Introduction
Japanese encephalitis (JE) is an acute and severe
viral infection of the human central nervous system that is caused
by JE virus (JEV) and is transmitted between animal hosts and
humans by mosquitoes (mostly of the genus Culex) (1). It is prevalent throughout Eastern
and South Eastern Asia and has expanded onto the Cape York
Peninsula of Northern Australia (2–4);
it has affected approximately 60% of the global population in
endemic countries at risk of exposure to JEV. JE primarily occurs
in children under 15 years of age and the elderly when protective
immunity decreases (2,5). Fortunately, most JEV infections are
subclinical with a symptomatic-to-asymptomatic ratio of 1:25-1,000
(6,7). With thebroad administration of
inactivated and attenuated live vaccines, the annual JE incidence
is estimated to range from 50,000–175,000 cases. Approximately
20–30% of cases are fatal, and approximately 30–50% of survivors
suffer serious neurologic, cognitive, or psychiatric complications
several years thereafter (8–10).
Therefore, JE remains a significant health threat to the public
worldwide.
The JE causal agent, JEV, is a member of the genus
Flavivirus, of the family Flaviviridae. The virion is
spherical, small in size (40–60 nm) and contains an electron dense
core ~30 nm in diameter that is surrounded by a lipid bilayer
envelope. The viral genome is composed of a positive-sense,
10,976-nucleotide, single-stranded RNA, which features a methylated
cap at the 5′ end and does not contain a 3′ poly (A) tail (11). Flanked by hundreds of nucleotides
in non-coding regions (NCRs) on both termini, the viral RNA encodes
a single long open reading frame (ORF) and translates into a large
polyprotein precursor that is subsequently processed into
structural (C, M and E) and non-structural (NS1, NS2A, NS2B, NS3,
NS4A, NS4B and NS5) proteins by cellular and viral proteinases
(12).
In viral encoded proteins, the envelope E protein on
the virion surface is the most crucial protein involved in
virus-host interactions, particularly during attachment to the cell
receptor, virus penetration, cell tropism and virulence, as well as
haemagglutination and neutralization in human protective immunity
(13). The E protein consists of
500 amino acid residues, appears as approximately 53 kDa, and is
N-glycosylated (14). An exact
crystal structure of the JEV E protein ectodomain (1–406) was
previously produced and the major conformational properties
previously suggested were confirmed based on a related flavivirus
[tick borne encephalitis virus (TBEV)] E protein counterpart
(15,16). In brief, the E protein is present
on the virion surface as an anti-parallel homodimer with a convex
external curvature and is anchored to the viral envelope (lipid
membrane) at the distal end. Each E protein subunit is composed of
3 domains based on antigenicity (13). Domain I is a discontinuous domain
composed of 3 fragments (1–51, 137–189 and 293–311 amino acids from
the amino terminal end) and is the central domain with an
8-stranded β-barrel located in the centre of the E protein
molecule. Domain II is the dimerization domain on the mature
virion, forms 2 loops from regions 52–136 and 190–289, projects
along the virus surface between the homodimer subunit transmembrane
regions, and contains a highly conserved fusion loop that likely
inserts into target cell membranes (17). Domain III (311–411) lies at the E
protein C-terminus, forms a constant β-barrel composed of 7
head-to-tail β-strands, and maintains an immunoglobulin-like fold
(18). It contains the
receptor-binding domain, which is connected to the central domain I
by a flexible region and is a major target of neutralizing
antibodies (19).
To inititiate JEV pathogenesis, the JEV E protein
and the cellular receptor on the surface of susceptible cells form
an initial interaction (20–24). Therefore, determining the cellular
receptor is a prerequisite for future investigations into precise
molecular events involved in JEV binding and entry into cells,
which has always been an intriguing basic virology field. Research
has advanced over the past few decades. Early in 1994, researchers
discovered that a 74 kDa molecule from Vero (African green monkey
kidney) cells can bind JEV without further characterization
(20). Later, researchers found
that a 57 kDa protein from BHK-21 (baby hamster kidney) cells
(21) and a GAG protein in
Chinese hamster ovary (CHO) cells (22) may be involved in JEV attachment or
entry. In previous studies from our laboratory, we generated
several pieces of evidence and, thus, hypothesised that heat shock
cognate protein 70 (HSC70) may act as a JEV binding receptor on
mosquito C6/36 cells (23). It
has been reported that heat shock protein (HSP)70, which is present
in the membrane fraction from mouse neuroblastoma cells (Neuro2a),
is the putative receptor for JEV (24). In addition, using a mouse
microglial cell line, researchers have demonstrated that the
extracellular matrix protein, laminin, and several other candidate
receptor molecules, including HSP70, HSP90, GRP78, CD14 and CD4,
are involved in JEV entry (25).
Nevertheless, these studies are limited to rodent or mosquito
cells, and the interacting cellular proteins from human or at least
primate cells are necessary to elucidate the functional role of E
protein with interacting cellular proteins. Herein, we discovered
that HSP90β in the monkey cell (Vero) membrane binds JEV
particles.
Materials and methods
JEV propagation, purification and
titration
The JEV strain SA14 (isolated in Xi'an, China in the
1950's, and stored in a refrigerator in our laboratory ever since)
was propagated in C6/36 cells (ATCC® CRL-1660™;
Institute for Microbial Epidemiology, Academy of Military Medical
Sciences, Beijing, China) cultured at 28°C in a closed incubator in
RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
containing 10% foetal calf serum (FCS; HyClone Laboratories Inc.,
Logan, UT, USA), 1% lactoalbumin hydrolysate (LH) and antibiotics
(penicillin and streptomycin). When CPE was apparent, the culture
supernatants were harvested following centrifugation at 500 × g at
4°C for 10 min. The supernatants were then received a serial
centrifugation in 10,000 × g for 20 min, 45,000 × g for 10 min and
70,000 × g for 4 h at 4°C (Optima L-100XP Ultracentrifuge, Beckman
Coulter, Brea, CA, USA). Viral particles were condensed in aliquots
and preserved at −80°C for use in subsequent experiments.
The condensed JEV was titrated by plaque formation
assay with a semi-solid overlay. BHK-21 cells (ATCC®
CCL-10™) were obtained from Ms. L. Jia (The National Institute for
the Control of Pharmaceutical and Biological Products, Beijing,
China). BHK-21 cells were used in JEV titration and JEV progeny
determination in RNAi-treated Vero cells, while attempt to identify
molecules capable of binding JEV by VOPBA using BHK-21 cells failed
(unpublished data). Briefly, BHK-21 cells (1×105
cells/well) were grown to a confluent monolayer in a 6-well plate
in culture medium [containing 60% RPMI-1640, 30% 0.5% LH, 10% FCS,
1% penicillin, 100 IU/ml streptomycin, 0.75% NaHCO3 (1%
in total) NaHCO3 and 3% glutamine (1% in total)] and
infected with 200 μl of 10-fold serially diluted JEV
aliquots dissolved in virus dilution solution [containing 96% LH,
2% FCS, and 0.75% NaHCO3 (2% in total)]. The cells
absorbed the virus at 37°C for 1 h with gentle circular tilting
every 15 min. The fluids were then pipetted out, and the monolayers
were washed 3 times with RPMI-1640 without serum and finally
covered with a semi-solid overlay [containing 1% methyl cellulose,
1/10 volume 10X RPMI-1640, 25% distilled water, 10% FCS, 1%
antibiotics (penicillin and streptomycin), 0.75% NaHCO3
(3% in total) and 3% glutamine (1% in total)]. Following incubation
at 37°C for 96 h, the overlay was withdrawn, and the cell monolayer
was stained with a 1% crystal violet solution containing 0.2%
formaldehyde. All plaque formation assay experiments were performed
in triplicate, and the plaques were counted and calculated in
PFU/ml.
Cell membrane protein extraction
Vero cells (ATCC® CCL-81™) were obtained
from Ms. L. Jia (The National Institute for the Control of
Pharmaceutical and Biological Products). Vero cell membrane
proteins were prepared as previously described (26) with minor modifications. Briefly,
the cells were cultured in RPMI-1640 medium containing 10% FCS in
an incubator (Hera Cell; Heraeus Holding GmbH, Hanau, Germany) with
5% CO2 at 37°C. Confluent cell monolayers were washed 3
times with PBS, treated with 1 mM EDTA in PBS (pH 7.2) for 3 min at
37°C, and subsequently resuspended in ice cold Buffer M (100 mM
NaCl, 20 mM Tris-HCl pH 8.0, 2 mM MgCl2, 1 mM EDTA and 1
mM β-mercaptoethanol) containing 1 mM phenylmethanesulfonyl
fluoride (PMSF; Sigma-Aldrich, St. Louis, MO, USA) on ice for 40
min with intermittent shaking, and lysed using 15–20 strokes and a
Dounce homogeniser. The nuclei and cell debris were removed by
centrifugation at 500 × g and 4°C for 10 min. The membrane-bound
proteins in the supernatant fraction were condensed through further
centrifugation at 36,000 × g and 4°C for 30 min. The pellet was
dissolved in Buffer M without β-mercaptoethanol. The concentrations
of the membrane proteins were determined using a BCA kit (Pierce;
Thermo Fisher Scientific Inc., Rockford, IL, USA).
Co-immunoprecipitation (Co-IP) and
SDS-PAGE
We thoroughly mixed 160 μg membrane protein
extracts with purified JEV (5×106 PFU/ml) in 1 ml
microcentrifuge tubes on ice and added an equal volume of 2X IP
buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, pH 8.0) containing
1% NP-40 overnight at 4°C. The complex was incubated with mouse
anti-JEV monoclonal antibody (mAb) 2H4 (created and produced in our
laboratory) (27) overnight at
4°C and separated through reacting with protein A/G agarose beads
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 2 h at 4°C in
a rocker on the lowest setting. The beads were washed 3 times with
1X IP buffer and centrifuged at 3,000 × g for 5 min at 4°C, and the
pellets with bound proteins were analysed through 10% SDS-PAGE
after boiling for 5 min. The protein bands in the gels were stained
by Coomassie brilliant blue (Amresco, Solon, OH, USA).
Mass spectrometry (MS) analysis
Novel stained bands of the Co-IP immunocomplex on
SDS-PAGE gels were excised for matrix-assisted laser desorption
ionization-time of flight MS (MALDI-TOF MS). Briefly, a gel with a
single band was minced into sections at approximately 1
mm3 and transferred into a sterile microcentrifuge tube.
The gel sections were immersed in a 500 μl preservation
solution (50% acetonitrile, 50% distilled water) and mailed to the
laboratory in Beijing (see Acknowledgements) at room temperature
for further MS processing protocol and analysis. Protein identity
was determined based on a score of >30 from the homology search
tool MASCOT, where the mass/charge values matched the information
available in primate databases (28).
Western blot analysis for HSP90β-JEV
binding
We routinely used western blot analysis to detect
JEV binding with HSP90β from the Vero membrane protein extracts
under non-reducing and non-boiling conditions. Due to the
unavailability of anti-monkey HSP90β antibodies and the high
homogeneity of HSP90β between humans and monkeys, a rabbit
anti-human HSP90β mAb (Product no. ab32568; Abcam, Cambridge, UK)
was used for blotting (1:500 in TBS containing 5% powdered milk).
We then used Odyssey™ IRDye680-labelled goat anti-rabbit IgG
(1:5,000; Cat. no. 926-68071; LI-COR Biosciences, Lincoln, NE,
USA). In a parallel blotting test, rabbit anti-human HSP90β mAb was
replaced by purified JEV (1×109 PFU/ml) followed by the
addition of anti-JEV mAb 2H4 (22 μg/ml) and Odyssey™
IRDye680-labelled goat anti-mouse IgG (1:5,000; Cat. no. 926-68070;
LI-COR Biosciences). The results were scanned and analysed by
GelDoc-It™ (Ultra-Violet Products Ltd., Cambridge, UK).
Confocal laser scanning microscopy
(CLSM)
Routine CLSM was performed (29) to detect the location of HSP90β-JEV
binding. Briefly, Vero cells were prepared on glass cover-slips,
fixed by paraformaldehyde, and incubated with JEV
(1.2×105 PFU/ml) at 37°C for 1 h. After washing 3 times
with PBS, a primary antibody (1:10 anti-JEV mAb 2H4, and 1:50
rabbit anti-human HSP90β mAb, respectively) was added followed by
incubation overnight at 4°C, and the secondary antibody [1:100
FITC-conjugated goat anti-mouse IgG (Sigma-Aldrich™ Cat. no.
F9006), and 1:100 Cy3-conjugated goat anti-rabbit IgG
(Sigma-Aldrich™ Cat. no. C2306), respectively (Sigma-Aldrich)] was
added followed by incubation in the dark at room temperature for 1
h followed by a PBS wash 3 times. The coverslips were then
thoroughly rinsed in deionised water, air dried, placed reversely
on a glass slide mounted with mixture of 50% glycerol and an equal
volume Hoechst (1:1,000; Beyotime Institute of Biotechnology,
Shanghai, China), and the samples were finally observed under an
Olympus FV-1000 microscope with the image analysis software package
FV1000 Viewer v1.4a (Olympus Corp; Tokyo, Japan).
Immunofluorescence assay (IFA)
When HSP90β-JEV binding on the Vero cell surface was
confirmed, a JEV infection inhibition test was performed under
impermeable conditions using an IFA with the rabbit anti-human
HSP90β mAb mentioned above. Well-grown Vero cells were treated with
3.5 mM EDTA instead of trypsin to avoid cell surface protein
digestion, washed with PBS, and transferred onto glass coverslips
(15 μl, 1×106/ml) in 24-well plates and grown
with 10% FCS RPMI-1640 in a humidified incubator with 5%
CO2 at 37°C. When the confluence reached 50–60%, the
cells were washed 3 times with PBS and were fixed in 4%
PBS-buffered formaldehyde for 30 min at room temperature. After a
PBS wash, the fixed cells were successively incubated with rabbit
anti-human HSP90β mAb (two concentrations of 10 μg/ml and 20
μg/ml) at 4°C for 2 h, titrated JEV (MOI=0.1) at 4°C
overnight, anti-JEV mAb 2H4 (1:10, primary antibody) at 4°C for 2 h
and FITC-conjugated goat anti-mouse IgG (1:100, secondary antibody;
Bioworld Technology, Inc., St. Louis Park, MN, USA) at room
temperature for 1 h. Immunofluorescence was observed under a BH-60
immunofluorescence microscope (Olympus Corp.) after glycerol
mounting.
Flow cytometry (FCM)
FCM was performed using a routine procedure.
Briefly, the Vero cells were treated with PBS containing 3.5 mM
EDTA, washed 3 times with FCM dilution, and resuspended (adjusting
concentration to 1×106/ml). Vero cells (50 μl per
tube) were consecutively incubated with rabbit anti-human HSP90β
mAb (1:100 and 1:50), JEV (MOI=1), anti-JEV mAb 2H4 (1:10) and
FITC-conjugated goat anti-mouse IgG (1:100) at 4°C for 1 h with
gentle shaking. Each incubation step was interspersed by washing 3
times with an FCM dilution solution and centrifugation at 1,000 rpm
and 4°C for 5 min. Finally, the cells were fixed in 300 μl
4% PBS-buffered formaldehyde, and immunofluorescence was detected
using a flow cytometer (Epics Elite ESP; Beckman Coulter Inc.,
Indianapolis, IN, USA). All detection experiments were performed in
quadruplicate.
Suppression of HSP90β expression in Vero
cells through siRNA
Several siRNA fragments were synthesised based on
the human HSP90β mRNA transcript sequence from the NCBI PDB
database. Following the general principles of siRNA design
(30), several siRNA fragments
were synthesised, subcloned into lentiviral vectors (Cat. no.
HPK-LvTR-20; GeneCopoeia, Rockville, MD, USA), packaged in the
293Ta lentiviral packaging cell line (Cat. no. Clv-PK-01;
GeneCopoeia), and the pseudoviruses thus produced infected intact
Vero cells in the presence of 5 μg/ml hexadimethrine
bromide. We considered HSP90β expression inhibition over 50%
effective as void of off-target effects, and the fragment was
selected for further experiments. Cells with reduced HSP90β
expression were then subjected to JEV infection (MOI=0.1), and
progeny virus yields were measured using plaque formation assay at
12, 24, 36 and 48 hours post-infection (hpi).
Statistical analysis
The quantitative measurement results are expressed
as the means ± standard deviation. One-way ANOVA t-test was used to
determine statistical differences between the means: a value of
P<0.05 was considered to indicate a statistically significant
difference. These t-tests were performed using Prism 5.0 software
(GraphPad Software Inc., La Jolla, CA, USA).
Results
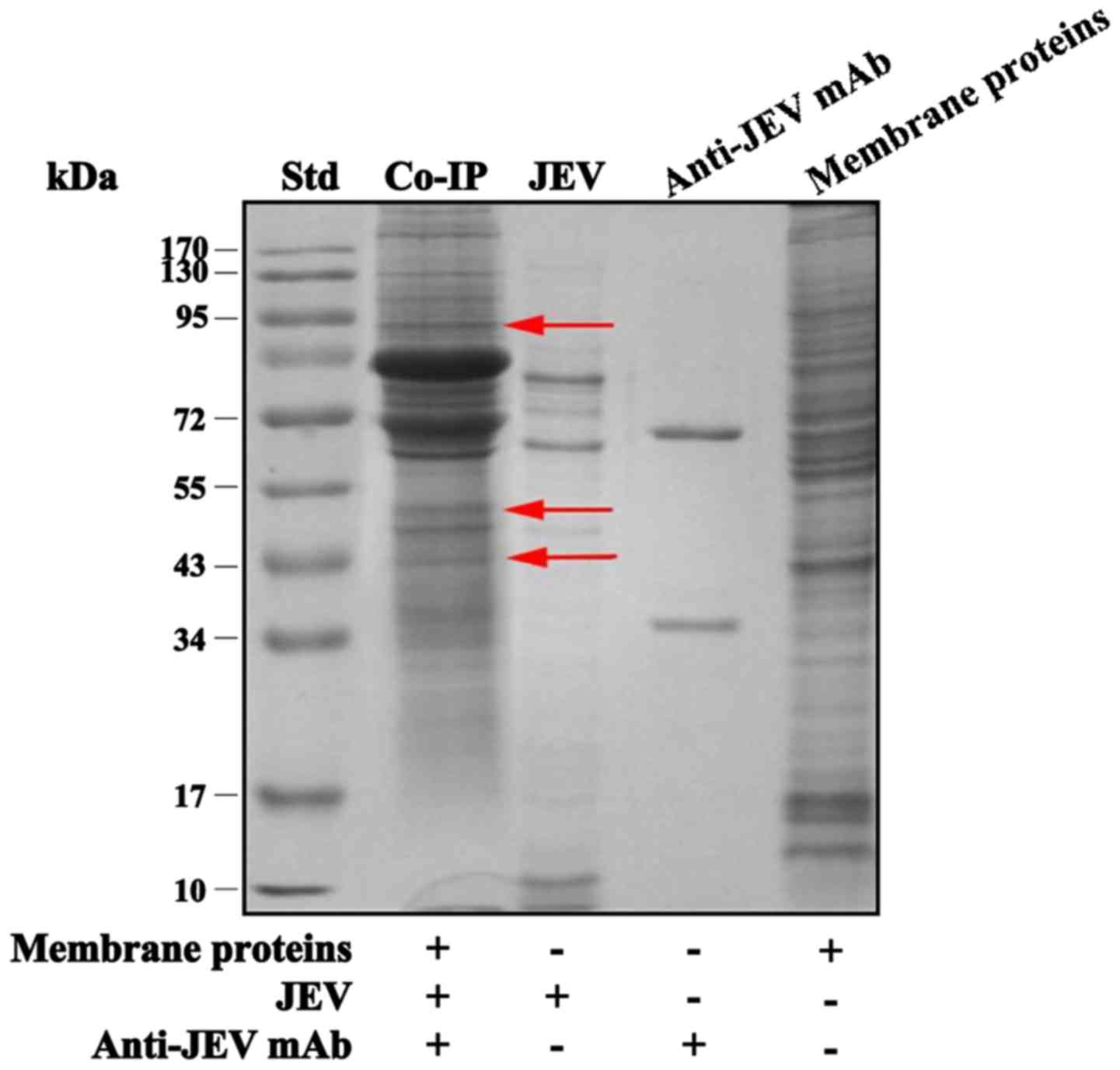
Results of Co-IP and MS analyses
The immunocomplexes, formed by membrane proteins,
JEV and anti-JEV mAb, were precipitated, washed, centrifuged and
subjected to SDS-PAGE (Fig. 1).
The results ostensibly indicated 3 novel protein bands at
approximately 43, 55 and 95 kDa, present in the membrane protein
sample but not present in the JEV and anti-JEV mAb controls. These
gel bands were cut off in sections for further MS analyses.
In MS experiments, the peptide sequences from the
gels were quested in the monkey database but generated no matches,
as the African green monkey (Cercopithecus aethiops)
database is not available. Therefore, the human database (Homo
sapiens) had to be used instead, and the 95-kDa-molecule was
probed as HSP90β, with a moderate score of 60.3 (Table I), and then selected as an entire
functioning protein for further investigation.
 | Table IResults of mass spectrometry analysis
of the novel bands. |
Table I
Results of mass spectrometry analysis
of the novel bands.
A, Monkey
(Macaca fascicularis) protein database
|
|---|
| Serial no. | Reference | Score | Accession | Peptides
(hits) |
|---|
| 1 | Unnamed protein
product | 80.5 | 67971184.0 | 8 (80,000) |
| 2 | Unnamed protein
product | 40.4 | 67971096.0 | 4 (40,000) |
|
B, Human (Homo
sapiens) protein database
|
| Serial no. | Reference | Score | Accession | Peptides
(hits) |
|
| 1 | Heat shock protein
90 kDa 1, β | 60.3 | 20149594.0 | 6 (60,000) |
| 2 | Unnamed protein
product A | 60.2 | 32486.0 | 6 (60,000) |
| 3 | chain A, the
3-dementional structure of glutathione S-transferase | 30.2 | 157831214.0 | 3 (30,000) |
| 4 | Aconitase 2
precursor | 30.2 | 4501867.0 | 3 (30,000) |
HSP90β from the Vero membrane binds JEV
on the Vero cell surface
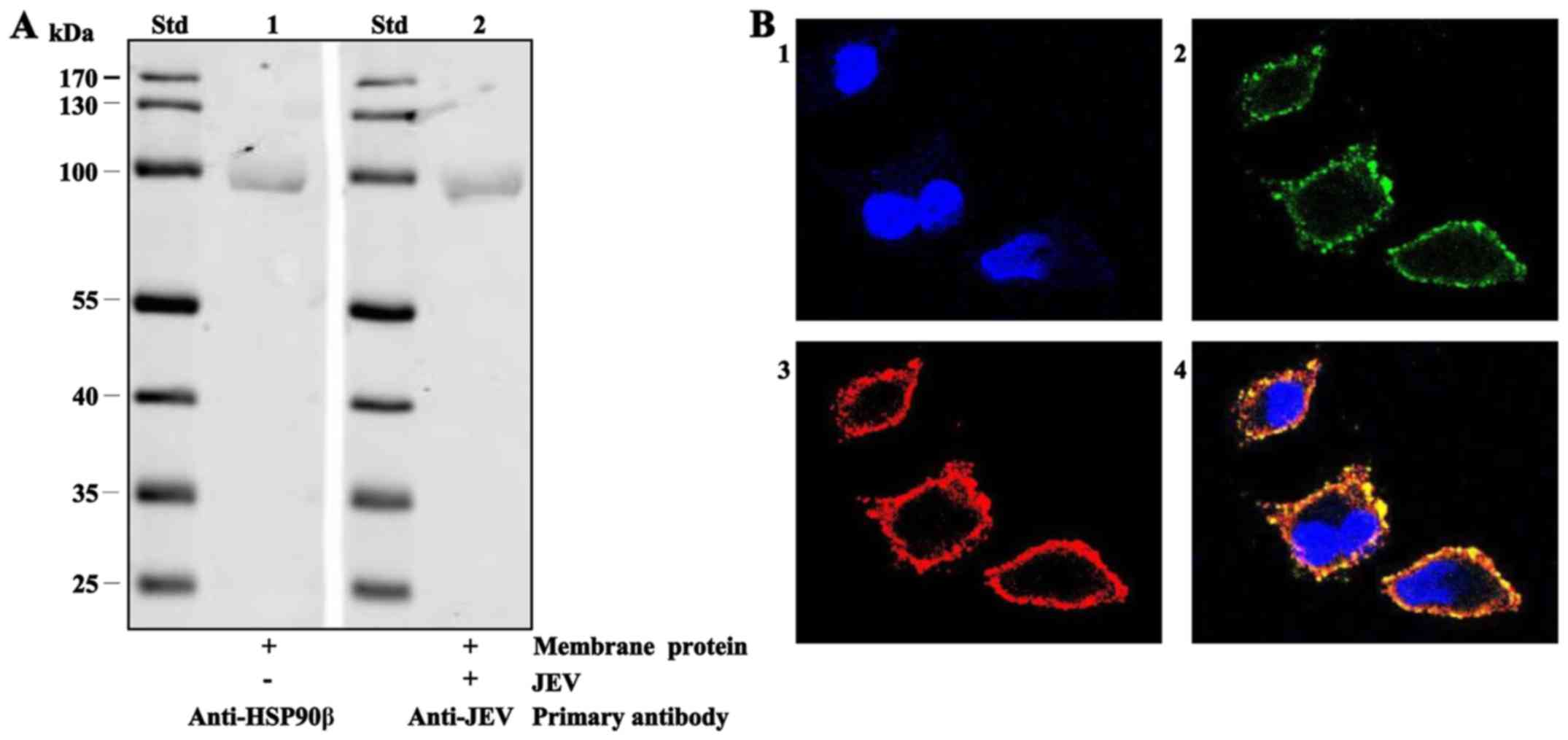
First, western blot analysis was used to test the
binding between JEV and HSP90β in the Vero membrane. Following
non-reducing SDS-PAGE, Vero membrane protein extract samples were
transferred onto PVDF membranes and reacted with rabbit anti-human
HSP90β mAb (as anti-monkey HSP90β mAbs are unavailable) and
indicated by fluorescent goat anti-rabbit IgG (Fig. 2A, lane 1). Vero membrane protein
samples also reacted alone with JEV and anti-JEV mAb 2H4 (Fig. 2A, lane 2) at the same 90 kDa
position, indicating that HSP90β binds JEV. Later, confocal laser
scanning microscopy was performed. As shown in Fig. 2B, JEV (green colour, Fig. 2B, panel 2) and HSP90β (red colour,
Fig. 2B, panel 3) merger
displayed yellow fluorescence on the cell surface (Fig. 2B, panel 4), evidently suggesting
that the presence of HSP90β in the cell membrane binds JEV exactly
on the cell surface.
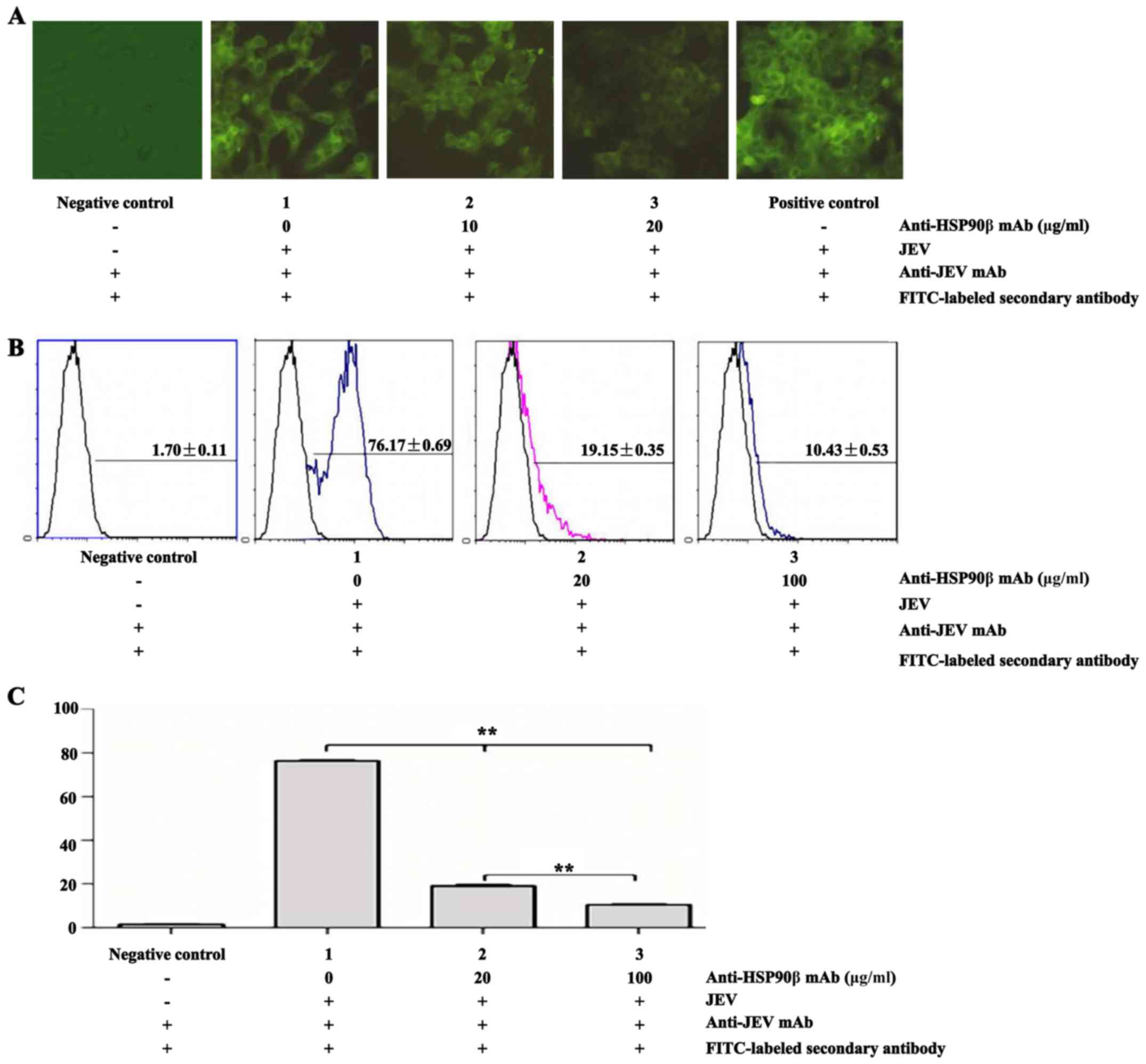
Specific mAb inhibits HSP90β-JEV binding
in a dose-dependent manner
When the binding of HSP90β and JEV was ascertained,
a virus infection inhibition experiment with rabbit anti-human
HSP90β mAb at various concentrations was conducted using
qualitative IFA and quantitative FCM. In the IFA experiment, green
fluorescence gradually decreased with an increase in the
anti-HSP90β mAb concentrations 0, 10 and 20 μg/ml (Fig. 3A). Moreover, the percentages of
HSP90β-JEV binding measured by FCM also significantly decreased
(P<0.01) from 76.17±0.69% (0 μg/ml anti-HSP90β mAb) to
19.15±0.35% (20 μg/ml anti-HSP90β mAb) and further to
10.43±0.53% (100 μg/ml anti-HSP90β mAb) compared with the
negative controls of 1.70±0.11% (Fig.
3B and C). These results clearly demonstrated that a specific
anti-HSP90β mAb inhibited HSP90β-JEV binding in a dose-dependent
manner.
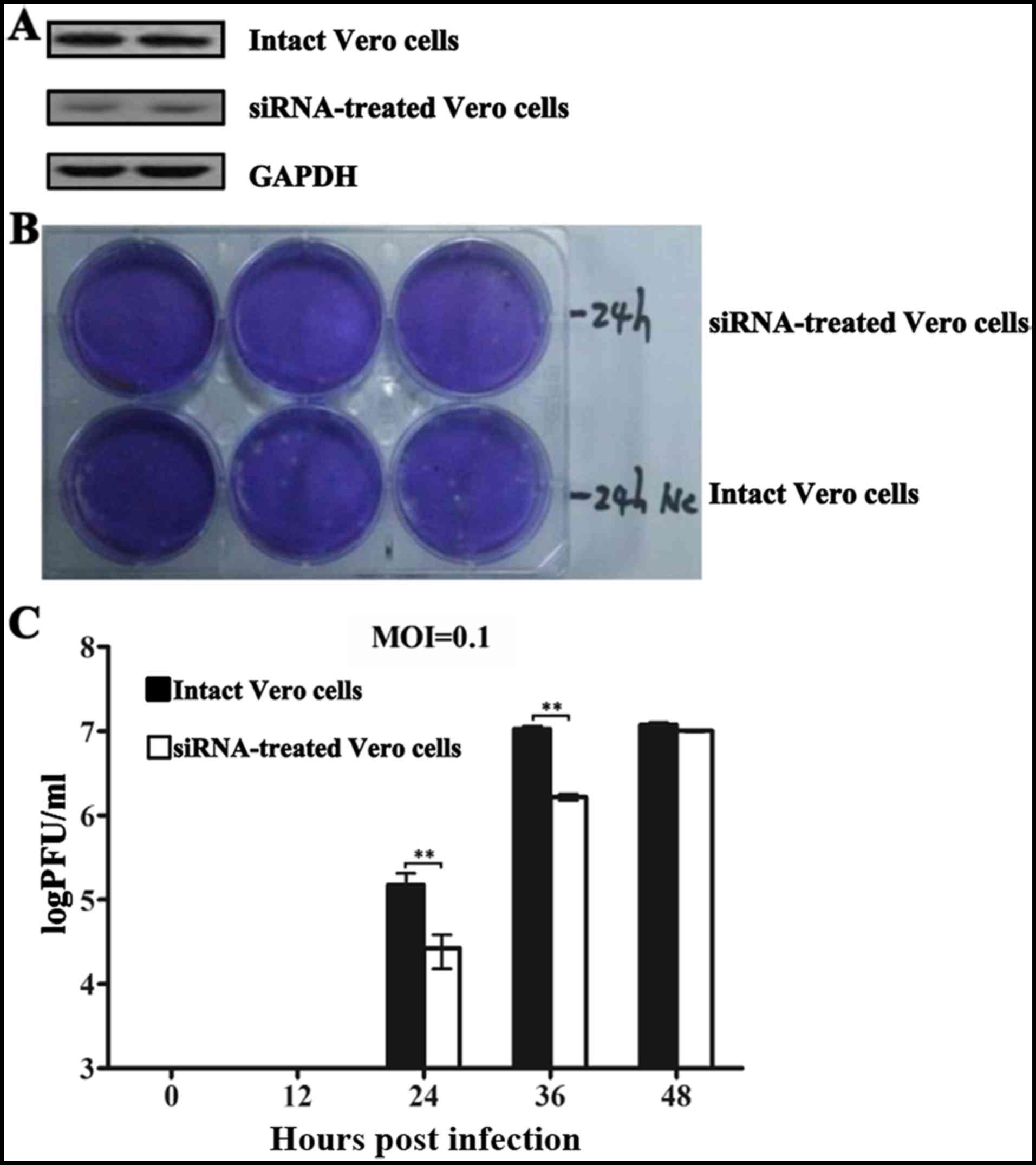
Decreased HSP90β expression in Vero cells
reduces JEV progeny yields
siRNAs subcloned in lentivirus vectors were
implemented to reduce HSP90β expression in Vero cells, and one
transfected cell line exhibited an over 50% steady inhibition of
HSP90β expression (Fig. 4A).
Subsequent sequencing data revealed that the 21-nt siRNA
(GGAUGACAGCGGUAAGGAUtt) was complementary to the human HSP90β mRNA
1012-1030 nt (GGATGACAGTGGTAA GGATAA), which is consistent with a
previous report (31). Overall,
the progeny virions in both intact and siRNA-treated Vero cell
culture supernatants were detected at 24 (Fig. 4B), 36 and 48 hpi. The commensurate
numbers of viral plaques at 36 and 48 hpi in the intact cells
indicated that fewer infectious viruses were released, implicating
that almost all cells became infected after 36 hpi when the MOI was
0.1. However, in the siRNA-treated cells, the virion yields were
significantly inhibited (P<0.01), particularly at 24 and 36 hpi,
by 6.5- and 5.6-fold, respectively (Fig. 4C).
Discussion
The conventional method for identifying virus
receptors is a virus overlay protein-binding assay (VOPBA), where
virions or virus attachment proteins (VAPs) are applied (20,32). In a VOPBA, cell membrane protein
extracts from virus-susceptible cells are first subjected to
SDS-PAGE, transferred onto a nitrocellulose membrane, then
incubated with virions or VAPs, and, finally, the specific
virus-binding protein is indicated by specific enzyme-conjugated
mAbs against the virus. However, VOPBA results cannot distinguish
the virus-binding protein but only its size (molecular weight)
(33,34). For a successful VOPBA experiment,
a single polypeptide must exhibit the tentative receptor activity,
bind viruses without other membrane components and maintain binding
activity in electrophoresis detergents (32). As it failed in a previous trial of
VOPBA on BHK-21 cells in our laboratory, we used a Co-IP assay
(35) instead to identify the
JEV-binding protein with the advantage of a direct virus-membrane
protein interaction under conditions that mimic physiological
conditions and more natural conformations to enhance the certainty
of protein binding. Due to the possible involvement of third party
proteins unrelated to JEV binding in the immunoprecipitates, MS was
necessarily introduced to probe the proteins in the precipitates
and provide clues to the candidate JEV receptor (JEVR) protein(s)
for further verification.
During the Co-IP experiments, unsurprisingly, more
than one protein precipitated, and the most notable 3 bands
(Fig. 1) compared with the
controls were subjected to MS analyses. The MS results (Table I) suggested that novel proteins at
approximately 95, 43 and 34 kDa are unnamed or hypothetical
proteins (scores over 30) based on a monkey protein database (genus
Macaca). Due to high genetic homogeneity between monkeys and
humans, the human protein database (Homo sapiens) was
quested, and the results revealed that the proteins were HSP90β, an
unnamed protein, the three-dimensional structure of glutathione
s-transferase, and aconitase 2 precursor. Possessing the highest
score of 60.3 among the four candidates, HSP90β is an entire,
functional protein and was then selected for further investigation,
though it is generally considered to be a cytoplasmic protein.
Considering the inefficient separation of the hydrophobic and
hydrophilic fractions during the cell membrane protein extraction
procedures, it was not surprising to discover that a few
hydrophilic proteins may be present in the hydrophobic fraction.
However, the hydrophobic proteins remained dominant in this
fraction. This finding is similar to our previous results when
using BHK-21 membrane proteins (36).
HSPs are a class of chaperone proteins that assist
other proteins in folding properly, stabilise proteins against heat
stress, aid in protein degradation and stabilise many proteins
required for tumour growth. They are the most highly conserved and
expressed cellular proteins across all species (37). As their name implies, HSPs protect
cells through increased expression from 1 to 2% of the total
proteins in unstressed cells to 4–6% in stressed cells upon
stimulation by elevated temperatures (38, and refs therein). HSP90
is one such heat-related chaperone protein and the '90' indicates
that it weighs approximately 90 kDa. It is found in bacteria and
all eukaryotes, but is absent in archaea (39), and its cytoplasmic counterpart is
essential for cell viability under all conditions in eukaryotes
(40).
Mammalian cells feature several HSP90 isoform
homologues (such as α1, α2 and β), which are defined by their
different coding genes (HSP90AA1, HSP90AA2 and HSP90AB1),
subcellular locations (cytosol, endoplasmic reticulum, and
mitochondria) (41), or
extracellular presence (42).
Human HSP90α is inducible, mostly forms anti-parallel homodimers,
and is 85% identical to the HSP90β amino acid sequence, which is
otherwise described as constitutive and a monomer due to several
single amino acid mutations in the C-terminal dimerization domain
(43–45).
No accurate HSP90β structure is available, even
though it shares significant protein homology with its isoform
HSP90α, which performs conservative functions in cells by forming
homodimers. The crystal structures indicate that HSP90α contains 3
functional domains: a highly conserved N-terminal domain (NTD) of
approximately 25 kDa, a middle domain (MD) of approximately 40 kDa
and a C-terminal domain (CTD) of approximately 12 kDa. The NTD
exhibits a common ATP binding pocket that is shared among HSP90
chaperone family members and is recognised by its natural
inhibitor, geldanamycin and its analogues (46,47), which target the adenosine
triphosphatase (ATPase activity) of the NTD region. The MD is
involved in client protein binding involving, for example, PKB/Akt1
and eNOS (48,49). The CTD features an alternative
ATP-binding site that is only accessible when the N-terminal
ATP-binding pocket is occupied (50). The CTD is also involved in protein
binding because it includes the tetratricopeptide repeat (TPR)
motif recognition site and conserved MEEVD pentapeptide that is
responsible for co-factor interactions, including the
immunophilins, stress-induced phosphoprotein 1 (Sti1/Hop),
cyclophilin-40, PP5 and Tom70 (51–53).
Over the past few years, researchers have reported
that several HSP family members (HSP90, HSP70, and HSC70) are
putative receptors to certain flaviviruses (23,24,26). Therefore, following the protein
clues suggested by the MS analyses, we testified the specific
binding of HSP90β in the Vero membrane protein extract and JEV
particles.
Using western blot analysis, we only produced
positive results under non-reducing and non-boiled conditions,
which is in contrast to the routine reducing method (data not
shown). The novel band at approximately 95 kDa from the cell
membrane protein extracts was reacted solely with either anti-human
HSP90β mAb recognizing the amino acid sequences 1–100 that
correspond to the human HSP90β N-terminus (see mAb manufacturer's
fact sheet; http://www.abcam.com/hsp90-beta-antibody-e296-ab32568.html)
or anti-JEV mAb against E protein at the same position around 90
kDa (Fig. 2A). These data suggest
that the HSP90β-JEV complex formed as it was recognised by two
different mAbs against the two respective complex components. In
addition, the non-reducing and non-boiled conditions imply that the
JEV binding activity of HSP90β may depend on the proper
conformation of its monomers, which is sensitive to the detergents
used in electrophoresis. This conformational requirement may
partially explain why the VOPBA produced good results for the
similar single peptide chain HSP70 (24,54), but did not demonstrate why HSP90β,
which is broadly expressed in mouse brain cells (55), did not bind JEV on Neuro2A cells
(24). For mouse microglial
(BV-2) cells, HSP family members, including HSP70, HSP90 and GRP78,
are also involved in JEV entry, but play a minor role in virus
internalization, which is in contrast to CD4 with a major entry
role (25). Furthermore, the
confocal laser scanning microscopic examination demonstrated that
HSP90β and JEV co-localised along the Vero cell surface (Fig. 2B), though HSP90β is generally
defined as a cytosolic protein.
When HSP90β-JEV binding was confirmed, an infection
inhibition test was conducted using IFA and FCM with a rabbit
anti-human HSP90β mAb at various concentrations. In the IFA
experiment, the HSP90β-JEV binding clearly decreased with the
increasing anti-HSP90β mAb concentrations, and the binding
percentages quantified by FCM also exhibited a significant decrease
(P<0.01) from 76.17±0.69% (0 μg/ml anti-HSP90β mAb) to
19.15±0.35% (20 μg/ml anti-HSP90β mAb) and further to
10.43±0.53% (100 μg/ml anti-HSP90β mAb) compared with the
negative control of 1.70±0.11% (Fig.
3B). These results clearly demonstrate that the specific
anti-HSP90β mAb inhibited HSP90β-JEV binding in a dose-dependent
manner. Moreover, it is reasonable to presume that the JEV-binding
domain of HSP90β is located at the N-terminus, which is recognised
by the anti-HSP90β mAb.
To elucidate the mechanisms through which HSP90β
influences JEV infection in Vero cells, siRNA technology was used
to knock down HSP90β expression instead of functional inhibitors,
such as geldanamycin, which interferes with HSP90β ATPase activity
(46). Several siRNA fragments
directed to the human HSP90β mRNA sequence were synthesised and
trans-fected into Vero cells via lentiviral vectors. We only
selected out one transfected cell line with over 50% steady
inhibition of HSP90β expression (Fig.
4A), whose siRNA sequence is identical to an effective one
reported earlier that corresponds to human HSP90β nt 1012–1030
(31). The plaque formation assay
results indicated that cells with reduced HSP90β expression
produced significantly less JEV progeny, particularly at 24 and 36
hpi (5.6-and 6.5-fold less, respectively).
Another report (56), stating that HSP90β is associated
JEV assembly, but not during the adsorption phase, confirms that
HSP90β-JEV interaction at the attachment stage is the predominant
factor for the decreased virus production.
In addition to interacting with JEV on the cell
surface, HSP90β facilitates enterovirus 71 viral particle assembly
in the human glioblastoma cell line SF268 (57). The different roles HSP90β plays
during the different viral replication stages may be another
intriguing field for investigation.
To summarise, we discovered that HSP90β in Vero cell
membranes bound JEV on the cell surface, and the HSP90β-JEV binding
as well as virus progeny were inhibited by anti-human HSP90β mAb
and siRNA. This is in contrast to the aforementioned reports, which
indicate that HSP70 family members are putative JEVR, HSP70 and its
counterparts do not react to JEV in Vero cells. Furthermore, JEV
seemed to prefer binding larger HSPs from HSC70 in mosquito cells
to HSP90 in primate cells. In other words, these high molecular
weight HSPs preserve the conservative conformation that distinctly
fits JEV, and shifts from HSC70, HSP70 and then to HSP90 over
millions years of evolution. To better support this presumption,
more information pertaining JEV binding molecules on human
susceptible cells is necessarily needed.
Acknowledgments
The authors gratefully acknowledge financial support
from the National Natural Scientific Foundation of China (no.
30872216 to T. D.). The funders did not play a role in the study
design, data collection and analysis, the decision to publish, or
the preparation of the manuscript. We would also thank the State
Key Laboratory of Biomembrane and Membrane Biotechnology, Institute
of Zoology, Chinese Academy of Sciences, Beijing for the MS
analyses.
References
|
1
|
Daep CA, Muñoz-Jordán JL and Eugenin EA:
Flaviviruses, an expanding threat in public health: Focus on
dengue, West Nile, and Japanese encephalitis virus. J Neurovirol.
20:539–560. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Misra UK and Kalita J: Overview: Japanese
encephalitis. Prog Neurobiol. 91:108–120. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yun SI and Lee YM: Japanese encephalitis:
The virus and vaccines. Hum Vaccin Immunother. 10:263–279. 2014.
View Article : Google Scholar
|
|
4
|
Mackenzie JS, Johansen CA, Ritchie SA, van
den Hurk AF and Hall RA: Japanese encephalitis as an emerging
virus: the emergence and spread of Japanese encephalitis virus in
Australasia. Curr Top Microbiol Immunol. 267:49–73. 2002.PubMed/NCBI
|
|
5
|
Tsai TF: New initiatives for the control
of Japanese encephalitis by vaccination: Minutes of a WHO/CVI
meeting, Bangkok, Thailand, 13–15 October 1998. Vaccine. 18:1–25.
2000. View Article : Google Scholar
|
|
6
|
Solomon T and Vaughn DW: Pathogenesis and
clinical features of Japanese encephalitis and West Nile virus
infections. Curr Top microbiol Immunol. 267:171–194.
2002.PubMed/NCBI
|
|
7
|
Solomon T and Winter PM: Neurovirulence
and host factors in flavivirus encephalitis-evidence from clinical
epidemiology. Arch Virol Suppl. 18:161–170. 2004.
|
|
8
|
Campbell GL, Hills SL, Fischer M, Jacobson
JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD and
Ginsburg AS: Estimated global incidence of Japanese encephalitis: a
systematic review. Bull World Health Organ. 89:766–774. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
No authors listed. Japanese Encephalitis
Vaccines: WHO position paper - February 2015. Wkly Epidemiol Rec.
90:69–87. 2015.
|
|
10
|
Ding D, Hong Z, Zhao SJ, Clemens JD, Zhou
B, Wang B, Huang MS, Zeng J, Guo QH, Liu W, et al: Long-term
disability from acute childhood Japanese encephalitis in Shanghai,
China. Am J Trop Med Hyg. 77:528–533. 2007.PubMed/NCBI
|
|
11
|
Sumiyoshi H, Mori C, Fuke I, Morita K,
Kuhara S, Kondou J, Kikuchi Y, Nagamatu H and Igarashi A: Complete
nucleotide sequence of the Japanese encephalitis virus genome RNA.
Virology. 161:497–510. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chambers TJ, Hahn CS, Galler R and Rice
CM: Flavivirus genome organization, expression, and replication.
Annu Rev Microbiol. 44:649–688. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McMinn PC: The molecular basis of
virulence of the encephalitogenic flaviviruses. J Gen Virol.
78:2711–2722. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Winkler G, Heinz FX and Kunz C: Studies on
the glycosylation of flavivirus E proteins and the role of
carbohydrate in antigenic structure. Virology. 159:237–243. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luca VC, AbiMansour J, Nelson CA and
Fremont DH: Crystal structure of the Japanese encephalitis virus
envelope protein. J Virol. 86:2337–2346. 2012. View Article : Google Scholar :
|
|
16
|
Rey FA, Heinz FX, Mandl C, Kunz C and
Harrison SC: The envelope glycoprotein from tick-borne encephalitis
virus at 2 A resolution. Nature. 375:291–298. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Allison SL, Schalich J, Stiasny K, Mandl
CW and Heinz FX: Mutational evidence for an internal fusion peptide
in flavivirus envelope protein E. J Virol. 75:4268–4275. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kolaskar AS and Kulkarni-Kale U:
Prediction of three-dimensional structure and mapping of
conformational epitopes of envelope glycoprotein of Japanese
encephalitis virus. Virology. 261:31–42. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu KP, Wu CW, Tsao YP, Kuo TW, Lou YC, Lin
CW, Wu SC and Cheng JW: Structural basis of a flavivirus recognized
by its neutralizing antibody: Solution structure of the domain III
of the Japanese encephalitis virus envelope protein. J Biol Chem.
278:46007–46013. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kimura T, Kimura-Kuroda J, Nagashima K and
Yasui K: Analysis of virus-cell binding characteristics on the
determination of Japanese encephalitis virus susceptibility. Arch
Virol. 139:239–251. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su CM, Liao CL, Lee YL and Lin YL: Highly
sulfated forms of heparin sulfate are involved in japanese
encephalitis virus infection. Virology. 286:206–215. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu SC, Chiang JR and Lin CW: Novel cell
adhesive glycosaminoglycan-binding proteins of Japanese
encephalitis virus. Biomacromolecules. 5:2160–2164. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren Junping LY, Wei Z, Jing Y and Chin Ma
W: Isolation and preliminary identification of 74×103 molecule as
putative Japanese encephalitis virus receptor. J Microbiol Immunol.
29:307–311. 2009.
|
|
24
|
Das S, Laxminarayana SV, Chandra N, Ravi V
and Desai A: Heat shock protein 70 on Neuro2a cells is a putative
receptor for Japanese encephalitis virus. Virology. 385:47–57.
2009. View Article : Google Scholar
|
|
25
|
Thongtan T, Wikan N, Wintachai P,
Rattanarungsan C, Srisomsap C, Cheepsunthorn P and Smith DR:
Characterization of putative Japanese encephalitis virus receptor
molecules on microglial cells. J Med Virol. 84:615–623. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chu JJ, Leong PW and Ng ML:
Characterization of plasma membrane-associated proteins from Aedes
albopictus mosquito (C6/36) cells that mediate West Nile virus
binding and infection. Virology. 339:249–260. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang MJ, Wang MX, Jiang SZ, Xiu ZZ and Ma
WY: Preparation and characterization of the monoclonal antibodies
against Japanese encephalitis virus. Acta Virol. 36:533–540.
1992.PubMed/NCBI
|
|
28
|
Perkins DN, Pappin DJ, Creasy DM and
Cottrell JS: Probability-based protein identification by searching
sequence databases using mass spectrometry data. Electrophoresis.
20:3551–3567. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Everett RD: Study of early events during
herpes simplex virus type 1 infection by confocal microscopy.
Methods. 55:144–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Henschel A, Buchholz F and Habermann B:
DEQOR: A web-based tool for the design and quality control of
siRNAs. Nucleic Acids Res. 32:Web Server. W113–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schulz R, Marchenko ND, Holembowski L,
Fingerle-Rowson G, Pesic M, Zender L, Dobbelstein M and Moll UM:
Inhibiting the HSP90 chaperone destabilizes macrophage migration
inhibitory factor and thereby inhibits breast tumor progression. J
Exp Med. 209:275–289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haywood AM: Virus receptors: Binding,
adhesion strengthening, and changes in viral structure. J Virol.
68:1–5. 1994.PubMed/NCBI
|
|
33
|
Salas-Benito JS and del Angel RM:
Identification of two surface proteins from C6/36 cells that bind
dengue type 4 virus. J Virol. 71:7246–7252. 1997.PubMed/NCBI
|
|
34
|
Martínez-Barragán JJ and del Angel RM:
Identification of a putative coreceptor on Vero cells that
participates in dengue 4 virus infection. J Virol. 75:7818–7827.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li W, Moore MJ, Vasilieva N, Sui J, Wong
SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough
TC, et al: Angiotensin-converting enzyme 2 is a functional receptor
for the SARS coronavirus. Nature. 426:450–454. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding T, Zhang W, Ma W and Ren J:
Identification of a mutated BHK-21 cell line that became less
susceptible to Japanese encephalitis virus infection. Virol J.
8:1152011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Csermely P, Schnaider T, Soti C, Prohászka
Z and Nardai G: The 90-kDa molecular chaperone family: Structure,
function, and clinical applications. A comprehensive review.
Pharmacol Ther. 79:129–168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crevel G, Bates H, Huikeshoven H and
Cotterill S: The Drosophila Dpit47 protein is a nuclear Hsp90
co-chaperone that interacts with DNA polymerase alpha. J Cell Sci.
114:2015–2025. 2001.PubMed/NCBI
|
|
39
|
Chen B, Zhong D and Monteiro A:
Comparative genomics and evolution of the HSP90 family of genes
across all kingdoms of organisms. BMC Genomics. 7:1562006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thomas JG and Baneyx F: Roles of the
Escherichia coli small heat shock proteins IbpA and IbpB in thermal
stress management: Comparison with ClpA, ClpB, and HtpG In vivo. J
Bacteriol. 180:5165–5172. 1998.PubMed/NCBI
|
|
41
|
Chen B, Piel WH, Gui L, Bruford E and
Monteiro A: The HSP90 family of genes in the human genome: Insights
into their divergence and evolution. Genomics. 86:627–637. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Correia AL, Mori H, Chen EI, Schmitt FC
and Bissell MJ: The hemopexin domain of MMP3 is responsible for
mammary epithelial invasion and morphogenesis through extracellular
interaction with HSP90β. Genes Dev. 27:805–817. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nemoto T, Ohara-Nemoto Y, Ota M, Takagi T
and Yokoyama K: Mechanism of dimer formation of the 90-kDa
heat-shock protein. Eur J Biochem. 233:1–8. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Barginear MF, Van Poznak C, Rosen N, Modi
S, Hudis CA and Budman DR: The heat shock protein 90 chaperone
complex: An evolving therapeutic target. Curr Cancer Drug Targets.
8:522–532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kobayakawa T, Yamada S, Mizuno A and
Nemoto TK: Substitution of only two residues of human Hsp90alpha
causes impeded dimerization of Hsp90beta. Cell Stress Chaperones.
13:97–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ochel HJ, Eichhorn K and Gademann G:
Geldanamycin: the prototype of a class of antitumor drugs targeting
the heat shock protein 90 family of molecular chaperones. Cell
Stress Chaperones. 6:105–112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kamal A, Thao L, Sensintaffar J, Zhang L,
Boehm MF, Fritz LC and Burrows FJ: A high-affinity conformation of
Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature.
425:407–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sato S, Fujita N and Tsuruo T: Modulation
of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA.
97:10832–10837. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fontana J, Fulton D, Chen Y, Fairchild TA,
McCabe TJ, Fujita N, Tsuruo T and Sessa WC: Domain mapping studies
reveal that the M domain of hsp90 serves as a molecular scaffold to
regulate Akt-dependent phosphorylation of endothelial nitric oxide
synthase and NO release. Circ Res. 90:866–873. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Söti C, Rácz A and Csermely P: A
Nucleotide-dependent molecular switch controls ATP binding at the
C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a
C-terminal binding pocket. J Biol Chem. 277:7066–7075. 2002.
View Article : Google Scholar
|
|
51
|
Young JC, Obermann WM and Hartl FU:
Specific binding of tetratricopeptide repeat proteins to the
C-terminal 12-kDa domain of hsp90. J Biol Chem. 273:18007–18010.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pearl LH and Prodromou C: Structure,
function, and mechanism of the Hsp90 molecular chaperone. Adv
Protein Chem. 59:157–186. 2001. View Article : Google Scholar
|
|
53
|
Wandinger SK, Richter K and Buchner J: The
Hsp90 chaperone machinery. J Biol Chem. 283:18473–18477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Daugaard M, Rohde M and Jäättelä M: The
heat shock protein 70 family: Highly homologous proteins with
overlapping and distinct functions. FEBS Lett. 581:3702–3710. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Loones MT, Chang Y and Morange M: The
distribution of heat shock proteins in the nervous system of the
unstressed mouse embryo suggests a role in neuronal and
non-neuronal differentiation. Cell Stress Chaperones. 5:291–305.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hung CY, Tsai MC, Wu YP and Wang RY:
Identification of heat-shock protein 90 beta in Japanese
encephalitis virus-induced secretion proteins. J Gen Virol.
92:2803–2809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang RY, Kuo RL, Ma WC, Huang HI, Yu JS,
Yen SM, Huang CR and Shih SR: Heat shock protein-90-beta
facilitates enterovirus 71 viral particles assembly. Virology.
443:236–247. 2013. View Article : Google Scholar : PubMed/NCBI
|