17a | 0.19±0.03a |
Of note, the most effective concentration was 20
μM caffeine in recovering cell viability and oxidative
stress intensity of the mercury-exposed cells, which were
pre-treated with IFN-γ in L-glutamine-containing medium
(P<0.05). The addition of IFN-γ to the glutamine-containing
medium aggravated average cell viability of the 24- plus 48-h
incubation periods by 15 and 22% in the 10 and 20 μM
caffeine-stimulated cells, respectively. These findings were in
accordance with the increase in the oxidative stress intensity
score with the IFN-γ stimulation of MeHg-exposed cells. Similarly,
when the mean values of the 24- and 48-h incubation periods were
considered, the elevation of Q was 37 and 31% in the 10 and 20
μM caffeine supplemented medium, respectively. The IFN-γ
stimulation of mercury-exposed SH-SY5Y cells in the
glutamine-containing medium reduced the protective effects of
caffeine.
Discussion
Glutamine is the primary precursor for the
biosynthesis of the neurotransmitters glutamate and γ-aminobutyric
acid. It is proposed that in vivo glutamine is synthesized
and released by astrocytes, and is then transported into the neuron
for subsequent conversion to neurotransmitters (35). The uptake of glutamine by neurons
is an integral step in the glutamate-glutamine cycle, and a major
pathway for the replenishment of neuronal glutamate (36). Besides, glutamatergic neurons
exhibit highly efficient transport systems to accumulate
L-glutamine, one of the major precursors of glutamate (37). Glutamine re-appears in neurons
before conversion back to glutamate by glutaminase (38,39). In this respect, without glutamine
influx, SH-SY5Y cells cannot produce glutamate in glutamine-free
medium (40). The neuroblastoma
cell line, SH-SY5Y, expresses a novel form of phosphate activated
glutaminase (PAG) which deamidates glutamine to glutamate and
ammonia at high rates (41).
Glutamate dyshomeostasis and oxidative stress have been identified
as two critical mechanisms mediating MeHg-induced neurotoxicity.
Glutamate/aspartate transporter (GLAST) and glutamate transporter-1
(GLT-1) appear to be inhibited by MeHg exposure (6) (Fig.
1).
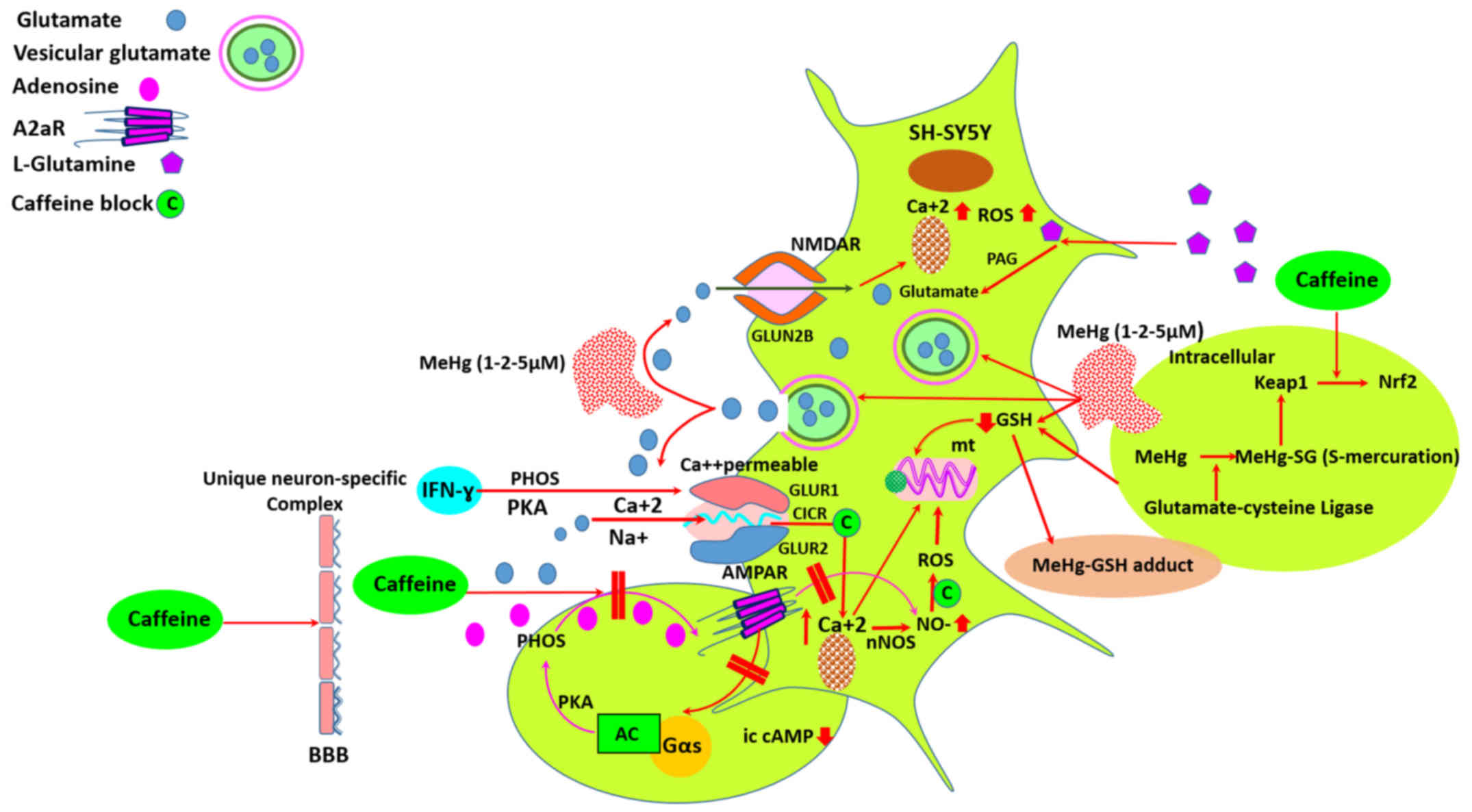 | Figure 1Mercury-induced neuronal death may
occur via the glutamate-mediated excitotoxicity through NMDARs.
Adenosine receptors blockade by caffeine equivalent doses of daily
coffee consumption may reduce the vulnerability to mercury
species-induced oxidative stress in L-glutamine contained medium.
IFN-γ sensitizes the mercury-exposed SH-SY5Y dopaminergic neurons
via AMPA receptor complex, and may diminish the neuroprotective
effect of caffeine in the presence of L-glutamine. MeHg, methyl
mercury; PAG, phosphate activated glutaminase; GSH, reduced
glutathione; NMDAR, N-methyl-D-aspartate receptor; AMPAR,
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
(iontotropic glutamate receptor); IFN-γ, interferon-γ; mt,
mitochondria; ROS, reactive oxygen species; nNOS, neuronal nitric
oxide synthase; PKA, protein kinase A; A2aR, adenosine A2a
receptor; PHOS, pphosphorylation of A2aR; AC, adenylyl cyclase;
Gαs, stimulatory G-protein subunit; ic cAMP, intracellular second
messenger cyclic adenosine monophosphate (cAMP); BBB, blood-brain
barrier; CICR, calcium-induced calcium release; Nrf2, nuclear
factor (erythroid-derived 2)-like 2; Keap1, Kelch-like
ECH-associated protein 1. |
In neurons, mitochondrial metabolism of exogenous
glutamine is mainly responsible for the net synthesis of glutamate,
which is a neurotransmitter, but it is also necessary for the
synthesis of glutathione, the main endogenous antioxidant (42). Thereby mitochondrial metabolic
activity is very important with respect to glutamatergic
neurotransmission and cell antioxidant capacity. The increased
activity of GSH/glutamate-cysteine ligase (GCL) in the cytoplasm
also leads to the concurrent elevation of GSH in the mitochondrial
compartment (Fig. 1). Kaur et
al demonstrated that treatment with 5 μM MeHg for 30 min
led to a significant increase in ROS generation and reduction in
GSH content (43). In a previous
study, SH-SY5Y cells treated for 24 h with MeHg exhibited a
significant reduction in glutathione peroxidase activity in the
brain. There was a concomitant significant decrease in cell
viability and an increase in apoptosis (44). In this context, MeHg may react
readily with GSH, leading to the formation of a MeHg-SG adduct that
is excreted into the extracellular space of SH-SY5Y human
neuroblastoma cells (45). As an
expected result, we found that imercury-induced oxidative stress
was not significantly affected in SH-SY5Y cells in glutamine-free
medium, except, 5 μM MeHg exposure, whereas the toxic
effects of mercury were significantly enhanced in
L-glutamine-containing medium, particularly during the second 24-h
incubation period.
Coffee consumption significantly reduced
mercury-related toxicity. Of note, caffeine acting through
adenosine receptors plays a prominent role in modulating
glutamatergic input to various neurons (Fig. 1). Similarly, glutamate acts on
both ionotropic and metabotropic G protein-coupled receptors.
Therefore, intense glutamatergic neurotransmission is known to
induce adenosine release (46).
On the other hand, caffeine blocks calcium-induced calcium release
(CICR) triggered by calcium influx through calcium permeable AMPA
receptors (47). In our study,
the addition of glutamine to the incubation medium increased the
oxidative stress intensity by 89–118% in the 5 μM
MeHg-exposed SH-SY5Y cells. Furthermore, caffeine supplementation
to L-glutamine-containing incubation medium reversed the
cytotoxicity of mercury compounds when compared with the
caffeine-free controls. Caffeine-mediated effects may occur with at
least two mechanisms: by directly blocking the glutamate activated
channels or by reducing postsynaptic glutamate receptor density. In
this respect, on the one hand caffeine decreases glutamatergic
excitatory post-synaptic currents amplitude by direct postsynaptic
block of glutamate-activated channels. On the other hand, caffeine
directly blocks AMPA receptor-mediated calcium currents responsible
for CICR (48). In daily life,
caffeine concentration in blood reaches approximately 20–30
μM after ingestion of the equivalent of 2 cups of coffee
(28). Thus, in our study, the
maximum dose supplemented to the incubation medium was 20
μM. Indeed, the blood-brain barrier is readily permeable to
caffeine, and thus the concentration in the brain is close to that
in the blood (49). Caffeine
inhibits glutamate receptors with an apparent IC50 value
of approximately 10 mM. Therefore, ingested caffeine is unlikely to
have any effect on ionotropic glutamate receptors. Instead,
caffeine likely produces stimulatory effects in humans through its
potent antagonism of the adenosine receptor (50). Caffeine-mediated glutamate
receptor blockade may only occur under extreme conditions of
toxicity (48).
Deletion of the A2A adenosine receptor reduces the
vulnerability to MeHg, consistent with the neuroprotective effects
of adenosine A2A receptor inactivation. Thus, MeHg toxicity can be
reduced by adenosine A1 and A2A receptor inactivation, either via
their genetic deletion or by treatment with their antagonist
caffeine (51). In this study, in
glutamine-free medium, caffeine did not block the toxicity of 5
μM MeHg. Thus, we observed a significant increase in the
oxidative stress intensity score and a marked decrease in
mitochondrial metabolic activity in the mercury-exposed SH-SY5Y
cells. Substantially, MeHg disrupts glutamate metabolism and
overexcites NMDA receptors of the neurons. At the same time, MeHg
reduces non-enzymatic and enzymatic antioxidants, enhances
neurocyte apoptosis, induces reactive oxygen species, and causes
DNA peroxidative damage in the neurons (52). However, in our study, caffeine
supplementation to glutamine-containing medium substantially
ameliorated matched doses of MeHg- and HgCl2-related
toxicity. Caffeine-inhibited currents are activated by the direct
application of glutamate to cortical neurons, confirming a
post-synaptic site of action. This unexpected form of inhibition
develops over tens of milliseconds and is independent of NMDA
receptors, consistent with non-NMDA receptor block (48). Furthermore, on human neuronal
SH-SY5Y cells, caffeine shows concentration-dependent non-enzymatic
antioxidant potential, decreases the basal levels of free radical
generation, and reduces both superoxide dismutase and catalase
activities (53). In addition,
chronic coffee or caffeine ingestion reduces the lipid peroxidation
in membranes of brain cells and increases the concentration of
reduced-glutathione (54). We
found that in glutamine-free medium, caffeine supplementation was
insufficient to control 5 μM MeHg-induced oxidative stress.
However, in glutamine-containing medium, caffeine inhibited
MeHg-induced-oxidative stress by approximately 58 and 69% at the
end of first and second incubation periods, respectively. These
results confirmed that the antioxidant potential of caffeine was
activated by glutamate, but was not mediated by NMDA receptor. In
our study, we also demonstrated that equivalent doses of caffeine
which were received during the daily coffee intake, substantially
inhibited mercury-induced oxidative stress. However, NMDA
receptor-mediated currents do not change in the presence of
caffeine. Collectively, caffeine is a non-selective adenosine A1
and A2A receptor antagonist that attenuates dopaminergic
neurotoxicity and neurodegeneration (55) (Fig.
1). It has been shown that pre-treatment with caffeine provides
a partial neuroprotection against severe striatal degeneration in
dopaminergic neurons and diminishes the extracellular glutamate in
the brain (56).
Whether the effect of caffeine was mediated by a
mechanism other than the NMDA receptor was examined by IFN-γ. IFN-γ
is a pro-inflammatory cytokine that plays a pivotal role in the
pathology of diseases in the CNS (20). Titze-de-Almeida et al
demonstrated that IFN-γ sensitized SH-SY5Y cells to
neurotoxin-induced injury, also causing an increase in ROS levels
(57). Furthermore, IFN-γ
directly induces neuronal dysfunction and enhances glutamate
neurotoxicity mediated by AMPA receptors, but not NMDA receptors
(20). Thus, in our study, IFN-γ
in the pure SH-SY5Y cell culture worked synergistically with
glutamate to promote neuronal excitotoxicity presumably through
AMPA receptor complex in SH-SY5Y cells (Fig. 1). At the second 24-h incubation
period, the addition of caffeine to IFN-γ-stimulated cells in
glutamine-free medium significantly enhanced the toxicity of 5
μM MeHg. This result is in accordance with the findings of
Titze-de-Almeida et al (57) and Vikman et al (58). Thus, Vikman et al indicated
that when the neurons were treated with IFN-γ, neurophysiological
alterations could be observed 48 h following exposure, when the
frequency of AMPA receptor-mediated spontaneous excitatory
post-synaptic currents are increased (58).
Caffeine supplementation could present a significant
protective effect against MeHg toxicity with glutamate
transmission. However, IFN-γ-stimulated neuronal cells were less
protected by caffeine in L-glutamine-containing medium.
Nevertheless, under the co-stimulation of SH-SY5Y cells with
glutamine and IFN-γ, caffeine decreased MeHg-induced average
oxidative stress by 50%. Glutamate seems to be an indispensable
mediator of the effects of both mercury-induced toxicity and
caffeine. Our results are in accordance with the findings of Bagga
et al, with respect to glutamatergic neuronal activity and
neurotransmission. Caffeine provides only partial neuroprotection
against mercury-induced toxicity in IFN-γ-stimulated SH-SY5Y
dopaminergic neurons (56).
In conclusion, these data suggest that
mercury-induced neuronal death may occur through glutamate-mediated
excitotoxicity. Adenosine receptor blockade by caffeine in
equivalent doses of daily coffee consumption reduced the
vulnerability to mercury-induced oxidative stress in
glutamine-containing medium. The IFN-γ stimulation of SH-SY5Y
dopaminergic neurons severely decreased cell viability and
increased oxidative stress in glutamine-free medium despite
caffeine supplementation. However, the addition of glutamine to the
medium increased cell viability by 62% and reduced MeHg-related
oxidative stress intensity by 62% in the presence of 10 μM
caffeine. It can thus be concluded that the IFN-γ stimulation of
mercury-exposed dopaminergic neurons in neuroinflammatory diseases
may diminish the neuroprotective effects of caffeine.
Acknowledgments
The study was partially supported by The Scientific
and Technological Research Council of Turkey (no. 214S112). This
study has been orally presented in the '35th Winter-Workshop on
Clinical, Chemical and Biochemical Aspects of Pteridines, February
23rd–26th, 2016, Innsbruck, Austria'.
References
|
1
|
Karimi R, Vacchi-Suzzi C and Meliker JR:
Mercury exposure and a shift toward oxidative stress in avid
seafood consumers. Environ Res. 146:100–107. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clarkson TW and Magos L: The toxicology of
mercury and its chemical compounds. Crit Rev Toxicol. 36:609–662.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lohren H, Blagojevic L, Fitkau R, Ebert F,
Schildknecht S, Leist M and Schwerdtle T: Toxicity of organic and
inorganic mercury species in differentiated human neurons and human
astrocytes. J Trace Elem Med Biol. 32:200–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aschner M, Yao CP, Allen JW and Tan KH:
Methylmercury alters glutamate transport in astrocytes. Neurochem
Int. 37:199–206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brookes N: In vitro evidence for the role
of glutamate in the CNS toxicity of mercury. Toxicology.
76:245–256. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu W, Xu Z, Deng Y, Xu B, Wei Y and Yang
T: Protective effects of memantine against methylmercury-induced
glutamate dyshomeostasis and oxidative stress in rat cerebral
cortex. Neurotox Res. 24:320–337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu F, Farkas S, Kortbeek S, Zhang F-X,
Chen L, Zamponi GW and Syed NI: Mercury-induced toxicity of rat
cortical neurons is mediated through N-Methyl-D-Aspartate
receptors. Mol Brain. 5:302012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Biessels GJ: Caffeine, diabetes,
cognition, and dementia. J Alzheimers Dis. 20(Suppl 1): S143–S150.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brown SJ, James S, Reddington M and
Richardson PJ: Both A1 and A2a purine receptors regulate striatal
acetylcholine release. J Neurochem. 55:31–38. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Penna S, Pocino M, Marval MJ, Lloreta J,
Gallardo L and Vila J: Modifications in rat testicular morphology
and increases in IFN-gamma serum levels by the oral administration
of subtoxic doses of mercuric chloride. Syst Biol Reprod Med.
55:69–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kempuraj D, Asadi S, Zhang B, Manola A,
Hogan J, Peterson E and Theoharides TC: Mercury induces
inflammatory mediator release from human mast cells. J
Neuroinflammation. 7:202010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu YJ, Guo DW, Tian L, Shang DS, Zhao WD,
Li B, Fang WG, Zhu L and Chen YH: Peripheral T cells derived from
Alzheimer's disease patients overexpress CXCR2 contributing to its
transendothelial migration, which is microglial
TNF-alpha-dependent. Neurobiol Aging. 31:175–188. 2010. View Article : Google Scholar
|
|
13
|
Man SM, Ma YR, Shang DS, Zhao WD, Li B,
Guo DW, Fang WG, Zhu L and Chen YH: Peripheral T cells overexpress
MIP-1alpha to enhance its transendothelial migration in Alzheimer's
disease. Neurobiol Aging. 28:485–496. 2007. View Article : Google Scholar
|
|
14
|
Minogue AM, Jones RS, Kelly RJ, McDonald
CL, Connor TJ and Lynch MA: Age-associated dysregulation of
microglial activation is coupled with enhanced blood-brain barrier
permeability and pathology in APP/S1 mice. Neurobiol Aging.
35:1442–1452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Popko B, Corbin JG, Baerwald KD, Dupree J
and Garcia AM: The effects of interferon-gamma on the central
nervous system. Mol Neurobiol. 14:19–35. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee M, McGeer E and McGeer PL: Neurotoxins
released from interferon-gamma-stimulated human astrocytes.
Neuroscience. 229:164–175. 2013. View Article : Google Scholar
|
|
17
|
Podolsky MA, Solomos AC, Durso LC, Evans
SM, Rall GF and Rose RW: Extended JAK activation and delayed STAT1
dephosphorylation contribute to the distinct signaling profile of
CNS neurons exposed to interferon-gamma. J Neuroimmunol. 251:33–38.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seifert HA, Leonardo CC, Hall AA, Rowe DD,
Collier LA, Benkovic SA, Willing AE and Pennypacker KR: The spleen
contributes to stroke induced neurodegeneration through interferon
gamma signaling. Metab Brain Dis. 27:131–141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kulkarni A, Ganesan P and O'Donnell LA:
Interferon Gamma: Influence on Neural Stem Cell Function in
Neurodegenerative and Neuroinflammatory Disease. Clin Med Insights
Pathol. 9(Suppl 1): 9–19. 2016.PubMed/NCBI
|
|
20
|
Mizuno T, Zhang G, Takeuchi H, Kawanokuchi
J, Wang J, Sonobe Y, Jin S, Takada N, Komatsu Y and Suzumura A:
Interferon-gamma directly induces neurotoxicity through a neuron
specific, calcium-permeable complex of IFN-gamma receptor and AMPA
GluR1 receptor. FASEB J. 22:1797–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee J, Kim SJ, Son TG, Chan SL and Mattson
MP: Interferon-gamma is up-regulated in the hippocampus in response
to intermittent fasting and protects hippocampal neurons against
excitotoxicity. J Neurosci Res. 83:1552–1557. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Werling U and Edelmann W: SLiCE:
A novel bacterial cell extract-based DNA cloning method. Nucleic
Acids Res. 40:e552012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosano GL and Ceccarelli EA: Recombinant
protein expression in Escherichia coli: Advances and challenges.
Front Microbiol. 5:1722014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Robichon C, Luo J, Causey TB, Benner JS
and Samuelson JC: Engineering Escherichia coli BL21(DE3) derivative
strains to minimize E. coli protein contamination after
purification by immobilized metal affinity chromatography. Appl
Environ Microbiol. 77:4634–4646. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abe R, Kudou M, Tanaka Y, Arakawa T and
Tsumoto K: Immobilized metal affinity chromatography in the
presence of arginine. Biochem Biophys Res Commun. 381:306–310.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Becker A and Soliman KFA: The role of
intracellular glutathione in inorganic mercury-induced toxicity in
neuroblastoma cells. Neurochem Res. 34:1677–1684. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Björkman L, Lundekvam BF, Laegreid T,
Bertelsen BI, Morild I, Lilleng P, Lind B, Palm B and Vahter M:
Mercury in human brain, blood, muscle and toenails in relation to
exposure: An autopsy study. Environ Health. 6:302007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cysneiros RM, Farkas D, Harmatz JS, von
Moltke LL and Greenblatt DJ: Pharmacokinetic and pharmacodynamic
interactions between zolpidem and caffeine. Clin Pharmacol Ther.
82:54–62. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Spyridopoulos I, Fichtlscherer S, Popp R,
Toennes SW, Fisslthaler B, Trepels T, Zernecke A, Liehn EA, Weber
C, Zeiher AM, et al: Caffeine enhances endothelial repair by an
AMPK-dependent mechanism. Arterioscler Thromb Vasc Biol.
28:1967–1974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Toimela T, Mäenpää H, Mannerström M and
Tähti H: Development of an in vitro blood-brain barrier
model-cytotoxicity of mercury and aluminum. Toxicol Appl Pharmacol.
195:73–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miranda KM, Espey MG and Wink DA: A rapid,
simple spectrophotometric method for simultaneous detection of
nitrate and nitrite. Nitric Oxide. 5:62–71. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Engin AB: Investigation of the effect of
C1Q and complement regulatory protein CD59 on the SH-SY5Y cell
viability in hyperglycemic conditions (unpublished PhD thesis).
Gazi University, Institute of Health Sciences; 2015
|
|
34
|
Sappington DR, Siegel ER, Hiatt G, Desai
A, Penney RB, Jamshidi-Parsian A, Griffin RJ and Boysen G:
Glutamine drives glutathione synthesis and contributes to radiation
sensitivity of A549 and H460 lung cancer cell lines. Biochim
Biophys Acta. 1860:836–843. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tamarappoo BK, Raizada MK and Kilberg MS:
Identification of a system N-like Na(+)-dependent glutamine
transport activity in rat brain neurons. J Neurochem. 68:954–960.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bröer S and Brookes N: Transfer of
glutamine between astrocytes and neurons. J Neurochem. 77:705–719.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su TZ, Campbell GW and Oxender DL:
Glutamine transport in cerebellar granule cells in culture. Brain
Res. 757:69–78. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Conti F and Minelli A: Glutamate
immunoreactivity in rat cerebral cortex is reversibly abolished by
6-diazo-5-oxo-L-norleucine (DON), an inhibitor of
phosphate-activated glutaminase. J Histochem Cytochem. 42:717–726.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pow DV and Crook DK: Direct
immunocytochemical evidence for the transfer of glutamine from
glial cells to neurons: Use of specific antibodies directed against
the d-stereoisomers of glutamate and glutamine. Neuroscience.
70:295–302. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Magistretti PJ and Pellerin L: Cellular
mechanisms of brain energy metabolism and their relevance to
functional brain imaging. Philos Trans R Soc Lond B Biol Sci.
354:1155–1163. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roberg BA, Torgner IA and Kvamme E:
Kinetics of a novel isoform of phosphate activated glutaminase
(PAG) in SH-SY5Y neuroblastoma cells. Neurochem Res. 35:875–880.
2010. View Article : Google Scholar
|
|
42
|
D'Alessandro G, Calcagno E, Tartari S,
Rizzardini M, Invernizzi RW and Cantoni L: Glutamate and
glutathione interplay in a motor neuronal model of amyotrophic
lateral sclerosis reveals altered energy metabolism. Neurobiol Dis.
43:346–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaur P, Aschner M and Syversen T:
Glutathione modulation influences methyl mercury induced
neurotoxicity in primary cell cultures of neurons and astrocytes.
Neurotoxicology. 27:492–500. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Franco JL, Posser T, Dunkley PR, Dickson
PW, Mattos JJ, Martins R, Bainy AC, Marques MR, Dafre AL and Farina
M: Methylmercury neurotoxicity is associated with inhibition of the
antioxidant enzyme glutathione peroxidase. Free Radic Biol Med.
47:449–457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yoshida E, Abiko Y and Kumagai Y:
Glutathione adduct of methylmercury activates the Keap1-Nrf2
pathway in SH-SY5Y cells. Chem Res Toxicol. 27:1780–1786. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ferré S, Karcz-Kubicha M, Hope BT, Popoli
P, Burgueño J, Gutiérrez MA, Casadó V, Fuxe K, Goldberg SR, Lluis
C, et al: Synergistic interaction between adenosine A2A and
glutamate mGlu5 receptors: Implications for striatal neuronal
function. Proc Natl Acad Sci USA. 99:11940–11945. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morton-Jones RT, Cannell MB and Housley
GD: Ca2+ entry via AMPA-type glutamate receptors triggers
Ca2+-induced Ca2+ release from ryanodine receptors in rat spiral
ganglion neurons. Cell Calcium. 43:356–366. 2008. View Article : Google Scholar
|
|
48
|
Vyleta NP and Smith SM: Fast inhibition of
glutamate-activated currents by caffeine. PLoS One. 3:e31552008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu X, Smith BJ, Chen C, Callegari E,
Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, et
al: Evaluation of cerebrospinal fluid concentration and plasma free
concentration as a surrogate measurement for brain free
concentration. Drug Metab Dispos. 34:1443–1447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Daly JW, Butts-Lamb P and Padgett W:
Subclasses of adenosine receptors in the central nervous system:
Interaction with caffeine and related methylxanthines. Cell Mol
Neurobiol. 3:69–80. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Björklund O, Kahlström J, Salmi P, Ogren
SO, Vahter M, Chen JF, Fredholm BB and Daré E: The effects of
methylmercury on motor activity are sex- and age-dependent, and
modulated by genetic deletion of adenosine receptors and caffeine
administration. Toxicology. 241:119–133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Feng S, Xu Z, Liu W, Li Y, Deng Y and Xu
B: Preventive effects of dextromethorphan on methylmercury-induced
glutamate dyshomeostasis and oxidative damage in rat cerebral
cortex. Biol Trace Elem Res. 159:332–345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeidán-Chuliá F, Gelain DP, Kolling EA,
Rybarczyk-Filho JL, Ambrosi P, Terra SR, Pires AS, da Rocha JB,
Behr GA and Moreira JC: Major components of energy drinks
(caffeine, taurine, and guarana) exert cytotoxic effects on human
neuronal SH-SY5Y cells by decreasing reactive oxygen species
production. Oxid Med Cell Longev. 2013:7917952013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Abreu RV, Silva-Oliveira EM, Moraes MFD,
Pereira GS and Moraes-Santos T: Chronic coffee and caffeine
ingestion effects on the cognitive function and antioxidant system
of rat brains. Pharmacol Biochem Behav. 99:659–664. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kalda A, Yu L, Oztas E and Chen JF: Novel
neuroprotection by caffeine and adenosine A(2A) receptor
antagonists in animal models of Parkinson's disease. J Neurol Sci.
248:9–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bagga P, Chugani AN and Patel AB:
Neuroprotective effects of caffeine in MPTP model of Parkinson's
disease: A (13)C NMR study. Neurochem Int. 92:25–34. 2016.
View Article : Google Scholar
|
|
57
|
Titze-de-Almeida SS, Lustosa CF, Horst CH,
Bel ED and Titze-de-Almeida R: Interferon Gamma potentiates the
injury caused by MPP(+) on SH-SY5Y cells, which is attenuated by
the nitric oxide synthases inhibition. Neurochem Res. 39:2452–2464.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vikman KS, Owe-Larsson B, Brask J,
Kristensson KS and Hill RH: Interferon-gamma-induced changes in
synaptic activity and AMPA receptor clustering in hippocampal
cultures. Brain Res. 896:18–29. 2001. View Article : Google Scholar : PubMed/NCBI
|














