Introduction
Atrial fibrillation (AF) is the most common
supraventricular arrhythmia in clinical practice and is considered
to be a growing cardiovascular epidemic (1). Emerging evidence demonstrates that
aging alone does not account for the standing increase in AF
prevalence (2). Obesity has been
characterized as a new risk factor contributing to AF (3). Obesity is a global pand emic with
more than 2/3 adults being overweight or obese (4,5).
It is well-documented that a long-term high calorie intake and
sedentary lifestyle are the underlying causes of the high
prevalence of obesity (6,7). Exposure to a chronic high-fat diet
(HFD) is not only linked to diabetes, it also has a strong
association with cardiovascular disease and stroke (7–9).
Although analysis of numerous studies over the last decades has
displayed the effect of obesity on atrial structural (10–14) and gap junctional [mainly focused
on connexin 43 (Cx43) expressed and distributed in ventricles]
remodeling (15,16), the mechanisms remain incompletely
elucidated. Despite the unequivocal linkage between obesity and AF,
it remains currently unknown whether structural and gap junctional
remodeling following a chronic HFD feeding and/or increased AF
risks are prevalent in the absence of HFD-induced obesity.
Therefore, our study analyzed the structural and gap junctional
electrophysiological alterations in the atria of female rats fed a
consistent HFD for 12 weeks, determined the impact of HFD on the
susceptibility to AF, investigated whether this effect may be the
same in both HFD-induced obesity (HFD-OB) and HFD-fed non-obesity
(HFD-NOB) rats, and aimed at identifying the underlying
mechanisms.
Materials and methods
Experimental animals
The female Sprague-Dawley (SD) rats used in this
study were obtained from the Laboratory Animal Center of Xi'an
Jiaotong University (Xi'an, China). Rats were housed in an animal
research facility with a 12-h light/dark cycle at 20–25°C room
temperature and given free access to food and water. Ethical
approval of this study was obtained from the Ethics Committee of
Xi'an Jiaotong University. Animal experiments involved in this
study were performed according to the guidelines of Animal Handling
and Experimentation (Xi'an, China).
HFD feeding and grouping
HFD-fed rats (n=21) were provided with an HFD
consisting of 60% of calories from fat (Research Diets, Inc., New
Brunswick, NJ, USA; Laboratory Animal Center of Xi'an Jiaotong
University) starting at 6–8 weeks of age for 12 weeks. Control rats
(CT, n=20) were fed a normal diet (ND) consisting of 4.5% fat.
Weight measure was performed and recorded per week. After 12 weeks
of HFD feeding, HFD-fed rats were divided into HFD-OB and HFD-NOB
according to the levels of weight increase.
Plasma lipid measurements
Rats were anaesthetized with 10% chloral hydrate.
Right after abdomen opening, 5 to 10 ml of blood was drawn from the
abdominal aortae and collected with heparinized anticoagulation
tubes and immediately centrifuged at 15,000 rpm for 30 min at 4°C,
and then the plasma was stored at −80°C. Samples were sent to the
Clinical Labo ratory Department of the First Affiliated Hospital of
Xi'an Jiaotong University to test the levels of total cholesterol
(TC), high-density lipoprotein cholesterol (HDL-C), low-density
lipoprotein cholesterol (LDL-C) and triglycerides (TGs).
Tissue harvesting and processing
After piercing the left ventricle and carving the
right atrium, the heart was perfused with phosphate-buffered saline
(PBS) injected into the left ventricle, vessels were rinsed
subsequently and both atria were harvested after thoroughly
removing aorta and fat. For histological analysis, atrial tissue
was fixed in 4% paraformaldehyde dissolved in PBS, dehydrated and
embedded in paraffin. For immunofluorescence assay, atrial tissue
was washed with PBS, fixed in 4% paraformaldehyde, dehydrated with
sucrose, embedded in optimal cutting temperature (OCT) compound,
and then kept at −80°C for long-term storage. For biochemical
examinations, atrial tissue was transiently frozen in liquid
nitrogen and stored at −80°C.
Hematoxylin and eosin (H&E) and
Masson's staining
Processing of rat atrial tissue
paraformaldehyde-fixing and paraffin-embedding has been previously
described. Sections (5-μm thick) from each atrial tissue
were stained with H&E or Masson's staining, and then sealed and
stored at 4°C. Atrial fibrosis was assessed using Image-Pro Plus
6.0 analysis software (Media Cybernetics, Inc., Rockville, MD,
USA).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from rat atrial tissue using
the TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) in accordance with the manufacturer's instructions, and then
reverse transcribed using the RevertAid First Strand cDNA synthesis
kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) for 60 min
at 42°C and 5 min at 80°C. Quantitative PCR (qPCR) was performed
using the FastStart Universal SYBR-Green Master (Rox) (Roche
Diagnostics, Indianapolis, IN, USA) on the iQ5™ Multicolor
Real-Time PCR detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The thermocycling parameters of PCR were 10 min
at 95°C, 40 cycles at 95°C for 15 sec and 60°C for 60 sec, followed
by a melting curve stage. Each atrial sample was performed in
triplicate. The primer sequences used for rat atrial Cx40 were
forward, 5′-CAG GCA GAT TTC CAG TGT GAT-3′ and reverse, 5′-AGT AGC
GGA TGT GAG AGA TGG-3′; rat atrial Cx43 forward, 5′-GCT GTG TCC TTG
GTG TCT CTT G-3′ and reverse, 5′-ACA GCT TTT GGA GGG GCT CAG T-3′;
rat atrial transforming growth factor-β1 (TGF-β1) forward, 5′-AAC
CCA CAA CGA AAT CTA TGA CAA-3′ and reverse, 5′-AGA GCA ACA CGG GTT
CAG GTA-3′; and rat atrial matrix metalloproteinase-2 (MMP-2)
forward, 5′-CAG GCT CTT CTC CTT TCA CAA C-3′ and reverse, 5′-AAG
CCA CGG CTT GGT TTT CCT C-3′. Target mRNAs mentioned above were
normalized to the GAPDH mRNA level.
Western blotting
The rat atrial tissue was sufficiently ground and
lysed within a tissue homogenizer supplemented with RIPA buffer and
protease inhibitors (100:1), laid on ice for 15 min, and then
centrifuged at 15,000 rpm for 30 min at 4°C. Supernatant liquid
removed from each sample was kept. The total protein concentration
was determined using BCA protein assay reagent (Pierce, Rockford,
IL, USA). After being standardized by SDS-PAGE sample loading
buffer and boiled for 5 min, equivalent amounts (30 μg) of
protein extracted from each sample were separated onto a 10%
SDS-PAGE gel for electrophoresis. Then proteins were transferred
onto polyvinylidene difluoride (PVDF) membranes (Millipore,
Bedford, MA, USA) under the condition of 200 mA for 1 h. The
blotted membranes were incubated in blocking buffer (5% defatted
milk in TBS containing 0.1% Tween-20) for 1 h at room temperature
(RT). Then the blots were probed overnight at 4°C with the
following primary antibodies: anti-TGF-β1 (ab92486; Abcam,
Cambridge, UK), anti-Cx43 (3512S), anti-MMP-2 (4022S) (Cell
Signaling Technology Inc., Danvers, MA, USA), anti-Cx40 (sc-20466),
and anti-GAPDH (sc-25778) (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). After completion of all the procedures mentioned
above, incubation with goat-anti-rabbit horseradish peroxidase
(HRP)-conjugated IgG as the secondary antibody was carried out for
1 h at RT. The blot bands were visualized using ECL and quantified
using Quantity One analysis software (both from Bio-Rad
Laboratories, Inc.).
Immunofluorescence and TUNEL
staining
Cryosections (6-μm thick) of atrial tissue
were performed after paraformaldehyde-fixing, dehydrating and
OCT-embedding, washed with PBS for 5 min, 3 times, followed by
incubation overnight at 4°C with the primary antibodies as follows:
goat polyclonal anti-Cx40 (1:200 to 1:500; ab16585; Abcam), rabbit
polyclonal anti-Cx43 (1:400; 3512S; Cell Signaling Technology,
Inc.). Secondary antibodies involved in the present study were
fluorescein isothiocyanate (FITC)-conjugated AffiniPure goat
anti-rabbit and donkey anti-goat IgG (H+L) (1:200; ZSGB-Bio Co.,
Ltd., Beijing, China). Nuclei were stained at RT for 15 min using
4′-6-diamidino-2-phenylindole (DAPI; Bioworld Technology, Inc., St.
Louis Park, MN, USA). Cell death was assessed using a terminal
deoxynucleotidyl transferase-mediated dUTP nick end labelling
(TUNEL) kit (Roche Diagnostics, Rotkreuz, Switzerland).
Statistical analysis
Statistical data are expressed as mean ± SD. To
compare the results between different groups, an unpaired Student's
t-test or analysis of variance (ANOVA) with post hoc Dunnett's test
was used. A P-value <0.05 was considered statistically
significant.
Results
Female SD rats fed an HFD exhibit
differences in weight increase
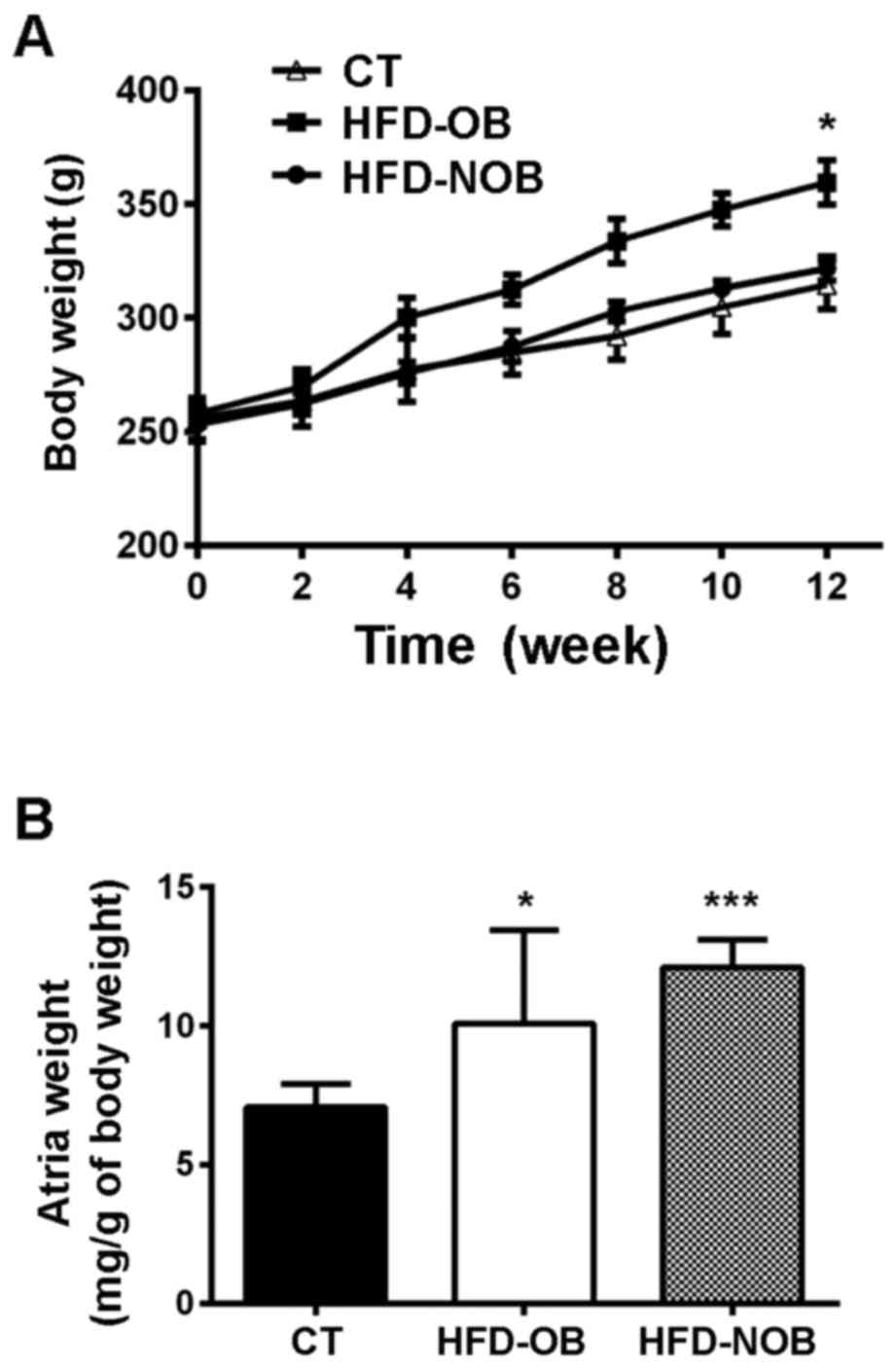
To determine the role of HFD in AF, we analyzed the
6- to 8-week-old SD rats fed an HFD or ND. The body weight showed
no significant difference among these groups until the 4th week.
After 12 weeks of feeding, only ~3/5 of the HFD-fed rats (HFD-OB,
n=13) displayed a significantly higher body weight increase than
the rest of the HFD-fed rats (HFD-NOB, n=8) and those fed with an
ND (CT, n=20) (equal as week 0; Fig.
1A). Furthermore, compared with the HFD-NOB rats, HFD-OB rats
were heavier which was not due to the differences in food intake
(data not shown). In comparison to the CT rats, no significant
difference in weight increase was shown in the NOB rats. Moreover,
the analysis revealed markedly increased atrial weight in the
HFD-fed rats (Fig. 1B). As shown
in Fig. 1B, there was a marked
increase in atrial weight in the HFD-NOB rats, as compared to that
noted in the HFD-OB rats.
Both HFD-OB and HFD-NOB rats exhibit
dyslipidemia
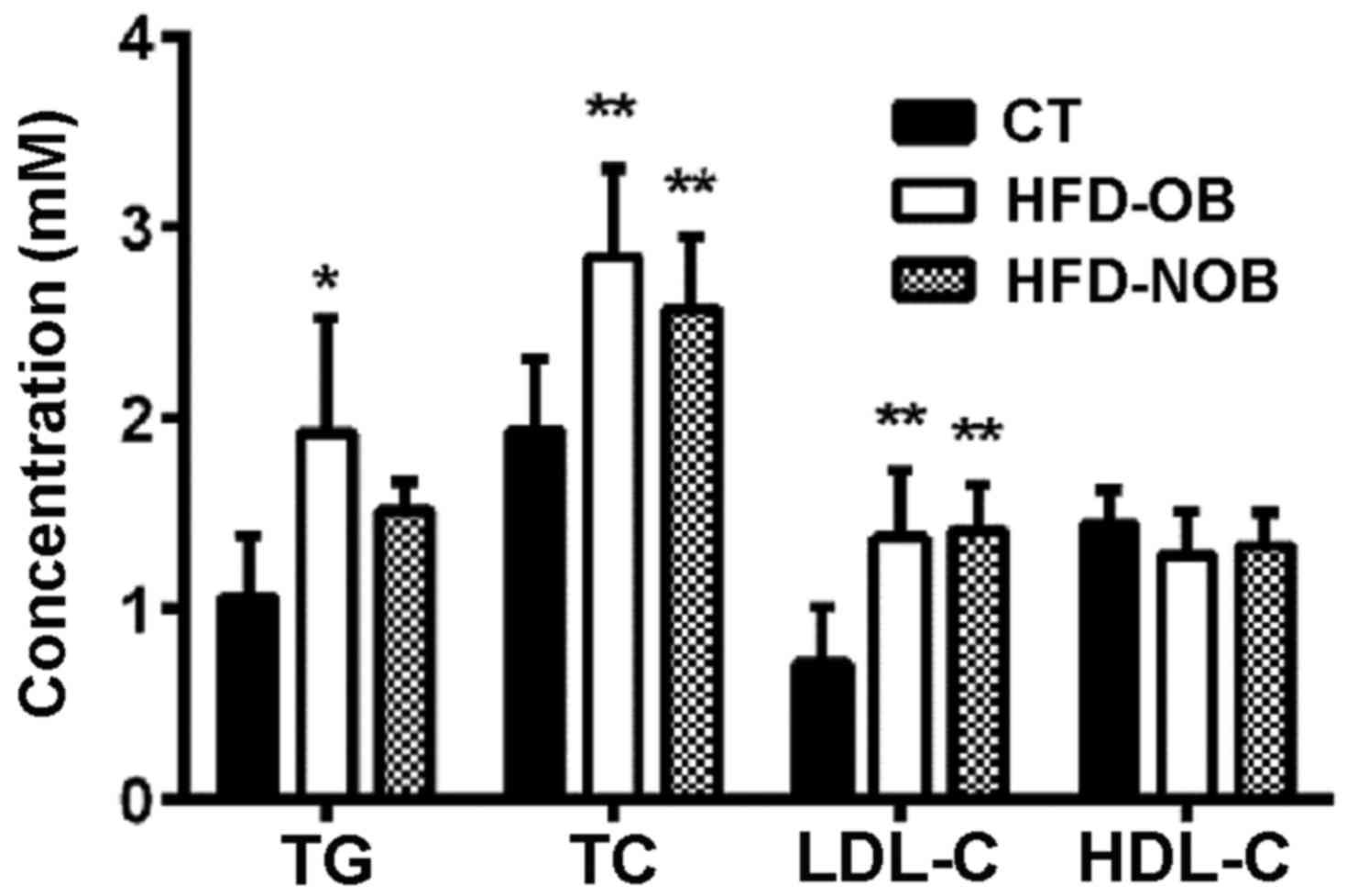
Metabolic disorders play a principal role in the
incidence, maintenance and recurrence of AF. To examine the status
of lipid metabolism in HFD-fed rats, we measured the plasma lipid
levels. HFD-NOB (n=8) rats displayed a marginal elevation in TG
level, as well as significantly upregulated levels of TC and LDL-C,
as compared to the CT rats (n=20) (Fig. 2). Although TG, TC and LDL-C levels
were all markedly increased in the HFD-OB rats (n=13), TG, a key
lipid metabolic factor, was not significantly elevated, suggesting
that not all factors participated in the alteration of the lipid
metabolism in the HFD-NOB rats. The HDL-C levels did not reveal any
difference among the three groups. The results indicated a
disturbed lipid homeostasis in the HFD-fed rats.
HFD-fed rats exhibit more extensive
atrial fibrosis
Atrial fibrosis is well described as an AF-promoting
condition and a potential predictor to recurrence (17). Tissue fibrosis is mediated by a
series of risk factors, such as age, sex, heart failure, stroke and
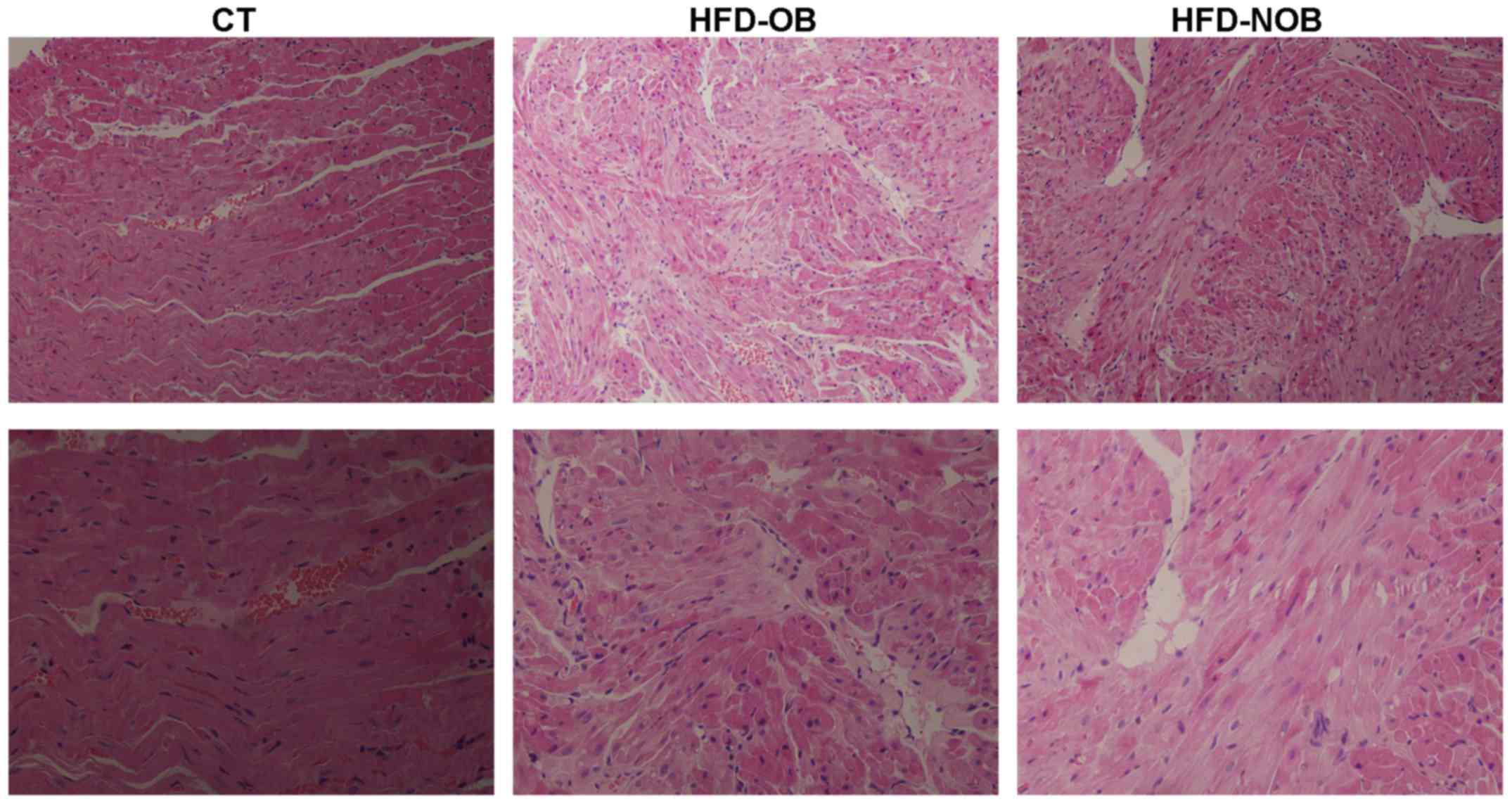
hypertension. We performed histological assay (H&E and Masson's
staining) to confirm the association between HFD and atrial
fibrosis. Indeed, the H&E staining of atrial paraffin sections
revealed more broadened interstitial space among the atrial
myofibers as well as distinctly disordered layout of atrial
myocytes in both the HFD-OB and HFD-NOB rats instead of a normal
myocardial structure (Fig. 3).
H&E staining of atrial tissue of the OB and NOB rats did not
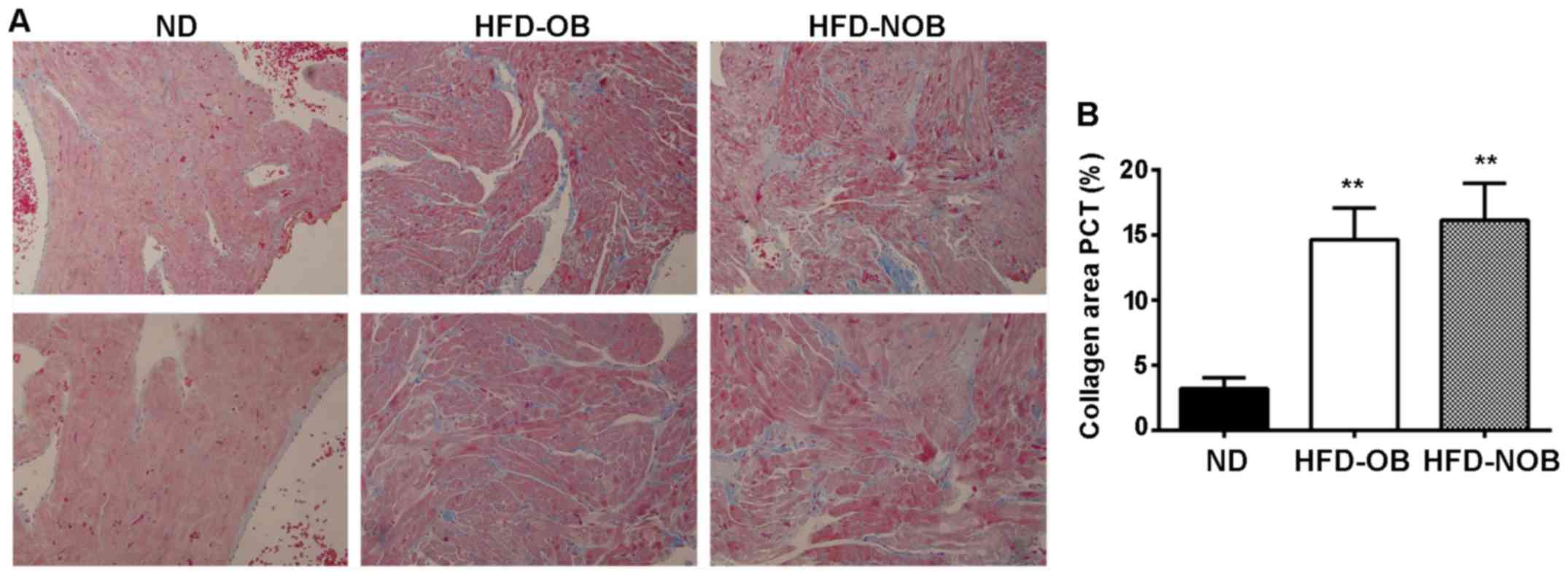
exhibit any difference. Significant fibrosis was shown in Masson's
staining of the HFD-OB and HFD-NOB rat atrial sections (Fig. 4). Extensive fibrosis was
particularly visible around atrial myocytes and was disorderly
distributed. In contrast, the tiny amounts of collagen fibers in
the CT rat atria were continuous and complete. As shown in Fig. 4B, compared with the HFD-OB rats,
the collagen area of the HFD-NOB rat atria was larger, while there
was no significant difference between these two groups. These
results above revealed that HFD may somehow induce and promote
atrial fibrosis. The impacts of HFD on atrial fibrosis exhibited in
the HFD-OB and HFD-NOB rats were similar.
HFD feeding upregulates expression of
mRNA and proteins related to atrial fibrosis
Previous studies demonstrated that TGF-β1, a
multifunctional cytokine participating in the regulation of cell
proliferation and differentiation, leads to atrial fibrosis and AF
(18). In addition, MMP-2,
playing a key role in extracellular matrix activity, promotes
atrial fibrosis and is upregulated during AF (19). We performed further biochemical
analyses to explore the association between HFD and atrial
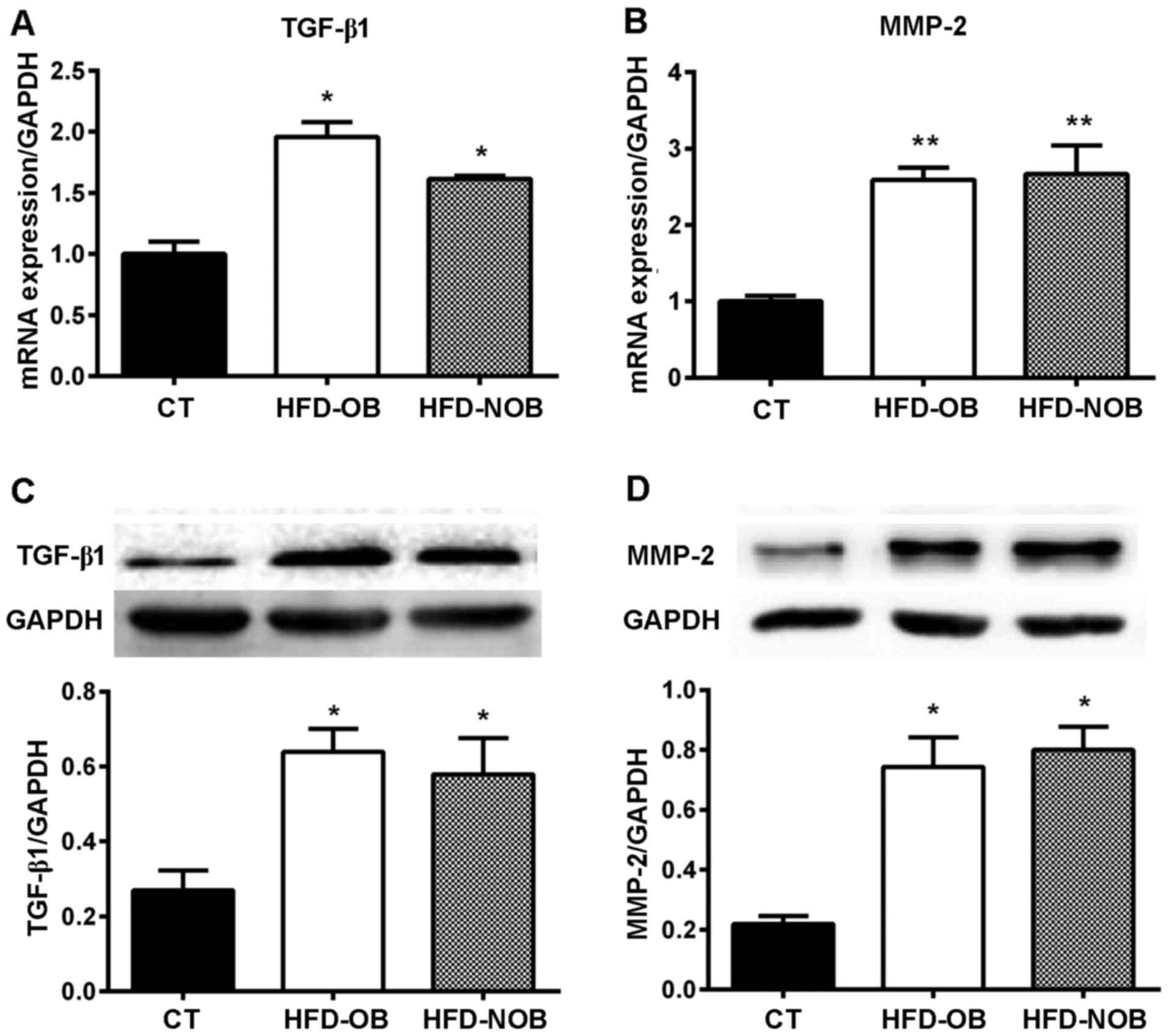
fibrosis. RT-qPCR analysis of TGF-β1 and MMP-2 mRNA revealed that
the expression of TGF-β1 and MMP-2 mRNA was significantly
upregulated in the HFD-OB and HFD-NOB rat atrial myocytes (Fig. 5A and B). No significant difference
was observed between these two groups. In accordance with the
results of the RT-qPCR analysis, western blotting bands showed a
significant increase in expression of TGF-β1 and MMP-2 protein in
the atria of rats fed an HFD as well (Fig. 5C and D). Similarly, the increases
showed no significant difference between the HFD-OB and HFD-NOB
groups. The marked increases in expression of TGF-β1 and MMP-2
proteins and mRNA further implied that HFD may induce tissue
fibrosis in the atria. In addition, the levels of atrial
fibrosis-promoting factors expressed in the HFD-OB and HFD-NOB rats
showed no significant difference.
HFD feeding downregulates expression of
gap junction mRNA and proteins
Abnormality in expression/distribution of gap
junction Cxs is one of the essential components of atrial
electrical remodeling. Cx40 and Cx43 are known to be the most
commonly expressed Cx types in atrial myocytes. Thus, for the
purpose of elucidating the potential mechanisms accounting for the
atrial remodeling-promoting effects of HFD, we measured the
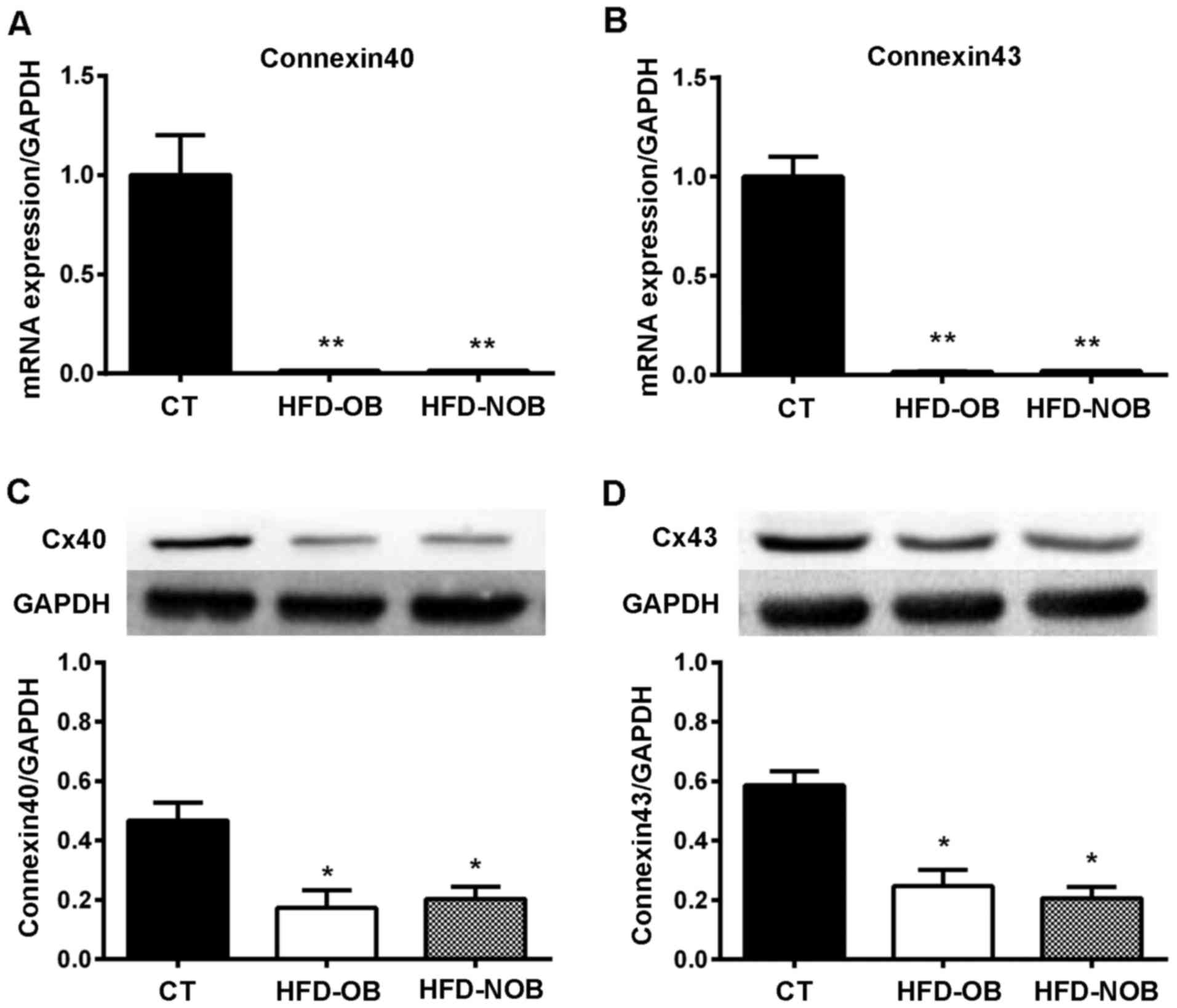
expression levels of Cx40 and Cx43 and their mRNA levels using
western blotting and RT-qPCR analysis respectively. RT-qPCR
analysis displayed a significant downregulation in the expression
of Cx40 and Cx43 mRNA in the HFD-fed rat atrial tissue (Fig. 6A and B). In addition, a similar
marked decrease in the protein expression of Cx40 and Cx43 was
observed by western blotting as well (Fig. 6C and D). Neither the RT-qPCR nor
the western blotting showed significant differences between the
HFD-OB and HFD-NOB groups. The altered expression levels of gap
junction Cxs suggest that an HFD could impact the electrical
remodeling to a certain extent, which may further provide the
substrate susceptible to AF.
HFD-fed rats show altered distribution of
gap junction in the atria
Previous studies have demonstrated an association
between the abnormal distribution of gap junction and
arrhythmogenesis (22,28). Further analysis using
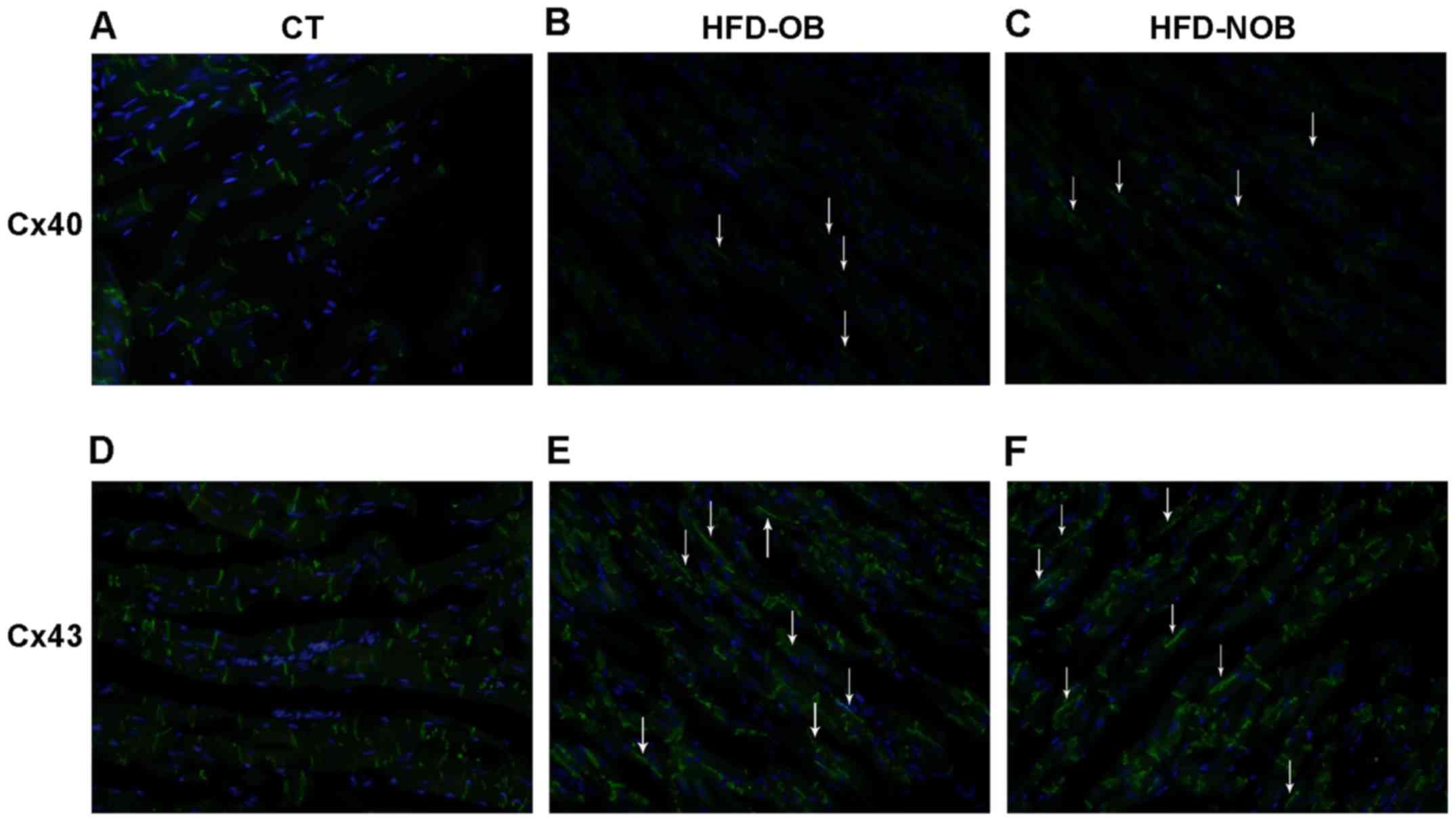
immunofluorescence exhibited altered expression and distribution of
Cx40 and Cx43 in atrial myocytes of the rats fed an HFD, as
compared to the ND-fed rats (Fig.
7). As shown in Fig. 7A and
D, Cx40 and Cx43 were mainly located at the intercalated discs
of atrial myocytes in the CT rats. Cx40 protein expression
displayed significant downregulation in the HFD rat atrial myocytes
via western blotting, supporting the results of the
immunofluorescence approach (Fig. 7B
and C). In addition, Cx40 was showed to be laterally
distributed along the longitudinal atrial myocyte membranes. Cx43
was markedly downregulated and laterally distributed in the HFD-OB
rat atria as well (Fig. 7E).
Nevertheless, in comparison to the CT rats, Cx43 signals seemingly
did not show a significant decrease in the HFD-NOB rats (Fig. 7F). Although lateralized
distribution of Cx43 was still observed in the HFD-NOB rat atrial
myocytes, these alterations in expression and distribution of gap
junction revealed that it may be more susceptible to AF via
HFD-induced Cx remodeling. In addition, HFD-OB and HFD-NOB rats
showed similar levels of gap junctional remodeling induced by
HFD.
HFD rat atrial tissue displays a tendency
of increased apoptotic cell death
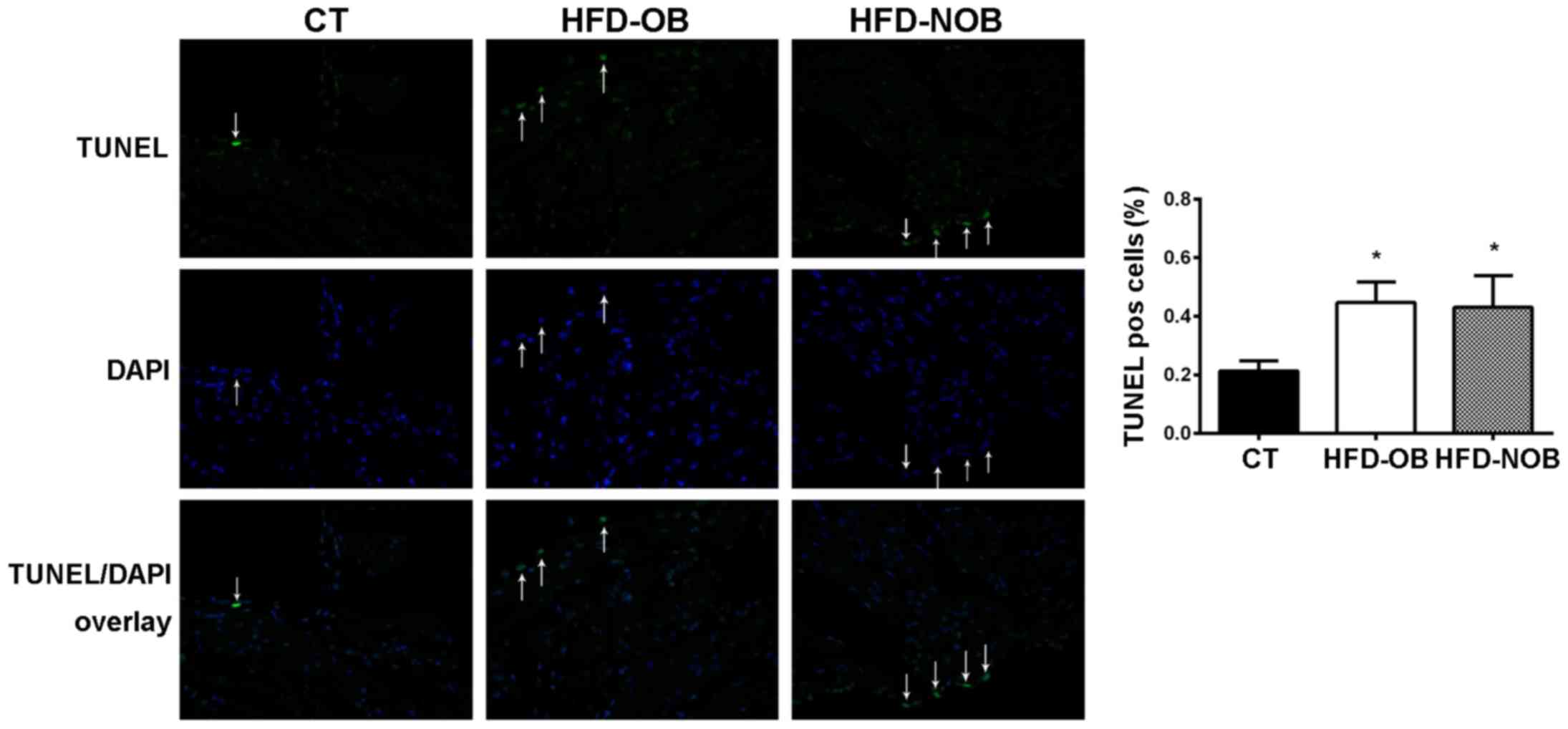
To highlight the effects of HFD on the atrial
substrate, we therefore quantified cell death in serial sections of
atrial tissue in the ND and HFD rats. Cell death was markedly lower
in the rats fed an ND, whereas there was no significant difference
observed between the HFD-OB and HFD-NOB rats (Fig. 8).
Taken together, these data demonstrated that whether
or not HFD induces obesity, HFD may affect lipid metabolism, alter
the expression of mRNA and proteins involved in atrial fibrosis and
promote the remodeling of gap junction in rat atrial tissue. The
impact of HFD on the atrial substrate may represent the underlying
cause for the incidence, maintenance and recurrence of AF.
Discussion
In the present study, 6- to 8-week-old female SD
rats were fed an HFD for 12 weeks, and subsequent atrial fibrosis
and gap junction remodeling were analyzed. Large numbers of
previous studies have reported significant increases in the body
weight of HFD-fed rats (20,21). However, our findings showed that
only approximately 3/5 of the HFD-fed rats were more susceptible to
HFD-induced obesity, while the other 2/5 were not (Fig. 1A). We considered that the
distinction in body weight increases may be due to the genetic
variations in female SD rats. Although the markedly disturbed lipid
homeostasis was observed in both HFD-OB and HFD-NOB rats (there
were some distinctions in the elevations of factors involved in
plasma lipid metabolism; Fig. 2),
the different alterations in body weight increases and plasma lipid
levels in the HFD-fed rats may partially explain why a certain
incidence of cardiovascular diseases and hyperlipidemia exists in
numerous individuals that are not overweight or obese as
encountered in clinical practice. The changes in lifestyle
(especially long-term high caloric intake), rather than only
nutritional obesity, may be a major cause for AF incidence.
Along with tissue fibrosis, atrial enlargement is
one of the two components of atrial structural remodeling (22). Atrial dimension has been
demonstrated to be a determinant factor of the persistence of AF
maintaining re-entry (23). In
the present study, although no significant increase in body weight
was observed in the HFD-NOB rats, the atrial weight was
significantly higher (Fig. 1B).
To some extent, this finding also suggested that chronic HFD
feeding may lead to atrial enlargement which made the rats more
susceptible to AF. In addition, distinctly broadened interstitial
space among atrial myofibers induced by chronic HFD feeding
(Fig. 3) may have contributed to
promotion of the atrial enlargement. It has been confirmed in a
previous study that tissue fibrosis could promote AF by
interrupting atrial myocardium continuity and disturbing local
conduction (24). Moreover, the
interactions between fibroblasts and cardiomyocytes may give rise
to changes in cardiomyocyte bioelectricity (25). Atrial fibrosis appears to be a
common endpoint for a variety of AF-promoting elements (17). It has been acknowledged that
TGF-β1 is the key regulating factor of atrial fibrosis (18). It causes atrial fibrosis by
activating downstream signaling pathways (26), and furthermore promotes the
incidence and maintenance of AF. Moreover, MMP-2 appears to be
another key factor involved in interstitial tissue alteration in
diseased atrial myocardium (19,27). MMP-2 not only plays an important
role in the degradation of the matrix, but also regulates the
synthesis of collagen. Normal collagen is degraded by increased
MMP-2, while it is replaced by fibrous interstitial that lacks
connective structure. Moreover, the altered MMP/TIMP equilibrium
could result in a loss of control of MMP-2 activity and thereby
contribute to an increase in MMP-2 activity in diseased atria.
Finally, the fibrotic response strengthens as the MMP-2 levels
increase. We analyzed Masson's staining, RT-qPCR and western
blotting data to determine whether HFD induces atrial fibrosis in
rats. In the present study, chronic HFD feeding increased fibrotic
atrial myocardium (Fig. 4),
upregulated expression of TGF-β1 and MMP-2 protein and mRNA
(Fig. 5), and these results were
in accordance with the studies mentioned above. Upregulation in the
expression of fibrosis-relevant factors revealed that the HFD
induced marked fibrosis which may create the atrial substrate for
re-entry. All these findings indicated that exposure to chronic HFD
may promote AF by impacting both of the components of atrial
structural remodeling, i.e., atrial enlargement and tissue
fibrosis.
The essential role of gap junction in atrial
conduction has been clearly described. Numerous studies in AF
patients (28) and rapid pacing
animal models (29,30) have revealed the strong association
between abnormal expression and heterogeneous distribution of Cxs
and AF. Cx40 and Cx43, which mediate cardiomyocyte-to-cardiomyocyte
electrical coupling, have been characterized as the major Cxs
expressed in murine atria (31).
Several animal studies (15,16,32) displayed abnormality in expression
and distribution of Cx43 in atria and ventricles following HFD
feeding. Nevertheless, there are few studies elucidating the
changes in expression and distribution of Cx40 in atria following
HFD feeding. In our study, regardless of whether HFD feeding
induced obesity, a significant decrease in expression of Cx40 and
Cx43 protein and mRNA were observed in the HFD-fed rats (Fig. 6). Although the expression of Cxs
in cardiac myocytes from animals fed an HFD have been studied
(15,16,32), discrepancies existed in these
results. Notably, similarly in the present study, we found that
Cx40 and Cx43 protein expression was distinctly upregulated in 1/5
of the OB rats and in 1/8 of the NOB rats which were opposite with
the total expression levels (no significant difference was
displayed between these two groups). However, no alteration in Cx40
and Cx43 expression was observed in any of the HFD rats. This
finding may also support the fact that chronic HFD feeding impacts
gap junction remodeling in atria. In addition, our analysis showed
that Cx40 and Cx43 were more localized along longitudinal atrial
myocyte membranes rather than the intercalated discs (Fig. 7). Spatial heterogeneities in
distributions of Cxs may disturb the atrial conduction via creating
microscopic obstacles altering the atrial action potential
(33), which then gives rise to
the incidence of AF. It therefore appears that HFD feeding may
promote gap junction remodeling in atria, affect atrial myocyte
action potential and atrial anisotropy conduction, thereby causing
AF.
Apart from the AF-promoting conditions mentioned
above, apoptosis cell death may be another potential mechanism
contributing to AF by participating in atrial remodeling (34,35). The effect of HFD-induced obesity
on apoptosis in ventricles has been well documented (20,21,36). In accordance with these results,
we found a robust increase in cell death in both HFD-OB and HFD-NOB
rat atria as detected by TUNEL assay (Fig. 8). The increase in the numbers of
TUNEL-positive nuclei exhibited no difference in these two groups.
We spec ulate that the alteration in cell apoptosis occurs in
atrial myocytes of HFD-fed rats in the absence of obesity as well,
thereby enhancing the atrial remodeling which may promote the
incidence of AF.
However, there are several limitations to the
present study. First, we measured atrial structure and gap junction
remodeling, which are considered to be the main causes of AF.
Although we determined the susceptibility to AF caused by chronic
HFD, the incidence of AF was not examined. We may further
investigate this aspect in the future. Second, the detailed
mechanisms and the crosstalk between TGF-β1, MMP-2 and gap
junctions need further investigation.
We conclude that, regardless of the presence of
HFD-induced obesity, an HFD promotes expression of factors involved
in atrial fibrosis, altering the expression and distribution of Cxs
in the atria, and inducing atrial myocyte apoptosis, which may
further contribute to the incidence and development of AF. Thus,
leading a healthy lifestyle by controlling calorie intake may be a
promising therapeutic strategy for AF.
Acknowledgments
This study was supported by the Clinical Research
Award of the First Affiliated Hospital of Xi'an Jiaotong
University, Xi'an, China (no. XJTU1AF-CRF-2015-007).
References
|
1
|
Braunwald E: Shattuck lecture -
cardiovascular medicine at the turn of the millennium: triumphs,
concerns, and opportunities. N Engl J Med. 337:1360–1369. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS,
Bailey KR, Abhayaratna WP, Seward JB and Tsang TSM: Secular trends
in incidence of atrial fibrillation in Olmsted county, Minnesota,
1980 to 2000, and implications on the projections for future
prevalence. Circulation. 114:119–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chamberlain AM, Agarwal SK, Ambrose M,
Folsom AR, Soliman EZ and Alonso A: Metabolic syndrome and
incidence of atrial fibrillation among blacks and whites in the
Atherosclerosis Risk in Communities (ARIC) study. Am Heart J.
159:850–856. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ogden CL, Carroll MD, Kit BK and Flegal
KM: Prevalence of childhood and adult obesity in the United States,
2011–2012. JAMA. 311:806–814. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng M, Fleming T, Robinson M, Thomson B,
Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF,
et al: Global, regional, and national prevalence of overweight and
obesity in children and adults during 1980–2013: a systematic
analysis for the Global Burden of Disease study 2013. Lancet.
384:766–781. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitchell S and Shaw D: The worldwide
epidemic of female obesity. Best Pract Res Clin Obstet Gynaecol.
29:289–299. 2015. View Article : Google Scholar
|
|
7
|
Hill JO and Peters JC: Environmental
contributions to the obesity epidemic. Science. 280:1371–1374.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Flegal KM, Graubard BI, Williamson DF and
Gail MH: Excess deaths associated with underweight, overweight, and
obesity. JAMA. 293:1861–1867. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kahn SE, Hull RL and Utzschneider KM:
Mechanisms linking obesity to insulin resistance and type 2
diabetes. Nature. 444:840–846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di Salvo G, Pacileo G, Del Giudice EM,
Natale F, Limongelli G, Verrengia M, Rea A, Fratta F, Castaldi B,
Gala S, et al: Atrial myocardial deformation properties in obese
nonhypertensive children. J Am Soc Echocardiogr. 21:151–156. 2008.
View Article : Google Scholar
|
|
11
|
Wong CY, O'Moore-Sullivan T, Leano R,
Byrne N, Beller E and Marwick TH: Alterations of left ventricular
myocardial characteristics associated with obesity. Circulation.
110:3081–3087. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang TJ, Parise H, Levy D, D'Agostino RB
Sr, Wolf PA, Vasan RS and Benjamin EJ: Obesity and the risk of
new-onset atrial fibrillation. JAMA. 292:2471–2477. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abed HS, Samuel CS, Lau DH, Kelly DJ,
Royce SG, Alasady M, Mahajan R, Kuklik P, Zhang Y, Brooks AG, et
al: Obesity results in progressive atrial structural and electrical
remodeling: implications for atrial fibrillation. Heart Rhythm.
10:90–100. 2013. View Article : Google Scholar
|
|
14
|
Mahajan R, Lau DH and Sanders P: Impact of
obesity on cardiac metabolism, fibrosis, and function. Trends
Cardiovasc Med. 25:119–126. 2015. View Article : Google Scholar
|
|
15
|
Aubin MC, Cardin S, Comtois P, Clément R,
Gosselin H, Gillis MA, Le Quang K, Nattel S, Perrault LP and
Calderone A: A high-fat diet increases risk of ventricular
arrhythmia in female rats: enhanced arrhythmic risk in the absence
of obesity or hyperlipidemia. J Appl Physiol. 108:933–940. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Axelsen LN, Calloe K, Braunstein TH,
Riemann M, Hofgaard JP, Liang B, Jensen CF, Olsen KB, Bartels ED,
Baandrup U, et al: Diet-induced pre-diabetes slows cardiac
conductance and promotes arrhythmogenesis. Cardiovasc Diabetol.
14:872015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oakes RS, Badger TJ, Kholmovski EG, Akoum
N, Burgon NS, Fish EN, Blauer JJE, Rao SN, DiBella EVR, Segerson
NM, et al: Detection and quantification of left atrial structural
remodeling with delayed-enhancement magnetic resonance imaging in
patients with atrial fibrillation. Circulation. 119:1758–1767.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verheule S, Sato T, Everett T IV, Engle
SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field
LJ and Olgin JE: Increased vulnerability to atrial fibrillation in
transgenic mice with selective atrial fibrosis caused by
overexpression of TGF-beta1. Circ Res. 94:1458–1465. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boixel C, Fontaine V, Rücker-Martin C,
Milliez P, Louedec L, Michel JB, Jacob MP and Hatem SN: Fibrosis of
the left atria during progression of heart failure is associated
with increased matrix metalloproteinases in the rat. J Am Coll
Cardiol. 42:336–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dhahri W, Drolet MC, Roussel E, Couet J
and Arsenault M: Chronic high-fat diet-induced obesity decreased
survival and increased hypertrophy of rats with experimental
eccentric hypertrophy from chronic aortic regurgitation. BMC
Cardiovasc Disord. 14:1232014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang HF, Lin PP, Chen CH, Yeh YL, Huang
CC, Huang CY and Tsai CC: Effects of lactic acid bacteria on
cardiac apoptosis are mediated by activation of the
phosphatidylinositol-3 kinase/AKT survival-signalling pathway in
rats fed a high-fat diet. Int J Mol Med. 35:460–470. 2015.
|
|
22
|
Nattel S and Harada M: Atrial remodeling
and atrial fibrillation: recent advances and translational
perspectives. J Am Coll Cardiol. 63:2335–2345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zou R, Kneller J, Leon LJ and Nattel S:
Substrate size as a determinant of fibrillatory activity
maintenance in a mathematical model of canine atrium. Am J Physiol
Heart Circ Physiol. 289:H1002–H1012. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Burstein B, Comtois P, Michael G, Nishida
K, Villeneuve L, Yeh YH and Nattel S: Changes in connexin
expression and the atrial fibrillation substrate in congestive
heart failure. Circ Res. 105:1213–1222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue L, Xie J and Nattel S: Molecular
determinants of cardiac fibroblast electrical function and
therapeutic implications for atrial fibrillation. Cardiovasc Res.
89:744–753. 2011. View Article : Google Scholar :
|
|
26
|
Rahmutula D, Marcus GM, Wilson EE, Ding
CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ and Olgin JE:
Molecular basis of selective atrial fibrosis due to overexpression
of transforming growth factor-β1. Cardiovasc Res. 99:769–779. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosenkranz S: TGF-beta1 and angiotensin
networking in cardiac remodeling. Cardiovasc Res. 63:423–432. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Patel P, Jones D and Dupont E: Remodeling
of human connexin43 expression in human atrial fibrillation. Eur
Heart J. 19:4652001.
|
|
29
|
Elvan A, Huang XD, Pressler ML and Zipes
DP: Radiofrequency catheter ablation of the atria eliminates
pacing-induced sustained atrial fibrillation and reduces connexin
43 in dogs. Circulation. 96:1675–1685. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van der Velden HM, Ausma J, Rook MB,
Hellemons AJ, van Veen TA, Allessie MA and Jongsma HJ: Gap
junctional remodeling in relation to stabilization of atrial
fibrillation in the goat. Cardiovasc Res. 46:476–486. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Delorme B, Dahl E, Jarry-Guichard T,
Briand JP, Willecke K, Gros D and Théveniau-Ruissy M: Expression
pattern of connexin gene products at the early developmental stages
of the mouse cardiovascular system. Circ Res. 81:423–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mahajan R, Brooks A, Manavis J, Wood J,
Finnie J, Sameul C, Lau D, Selvanayagam J, Roberts-Thomson K and
Sanders P: Atrial fibrillation and obesity: impact of weight
reduction on the atrial substrate. Heart Lung Circ. 22:S1–S2. 2013.
View Article : Google Scholar
|
|
33
|
Allessie M, Ausma J and Schotten U:
Electrical, contractile and structural remodeling during atrial
fibrillation. Cardiovasc Res. 54:230–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gasparovic H, Cikes M, Kopjar T, Hlupic L,
Velagic V, Milicic D, Bijnens B, Colak Z and Biočina B: Atrial
apoptosis and fibrosis adversely affect atrial conduit, reservoir
and contractile functions. Interact Cardiovasc Thorac Surg.
19:223–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu G, Cao Y, Lu J, Li J, Liu L, Wang H, Su
F and Zheng Q: Programmed cell death-1 deficiency results in atrial
remodeling in C57BL/6 mice. Int J Mol Med. 31:423–429. 2013.
|
|
36
|
Shi Z, Fu F, Yu L, Xing W, Su F, Liang X,
Tie R, Ji L, Zhu M, Yu J, et al: Vasonatrin peptide attenuates
myocardial ischemia-reperfusion injury in diabetic rats and
underlying mechanisms. Am J Physiol Heart Circ Physiol.
308:H281–H290. 2015. View Article : Google Scholar
|