Introduction
Accumulating evidence indicates that hyperglycemia
is recognized as the most important factor inducing almost all
cardiovascular complications associated with chronic diabetes, such
as diabetic cardiomyopathy (DCM) (1–4).
Multiple factors have been demonstrated to participate in
hyperglycemia-induced cardiac injury, such as reactive oxygen
species (ROS) generation (3–5),
apoptosis (3,6–8)
and the activity of several signaling molecules, including
mitogen-activated protein kinase (MAPK) (3,4,6,9),
p53 (7,10) and nuclear factor-κB (NF-κB)
(11,12). In addition, inflammation is also
involved in high glucose (HG)-induced cardiomyocyte injury
(13). More recently, we
indicated that the NF-κB and interleukin-1β (IL-1β) pathways are
implicated in the HG-elicited injury and inflammation in H9c2
cardiac cells (13). However, the
mechanisms responsible for the deteriorative effects of
hyperglycemia on cardiomyocytes are complex, and are not yet fully
understood. Thus, to explore the detailed mechanisms underlying
hyperglycemia-induced cardiomyocyte injury is important for the
prevention and treatment of diabetic cardiovascular
complications.
Recently, a novel mechanism known as 'programmed
necrosis' or necroptosis has been considered as another important
mediator of cell death in the heart (14). Similar to apoptosis, necroptosis
is tightly regulated by distinct molecules, but leads to the
typical morphological characteristics of necrosis, such as defects
in membrane integrity and inflammation, thus combining the features
of both mechanisms (14–16). In vitro studies have
indicated that the tumor necrosis factor-α (TNF-α)-dependent
formation of a complex between receptor-interacting protein (RIP)1
and another kinase, RIP3 is an essential step for inducing
necroptosis (15,17,18). In this process, RIP3 appears to
play an important role, controlling RIP1 phosphorylation, a
necessary step in necroptosis (15,18).
Increasing evidence has demonstrated that
necroptosis is involved in a number of pathological processes in
cardiovascular diseases (19–27). In hearts affected by
ischemia/reperfusion (I/R), RIP1 and RIP3 expression and
phosphorylation have been shown to be increased, and the
necroptosis inhibitor, necrostatin-1 (Nec-1) reduces the infarct
size (23–25). RIP3 expression has also been shown
to be enhanced in hearts affected by ischemia and RIP3 deficiency
protects mouse heart function (26). In addition, Luedde et al
revealed that RIP3 mediates the inflammatory response in mice with
myocardial infarction (26).
Collectively, the above-mentioned studies suggest that necroptosis
is implicated in ischemic cardiac lesions (23–26) and inflammation (27). However, the exact role of
necroptosis in diabetic cardiac injury and inflammation remains
unclear.
ROS are highly reactive molecules that have been
considered to function both as second messengers of TNF-α-elicited
cell death and modulators of signaling pathways (28,29). Since both ROS and necroptosis have
been reported to be involved in cell death and inflammation, the
interaction between ROS and necroptosis has recently attracted
attention. Classically, the execution of necroptosis is believed to
involve the generation of ROS and mitochondrial dysfunction
(30,31). On the other hand, RIP3 has been
demonstrated to be a key regulator in energy metabolism-associated
ROS generation, which partially accounts for the ability of RIP3 to
promote necrosis (16,32). In addition, RIP3 has been
repeatedly reported to regulate ROS production (18,26,33). Of note, a more recent study
demonstrated that in BV6/TNF-α-treated Jurkat cells, there was a
positive interaction between necroptosis and ROS, as on the one
hand, radical scavengers reduced necroptosis, but on the other
hand, ROS generation was decreased by the knockdown of RIP1 or RIP3
(34). Since both necroptosis and
ROS play critical roles in cell death in the heart (14), it would of interest to explore
whether there is a positive interaction between necroptosis and ROS
in hyperglycemia-induced cardiac injury and inflammation in order
to provide a novel mechanistic explanation for diabetic cardiac
lesions.
In this study, we report that HG induces
necroptosis-dependent cardiac injury and inflammation. Furthermore,
there was a positive interaction between necroptosis and ROS
generation, which plays important roles in HG-induced injury and
inflammation in H9c2 cardiac cells.
Materials and methods
Materials
Anti-RIP3 antibody (cat. no. ab152130) was purchased
from Abcam (Cambridge, MA, USA); anti-GAPDH antibody (cat. no.
10494-1-AP) was purchased from Proteintech Group, Inc. (Wuhan,
China). Dulbecco's modified Eagle's medium (DMEM) medium and fetal
bovine serum (FBS) were purchased from Gibco-BRL (Grand Island, NY,
USA). The BCA protein quantification kit and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit secondary antibody were obtained
from KangChen Bio-tech (Shanghai, China). N-acetyl-L-cysteine
(NAC), rhodamine 123 (Rh123), Nec-1 and 2′,7′-dichlorofluorescein
diacetate (DCFH-DA) were obtained from Sigma-Aldrich (St. Louis,
MO, USA). Enhanced chemiluminescence (ECL) solution was purchased
from KeyGen Biotech Co., Ltd. (Nanjing, China). The Cell Counting
Kit-8 (CCK-8) was offered by Dojindo Laboratories (Kumamoto,
Japan). IL-1β and TNF-α enzyme-linked immunosorbent assay (ELISA)
kits were purchased from Cusabio Biotech Co., Ltd. (Wuhan, China).
The H9c2 cardiac cells were supplied by the Sun Yatsen University
Experimental Animal Center (Guangzhou, China).
Cell culture and treatments
H9c2 cardiac cells, derived from rat embryonic
ventricular cardiomyocytes, were maintained in DMEM, supplemented
with 10% FBS in a humidified incubator with 95% air and 5%
CO2 at 37°C. The culture medium was replaced with fresh
medium every 2–3 days. When the cells grew to approximately 80%
confluency, they were expanded to new culture plates.
In the control group, H9c2 cardiac cells were
incubated with 5.5 mM glucose. To observe the effects of glucose at
35 mM glucose (HG) on the expression level of RIP3, the cells were
exposed to HG for 3, 6, 9, 12 and 24 h (Fig. 1). In order to examine the effect
of necroptosis on HG-induced injury, the H9c2 cells were
co-processed with different concentrations (75, 100, 200, 400, 600
and 800 µM) of Nec-1 (a specific inhibitor of necroptosis)
and HG for 24 h. To determine whether there was an interaction
between necroptosis and ROS, the H9c2 cells were treated with 1 mM
NAC (a scavenger of ROS) for 60 min prior to HG exposure.
Western blot analysis
After being subjected to the indicated treatments,
the H9c2 cardiac cells were harvested and lysed with RIPA buffer
containing 1 mM phenylmethanesulfonyl fluoride (PMSF) at 4°C for 30
min. The protein concentration was determined using the BCA protein
quantification kit. Loading buffer was added to the cytosolic
extracts and after boiling for approximately 5 min, equal amounts
of supernatant from each sample were subjected to 10% sodium
dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). The
separated proteins were transferred onto polyvinylidene difluoride
(PVDF) membranes followed by the blocking of the membranes with
fresh blocking buffer [0.1% Tween-20 in Tris-buffered saline
(TBS-T) containing 5% fat-free milk] for approximately 90 min at
room temperature. The membranes were then incubated with either
anti-RIP3 or anti-GAPDH (1:1,000 dilution) antibody in freshly
prepared TBS-T with 3% fat-free milk overnight with slow agitation
at 4°C temperature. Following 3 washes with TBS-T, the membranes
were incubated with HRP-conjugated goat anti-rabbit secondary
antibody (1:2,500 dilution) in TBS-T with 3% fat-free milk for 90
min at room temperature. The membranes were then washed 3 times
with TBS-T solution for 15 min. The immunoreactive signals were
visualized by using ECL detection. In order to quantify the protein
expression, the X-ray films were scanned and analyzed using ImageJ
1.47i software. The experiment was repeated 5 times.
Cell viability assay
CCK-8 assay was applied to detect the viability of
the cells. The H9c2 cells were digested and seeded in a 96-well
growth-medium plate at a concentration of 1×104 cells/ml
and incubated at 37°C. After the indicated treatments, the cells
were washed twice with phosphate-buffered saline (PBS).
Subsequently, 10 µl CCK-8 test solution and 90 µl
DMEM were added to each well, and the cells were incubated at 37°C
for 2.5 h. The absorbance value (OD value) at the 450 nm wavelength
was measured using a microplate reader (Molecular Devices,
Sunnyvale, CA, USA). The means of the optical density (OD) of 3
wells in the indicated groups were used to calculate the percentage
of cellular activity according to the following formula: cell
viability (%) = (ODtreatment group/ODcontrol
group) ×100%. The experiment was repeated 5 times.
Measurement of the secretion levels of
IL-1β and TNF-α by ELISA
The H9c2 cells were seeded in 96-well growth-medium
plates. After the indicated treatments, the levels of IL-1β and
TNF-α in the culture supernatant were evaluated by ELISA according
to the manufacturer's instructions (Cusabio Biotech Co., Ltd.). The
experiment was performed 5 times.
Measurement of the intracellular ROS
level
The intracellular level of ROS was detected using
the redox-sensitive fluorescent dye, DCFH-DA. Briefly, the culture
medium was removed and the cells were washed 3 times with PBS. The
cells were incubated with DCFH-DA (10 µM) which was diluted
by serum-free medium at 37°C during the last 20 min. The cells were
then washed 5 times with PBS and the relative amount of fluorescent
product was captured using a fluorescence microscope connected to
an imaging system (BX50-FLA; Olympus, Tokyo, Japan). ImageJ 1.47i
software was applied to analyze the mean fluorescence intensity
(MFI) of DCFH-DA, which indirectly showed the level of cell ROS.
The experiment was carried out 5 times.
Measurement of mitochondrial membrane
potential (MMP)
MMP was assessed using a fluorescent dye, Rh123, an
indicator of mitochondrial polarization that preferentially enters
the mitochondria based on the highly negative MMP. The
depolarization of MMP leads to the loss of Rh123 from the
mitochondria and a decrease in intracellular green fluorescence.
The H9c2 cardiac cells were plated in 24-well plates. After the
indicated treatments, the cells were washed 3 times with PBS. The
H9c2 cells were incubated with 1 µM Rh123 at 37°C for 30 min
in an incubator and washed 3 times with PBS. The Rh123 fluorescence
was then measured over the entire field of vision using a
fluorescence microscope connected to an imaging system (BX50-FLA;
Olympus). The MFI of Rh123 from 5 random fields was analyzed using
ImageJ 1.47i software and was regard as an index of the level of
MMP. The experiment was carried out 5 times.
Statistical analysis
All data are expressed as the means ± SEM.
Differences between groups were determined by one-way analysis of
variance (ANOVA) using SPSS 13.0 software (SPSS, Inc., Chicago, IL,
USA) followed by the least significant difference (LSD) post hoc
comparison test. Differences were considered statistically
significant at a P-value <0.05.
Results
Inhibitor of necroptosis attenuates the
HG-induced upregulation of RIP3 expression in H9c2 cardiac
cells
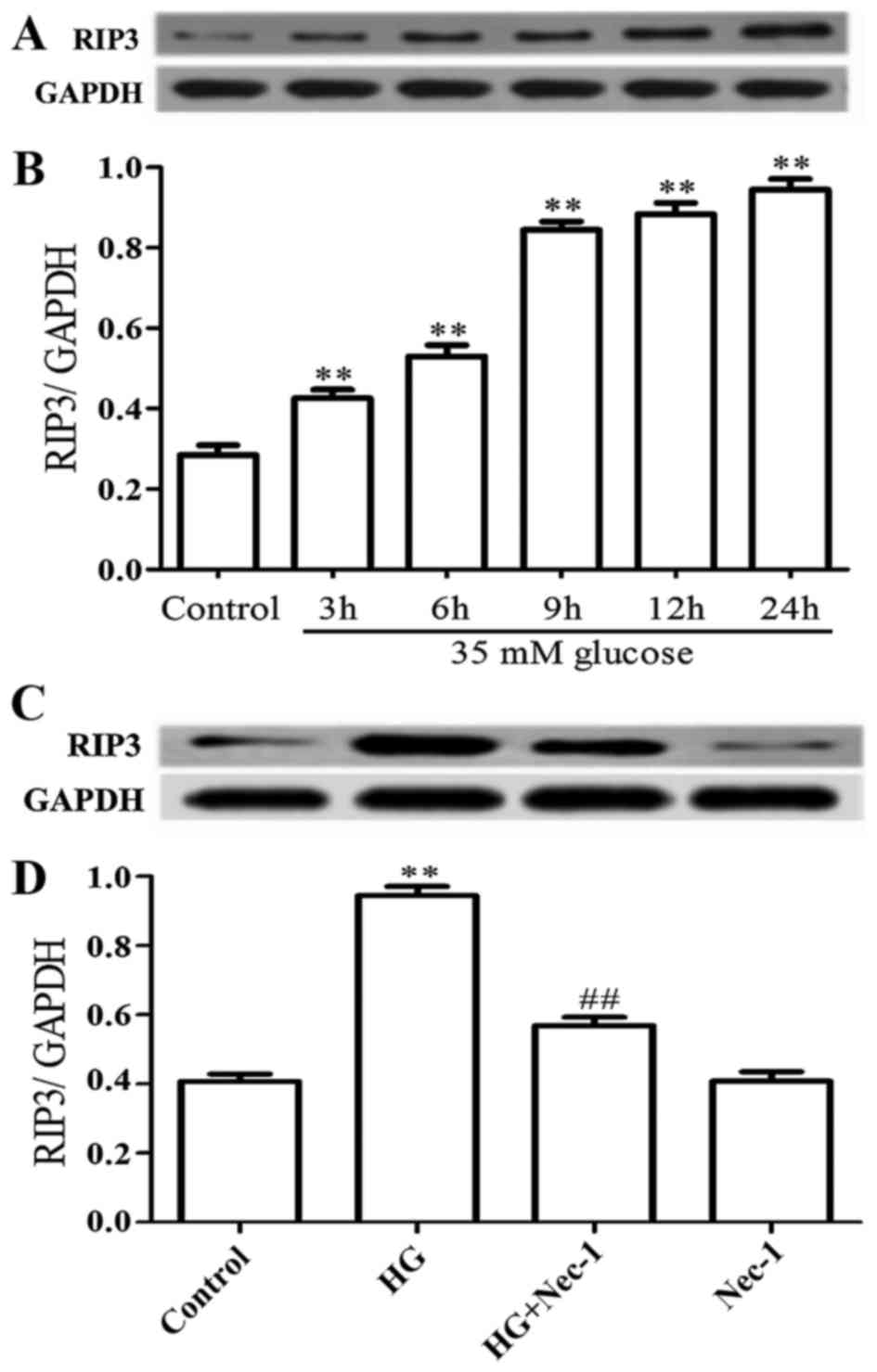
Based on the results from our primary dose-response
experiment (data not shown), 35 mM glucose was selected as an
effective injury-inducing concentration for H9c2 cardiac cells in
our recent studies (3,13). In this study, to examine the
effect of HG (35 mM glucose) on the protein expression of RIP3 in
H9c2 cardiac cells, a time-response experiment to evaluate the
protein expression level of RIP3 was performed. As shown in
Fig. 1A and B, after the cells
were exposed to HG for 3, 6, 9, 12 and 24 h, the protein expression
level of RIP3 was markedly increased (P<0.01), reaching the
maximum level at 24 h.
Of note, co-treatment of the H9c2 cardiac cells with
100 µM Nec-1 (a specific inhibitor of necroptosis) and HG
for 24 h considerably blocked the upregulation of RIP3 expression
induced by HG (Fig. 1C and D;
P<0.01). Alone 100 µM Nec-1 did not alter the basal
expression level of RIP3.
Scavenger of ROS ameliorates the
HG-induced upregulation of RIP3 expression in H9c2 cardiac
cells
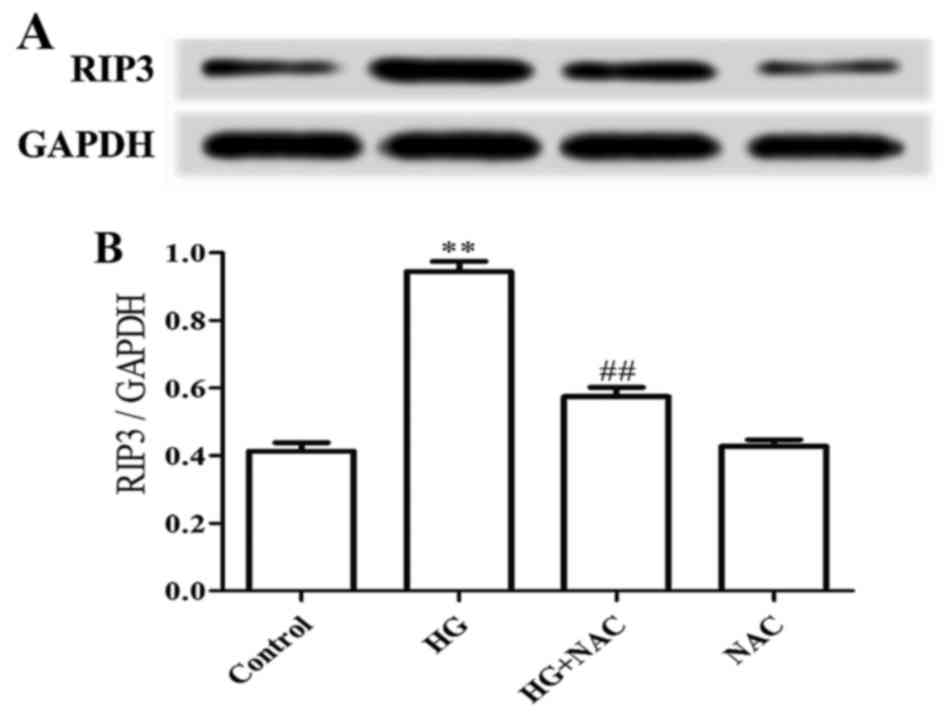
Since Schenk et al (34) have indicated that ROS is involved
in the regulation of BV6/TNF-α-induced necroptosis in Jurkat cells,
in this study, we evaluated the role of ROS in the HG-induced
upregulation of RIP3 expression in H9c2 cardiac cells. As shown in
Fig. 2, treatment of the cells
with 1 mM NAC (a scavenger of ROS) for 60 min prior to exposure to
HG for 24 h markedly inhibited the increased protein expression
level of RIP3. NAC at 1 mM alone did not affect the basal
expression level of RIP3 in H9c2 cardiac cells. The above-mentioned
results indicate that ROS participates in the HG-induced
upregulation of RIP3 expression.
Necroptosis is involved in HG-induced
oxidative stress in H9c2 cardiac cells
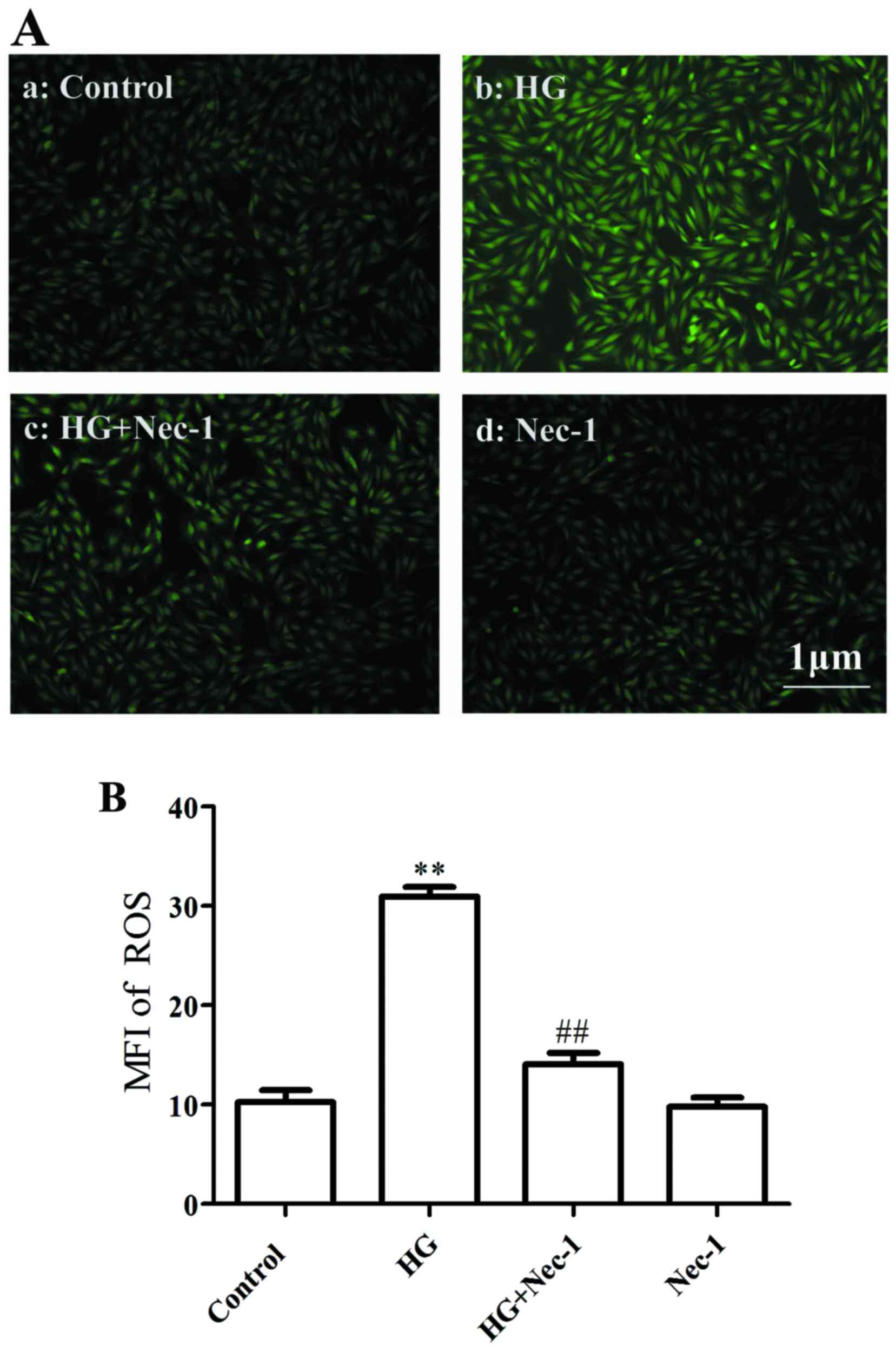
As shown in Fig. 3A–b
and B, exposure of the cells to 35 mM glucose (HG) for 24 h
significantly increased the intracellular generation of ROS.
However, co-treatment of the cells with 100 µM Nec-1 and HG
for 24 h markedly attenuated the increased generation of ROS
(Fig. 3A, panel c and B). Alone 100 µM Nec-1 did not
affect the basal intracellular generation of ROS. These results
indicated that necroptosis contributes to the HG-induced
overproduction of ROS in cardiac cells.
Necroptosis and ROS are implicated in
HG-induced cytotoxicity to H9c2 cardiac cells
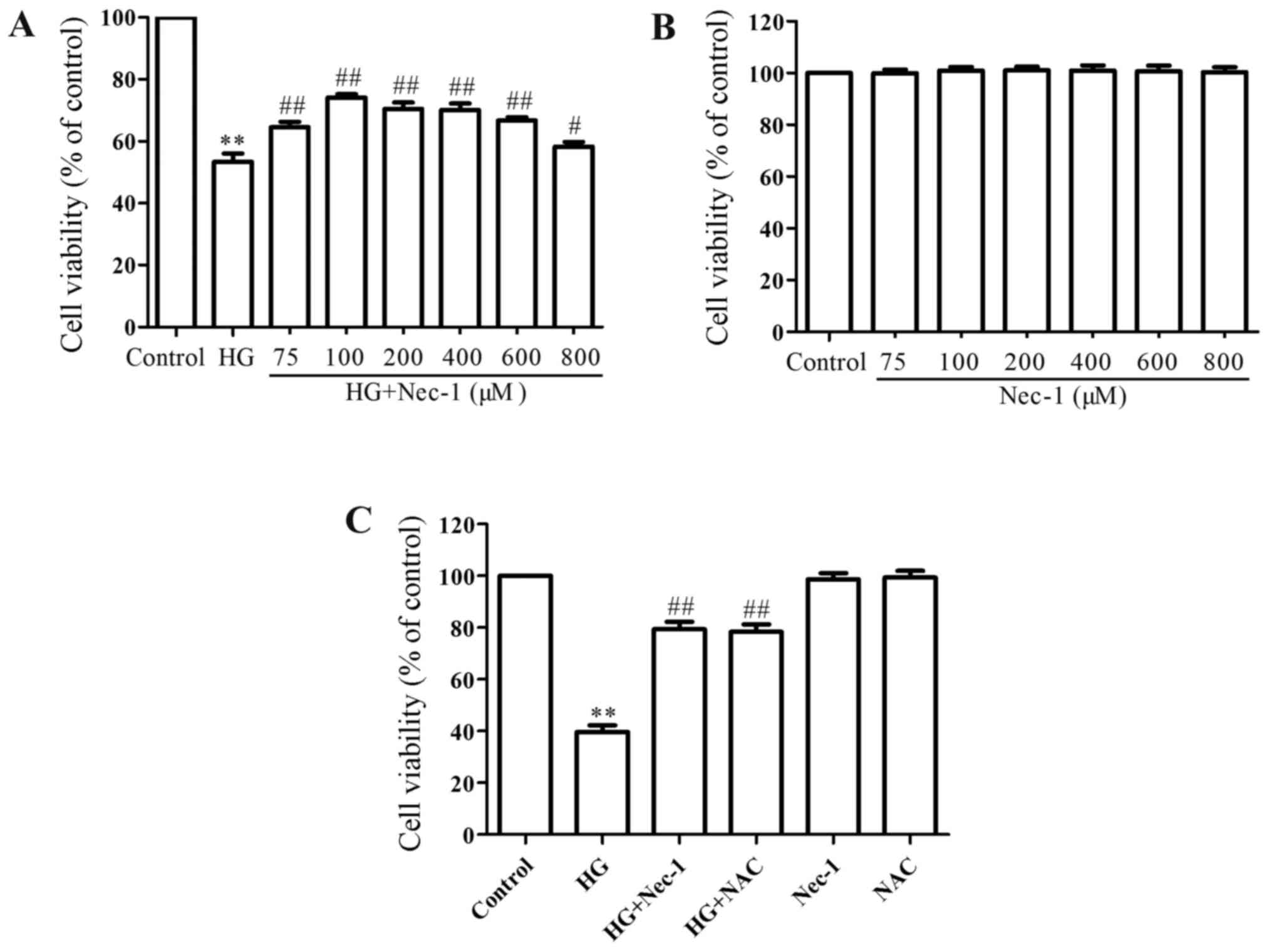
Consistent with previous studies (3,9,13),
the exposure of H9c2 cardiac cells to HG for 24 h markedly induced
cytotoxicity, leading to a decrease in cell viability. Co-treatment
of the cells with HG and 75, 100, 200, 400, 600, 800 µM of
Nec-1 for 24 h considerably reduced HG-induced cytotoxicity, as
evidence by an increase in cell viability (Fig. 4A), and at the concentration of 100
µM, Nec-1 exhibited the most prominent anti-cytotoxic
effect. Thus, 100 µM was used as the effective concentration
of Nec-1 in the following experiments. Alone, Nec-1 at 75, 100,
200, 400, 600 and 800 µM did not significantly affect cell
viability (Fig. 4B). Similar to
the protective effects of Nec-1 against HG-induced cytotoxicity,
pre-treatment of the H9c2 cardiac cells with 1 mM NAC also
antagonized the HG-induced cytotoxicity, leading to an increase in
cell viability (Fig. 4C). Alone,
1 mM NAC did not significantly alter cell viability. The
above-mentioned data suggest that necroptosis and ROS mediate
cytotoxicity in HG-exposed H9c2 cardiac cells.
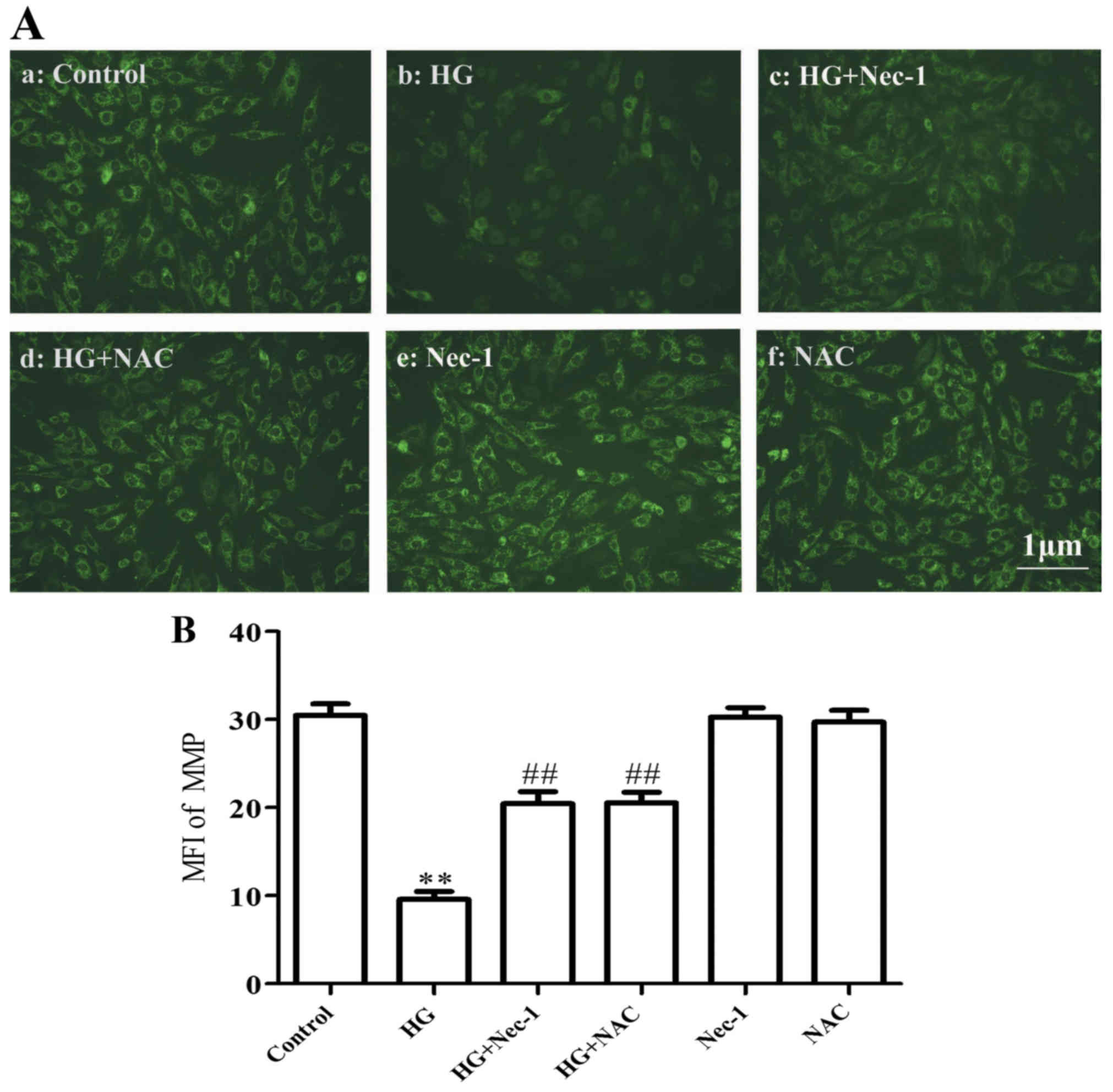
Necroptosis and ROS are linked to
HG-induced mitochondrial insults in H9c2 cardiac cells
Exposure of the cells to HG for 24 h markedly
induced mitochondrial damage, leading to a loss of MMP (Fig. 5A, panel b and B). However, co-treatment of the cells
with 100 µM Nec-1 and HG for 24 h or treatment of the cells
with 1 mM NAC for 60 min prior to exposure to HG for 24 h markedly
attenuated the HG-induced dissipation of MMP (Fig. 5A, panels c and d, and B). Alone, 100 µM Nec-1 and 1 mM
NAC did not significantly affect the MMP of the cells. These
results indicate that necroptosis and ROS are involved in
HG-induced mitochondrial damage.
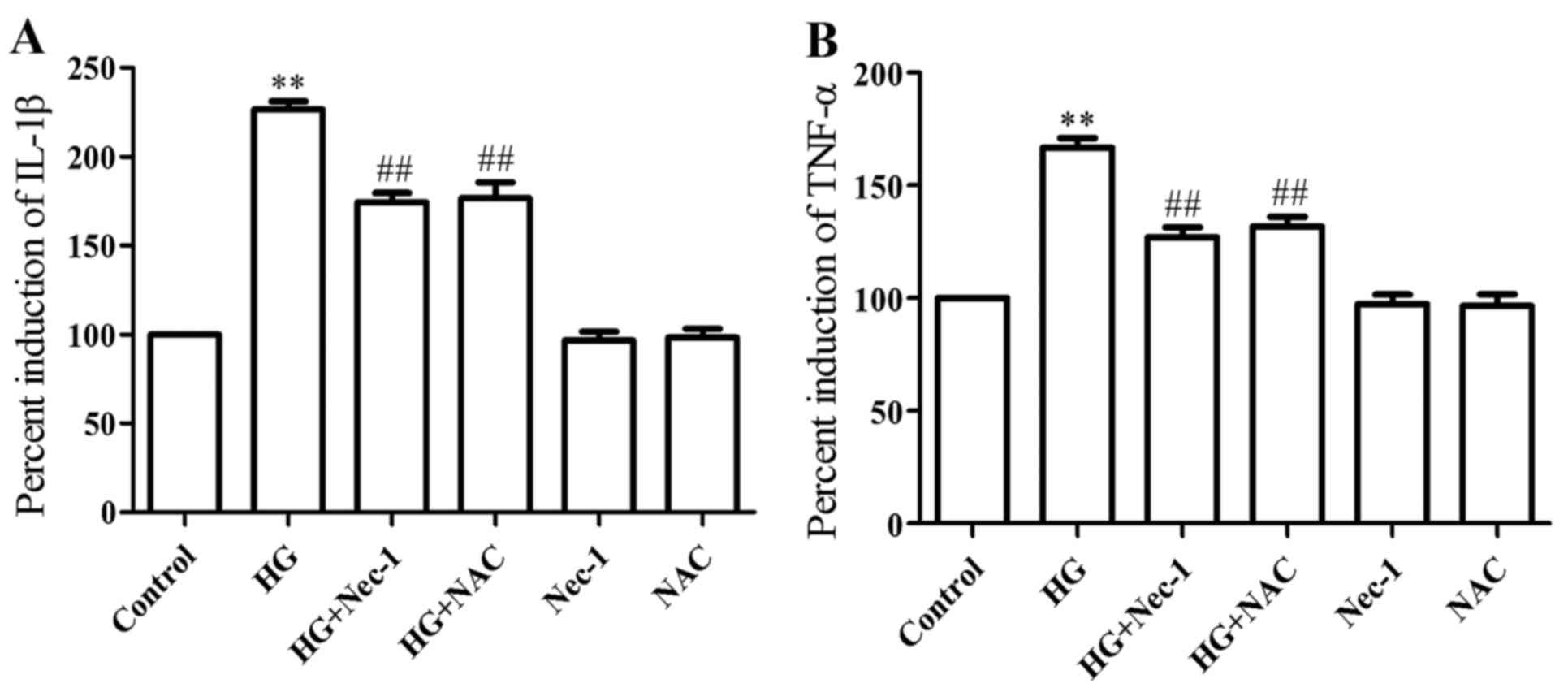
Necroptosis and ROS mediate the
HG-induced secretion of pro-inflammatory cytokines in H9c2 cardiac
cells
As shown in Fig.
6, after the cells were treated with HG for 24 h, the secretion
levels of IL-1β (Fig. 6A) and
TNF-α (Fig. 6B) were markedly
increased, as compared with the control group (P<0.01). However,
co-treatment of the cells with 100 µM Nec-1 and HG for 24 h
or treatment of the cells with 1 mM NAC for 60 min prior to
exposure to HG for 24 h markedly alleviated the increased
production of IL-1β and TNF-α, revealing that necroptosis and ROS
contribute to the HG-induced inflammatory response in H9c2 cardiac
cells.
Discussion
To date, four forms of cell death have been defined
and confirmed: necrosis, apoptosis, autophagy and necroptosis
(35–37). Among these, apoptosis, necrosis
and autophagy have been demonstrated to contribute to the
development of DCM (3,6,9,22,38,39). However, the role of necroptosis in
hyperglycemia-induced cardiac injury remains incompletely
understood, although Liu et al reported that the expression
level of RIP3, a kinase promoting necroptosis, was upregulated in
diabetic rats (22). In this
study, to the best of our knowledge, we demonstrate for the first
time that necroptosis plays important roles in HG-induced cardiac
injury (cytotoxicity, oxidative stress and dissipation of MMP) and
inflammation. Therefore, necroptosis may represent a promising
novel target for therapeutic strategies in DCM. Moreover, the
findings of this study suggested that there is a positive
interaction between necroptosis and ROS production, which may be a
novel mechanism underlying HG-elicited cardiac injury and
inflammation.
Necroptosis (also known as programmed necrosis)
represents a newly indentified mechanism of cell death combining
the features of both apoptosis and necrosis. Several types of
stimuli, including ligands of death receptors (such as Fas, TRAIL
and TNF-α), viral infection and anticancer agents, can induce
necroptosis (40). In recent
years, necroptosis has been demonstrated to be an important
mediator of cell death in the heart (14,23–26). Several studies have indicated that
I/R induces an increase in the expression levels of cardiac RIP1
and RIP3, and that Nec-1, an inhibitor of necroptosis, leads to a
reduction in myocardial infarct size (23–25). Therefore, necroptosis may be a
novel mechanism responsible for cardiac lesions. Recently, the
effect of hyperglycemia on necroptosis has attracted attention. A
more recent study by Liu et al demonstrated that RIP3
expression was enhanced in diabetic rats; however, the roles of
necroptosis in hyperglycemia-induced cardiac injury were not
determined (22).
In order to clarify this issue, in this study, we
first observed the effects of HG on the expression level of RIP3 in
cardiomyocytes. Consistent with the results reported by Liu et
al (22), we found that the
expression level of RIP3 was upregulated in HG-exposed H9c2 cardiac
cells. Combining our results and the ones reported by Liu et
al, it is suggested that hyperglycemia is a strong stimuli for
inducing necroptosis. Second, we examined the effects of Nec-1 on
HG-induced cardiac injury (including cytotoxicity, ROS generation
and dissipation of MMP). The findings of the present study
indicated that Nec-1 markedly attenuated the increased expression
of RIP3 by HG, along with the inhibitory effects on HG-induced
cardiac injury, as evidenced by an increase in cell viability, a
decrease in ROS generation and the attenuation of the dissipation
of MMP. These results provide novel evidence that necroptosis
contributes to HG-induced cardiomyocyte injury, and extend the
findings reported by Liu et al (22).
Another important result of this study relates to
the role of necroptosis in HG-induced cardiomyocyte inflammation.
Chronic mild inflammation has been considered as one of the
features of DCM in humans (41,42). Moreover, in a mouse model of
streptozotocin-induced type 1 diabetes, anti-inflammatory therapy
represented a potential approach for the therapy of diabetes and
its complications (43).
Therefore, the further exploration of the mechanisms and the
identification of novel therapeutic targets of HG-induced
inflammatory response has a promising future. Inflammatory
signaling molecules, such as TNF-α, Fas and TRAIL, have been
reported to be initiators of necroptosis (40,44). On the other hand, necroptosis has
been found to trigger intestinal inflammation, acute pancreatitis,
experimental sepsis, salmonella infection and inflammation in
atherosclerosis (45,46). Our results demonstrated that the
exposure of H9c2 cardiac cells to HG induced an obvious
inflammatory response, as evidenced by the increased secretion
levels of IL-1β and TNF-α, whichwasis similar to the results of our
previous study (13). However,
the increased secretion of IL-1β and TNF-α was ameliorated by
Nec-1, indicating the involvement of necroptosis in HG-induced
inflammation. Of note, necroptosis was triggered by TNF-α (40), and we demonstrated the
contribution of necroptosis to the HG-induced increase in TNF-α
secretion; thus, we speculated that there was a positive feedback
loop between necroptosis and TNF-α in the HG-treated H9c2 cardiac
cells. To confirm this hypothesis, further studies are
required.
Importantly, it has been shown that there is a
positive interaction between necroptosis and ROS generation in
BV6/TNF-α-treated Jurkat cells (34). This led us to explore whether
there was a similar interaction between necroptosis and ROS
generation in HG-exposed cardiac cells. Our results demonstrated
that NAC, a ROS scavenger, markedly ameliorated the HG-induced an
increase in RIP3 expression, accompanied by the inhibition of the
HG-induced cardiac injury and inflammation, as indicated by an
increase in cell viability and a decrease in ROS generation, the
attenuation of MMP dissipation and a derease in the secretion
levels of IL-1β and TNF-α induced by HG. These results clearly
demonstrate that a positive feedback loop between necroptosis and
ROS production exists in HG-exposed H9c2 cardiac cells, which plays
important roles in cardiac injury and inflammation induced by HG.
Since the roles of necroptosis and ROS in diabetic cardiac injury
have been emphasized by us and others, further experiments using
conditional RIP3-knockout mice are warranted in order to clarify
the mutual interaction between necroptosis and ROS generation in
vivo.
In conclusion, revealing the contribution of
necroptosis to HG-induced cardiac injury and inflammation, the
present study provides further insight into the mechanisms
underlying diabetic cardiovascular complications, such as DCM.
Considering the significance of the positive interaction between
necroptosis and ROS generation in HG-induced cardiac injury and
inflammation, a better understanding of the molecular mechanisms of
this interaction will likely have important implications for the
development of novel strategies to interfere with necroptosis and
ROS generation in patients with diabetes.
Acknowledgments
The present study was supported by grants from
Guangdong Natural Science Foundation (no. 2015A030313690).
References
|
1
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren J and Davidoff AJ: Diabetes rapidly
induces contractile dysfunctions in isolated ventricular myocytes.
Am J Physiol. 272:H148–H158. 1997.PubMed/NCBI
|
|
3
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925.
2013.PubMed/NCBI
|
|
4
|
Soetikno V, Sari FR, Sukumaran V,
Lakshmanan AP, Mito S, Harima M, Thandavarayan RA, Suzuki K, Nagata
M, Takagi R, et al: Curcumin prevents diabetic cardiomyopathy in
streptozotocin-induced diabetic rats: possible involvement of
PKC-MAPK signaling pathway. Eur J Pharm Sci. 47:604–614. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen J, Mo H, Guo R, You Q, Huang R and Wu
K: Inhibition of the leptin-induced activation of the p38 MAPK
pathway contributes to the protective effects of naringin against
high glucose-induced injury in H9c2 cardiac cells. Int J Mol Med.
33:605–612. 2014.PubMed/NCBI
|
|
7
|
Huang H and Wu K, You Q, Huang R, Li S and
Wu K: Naringin inhibits high glucose-induced cardiomyocyte
apoptosis by attenuating mitochondrial dysfunction and modulating
the activation of the p38 signaling pathway. Int J Mol Med.
32:396–402. 2013.PubMed/NCBI
|
|
8
|
Cai L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hyperglycemia-induced apoptosis in mouse myocardium:
mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen J, Guo R, Yan H, Tian L, You Q, Li S,
Huang R and Wu K: Naringin inhibits ROS-activated MAPK pathway in
high glucose-induced injuries in H9c2 cardiac cells. Basic Clin
Pharmacol Toxicol. 114:293–304. 2014. View Article : Google Scholar
|
|
10
|
Fiordaliso F, Leri A, Cesselli D, Limana
F, Safai B, Nadal-Ginard B, Anversa P and Kajstura J: Hyperglycemia
activates p53 and p53-regulated genes leading to myocyte cell
death. Diabetes. 50:2363–2375. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fei L, Yong-Jun H, Zhang-Min M, Bing X,
Shuang W, Qian-qian S and Jun L: Rosiglitazone attenuates memory
impairment in aged rat with diabetes by inhibiting NF-kappa B
signal pathway activation. Exp Clin Endocrinol Diabetes.
123:536–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thandavarayan RA, Giridharan VV, Sari FR,
Arumugam S, Veeraveedu PT, Pandian GN, Palaniyandi SS, Ma M, Suzuki
K, Gurusamy N, et al: Depletion of 14-3-3 protein exacerbates
cardiac oxidative stress, inflammation and remodeling process via
modulation of MAPK/NF-κB signaling pathways after
streptozotocin-induced diabetes mellitus. Cell Physiol Biochem.
28:911–922. 2011. View Article : Google Scholar
|
|
13
|
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W,
Feng J, Wu W and Zheng D: Exogenous H S protects H9c2 cardiac cells
against high glucose-induced injury 2 and inflammation by
inhibiting the activation of the NF-κB and IL-1β pathways. Int J
Mol Med. 35:177–186. 2015.
|
|
14
|
Kung G, Konstantinidis K and Kitsis RN:
Programmed necrosis, not apoptosis, in the heart. Circ Res.
108:1017–1036. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Declercq W, Vanden Berghe T and
Vandenabeele P: RIP kinases at the crossroads of cell death and
survival. Cell. 138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Linkermann A, Hackl MJ, Kunzendorf U,
Walczak H, Krautwald S and Jevnikar AM: Necroptosis in immunity and
ischemia-reperfusion injury. Am J Transplant. 13:2797–2804. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dmitriev YV, Minasian SM, Demchenko EA and
Galagudza MM: Study of cardioprotective effects of necroptosis
inhibitors on isolated rat heart subjected to global
ischemia-reperfusion. Bull Exp Biol Med. 155:245–248. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koshinuma S, Miyamae M, Kaneda K, Kotani J
and Figueredo VM: Combination of necroptosis and apoptosis
inhibition enhances cardioprotection against myocardial
ischemia-reperfusion injury. J Anesth. 28:235–241. 2014. View Article : Google Scholar
|
|
22
|
Liu YS, Huang ZW, Wang L, Liu XX, Wang YM,
Zhang Y and Zhang M: Sitagliptin alleviated myocardial remodeling
of the left ventricle and improved cardiac diastolic dysfunction in
diabetic rats. J Pharmacol Sci. 127:260–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lim SY, Davidson SM, Mocanu MM, Yellon DM
and Smith CC: The cardioprotective effect of necrostatin requires
the cyclophilin-D component of the mitochondrial permeability
transition pore. Cardiovasc Drugs Ther. 21:467–469. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith CC, Davidson SM, Lim SY, Simpkin JC,
Hothersall JS and Yellon DM: Necrostatin: a potentially novel
cardioprotective agent? Cardiovasc Drugs Ther. 21:227–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luedde M, Lutz M, Carter N, Sosna J,
Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F,
et al: RIP3, a kinase promoting necroptotic cell death, mediates
adverse remodelling after myocardial infarction. Cardiovasc Res.
103:206–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meng L, Jin W and Wang X: RIP3-mediated
necrotic cell death accelerates systematic inflammation and
mortality. Proc Natl Acad Sci USA. 112:11007–11012. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han D, Ybanez MD, Ahmadi S, Yeh K and
Kaplowitz N: Redox regulation of tumor necrosis factor signaling.
Antioxid Redox Signal. 11:2245–2263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morgan MJ and Liu ZG: Reactive oxygen
species in TNFalpha-induced signaling and cell death. Mol Cells.
30:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fortes GB, Alves LS, de Oliveira R, Dutra
FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher
M, Golenbock D, et al: Heme induces programmed necrosis on
macrophages through autocrine TNF and ROS production. Blood.
119:2368–2375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ye YC, Wang HJ, Yu L, Tashiro S, Onodera S
and Ikejima T: RIP1-mediated mitochondrial dysfunction and ROS
production contributed to tumor necrosis factor alpha-induced L929
cell necroptosis and autophagy. Int Immunopharmacol. 14:674–682.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu X, Deng Q, Li W, Xiao L, Luo X, Liu X,
Yang L, Peng S, Ding Z, Feng T, et al: Neoalbaconol induces cell
death through necroptosis by regulating RIPK-dependent autocrine
TNFα and ROS production. Oncotarget. 6:1995–2008. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kikuchi M, Kuroki S, Kayama M, Sakaguchi
S, Lee KK and Yonehara S: Protease activity of procaspase-8 is
essential for cell survival by inhibiting both apoptotic and
nonapoptotic cell death dependent on receptor-interacting protein
kinase 1 (RIP1) and RIP3. J Biol Chem. 287:41165–41173. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schenk B and Fulda S: Reactive oxygen
species regulate Smac mimetic/TNFα-induced necroptotic signaling
and cell death. Oncogene. 34:5796–5806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nunes T, Bernardazzi C and de Souza HS:
Cell death and inflammatory bowel diseases: apoptosis, necrosis,
and autophagy in the intestinal epithelium. BioMed Res Int.
2014:2184932014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fenton K: The effect of cell death in the
initiation of lupus nephritis. Clin Exp Immunol. 179:11–16. 2015.
View Article : Google Scholar
|
|
37
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fang ZY, Prins JB and Marwick TH: Diabetic
cardiomyopathy: evidence, mechanisms, and therapeutic implications.
Endocr Rev. 25:543–567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shimomura H, Terasaki F, Hayashi T,
Kitaura Y, Isomura T and Suma H: Autophagic degeneration as a
possible mechanism of myocardial cell death in dilated
cardiomyopathy. Jpn Circ J. 65:965–968. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vanlangenakker N, Vanden Berghe T and
Vandenabeele P: Many stimuli pull the necrotic trigger, an
overview. Cell Death Differ. 19:75–86. 2012. View Article : Google Scholar :
|
|
41
|
Boudina S, Sena S, Theobald H, Sheng X,
Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, et al:
Mitochondrial energetics in the heart in obesity-related diabetes:
direct evidence for increased uncoupled respiration and activation
of uncoupling proteins. Diabetes. 56:2457–2466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Agrawal NK and Kant S: Targeting
inflammation in diabetes: newer therapeutic options. World J
Diabetes. 5:697–710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fang Q, Wang J, Wang L, Zhang Y, Yin H, Li
Y, Tong C, Liang G and Zheng C: Attenuation of inflammatory
response by a novel chalcone protects kidney and heart from
hyperglycemia-induced injuries in type 1 diabetic mice. Toxicol
Appl Pharmacol. 288:179–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu X, Deng Q, Bode AM, Dong Z and Cao Y:
The role of necroptosis, an alternative form of cell death, in
cancer therapy. Expert Rev Anticancer Ther. 13:883–893. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang Y, Jiang G, Zhang P and Fan J:
Programmed cell death and its role in inflammation. Mil Med Res.
2:122015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu
BQ, Li JH, Jing L, Jiang JL, Tang J, et al: Necrostatin-1 reduces
intestinal inflammation and colitis-associated tumorigenesis in
mice. Am J Cancer Res. 5:3174–3185. 2015.PubMed/NCBI
|