Introduction
Stroke is a serious threat to human health (1), and ischemia/reperfusion (I/R) injury
(IRI) is an important pathophysiological mechanism of ischemic
stroke (2). Although the
mechanisms of IRI are complex, increasing evidence indicates that
the immune inflammatory response plays an important role in this
process (2,3). Microglial cells are residential
macrophages in the central nervous system (CNS). As the first
defensive line against pathogenic microorganisms, microglial cells
provide innate immune signaling and adaptive immune responses
(4). Microglial activation is
considered to be a hallmark of neuroinflammation. It is well
documented that the activation of microglia in various
neurodegenerative diseases contributes to neuroinflammation through
the release of large amounts of pro-inflammatory cytokines. Some of
these are neurotoxic, suggesting that activated microglia
participate in acute brain injury and cerebral ischemia (5,6).
Toll-like receptors (TLRs) are a family of
transmembrane receptors that are able to recognize
pathogen-associated molecular patterns. As essential components of
the microglial innate immune response, TLRs induce the production
of neurotoxic factors from microglia, contributing to neuronal
damage (7,8). The activation of TLR2 leads to the
activated microglial expression of pro-inflammatory cytokines,
including interleukin (IL)-23, IL-17, IL-1β and tumor necrosis
factor-α (TNF-α) in various neuroimmunological diseases, such as
multiple sclerosis, experimental autoimmune encephalomyelitis and
IRI (4,9–12).
Sphingosine-1-phosphate (S1P) can regulate cell
proliferation, survival, apoptosis, migration and Ca2+
osmotic balance (13). The S1P
signaling pathway in the CNS plays a role in important processes,
such as neurotransmitter release, proliferation and cell survival
(14). As an intracellular second
messenger and an extracellular ligand that interacts with G
protein-coupled receptors (15,16), S1P regulates peripheral
macrophages and immune cell function (17–19), which are involved in major
pathophysiological mechanisms in autoimmune diseases of the CNS
(20,21). Sphingosine kinase (Sphk) is a key
enzyme in S1P synthesis (22,23) and plays an important role in human
immune cell chemotaxis and wound healing processes. It has two
isoforms, Sphk1 and Sphk2 (20,24). Sphk1 is the main source of Sphk
activity in brain tissue (25,26). BV2 microglial cells and purified
microglia from primary cultures have been shown to express some or
all of the five S1P receptors (27). The Sphk1/S1P signaling pathway has
been shown to be involved in the inflammation of peripheral immune
cells and BV2 cells through autocrine/paracrine pathways and to
promote the production of TNF-α, IL-1β and nitric oxide (NO)
through the nuclear factor-κB (NF-κB) pathway (20,28,29). The Sphk1-specific inhibitor,
N,N-dimethylsphingosine (DMS), or Sphk1 knockout can
inhibit microglial cells expressing NF-κB, TNF-α, IL-1β and
inducible nitric oxide synthase (iNOS), as well as reduce TNF-α and
NO release (28,29). Resting microglia typically express
very low levels of sphingolipid metabolities, including Sphk1
(30). Exposure to
Lipopolysaccharide (LPS) or hypoxia upregulates Sphk1 expression in
amoeboid microglia and BV2 cells, and Sphk1 plays a crucial role in
the early stages of CNS inflammation (28,29).
It has been suggested that TLR2 in microglia
mediates the activation of the innate immune system by the
production of pro-inflammatory cytokines in cerebral I/R (12). Sphk1 in microglia is involved in
hypoxic brain damage and inflammation (29). Moreover, TLR2 signaling results in
NF-κB formation and then induces the production of pro-inflammatory
signals, such as IL-1β (31–34). Sphk1 is involved in LPS-induced
NF-κB activation, and DMS can block LPS-stimulated NF-κB expression
(35). Based on this evidence, we
hypotheseized that TLR2 is closely associated with Sphk1 in
activated microglial cells and is involved in the inflammatory
response following cerebral I/R. In the present study, we assessed
the TLR2 and Sphk1 expression levels in activated microglia during
the course of cerebral I/R. We then examined the effect of the TLR2
and Sphk1 on the release of the pro-inflammatory cytokines, IL-1β,
TNF-α, IL-17 and IL-23. We also explored the crosstalk between TLR2
and Sphk1.
Materials and methods
Animals
Clean grade healthy male C57BL/6 mice (10–12 weeks
old, weighing 25–30 g) were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice
were housed in an animal room that was sealed and sterilized by
ultraviolet (UV) light irradiation, room temperature 27°C and 40%
humidity. Mouse feed, litter and drinking water were autoclaved
(120°C, 40 min). The mice were randomly divided into a
sham-operated group (n=54), an I/R group (n=54), an I/R + TLR2
antibody group (n=36), and an I/R + DMS group (n=36). Each of these
groups was randomly divided into subgroups of ischemia for 1 h
followed by reperfusion for 12, 24 or 48 h. In the sham-operated
and I/R groups, the mice in each subgroup (I/R 12-h subgroup, n=18;
I/R 24-h subgroup, n=18; I/R 48-h subgroup, n=18) were separately
used for immunohistochemistry (n=6), fluorescence quantitative
polymerase chain reaction (PCR) (n=6) and enzyme-linked
immunosorbent assay (ELISA) (n=6). In the I/R + TLR2 antibody and
I/R + DMS groups, the mice in each sub-group (I/R 12-h subgroup,
n=12; I/R 24-h subgroup, n=12; I/R 48-h subgroup, n=12) were
separately used for fluorescence quantitative PCR (n=6) and ELISA
(n=6). This study was approved by the Ethics Board of the First
Affiliated Hospital of Harbin Medical University, Harbin, China and
all animal experiments were performed in compliance with the
principles and procedures outlined in the Care and Use of
Laboratory Animals guidelines, which is published by the China
National Institute of Health and approved by the Institutional
Animal Care and Use Committee.
Reagents and antibodies
Mouse anti-TLR2 monoclonal antibody [T2.5]
(ab59711), mouse anti-Iba1 monoclonal antibody (ab15690; used as
marker for microglia) and mouse anti-glial fibrillary acidic
protein (GFAP) monoclonal antibody (ab10062) were purchased from
Abcam (Cambridge, UK); rabbit anti Sphk1 polyclonal antibody
(sc-48825) and rabbit anti-TLR2 polyclonal antibody (sc-10739) were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA); fluorescence fluorescein isothiocyanate (FITC)-labeled goat
anti-mouse secondary antibody (ZF-0312) and fluorescent
rhodamine-labeled goat anti-rabbit secondary antibody (ZF-0316)
were purchased from Beijing Zhongshan Golden Bridge Technology Co.
(Beijing, China); the Ultrapure RNA extraction kit (CW0581),
HiFi-MMLV cDNA First Strand synthesis kit (CW0744), UltraSYBR
Mixture (with Rox) (CW0956), and DNase I (CW2090A) were purchased
from Beijing Kangwei Century Biotechnology Co. (Beijing, China);
immunoassay kits (IL-23, IL-17, IL-1, TNF-α, no. 870257) were
purchased from LightArray Biotech Co. (Shanghai, China); and DMS
(62575) was purchased from Cayman Chemical (Ann Arbor, MI,
USA).
Establishment of focal cerebral I/R
model
The suture method was used to occlude the cerebral
middle artery of the mice to establish a focal cerebral I/R model.
Briefly, 10% chloral hydrate (intraperitoneal injection of 3.5
ml/kg body weight) was used to anesthetize the mice, and the
carotid artery, external carotid artery and internal carotid artery
were then isolated and exposed. A monofilament nylon wire coated
with poly-D-lysine (diameter 0.2 mm) was inserted into the internal
carotid artery through the external carotid artery for
approximately 10–12 mm to induce ischemia in the middle cerebral
artery of the brain. The wire was removed after 1 h to achieve
reperfusion. Following 45 min of ischemia, the mice in the I/R +
TLR2 antibody group were injected with TLR2 antibody [T2.5] (0.05
μg/mouse) through the tail vein, and the mice in the I/R +
DMS group received intraperitoneal injections of DMS (400
μg/kg) (29,64). In the sham-operated group, a wire
was inserted into the carotid artery, but was immediately pulled
out after encountering any resistance. A thermal blanket was used
to maintain rectal temperature within 37±1°C.
Immunohistochemistry
The double-label immunofluorescence method was used
to observe the expression of TLR2 and Sphk1 in microglial cells
during the process of cerebral I/R. Each group of mice were
reperfused for 12, 24 or 48 h, then anesthetized with an
intraperitoneal injection of 10% chloral hydrate (3.5 ml/kg body
weight) before they were perfused with 4% paraformaldehyde (pH
7.4). The brain tissues were removed and stored at −80°C. A
microtome cryostat was used to prepare continuous sections, and the
slice thickness was approximately 4 μm. The slices were
mounted on glass plates pre-coated with 2% APES, fixed with acetone
for 1 min, then placed in a 4°C thermostatic showcase for storage.
The sections were then washed in 0.01 mol/l phosphate-buffered
saline (PBS, pH 7.4) for 5 min, and goat serum was used to prevent
non-specific antibody binding to the tissue. The slices were placed
under room temperature in a wet box for 30 min, followed by
overnight incubation with primary antibodies at 4°C. The
corresponding secondary antibodies were incubated after washing
with PBS. Images of the tissue sections were acquired using a
fluorescence microscope (DMI6000B; Leica, Wetzlar, Germany) at a
magnification of ×200.
Fluorescence quantitative PCR
A fluorescence quantitative PCR method was used to
measure the mRNA levels of TLR2 and Sphk1 in ischemic brain tissue.
Each group of mice after 12, 24 or 48 h of reperfusion were then
anesthetized with 10% chloral hydrate (3.5 ml/kg body weight) by
intraperitoneal injection. The ischemic side brain tissue was
storeed at −80°C in a low temperature freezer for future use.
Primers for the real-time PCR detection of the target gene were
designed and synthesized by Beijing Kangwei Century Biotechnology
Co. and were as follows: TLR2 forward, CAGTCCCAAAGTCT AAAGTC and
reverse, CTACGGGCAGTGGTGAAAAC, amplification product 166 bp; Sphk1
forward, GGAACC AGTAGAATGCCCTC and reverse, GGTTCTTCCGTTCGG TGAGT,
amplification product 173 bp. An ultrapure RNA extraction kit (cat
no. CW0581; CW Bio. Co., Ltd., Beijing, China) was used to extract
total RNA amount from the tissue. Samples containing 5 μl
RNA were assessed with 1% agarose gel electrophoresis, and a
HiFi-MMLV cDNA first-strand synthesis kit (cat no. CW0744; CW Bio.
Co., Ltd.) was used to perform reverse transcription. Using an
LC-480II quantitative PCR machine, the 2−ΔΔCT method was
used for the quantitative analysis of related data. Each sample was
analyzed in duplicate.
ELISA
A multi-factor protein biomarker detection system
(purchased from LightArray Biotech Co.) was used to measure the
protein contents of IL-23, IL-17, IL-1β and TNF-α in the ischemic
brain tissue. Following reperfusion for 12, 24 or 48 h, each group
of mice was anesthetized with 10% chloral hydrate (3.5 ml/kg body
weight) by intraperitoneal injection, and the ischemic brain tissue
was collected and stored at −80°C. Mouse brain tissue was
homogenized, then 50 μl standard sample or 50 μl test
samples were incubated under 20–25°C (200 rpm for 3 h).
Subsequently, 50 μl biotinylated antibody reagent were added
to each well and incubated at 20–25°C (200 rpm for 30 min). This
was followed by the addition of 50 μl
streptavidin-biotin-horseradish peroxidase reagent and incubated
the mixtures at 20–25°C (200 rpm for 30 min). Finally, 50 μl
peroxide and SuperSignal Luminol/Enhancer mixtures were added to
each well and the fluorescence was measured using a CCD camera. A
Cirascan scanning and analysis system (Aushon BioSystems, Inc.,
Billerica, MA, USA) was used to scan the images, and CIRAS multiple
analysis system software was employed to analyze and calculate the
light signal density in the image. Each sample was analyzed in
duplicate.
Statistical analysis
In this study, the immunofluorescence quantitative
PCR and ELISA results are expressed as mean ± SD. Statistical
analysis was evaluated using one-way analysis of variance (ANOVA;
SPSS version 19.0; IBM, Armonk, NY, USA). A value of p<0.05 was
considered to indicate a statistically significant difference.
Results
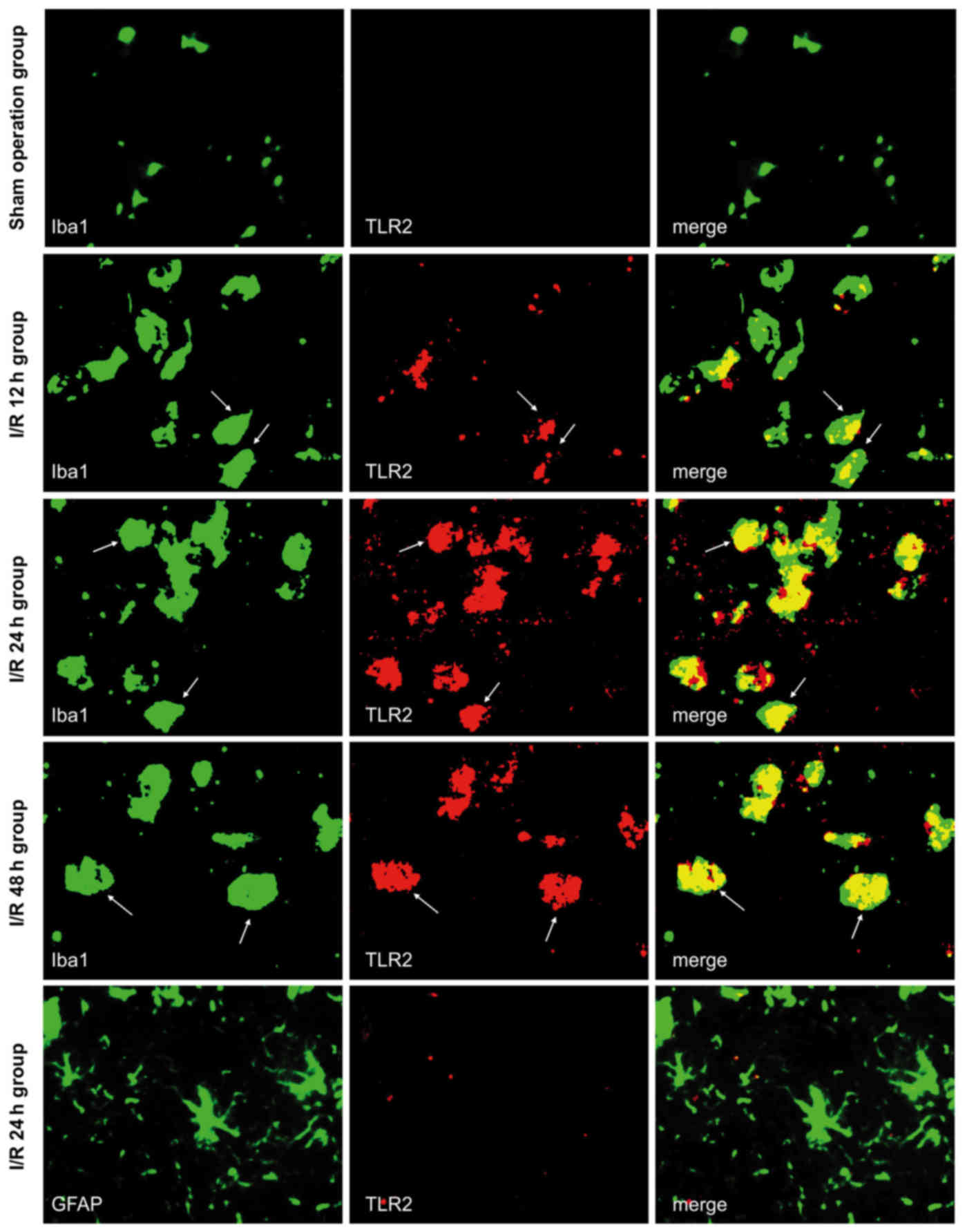
TLR2 is expressed in microglia in
response to cerebral I/R and is unaffected by Sphk1
Double immunofluorescence labeling was used to
observe the co-expression of TLR2/Iba1 and TLR2/GFAP in the brain
tissue of the sham-operated and I/R groups. Both of these exhibited
green fluorescence, as shown in Fig.
1, whereas TLR2 exhibited red fluorescence. All the images were
taken from the peri-infarct cortex at 12, 24, and 48 h following
cerebral I/R, and the same area was assessed in the sham-operated
group. The majority of TLR2-positive cells also expressed Iba1
(marker for microglia), while TLR2 did not co-localize with GFAP in
any subgroup of the sham-operated or I/R groups. Although
Iba1-positive cells were present, no TLR2 signal was observed in
sham-operated mice. In the I/R group, TLR2-positive cells were
found in the peri-infarct tissue. Fewer were observed in the I/R
12-h subgroup, higher levels were noted in the I/R 24-h subgroup,
and slightly lower levels were noted in the I/R 48-h subgroup.
These results suggested that cerebral I/R may activate microglia
and sequentially induce the expression of TLR2, which is important
for inflammatory responses.
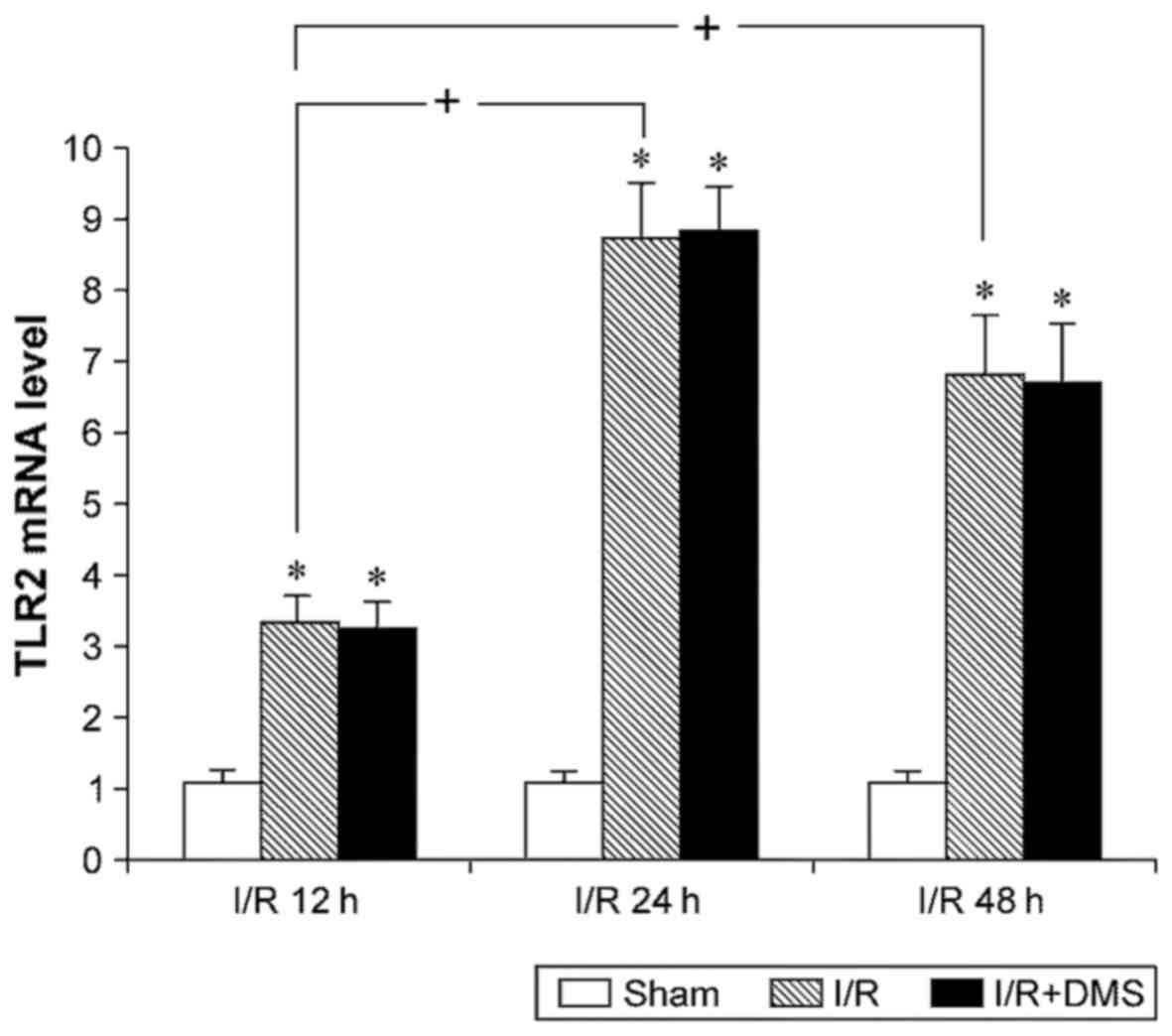
The results of fluorescence quantitative PCR for
TLR2 revealed that I/R upregulated the mRNA expression level of
TLR2. Compared with sham-operated mice, the TLR2 mRNA level in the
I/R brain tissue was higher at all time points (p<0.05).
Moreover, the TLR2 mRNA levels in both the I/R 24- and 48-h
subgroups were higher than those in the 12-h subgroup (p<0.05),
although no significant differences were observed between them at
24 and 48 h (p>0.05). The Sphk1-specific inhibitor, DMS, was
used to inhibit Sphk1 activity and to examine the effects of Sphk1
on TLR2. The data demonstrated that DMS did not affect the TLR2
levels in the I/R group (p>0.05) (Fig. 2).
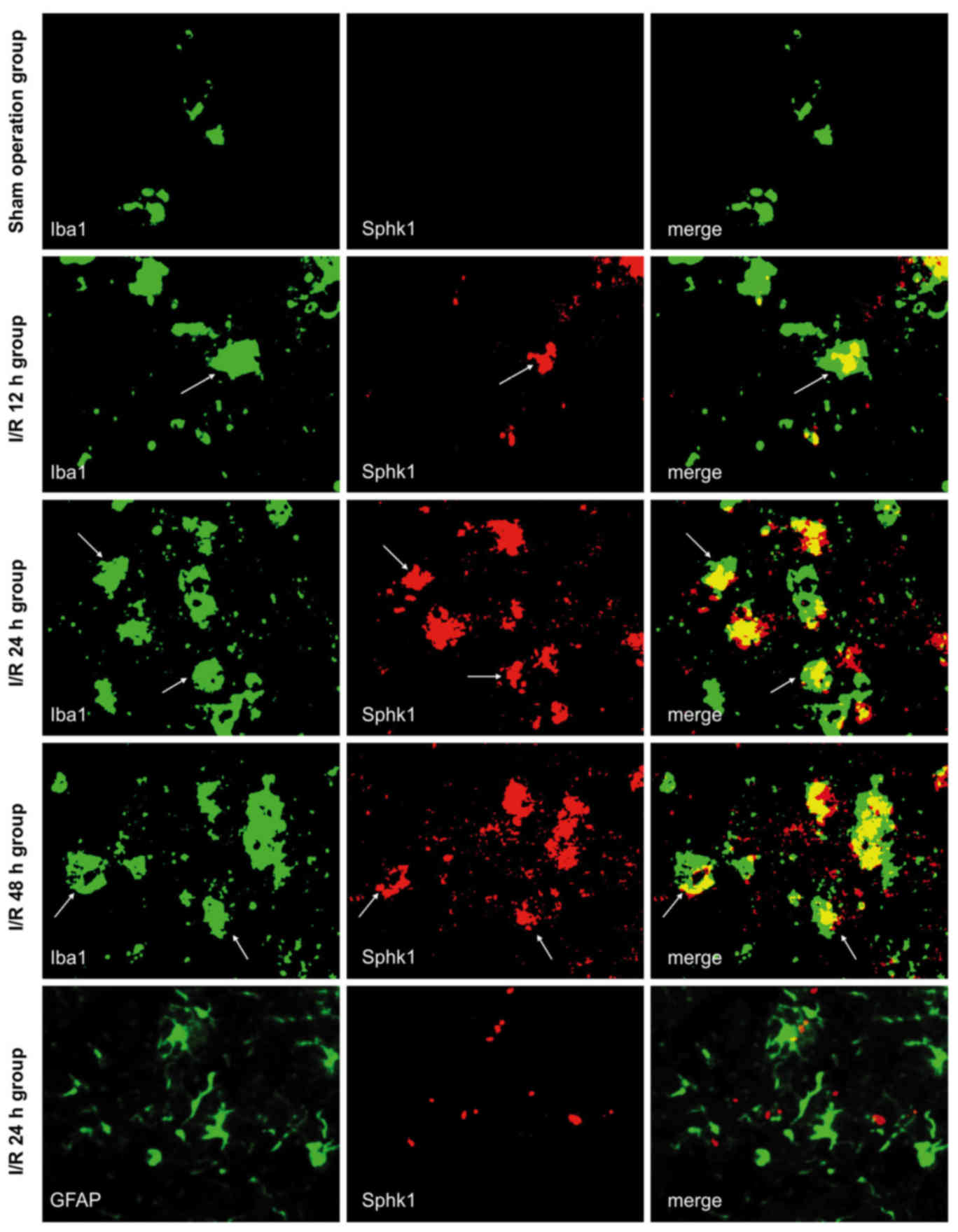
Sphk1 is expressed in microglia in
response to cerebral I/R and is dependent on TLR2
Immunofluorescence double labeling was used to
observe the co-expression of Sphk1/Iba1 and Sphk1/GFAP in brain
tissue from each group (Fig. 3).
All the images were taken from the peri-infarct cortex of I/R mice,
and the same area was observed in the sham-operated group. The
results were similar to those obtained for TLR2; the majority of
Sphk1-positive cells also expressed Iba1, while Sphk1 did not
co-localize with GFAP in any subgroup of the sham-operated or I/R
groups. Although the Iba1-positive cells were present, no Sphk1 was
observed in the sham-operated mice. In the I/R group,
Sphk1-positive cells appeared after 12 h, peaked at 24 h and
slightly decreased at 48 h. These results suggested that cerebral
I/R may activate microglia and sequentially induce the expression
of Sphk1, which showed temporal dynamics consistent with TLR2.
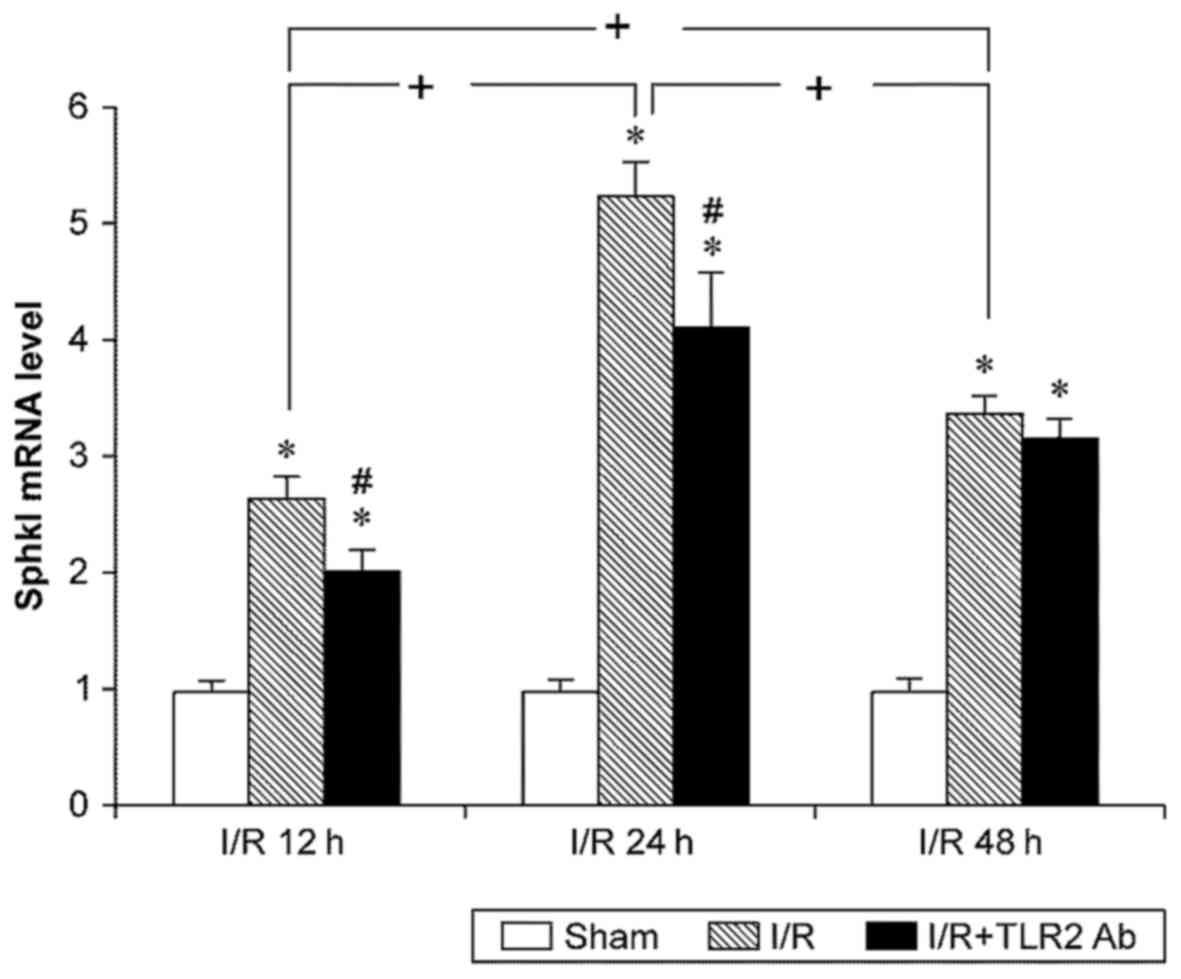
The data of fluorescence quantitative PCR for Sphk1
indicated that I/R led to an increase in Sphk1 mRNA expression.
Similar to the results of TLR2, the Sphk1 mRNA levels in sI/R mice
were higher than those in the sham operation group at any
time-point (p<0.05). TLR2 mRNA increased after 12 h reperfusion,
peaked at 24 h, and slightly decreased at 48 h compared with 24 h.
Anti-TLR2 monoclonal antibody was used to inhibit TLR2 activity and
to examine the effects of TLR2 on Sphk1. Notably, TLR2
downregulated the Sphk1 mRNA levels both in the 12- and 24-h I/R
subgroups (p<0.05). However, TLR2 did not have any significant
effects on the Sphk1 levels in the 48-h I/R subgroup (p>0.05)
(Fig. 4).
Suppression of either TLR2 or Sphk1
inhibits the generation of pro-inflammatory cytokines following
I/R
To further confirm the roles of TLR2 and Sphk1 in
I/R-induced inflammation, the IL-β, TNF-α, IL-17 and IL-23 contents
in brain tissue from each group were determined. The results
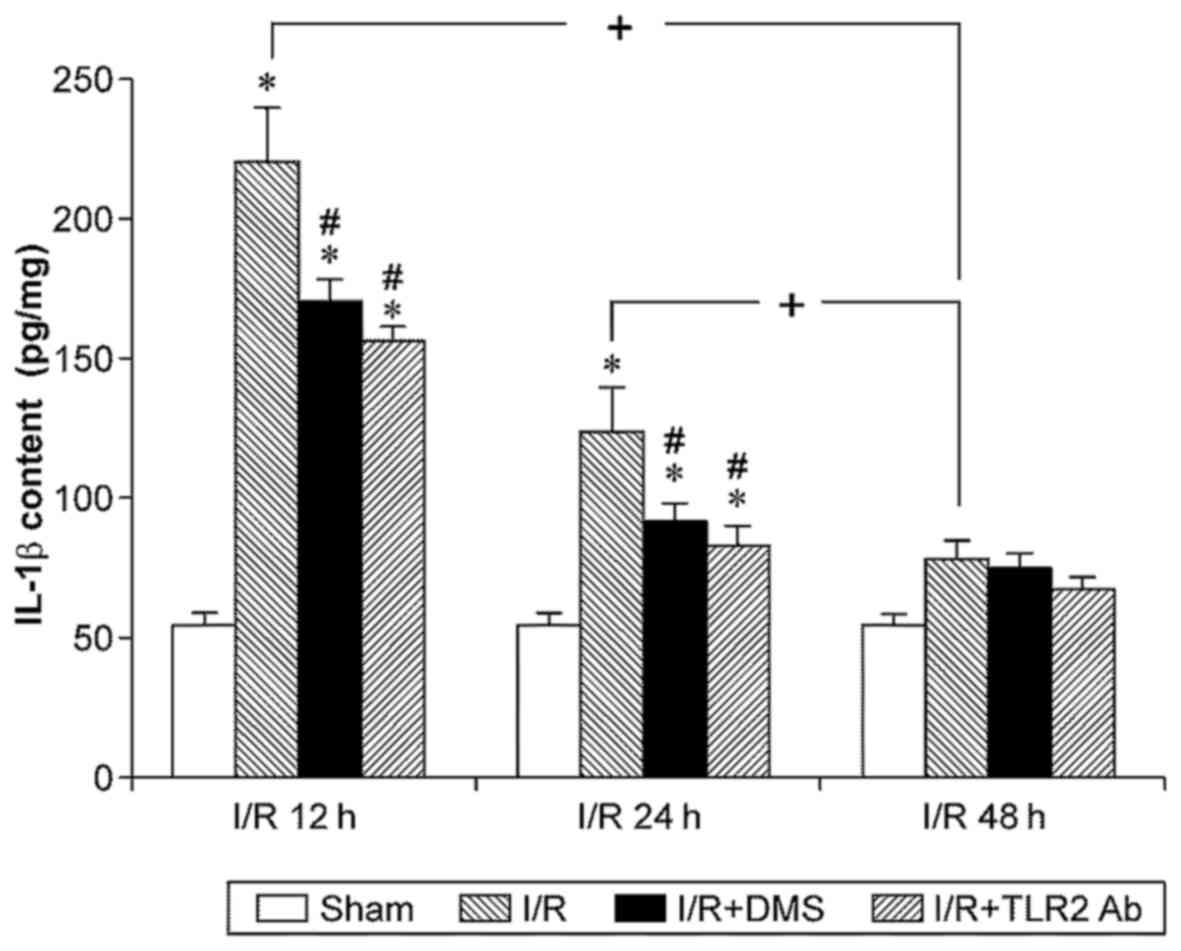
revealed that IL-1β was expressed at significantly higher levels in
the ischemic brain tissue after 12 or 24 h of reperfusion compared
with the sham-operated group (p<0.05). However, no significant
differences were observed between the sham-operated group and the
48-h I/R subgroup (p>0.05). Although the IL-1β level in the 12-h
I/R subgroup appeared higher than that in the 24-h I/R subgroup,
the difference was not significant (p>0.05). Importantly,
treatment with TLR2 antibody or DMS significantly decreased the
IL-1β content in the samples from the the 12- and 24-h I/R
subgroups (p<0.05) (Fig.
5).
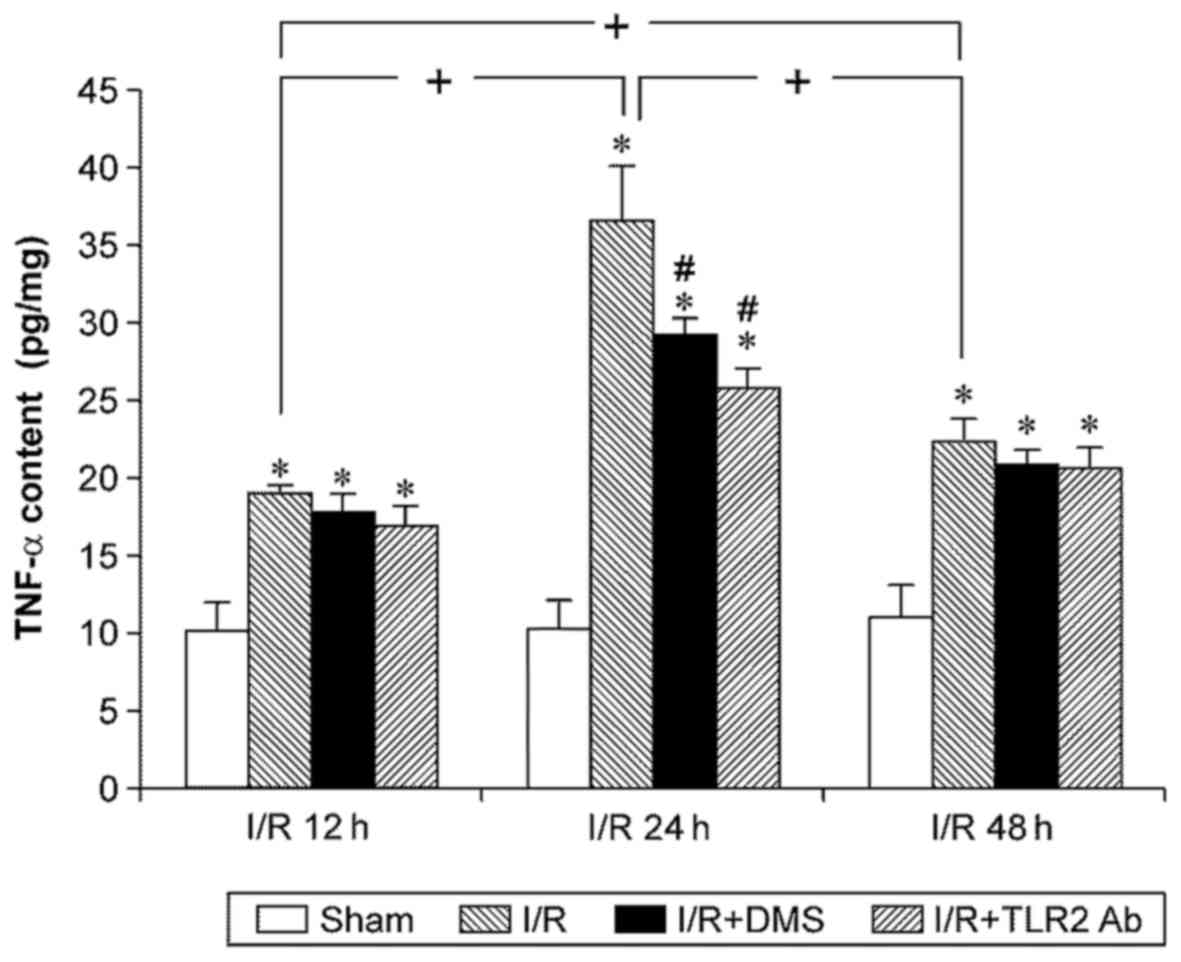
Cerebral I/R significantly increased the expression
of TNF-α at any time point (p<0.05). The TNF-α level in the I/R
group increased at 12 h of reperfusion, peaked at 24 h of
reperfusion, and began to decrease at 48 h after reperfusion.
Treatment with both TLR2 antibody and DMS reduced the TNF-α level
in the ischemic brain tissue in the I/R group at 24 h (p<0.05).
Nonetheless, neither TLR2 antibody nor DMS had any effects on the
TNF-α level in the 12-h subgroup or the 48-h subgroup (Fig. 6).
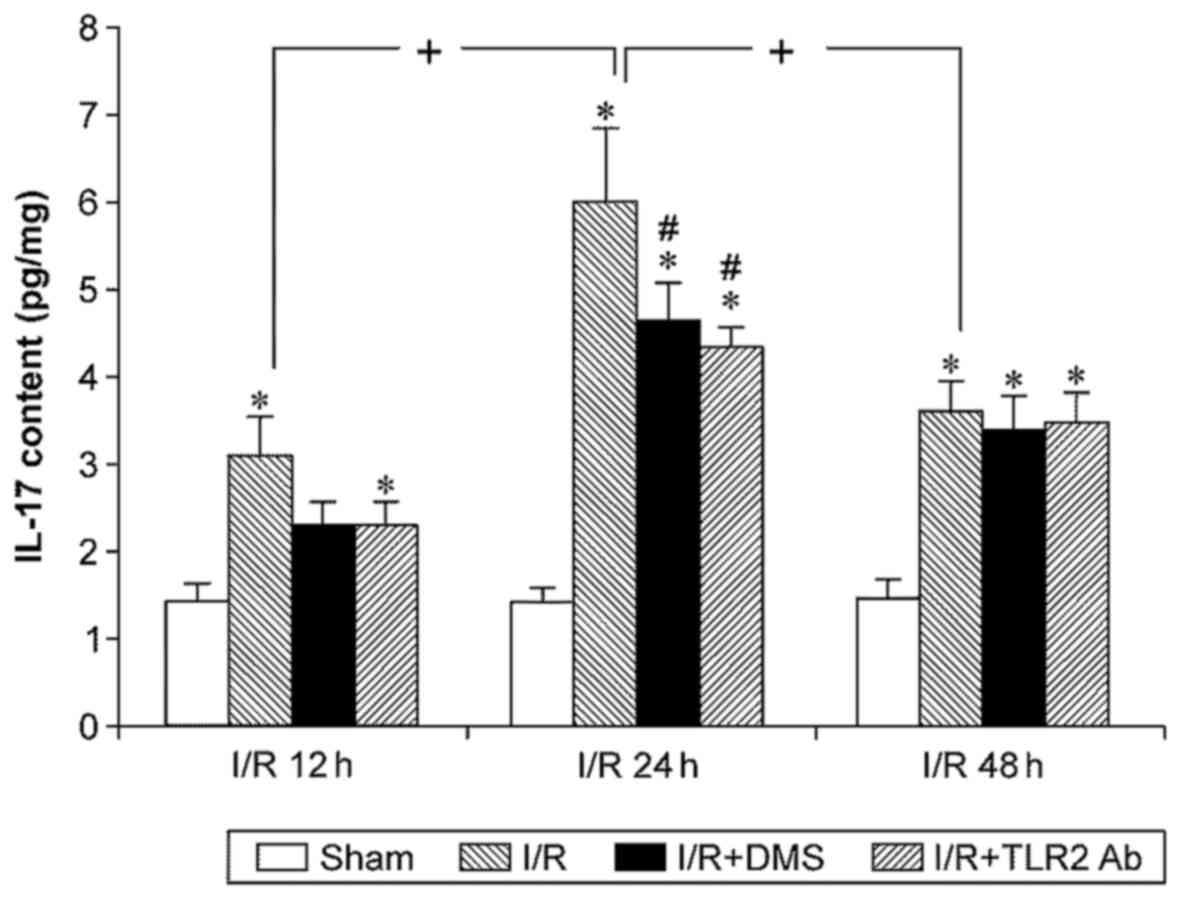
IL-17 expression levels in mouse brain tissue were
measured using ELISA, and the results revealed that I/R
significantly increased IL-17 expression (p<0.05). The
upregulated IL-17 expression appeared after 12 h, peaked at 24 h,
and the levels at 48 h were similar to those at 12 h (p<0.05).
Samples from the mice treated with TLR2 antibody or DMS showed
significantly decreased levels of IL-17 at 24 h (p<0.05).
Conversely, neither TLR2 antibody nor DMS altered IL-17 expression
at 12 or 48 h (p>0.05) (Fig.
7).
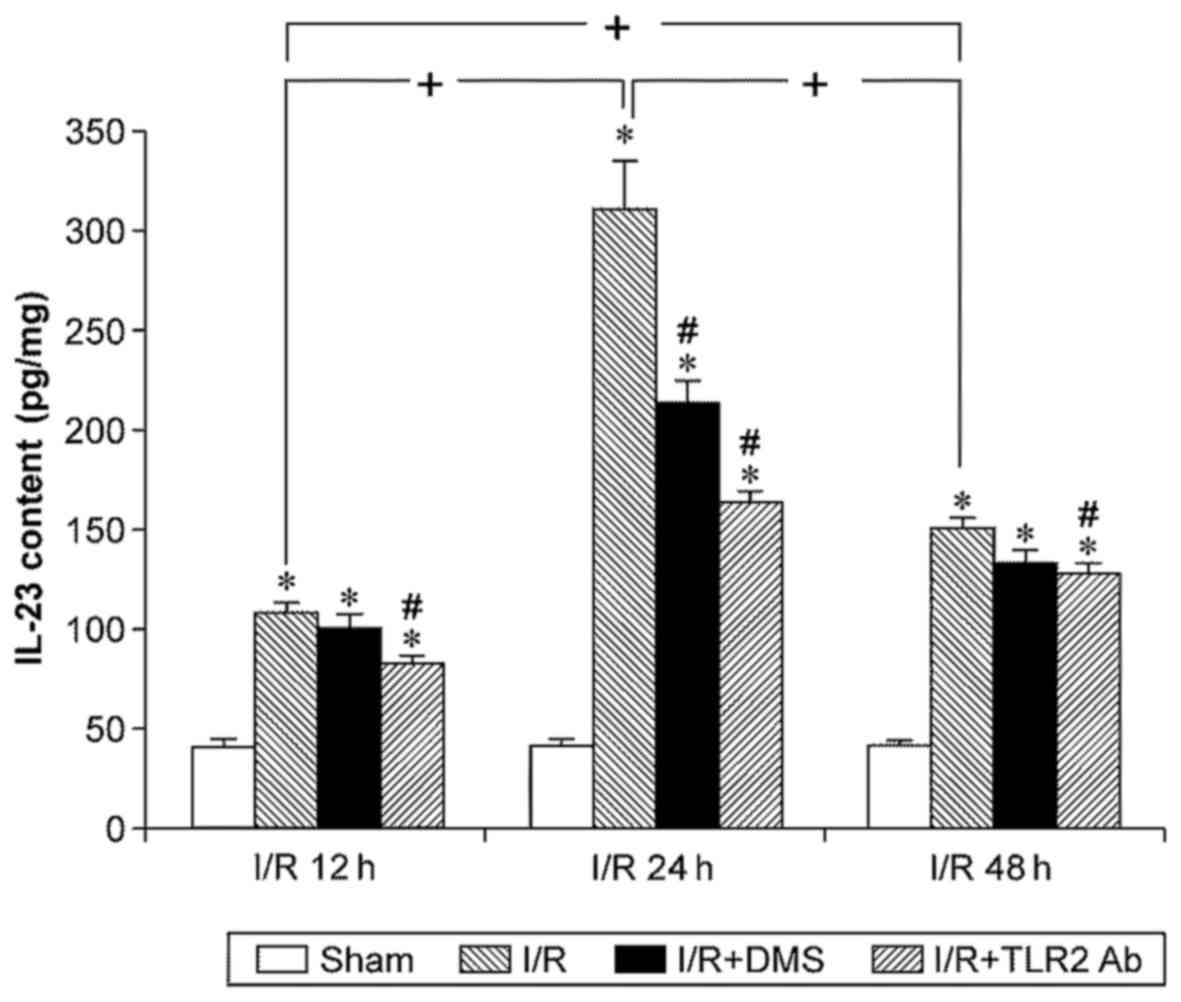
The IL-23 content was low in the brain tissue of the
sham-operation group and was upregulated significantly by I/R
(p<0.05). Similar to IL-17, the IL-23 levels increased at 12 h
of reperfusion, peaked at 24 h, and began to decline at 48 h of
reperfusion (p<0.05). TLR2 antibody downregulated the IL-23
levels in each subgroup (p<0.05). Moreover, DMS only reduced the
IL-23 content in the 24-h subgroup (p<0.05) (Fig. 8).
Discussion
The primary aim of this study was to determine the
interaction between TLR2 and Sphk1 in microglia following cerebral
I/R. We confirmed the effects of TLR2 and Sphk1 on the inflammatory
response induced by I/R. Both TLR2 and Sphk1 were primarily
expressed in microglia and were upregulated by cerebral I/R. In
addition, TLR2 and Sphk1 elevated the levels of pro-inflammatory
cytokines, including IL-β, TNF-α, IL-17 and IL-23. Furthermore, as
the upstream factor of Sphk1, TLR2 regulated the expression of
microglial Sphk1 in cerebral I/R. Therefore, activated microglial
cells may be involved in cerebral IRI through the
TLR2/Sphk1/pro-inflammatory cytokine pathway.
Microglia are currently considered a double-edged
sword. Activated microglia swallow infiltrating neutrophils to
protect neurons through the release of neurotrophic factors and
anti-inflammatory cytokines (7,36).
In neuroinflammatory diseases, excessive activation of microglial
cells can result in the release of large amounts of cytotoxic
factors, such as peroxides, NO, TNF-α, free radicals, adhesion
molecules and cytokines, which induce neuronal damage (37–41). Microglial activation is an early
response to brain ischemia. Microglia proliferate and migrate to
the sites of injury and induce a non-specific innate immune
response that may exacerbate acute ischemic injury (42). In this study, activated microglia
infiltrated the ischemic brain tissue and expressed TLR2 and Sphk1.
Both TLR2 and Sphk1 separately induced the release of
pro-inflammatory cytokines, including IL-β, TNF-α, IL-17 and IL-23
(Figs. 5Figure 6Figure 7–8). These results suggest that cerebral
I/R induces microglial activation, which may mediate an
inflammatory response to aggravate cerebral IRI.
The ischemia-induced damage of adjacent cells is
detected by microglial receptors, such as TLRs. The activation of
microglia leads to the release of important pro-inflammatory
cytokines, including TNF-α (43)
and various ILs (44,45). While mRNA for all TLRs can be
detected in the brain, TLR2 and TLR4 are most highly expressed
(46). It is increasingly clear
that post-stroke neuroinflammation due to TLR4 signaling aggravates
stroke outcome as measured by infarct volumes, neurological
function and inflammatory markers (47–49). Several models of cerebral ischemia
have identified the role of TLR4 signaling in neuroinflammation and
exacerbated stroke injury. TLR4-deficient mice exhibit improved
neurological and/or behavioral outcomes in various models of
cerebral infarction (50,51). It has been shown that TLR2 is
responsible for part of the damaging inflammatory response that
follows cerebral ischemia (52).
Consistent with previous studies, our results revealed that TLR2
was responsible for the production of pro-inflammatory cytokines
from microglia in cerebral I/R.
Recently, the role of both Sphks in immunity and
inflammation has been the focus of multiple studies (20,35,53,54), which suggest a broader role for
Sphks. A study by Puneet and colleagues demonstrated that Sphk1
inhibition led to decreased phagocyte production of
endotoxin-induced pro-inflammatory cytokines (55). Other studies revealed that the
inhibition of Sphk1 by its inhibitor and/or siRNA decreased the
expression of pro-inflammatory cytokines, such as TNF-α, IL-1β and
iNOS; when activated by LPS, microglial cells release these
chemokines (28). In this study,
Sphk1 expression in microglia was induced by cerebral I/R,
triggering the inflammatory response in the process.
TNF-α and IL-1β are probably the most extensively
studied cytokines in experimental stroke and are upregulated early
in peri-infarct microglia along with IL-17 and IL-23 (12,56). However, microglial activation
requires hours to days to fully develop in brain ischemic injury
(42), and the increases in
cytokine mRNA and protein levels are at most moderate during the
first 4.5 h after ischemia (56).
Based on the results of our experiments, the protein expression of
pro-inflammatory cytokines, including TNF-α, IL-17 and IL-23,
displayed almost the same patterns as TLR2 and Sphk1 in cerebral
I/R. IL-1β seemed to increase earlier than the other cytokines, and
the elevated level of IL-1β remained from 12 to 24 h after
reperfusion. The above-mentioned results indicate that it takes at
least 12 to 24 h for microglial activation to progress and release
pro-inflammatory cytokines. Moreover, higher levels of cytokines
may continue within 48 h from ischemia, which is in accordance with
previous reports (12,56).
Previous research has indicated that cerebral
ischemia upregulates TLR2 and TLR4 expression (57–59), and the increase of TLR2 is more
significant than that of TLR4 (9,60,61). TLRs activate the endogenous immune
system to release pro-inflammatory factors (e.g., IL-β) through the
NF-κB pathway (31–34). The expression of Sphk1 in amoeboid
microglia has been shown to be upregulated in post-natal rats under
hypoxic conditions, which also promotes IL-β and TNF-α production
via the NF-κB pathway (29).
Therefore, a common downstream pathway is activated following TLR2
and Sphk1 expression under ischemic conditions. In addition,
studies on cultured human gingival epithelial cells, RAW264.7
macrophages, 293 cells, and human bone marrow-derived macrophages
(THP-1) have revealed a close association between TLRs and Sphk1.
Ligands of either TLR2 or TLR4 are able to activate Sphk1 by
elevating transcription levels of Sphk1, increasing its activity,
and inducing membrane translocation (62–65). Therefore, it is reasonable to
infer that cerebral ischemia can increase TLR2 expression, which
further activates Sphk1 to promote the release of IL-β, TNF-α, and
other pro-inflammatory cytokines via the NF-κB pathway. In this
study, the Sphk1 levels were decreased by anti-TLR2 antibody, which
downregulated TLR2 activity. However, DMS did not affect TLR2
expression by inhibiting Sphk1 activity. Our results confirm the
role of TLR2 as a positive regulator of Sphk1 in cerebral I/R.
In conclusion, to the best of our knowledge, this is
the first study to describe the involvement of microglial cells in
inflammatory reactions via a TLR2→Sphk1→pro-inflammatory cytokine
(IL-23, IL-17, IL-1β and TNF-α) pathway in cerebral IRI. Microglial
activation presents a target for therapeutic intervention with a
much longer window of opportunity than can be achieved with current
treatments. Effective agents are now available for blocking both
the activation of TLR2 and the response of Sphk1 that drive the
inflammatory response after stroke. Effective agents are also
available for targeting the signal transduction mechanisms between
TLR2 and Sphk1 or between TLR2/Sphk1 and pro-inflammatory
cytokines. However, the innate immune response can exert both
beneficial and deleterious effects on stroke outcome, and it will
be challenging to find ways to selectively suppress the deleterious
effects of microglial activation after stroke without compromising
neurovascular repair and remodeling.
Acknowledgments
This study was supported by the Postdoctoral
Foundation of Heilongjiang Province of China (no. LBH-Z10065) and
the Open Project of Neurobiology Key Laboratory in General
Institutes of Higher Education of Heilongjiang Province of
China.
References
|
1
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 371:1612–1623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang Y, Rabb H and Womer KL:
Ischemia-reperfusion and immediate T cell responses. Cell Immunol.
248:4–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muir KW, Tyrrell P, Sattar N and Warburton
E: Inflammation and ischaemic stroke. Curr Opin Neurol. 20:334–342.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olson JK and Miller SD: Microglia initiate
central nervous system innate and adaptive immune responses through
multiple TLRs. J Immunol. 173:3916–3924. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dirnagl U, Iadecola C and Moskowitz MA:
Pathobiology of ischaemic stroke: An integrated view. Trends
Neurosci. 22:391–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Block ML and Hong JS: Microglia and
inflammation-mediated neurodegeneration: Multiple triggers with a
common mechanism. Prog Neurobiol. 76:77–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: Uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar
|
|
8
|
Lehnardt S: Innate immunity and
neuroinflammation in the CNS: The role of microglia in Toll-like
receptor-mediated neuronal injury. Glia. 58:253–263. 2010.
|
|
9
|
Lehnardt S, Henneke P, Lien E, Kasper DL,
Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT and
Vartanian T: A mechanism for neurodegeneration induced by group B
streptococci through activation of the TLR2/MyD88 pathway in
microglia. J Immunol. 177:583–592. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Chu N, Hu A, Gran B, Rostami A and
Zhang GX: Increased IL-23p19 expression in multiple sclerosis
lesions and its induction in microglia. Brain. 130:490–501. 2007.
View Article : Google Scholar
|
|
11
|
Li Y, Chu N, Hu A, Gran B, Rostami A and
Zhang GX: Inducible IL-23p19 expression in human microglia via p38
MAPK and NF-kappaB signal pathways. Exp Mol Pathol. 84:1–8. 2008.
View Article : Google Scholar
|
|
12
|
Lv M, Liu Y, Zhang J, Sun L, Liu Z, Zhang
S, Wang B, Su D and Su Z: Roles of inflammation response in
microglia cell through Toll-like receptors
2/interleukin-23/interleukin-17 pathway in cerebral
ischemia/reperfusion injury. Neuroscience. 176:162–172. 2011.
View Article : Google Scholar
|
|
13
|
Alemany R, van Koppen CJ, Danneberg K, Ter
Braak M and Meyer Zu Heringdorf D: Regulation and functional roles
of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol.
374:413–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okada T, Kajimoto T, Jahangeer S and
Nakamura S: Sphingosine kinase/sphingosine 1-phosphate signalling
in central nervous system. Cell Signal. 21:7–13. 2009. View Article : Google Scholar
|
|
15
|
Ozaki H, Hla T and Lee MJ:
Sphingosine-1-phosphate signaling in endothelial activation. J
Atheroscler Thromb. 10:125–131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosen H and Goetzl EJ: Sphingosine
1-phosphate and its receptors: An autocrine and paracrine network.
Nat Rev Immunol. 5:560–570. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alvarez SE, Milstien S and Spiegel S:
Autocrine and paracrine roles of sphingosine-1-phosphate. Trends
Endocrinol Metab. 18:300–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu
J, Griffiths R, Barbour SE, Milstien S and Spiegel S: Apoptosis
induces expression of sphingosine kinase 1 to release
sphingosine-1-phosphate as a 'come-and-get-me' signal. FASEB J.
22:2629–2638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hammad SM, Crellin HG, Wu BX, Melton J,
Anelli V and Obeid LM: Dual and distinct roles for sphingosine
kinase 1 and sphingosine 1 phosphate in the response to
inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid
Mediat. 85:107–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Melendez AJ: Sphingosine kinase signalling
in immune cells: Potential as novel therapeutic targets. Biochim
Biophys Acta. 1784:66–75. 2008. View Article : Google Scholar
|
|
21
|
Haghikia A and Gold R:
Sphingosine-1-phosphate and its receptors as a possible therapeutic
target in autoimmune diseases of the nervous system. J
Neuroimmunol. 218:1–2. 2010. View Article : Google Scholar
|
|
22
|
Bryan L, Kordula T, Spiegel S and Milstien
S: Regulation and functions of sphingosine kinases in the brain.
Biochim Biophys Acta. 1781:459–466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hait NC, Oskeritzian CA, Paugh SW,
Milstien S and Spiegel S: Sphingosine kinases, sphingosine
1-phosphate, apoptosis and diseases. Biochim Biophys Acta.
1758:2016–2026. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ibrahim FB, Pang SJ and Melendez AJ:
Anaphylatoxin signaling in human neutrophils. A key role for
sphingosine kinase. J Biol Chem. 279:44802–44811. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blondeau N, Lai Y, Tyndall S, Popolo M,
Topalkara K, Pru JK, Zhang L, Kim H, Liao JK, Ding K, et al:
Distribution of sphingosine kinase activity and mRNA in rodent
brain. J Neurochem. 103:509–517. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukuda Y, Kihara A and Igarashi Y:
Distribution of sphingosine kinase activity in mouse tissues:
Contribution of SPHK1. Biochem Biophys Res Commun. 309:155–160.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tham CS, Lin FF, Rao TS, Yu N and Webb M:
Microglial activation state and lysophospholipid acid receptor
expression. Int J Dev Neurosci. 21:431–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nayak D, Huo Y, Kwang WX, Pushparaj PN,
Kumar SD, Ling EA and Dheen ST: Sphingosine kinase 1 regulates the
expression of proinflammatory cytokines and nitric oxide in
activated microglia. Neuroscience. 166:132–144. 2010. View Article : Google Scholar
|
|
29
|
Lin H, Baby N, Lu J, Kaur C, Zhang C, Xu
J, Ling EA and Dheen ST: Expression of sphingosine kinase 1 in
amoeboid microglial cells in the corpus callosum of postnatal rats.
J Neuroinflammation. 8:132011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maceyka M, Payne SG, Milstien S and
Spiegel S: Sphingosine kinase, sphingosine-1-phosphate, and
apoptosis. Biochim Biophys Acta. 1585:193–201. 2002. View Article : Google Scholar
|
|
31
|
Aliprantis AO, Yang RB, Weiss DS, Godowski
P and Zychlinsky A: The apoptotic signaling pathway activated by
Toll-like receptor-2. EMBO J. 19:3325–3336. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Karikó K, Weissman D and Welsh FA:
Inhibition of Toll-like receptor and cytokine signaling - a
unifying theme in ischemic tolerance. J Cereb Blood Flow Metab.
24:1288–1304. 2004. View Article : Google Scholar
|
|
33
|
Kielian T: Toll-like receptors in central
nervous system glial inflammation and homeostasis. J Neurosci Res.
83:711–730. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang G and Ghosh S: Toll-like
receptor-mediated NF-kappaB activation: A phylogenetically
conserved paradigm in innate immunity. J Clin Invest. 107:13–19.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu W, Mosteller RD and Broek D:
Sphingosine kinase protects lipopolysaccharide-activated
macrophages from apoptosis. Mol Cell Biol. 24:7359–7369. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Neumann J, Sauerzweig S, Rönicke R, Gunzer
F, Dinkel K, Ullrich O, Gunzer M and Reymann KG: Microglia cells
protect neurons by direct engulfment of invading neutrophil
granulocytes: A new mechanism of CNS immune privilege. J Neurosci.
28:5965–5975. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Colton CA and Gilbert DL: Production of
superoxide anions by a CNS macrophage, the microglia. FEBS Lett.
223:284–288. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SC, Liu W, Dickson DW, Brosnan CF and
Berman JW: Cytokine production by human fetal microglia and
astrocytes. Differential induction by lipopolysaccharide and IL-1
beta. J Immunol. 150:2659–2667. 1993.PubMed/NCBI
|
|
39
|
Moss DW and Bates TE: Activation of murine
microglial cell lines by lipopolysaccharide and interferon-gamma
causes NO-mediated decreases in mitochondrial and cellular
function. Eur J Neurosci. 13:529–538. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu B, Gao HM, Wang JY, Jeohn GH, Cooper
CL and Hong JS: Role of nitric oxide in inflammation-mediated
neurodegeneration. Ann NY Acad Sci. 962:318–331. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sawada M, Kondo N, Suzumura A and
Marunouchi T: Production of tumor necrosis factor-alpha by
microglia and astrocytes in culture. Brain Res. 491:394–397. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yenari MA, Kauppinen TM and Swanson RA:
Microglial activation in stroke: Therapeutic targets.
Neurotherapeutics. 7:378–391. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lambertsen KL, Meldgaard M, Ladeby R and
Finsen B: A quantitative study of microglial-macrophage synthesis
of tumor necrosis factor during acute and late focal cerebral
ischemia in mice. J Cereb Blood Flow Metab. 25:119–135. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Minami M, Kuraishi Y, Yabuuchi K, Yamazaki
A and Satoh M: Induction of interleukin-1 beta mRNA in rat brain
after transient forebrain ischemia. J Neurochem. 58:390–392. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Block F, Peters M and Nolden-Koch M:
Expression of IL-6 in the ischemic penumbra. Neuroreport.
11:963–967. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nishimura M and Naito S: Tissue-specific
mRNA expression profiles of human Toll-like receptors and related
genes. Biol Pharm Bull. 28:886–892. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Caso JR, Pradillo JM, Hurtado O, Lorenzo
P, Moro MA and Lizasoain I: Toll-like receptor 4 is involved in
brain damage and inflammation after experimental stroke.
Circulation. 115:1599–1608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Abate W, Alghaithy AA, Parton J, Jones KP
and Jackson SK: Surfactant lipids regulate LPS-induced
interleukin-8 production in A549 lung epithelial cells by
inhibiting translocation of TLR4 into lipid raft domains. J Lipid
Res. 51:334–344. 2010. View Article : Google Scholar :
|
|
49
|
Tasaki K, Ruetzler CA, Ohtsuki T, Martin
D, Nawashiro H and Hallenbeck JM: Lipopolysaccharide pre-treatment
induces resistance against subsequent focal cerebral ischemic
damage in spontaneously hypertensive rats. Brain Res. 748:267–270.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hua F, Ma J, Ha T, Xia Y, Kelley J,
Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, et
al: Activation of Toll-like receptor 4 signaling contributes to
hippocampal neuronal death following global cerebral
ischemia/reperfusion. J Neuroimmunol. 190:101–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cao CX, Yang QW, Lv FL, Cui J, Fu HB and
Wang JZ: Reduced cerebral ischemia-reperfusion injury in Toll-like
receptor 4 deficient mice. Biochem Biophys Res Commun. 353:509–514.
2007. View Article : Google Scholar
|
|
52
|
Ziegler G, Harhausen D, Schepers C,
Hoffmann O, Röhr C, Prinz V, König J, Lehrach H, Nietfeld W and
Trendelenburg G: TLR2 has a detrimental role in mouse transient
focal cerebral ischemia. Biochem Biophys Res Commun. 359:574–579.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lai WQ, Goh HH, Bao Z, Wong WS, Melendez
AJ and Leung BP: The role of sphingosine kinase in a murine model
of allergic asthma. J Immunol. 180:4323–4329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Snider AJ, Kawamori T, Bradshaw SG, Orr
KA, Gilkeson GS, Hannun YA and Obeid LM: A role for sphingosine
kinase 1 in dextran sulfate sodium-induced colitis. FASEB J.
23:143–152. 2009. View Article : Google Scholar :
|
|
55
|
Puneet P, Yap CT, Wong L, Lam Y, Koh DR,
Moochhala S, Pfeilschifter J, Huwiler A and Melendez AJ: SphK1
regulates proinflammatory responses associated with endotoxin and
poly-microbial sepsis. Science. 328:1290–1294. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lambertsen KL, Biber K and Finsen B:
Inflammatory cytokines in experimental and human stroke. J Cereb
Blood Flow Metab. 32:1677–1698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lehnardt S, Lehmann S, Kaul D, Tschimmel
K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A and Weber JR:
Toll-like receptor 2 mediates CNS injury in focal cerebral
ischemia. J Neuroimmunol. 190:28–33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Caso JR, Pradillo JM, Hurtado O, Leza JC,
Moro MA and Lizasoain I: Toll-like receptor 4 is involved in
subacute stress-induced neuroinflammation and in the worsening of
experimental stroke. Stroke. 39:1314–1320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kilic U, Kilic E, Matter CM, Bassetti CL
and Hermann DM: TLR-4 deficiency protects against focal cerebral
ischemia and axotomy-induced neurodegeneration. Neurobiol Dis.
31:33–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hoffmann O, Braun JS, Becker D, Halle A,
Freyer D, Dagand E, Lehnardt S and Weber JR: TLR2 mediates
neuroinflammation and neuronal damage. J Immunol. 178:6476–6481.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mishra BB, Mishra PK and Teale JM:
Expression and distribution of Toll-like receptors in the brain
during murine neurocysticercosis. J Neuroimmunol. 181:46–56. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Benakanakere MR, Zhao J, Galicia JC,
Martin M and Kinane DF: Sphingosine kinase-1 is required for toll
mediated beta-defensin 2 induction in human oral keratinocytes.
PLoS One. 5:e115122010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pitson SM, Moretti PA, Zebol JR, Lynn HE,
Xia P, Vadas MA and Wattenberg BW: Activation of sphingosine kinase
1 by ERK1/2-mediated phosphorylation. EMBO J. 22:5491–5500. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Spiegel S and Milstien S:
Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol
Cell Biol. 4:397–407. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pchejetski D, Nunes J, Coughlan K, Lall H,
Pitson SM, Waxman J and Sumbayev VV: The involvement of sphingosine
kinase 1 in LPS-induced Toll-like receptor 4-mediated accumulation
of HIF-1α protein, activation of ASK1 and production of the
pro-inflammatory cytokine IL-6. Immunol Cell Biol. 89:268–274.
2011. View Article : Google Scholar
|