Introduction
Acute lung injury (ALI) is clinical syndrome of
acute respiratory failure, which is caused primarily by acute lung
inflammation, damaging the alveolar capillary barrier (1), followed by protein-rich edema fluid
accumulation in the airway, causing diffuse pulmonary interstitial
edema and abnormal gas exchange (2). The injury factors have been
described as follows: Sepsis, pneumonia, aspiration of gastric
contents, severe trauma with shock, severe acute pancreatitis
(3-5). ALI is a life-threatening disease
with high morbidity and mortality rates, and to the best of our
knowledge, no specific pharmacological treatment has been
established (2).
Lipopolysaccharide (LPS), a key component of the
cell wall in gram-negative bacteria, is a common factor that leads
to ALI (6). Toll-like receptor
(TLR) signaling serves a key role in the pathogenesis of ALI.
Following binding with LPS, TLR4 may induce the activation of
myeloid differentiation factor 88-dependent interleukin (IL)-1
receptor activated kinase 1/4, and promote mitogen activated
protein kinase (MAPK) activation and nuclear factor
kappa-light-chain-enhancer of activated B cells transcription
factor p65 subunit (NF-κB p65) translocation to the nucleus
(7). The activation of NF-κB has
been demonstrated to regulate the expression of a series of genes,
including pro-inflammatory cytokines tumor necrosis factor-α
(TNF-α), IL-1β and IL-6 (8),
which may lead to lung tissue damage (9). p38 MAPKs are involved in the
occurrence and development of ALI caused by various etiologies
(10,11). Inhibition of p38 MAPKs may block
the activation of NF-κB, and effectively inhibit lung injury
induced by LPS, including neutrophil recruitment in the lung,
protein exudation and apoptosis in bronchoalveolar lavage fluid
(BALF) (12). Therefore,
inhibiting the activity of the TLR4/NF-κB and p38 MAPKs signaling
pathways has potential therapeutic value in LPS-induced ALI.
Penehyclidine hydrochloride (PHC) is a novel
anticholinergic drug, which may selectively inhibit muscarinic
acetylcholine receptor M1 (M1), muscarinic acetylcholine receptor
(M3) and nicotinic acetylcholine receptors N1 and N2 (N1 and N2),
with almost no cardiovascular side effects associated with
muscarinic acetylcholine receptor M2 (M2) receptors (13). It has been demonstrated that PHC
has a protective effect in a rat model of LPS-induced ALI, and the
effect may involve the inhibition of the p38 MAPKs signaling
pathway and NF-κB activation (14). However, the underlying mechanism
of PHC inhibition in the activation of these signaling pathways
remains unclear.
Caveolin-1 (Cav-1) is the principal structural
component of caveolae, which are omega-shaped invaginations of
plasma membrane (15-17). Previous studies have suggested
that Cav-1 serves a crucial role in the pathogenesis of ALI, which
may regulate acute inflammation and capillary leakage during lung
injury (18). Wang et al
(19) observed that Cav-1 in
murine peritoneal and alveolar macrophages had the ability to
regulate LPS-induced pro-inflammatory cytokine TNF-α and IL-6
production. This effect may be due to the fact that Cav-1 inhibits
LPS-induced p38 MAPKs phosphorylation and NF-κB signaling pathway
activation (20).
The aim of the present study was to observe the
involvement of Cav-1 in the PHC-based inhibition of LPS-induced ALI
in vivo and in vitro and the potential underlying
mechanisms.
Materials and methods
Reagents
LPS (E coli 0111:B4) was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). ELISA kits were
purchased from R&D Systems, Inc. (Minneapolis, MN, USA), mouse
TNF-α (PMTA00B), IL-6 (PM6000B) and IL-1β (PMLB00C) Quantikine
ELISA kit, rat TNF-α (PRTA00), IL-6 (PR6000B) and IL-1β (PRLB00)
Quantikine ELISA kit. The myeloperoxidase (MPO) kit was obtained
from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
PHC was purchased from Chengdu List Pharmaceutical Co. Ltd.
(Chengdu, China).
Animal model
Male Sprague-Dawley (SD) rats (170-190 g; 8-10
weeks-old) were purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd (Beijing, China; certificate no.
SCXK2016-0006). The rats were housed in a specific pathogen-free,
laminar-flow atmosphere under controlled temperature (25±2°C),
humidity (50±10%), and 12 h light: Dark cycle, with ad
libitum access to food and water. The animals acclimated to
this environment for 1 week prior to the experiment. The present
study was approved by Medical Ethics Committee of Renmin Hospital
of Wuhan University and was performed in accordance with the
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals (21).
LPS-induced ALI in rats
Male SD rats were randomly divided into 3 groups: i)
The control group (C group); ii) ALI model group (LPS group); and
iii) ALI + PHC treatment group (P + LPS group). The ALI model was
established using a protocol based on the study by Shen et
al (14), by intratracheal
instillation of LPS. A total of 0.2 ml LPS (5 mg/kg) was
administered to rats in the LPS and P + LPS groups by intratracheal
instillation, while rats in the C group received an equal volume of
normal saline. A total of 30 min later, the rats in the P + LPS
group received an intraperitoneal (i.p.) injection of 0.5 ml PHC (2
mg/kg), while C and LPS groups received 0.5 ml normal saline. The
doses of PHC were selected based on data from our previous studies
(22,23). At 24 h after LPS treatment, the
rats were anesthetized by an i.p. injection of pentobarbital (50
mg/kg; Sigma-Aldrich; Merck KGaA), and arterial blood, BALF and
lung tissue samples were collected. Animal death was confirmed by
observation of apnea and asystole. Lung tissues were snap-frozen in
liquid nitrogen, and stored at −80°C for subsequent analysis.
Arterial blood gas analysis
Following anesthesia, arterial blood samples were
collected from the rats with a heparinized syringe from the carotid
artery, followed by thoracotomy and alveolar lavage and removal of
lung tissues. The arterial blood samples were immediately injected
into an ABL700 Radiometer (Radiometer America Inc, Brea, CA, USA)
to measure pH value, partial gas pressures of oxygen
(PaO2) and carbon dioxide (PaCO2), and lactic
acid (Lac).
Histopathological lung examination
The right lung lobes were excised, washed and fixed
in 4% (v/v) paraformaldehyde (4°C, 12 h). Lung tissues were
embedded in paraffin, sectioned at 4 µm thickness, and
stained with hematoxylin and eosin (H&E) solution
(Sigma-Aldrich; Merck KGaA) (0.2% hematoxylin staining for 10 min,
and 0.5% eosin staining for 2 min; each at room temperature) to
estimate inflammation in the peribronchial and alveolar lesions.
The stained slides were then observed with a light microscope
(BX51; Olympus Corporation, Tokyo, Japan; magnification, ×100) and
the digital micrographs were captured for analysis. The histologic
injury scores were evaluated from 0 to 4, as described previously
(24). Briefly, the degree of
lung injury was graded according to the following scoring system:
0, no injury and appears normal; 1, minimal (injury up to 25% of
the field); 2, mild (injury between 25-50% of the field); 3,
moderate (injury between 50-75% of the field); and 4, severe
(>75%, diffuse injury). Tissue sections were examined by a
pathologist blinded to the experiment.
Ultrastructural changes of lung tissues
under transmission electron microscopy
The lungs were isolated and cut into 1-2
mm3 cubes. Lung tissue samples were fixed by immersion
in a 2.5% buffer of glutaraldehyde for 24 h at 4°C, washed by PBS
solution 3 times, post-fixed for 1 h at room temperature in 1%
osmium tetroxide, dehydrated in graded concentrations of ethyl
alcohol (30, 50, 70, 90 and 100%), and embedded in epoxy resin.
Ultrathin sections (70 nm) were double-stained with 1% uranyl
acetate for 30 min at room temperature and lead citrate for 15 min
at room temperature. Subsequently the sections were examined under
a transmission electron microscope (JEOL 100 CX; JEOL, Ltd., Tokyo,
Japan).
BALF collection and inflammatory cell
counting
To obtain the BALF, ice-cold PBS (2 ml) was infused
into the lungs of the rats 3 times and withdrawn each time using a
tracheal cannula (a total volume of 6 ml). The collected BALF was
then centrifuged at 200 × g for 15 min at 4°C. Following
centrifugation, the supernatant was stored at −80°C for subsequent
assays. Following exclusion of dead cells by trypan blue staining
(0.5 ml 0.4% trypan blue solution was transferred to the test tube.
Subsequently, 0.3 ml Hanks balanced salt solution (HBSS) and 0.2 ml
cell suspension were added and mixed sufficiently and allowed to
stand for 5 to 15 min. All the procedures were performed at room
temperature. The stained cell suspension was absorbed and
transferred to the blood cell count board, and the number of living
and dead cells were then counted under a light microscope (BX51;
Olympus, Tokyo, Japan; magnification, ×400). The total inflammatory
cells in BALF were determined by counting cells with a
hemocytometer (cat. no. 3100; Hausser Scientific, Horsham, PA,
USA). In order to analyze differential cell counting, 100 µl
BALF was centrifuged (200 × g for 10 min at 4°C) onto slides using
a Cytospin (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Subsequent to natural drying of the slides, the cells were fixed,
and stained using Wright's Stain solution (Sigma-Aldrich; Merck
KGaA) according to the manufacturer's protocol. Several drops of
Wright's Stain were added to cover the slides followed by
incubation for approximately 1 min at room temperature. Then we
added the same amount of phosphate buffer (pH 6.4) and shaked the
slides gently, and allowed to stand for 5 min. The slides were then
rinsed gently with ultrapure water 3 times. After the slides were
dried naturally, they were examined under a light microscope
(magnification, ×1,000; oil immersion lens). The number of
polymorphonuclear neutrophils (PMNs) was classified to obtain the
percentage of neutrophils.
Protein content determination in
BALF
The frozen BALF supernatant was thawed and
thoroughly mixed. The total protein concentration was determined
using a bicinchoninic acid method.
Lung wet/dry (W/D) weight ratio
The water content of the lungs was determined by
calculating the W/D weight ratio of the lung tissues. The left lobe
of the lung was excised, washed with PBS, blotted and then weighed
to obtain the 'wet' weight. The lung was then dried at 65°C for 48
h to obtain the 'dry' weight. The W/D ratio was calculated to
assess the degree of pulmonary edema.
MPO activity assay
Radioimmunoprecipitation assay lysis buffer
(Sigma-Aldrich; Merck KGaA) was used for lysing tissues, and 10 mg
tissue was obtained for each sample. Following washing with cold
PBS, the tissues were resuspended in 4 volumes of MPO Assay buffer
(included with the Myeloperoxidase Activity Assay kit), and then
centrifuged at 4°C and 13,000 × g for 10 min. The supernatant was
collected and transferred to a clean tube, which was then placed on
ice. The MPO activity was assayed using Myeloperoxidase Activity
Assay kit (cat. no. ab105136; Abcam, Cambridge, MA, USA), by
measuring the absorbance of the sample at 460 nm using a microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
specific MPO activity in the lungs was measured as units/mg
protein.
Measurement of pro-inflammatory cytokines
in serum
Blood was collected from the carotid artery, and the
serum was obtained following centrifugation at 1,000 × g for 10 min
at 4°C and used for ELISA analysis. The levels of TNF-α, IL-6 and
IL-1β in serum were determined using ELISA kits according to the
manufacturer's instructions (R&D Systems, Inc., Minneapolis,
MN, USA). The absorbance was measured at 450 nm using an ELISA
reader (BioTek Instruments, Inc., Winooski, VT, USA).
Western blot analysis
A total of 24 h after LPS injection, the lung
tissues were harvested and snap-frozen in liquid nitrogen until
homogenization. The lung tissues were homogenized using a
homogenizer with tissue nuclear and cytoplasmic protein extraction
reagents (Sigma-Aldrich; Merck KGaA), according to the
manufacturer's protocol. Protein concentrations were determined
using a BCA protein assay kit. Equal amounts of protein (40
µg) were loaded per well on 12% SDS-PAGE gels and
transferred onto polyvinylidene difluoride membranes. The resulting
membranes were blocked by incubation with 5% skim milk in TBS +
Tween-20 (TBST) (0.1% Tween-20) at room temperature for 2 h on a
rotary shaker, followed by washing with TBST. Subsequently, the
membranes were incubated with specific primary antibodies at 4°C
overnight: Rabbit anti-TLR4 (1:300; cat. no. ab217274; Abcam,
Cambridge, UK Abcam, Cambridge, UK); Cav-1 (1.2 µg/ml; cat.
no. ab2910); p38 MAPKs (1.8 µg/ml; cat. no. ab27986);
phosphorylated (p)-p38 MAPKs (1:600; cat. no. ab47363); NF-κB p65
(0.5 µg/ml; cat. no. ab16502; all Abcam); GAPDH (1:1,000;
cat. no. 5174; Cell Signaling Technology, Inc., Danvers, MA, USA);
and histone H3 antibody (1:2,000; cat. no. 4499; Cell Signaling
Technology, Inc., Danvers, MA, USA). The membranes were then washed
with TBST followed by incubation with the horseradish peroxidase
(HRP)-conjugated secondary antibody (1:2,000; cat. no. sc-2004;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at room
temperature for 1 h. The blots were washed with TBST and detected
using an enhanced chemiluminescence (ECL) western blotting
detection kit (32132; Thermo Fisher Scientific, Inc. Waltham, MA,
USA). The proteins bands were then observed using an ECL western
blotting analysis system (Bio-Rad Laboratories, Inc.) and
quantified by densitometry using Image Lab v5.2.1 software
(Informer Technologies, Inc.).
J774A.1 cells culture and treatment
The mouse monocyte/macrophage J774A.1 cell line was
purchased from the American Type Culture Collection (Rockville, MD,
USA). The J774A.1 cells were seeded (desity, 2×105/ml)
onto a 6 cm dish and were cultured in Dulbecco's modified Eagle's
medium (DMEM; 11965092) supplemented with 10% fetal bovine serum
(26400044; both Thermo Fisher Scientific, Inc.), 50 U/ml penicillin
G and 50 µg/ml streptomycin, and maintained at 37°C in a
humidified atmosphere containing 5% CO2. The cells were
grown until 50-70% confluence prior to treatment. The cells were
divided into 5 groups, as follows: i) The scramble (Scr)-short
interfering RNA (siRNA) control (S group); ii) the Scr-siRNA + LPS
(LPS group); iii) the Scr-siRNA + PHC + LPS (P + LPS group); iv)
the SMARTpool Cav-1-siRNA + LPS (C + LPS group); and the SMARTpool
Cav-1- siRNA + PHC + LPS (C + P + LPS group). According to the
manufacturer's protocol, Scr-siRNA (D-001810-10-20) and SMARTpool
Cav-1-siRNAs (12389) were added to the cells (50-70% confluence)
using DharmaFECT Transfection Reagent (T-2001-03; all Dharmacon,
Lafayette, CO, USA). The siRNA sequences against CAV-1 (CAV1-1
siRNA) were 5′-AGA CGA GCU GAG CGA GAA GCA-3′ (sense) and 5′-UGC
UUC UCG CUC AGC UCG UCU-3′ (antisense) and (CAV1-2 siRNA)
5′-CAU-CUA CAA GCC CAA CAA C-3′ (sense) and 5′-GUU GUU GGG CUU GUA
GAU G-3′ (antisense). The final siRNA concentrations were 50 nM.
Cells in the S, LPS and P + LPS groups were transfected with
Scr-siRNA for 48 h, and cells in the C + LPS and C + P + LPS groups
were transfected with SMARTpool Cav-1-siRNAs for 48 h. Following
transfection, the cells in all groups, with the exception of the S
group, were incubated in the presence of LPS (1 µg/ml) at
37°C for 2 h, and then cells in the P + LPS and C + P + LPS groups
were incubated with PHC (2 µg/ml) at 37°C for an additional
2 h. The protein expression levels of Cav-1, TLR4, p38 MAPKs, p-p38
MAPKs and nuclear NF-κB p65 were the determined by western blot
analysis, as described in the above paragraph. The primary
antibodies used were: Rabbit anti-TLR4 (1:500; cat. no. ab13556);
Cav-1 (1.5 µg/ml; cat. no. ab2910); p38 MAPKs (1.5
µg/ml; cat. no. ab27986; all Abcam); p-p38 MAPKs (1:800;
cat. no. ab47363); NF-κB p65 (0.5 µg/ml; cat. no. ab16502;
both Abcam, Cambridge, UK); GAPDH (1:1,000; cat. no. 5174); and
histone H3 antibody (1:2,000; cat. no. 4499; both Cell Signaling
Technology, Inc.). HRP-conjugated secondary antibody (1:2,000; cat.
no. sc-2004; Santa Cruz Biotechnology, Inc.) was selected as the
secondary antibody. The levels of TNF-α, IL-6 and IL-1β in cell
culture supernatant were determined using Quantikine ELISA kits.
The MPO activity was measured by colorimetry, as mentioned
above.
Statistical analysis
The data are expressed as the mean ± standard error
of the mean. The statistical analysis was performed using GraphPad
Prism (version 7.0; GraphPad Software, Inc., La Jolla, CA, USA),
and groups were compared using a one-way analysis of variance
followed by Dunnett's least significant difference post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
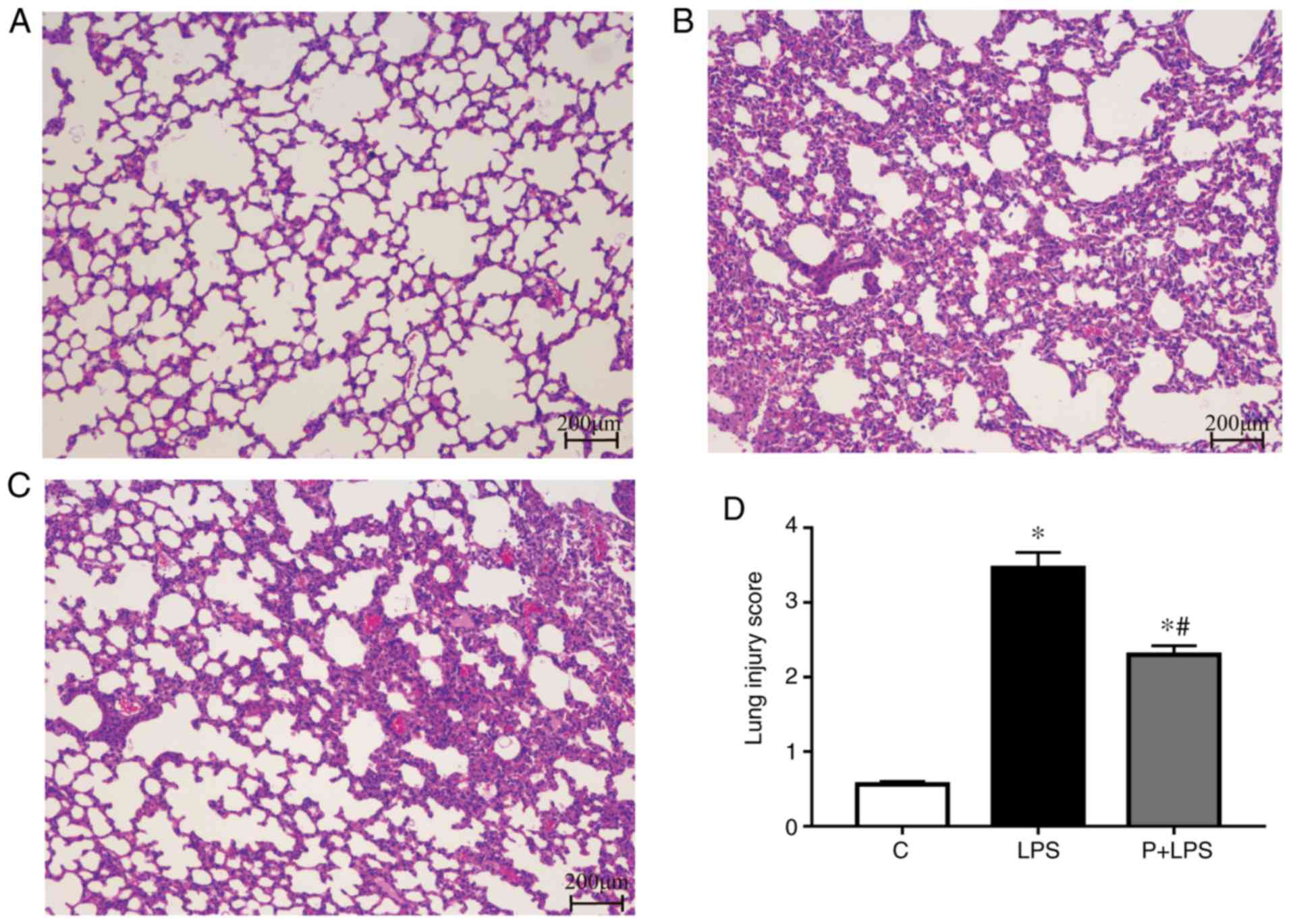
Effect of PHC on LPS-mediated lung
histopathological changes
To evaluate the histological changes of the lung
tissues following LPS treatment, tissues were harvested at 24 h
following LPS stimulation and subjected to H&E staining. No
histological changes were observed in the lung tissues of rats in
the C group (Fig. 1A).
Significant pathological changes were observed in lung tissues of
LPS-treated rats, including pulmonary capillary congestion,
pulmonary interstitial edema, mass inflammatory cell infiltration
and alveolar wall thickening (Fig.
1B). Treatment with PHC significantly decreased the
histopathological changes induced by LPS (Fig. 1C). In addition, as indicated in
Fig. 1D, quantitative scoring of
histological lung injury in the ALI rats was increased compared
with the C group at 24 h following LPS administration. However, PHC
pre-treatment decreased the pathological scores of the lung
tissues.
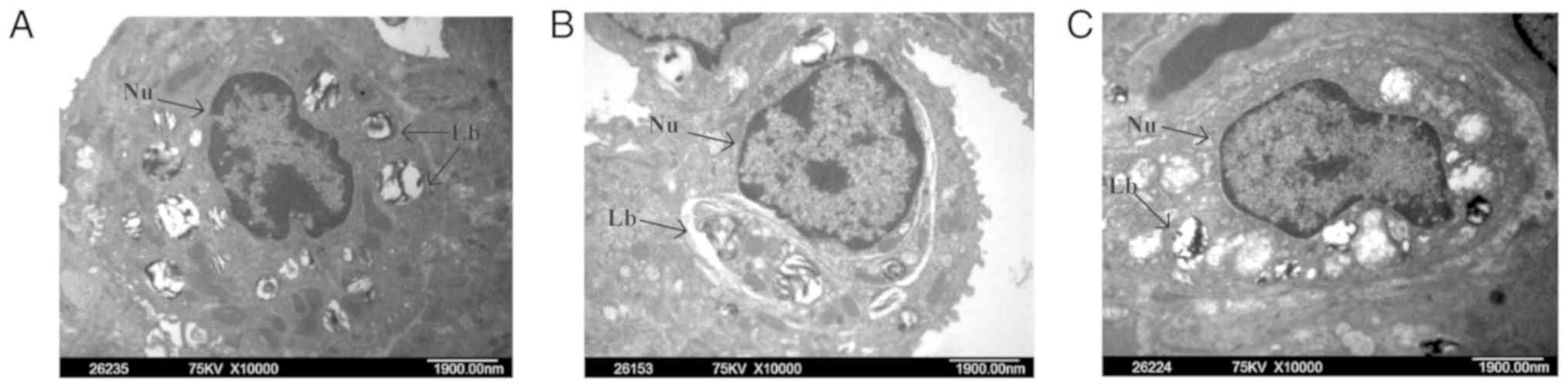
Effect of PHC on LPS-mediated lung
ultrastructural changes
Transmission electron microscopy was used to examine
the ultrastructural changes in lung tissues. Lung tissues from the
control group indicated normal structure (Fig. 2A). Lung tissues from LPS-treated
rats exhibited enlarged mitochondria and disrupted mitochondrial
cristae in the alveolar epithelial cells, lamellar corpuscle
vacuole-like degeneration and degranulated rough endoplasmic
reticulum membrane (Fig. 2B). In
the P + LPS group, which received PHC pre-treatment, the level of
pathological damage was significantly decreased compared with the
LPS group (Fig. 2C).
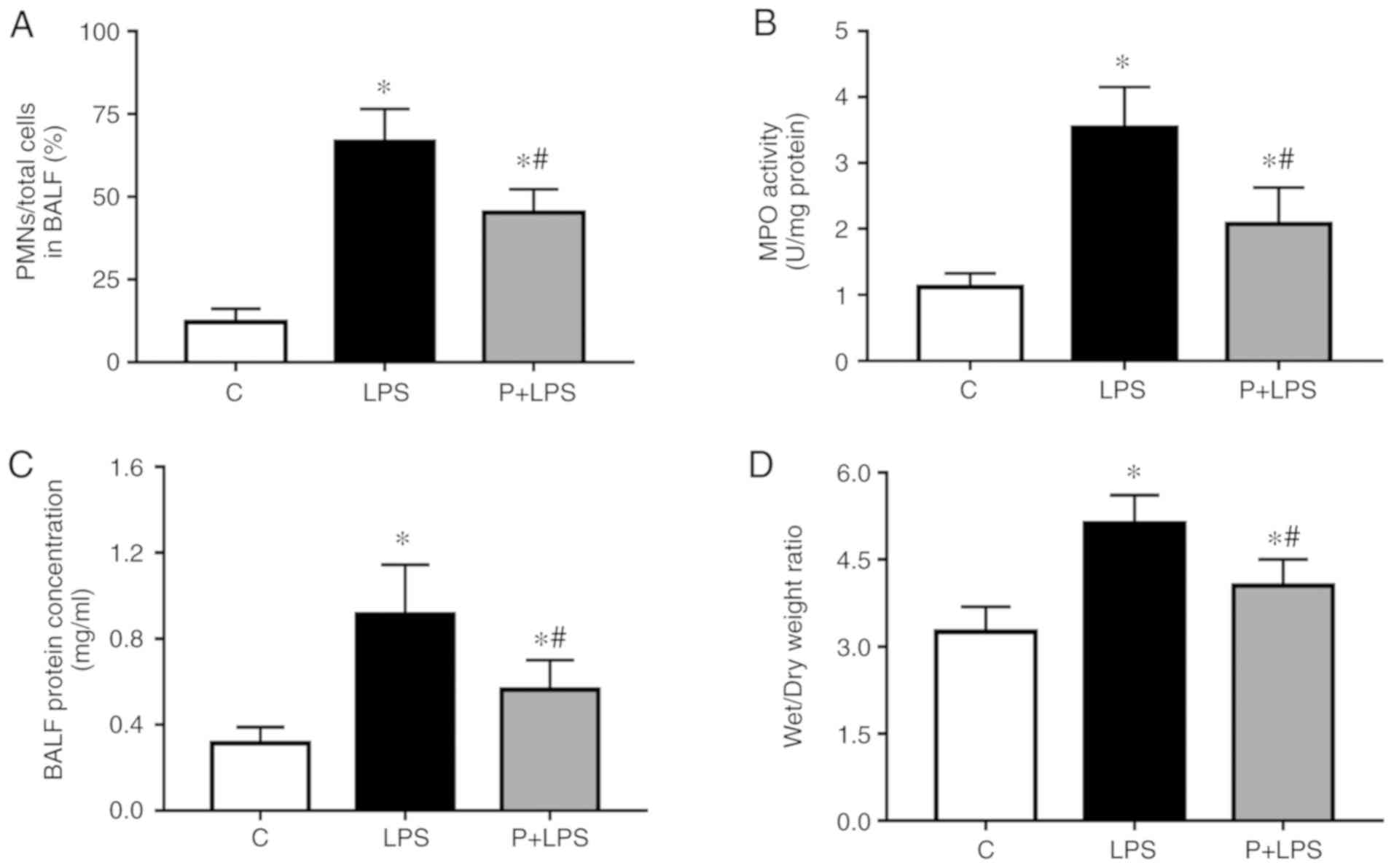
Effect of PHC on pulmonary inflammation
and vascular permeability during LPS-induced ALI
To examine the effects of PHC on LPS-induced
pulmonary inflammation and vascular permeability, the ratio of PMNs
to the total number of cells in the BALF, MPO activity, BALF
protein content and lung W/D ratio were analyzed at 24 h following
LPS injection. As indicated in Fig.
3, following LPS stimulation, the ratio of PMNs to total cells
in the BALF (Fig. 3A), MPO
activity (Fig. 3B), BALF protein
content (Fig. 3C) and lung W/D
ratio (Fig. 3D) were
significantly increased compared with the C group. However, the
increase in lung injury index was decreased by pre-treatment with
PHC.
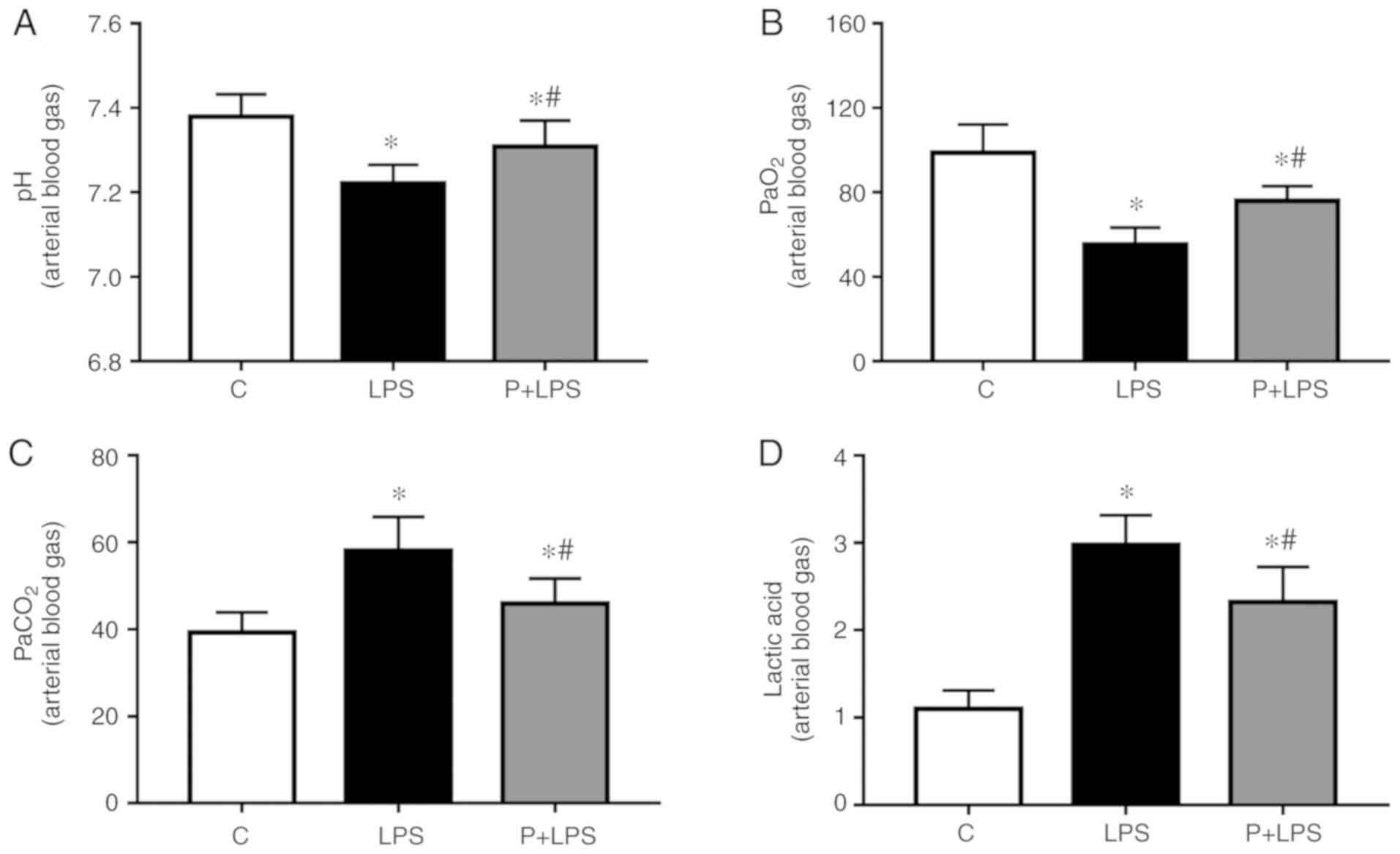
Effects of PHC on the arterial blood gas
of rats with LPS-induced lung injury
As presented in Fig.
4, compared with the control group, the arterial blood gas
analysis of rats receiving LPS treatment indicated a significant
change, as pH (Fig. 4A) and
PaO2 (Fig. 4B)
decreased, and the levels of PaCO2 (Fig. 4C) and Lac (Fig. 4D) increased. Compared with the LPS
group, the pH and PaO2 were increased in the P + LPS
group, while the levels of PaCO2 and Lac were
decreased.
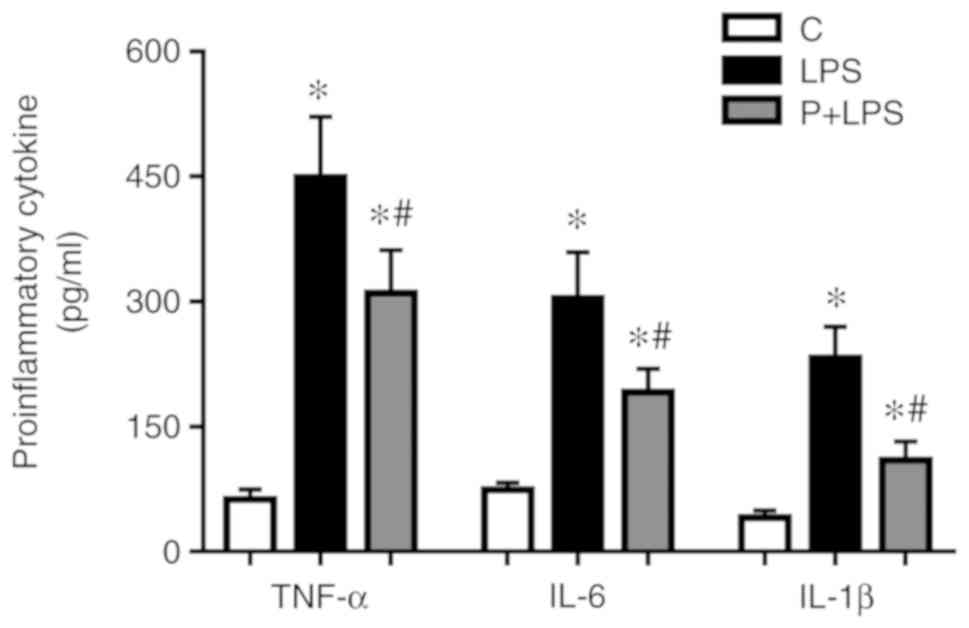
Effect of PHC on pro-inflammatory
cytokine production in the sera isolated from rats with LPS-induced
ALI
TNF-α, IL-6 and IL-1β expression levels in the sera
were significantly increased in LPS-treated rats compared with the
control group. By contrast, PHC-treated rats exhibited a
significant decrease in TNF-α, IL-6 and IL-1β expression levels
compared with the LPS group (Fig.
5).
Effect of PHC on protein expression in
the lung tissue of rats with LPS-induced ALI
Rats with LPS-induced ALI exhibited increased TLR4,
p-p38 MAPKs and nuclear NF-κB p65 expression levels, and decreased
Cav-1 expression levels in the lung tissue samples compared with
the control group. However, there was a significant decrease in
TLR4, p-p38 MAPKs and nuclear NF-κB p65 expression levels, and an
increase in Cav-1 expression levels in the lung tissue of
PHC-treated rats compared with the LPS-treated rats. There was no
statistically significant difference in p38 MAPKs protein
expression among the different groups (Fig. 6).
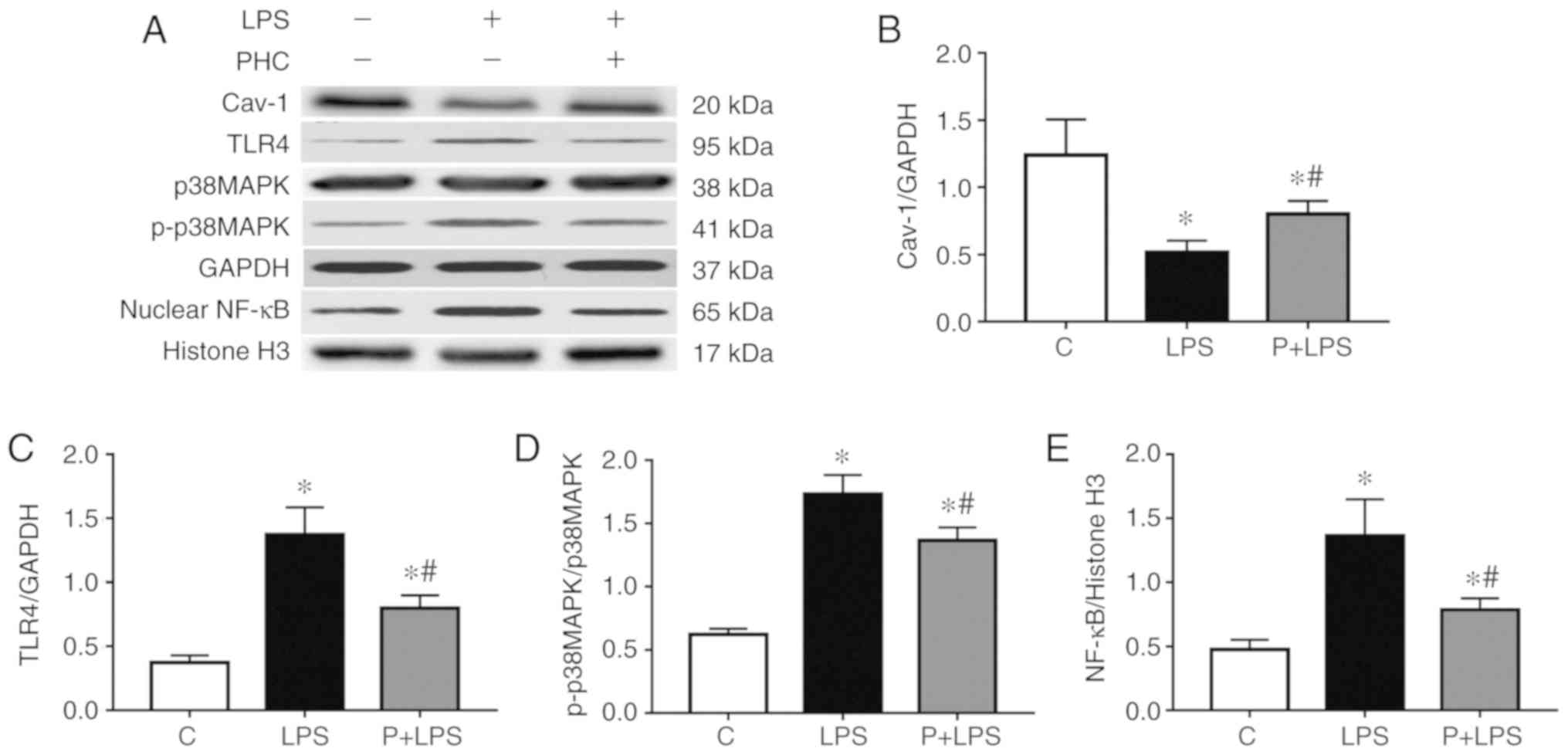 | Figure 6Effect of PHC on protein expression
in the lung tissue of rats with LPS-induced acute lung injury. (A)
Western blot analysis of Cav-1, TLR4, p38 MAPK, p-p38 MAPKs and
nuclear NF-κB p65 protein expression. Relative protein expression
levels of (B) Cav-1, (C) TLR4, (D) p-p38 MAPKs (p-p38 MAPKs/p38
MAPKs) and (E) nuclear NF-κB p65. The data are presented as the
mean ± standard error of the mean (n=10). *P<0.05 vs.
C group. #P<0.05 vs. LPS group. PHC/P, penehyclidine
hydrochloride; LPS, lipopolysaccharide; Cav-1, caveolin-1; TLR4,
toll-like receptor 4; p, phosphorylated; p38 MAPKs, p38
mitogen-activated protein kinases; NF-κB p65, nuclear factor
kappa-light-chain-enhancer of activated B cells transcription
factor p65 subunit; C, control. |
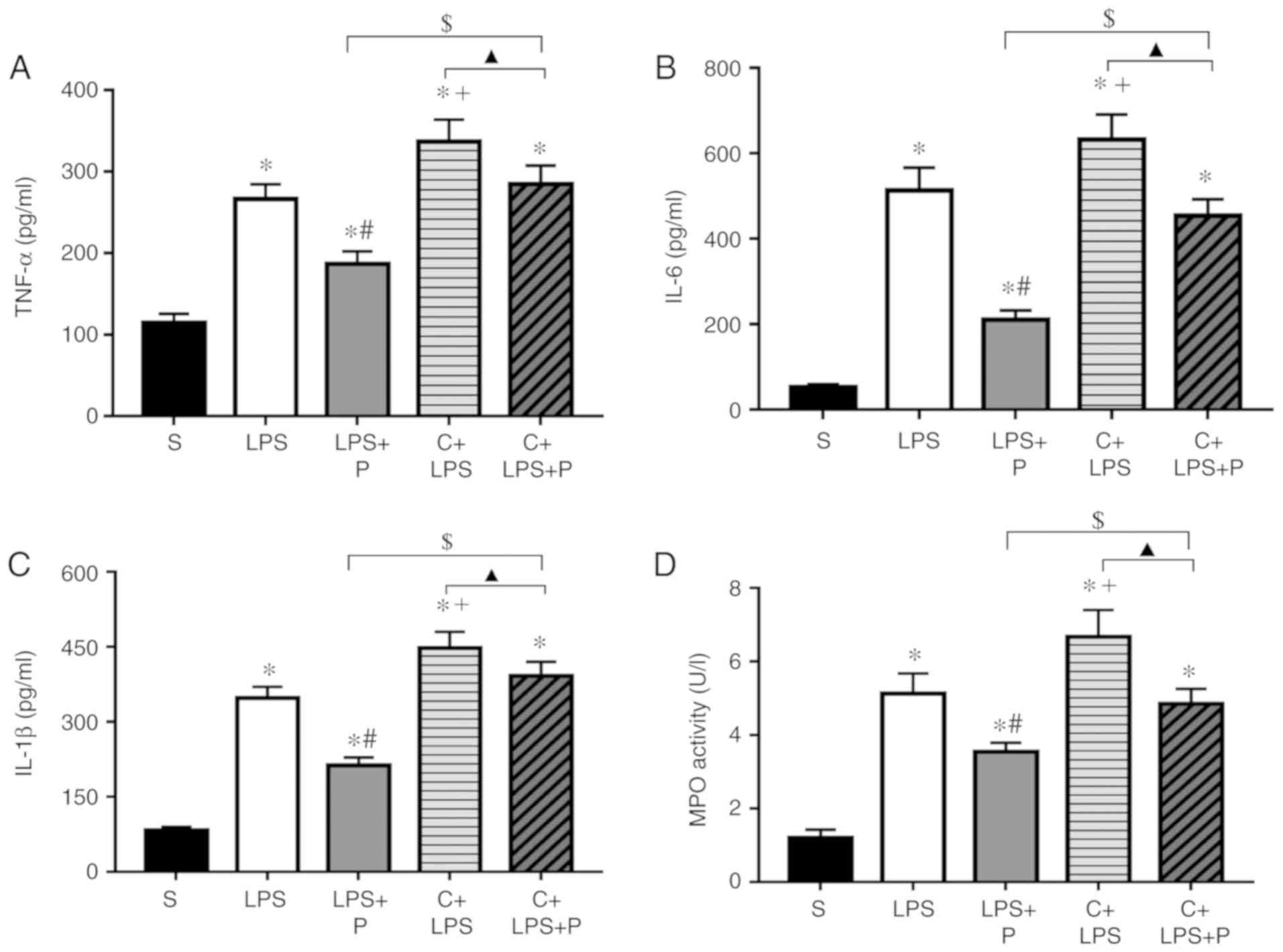
Effect of PHC on pro-inflammatory
cytokine production in cell culture supernatant in LPS-stimulated
J774A.1 cells
Supernatants were collected from the LPS/PHC treated
J744.A1 cells in the presence of Scr-siRNA or SMARTpool
Cav-1-siRNA. As presented in Fig.
7, treatment with LPS resulted in a significant increase in
pro-inflammatory cytokines production in supernatants compared with
the negative control group. Compared with the LPS group, the
expression levels of TNF-α, IL-6 and IL-1β in the cell culture
supernatant were decreased in the LPS + P group; however, the C +
LPS group with Cav-1 knockdown exhibited the opposite result. The
expression levels of TNF-α, IL-6 and IL-1β in the supernatant in
the C + LPS + P group were increased compared with the LPS + P
group, however, they were decreased compared with the C + LPS
group.
Effect of PHC on MPO activity in
LPS-stimulated J774A.1 cells
Compared with the negative control group, MPO
activity was significantly increased in LPS-stimulated J774A.1
cells. Compared with the LPS group, MPO activity was decreased in
the LPS + P group; however, the C + LPS group with Cav-1 knockdown
exhibited the opposite result. MPO activity in the group C + LPS +
P was increased compared with the LPS + P group, however, it was
decreased compared with the C + LPS group (Fig. 7D).
Effect of PHC on protein expression
levels in LPS-stimulated J774A.1 cells
The involvement of Cav-1 signaling in macrophages
was examined directly in cross-protection experiments, in which
Cav-1 expression was knocked down using siRNA. J744.A1 cells were
transfected with Cav-1 siRNA and a control siRNA, which targeted a
non-specific gene. The results of the western blot analysis
indicated that Cav-1 siRNA transfection successfully knocked down
Cav-1 in J744.A1 cells (Fig. 8A).
As indicated in Fig. 8, TLR4,
p-p38 MAPKs and nuclear NF-κB p65 protein expression levels were
significantly increased in LPS-stimulated J774A.1 cells, while the
Cav-1 protein expression level was significantly decreased, with
the exception of the LPS + P group. Compared with the LPS group,
TLR4, p-p38 MAPKs and nuclear NF-κB p65 protein expression levels
were decreased and Cav-1 protein level was increased in the LPS + P
group; however, the C + LPS group with Cav-1 knockdown exhibited
the opposite result. Compared with the LPS + P group, TLR4, p-p38
MAPKs and nuclear NF-κB p65 protein expression levels were
increased, whereas Cav-1 expression protein level was decreased in
the C + LPS + P group. Cells in the C + LPS + P group exhibited
significantly decreased TLR4, p-p38 MAPKs and nuclear NF-κB p65
expression levels and increased Cav-1 expression compared with the
C + LPS group. There was no statistical difference in p38 MAPKs
protein expression among the different groups.
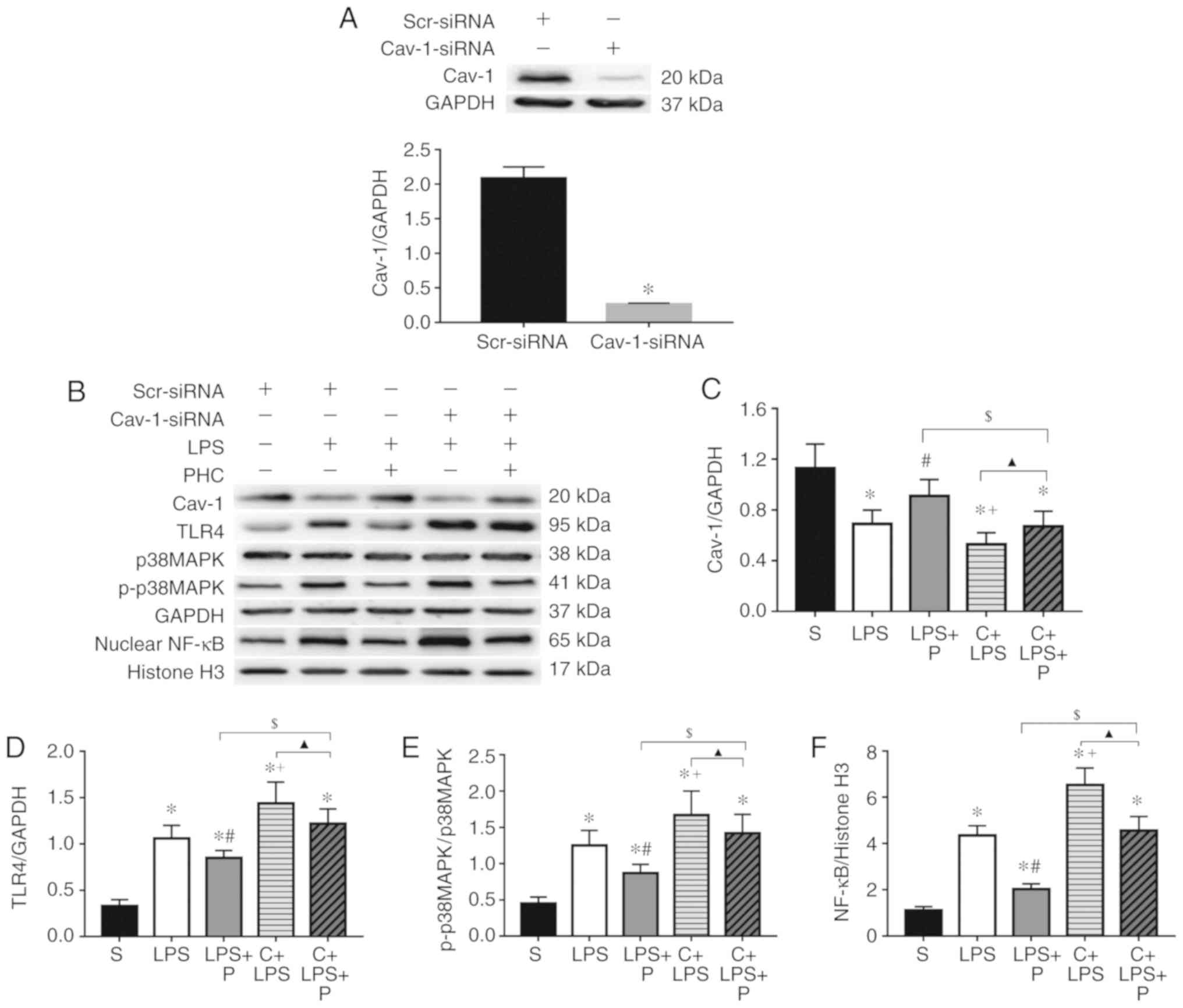 | Figure 8Effect of PHC on protein expression
levels in LPS-stimulated J774A.1 cells. (A) Western blot analysis
of Cav-1 and relative protein expression levels of Cav-1 in J774A.1
cells transfected with Scr-siRNA or Cav-1-siRNA. Data are presented
as the mean ± standard error of the mean (n=5).
*P<0.05 vs. Scr-siRNA group. (B) Western blot
analysis of Cav-1, TLR4, p38 MAPK, p-p38 MAPKs and nuclear NF-κB
p65 protein expression. Relative protein expression levels of (C)
Cav-1, (D) TLR4, (E) p-p38 MAPKs (p-p38 MAPKs/p38 MAPKs) and (F)
nuclear NF-κB p65. Data are presented as the mean ± standard error
of the mean (n=5). *P<0.05 vs. S group.
#P<0.05 vs. LPS group. +P<0.05 vs. LPS
group. $P<0.05 vs. LPS + P group.
▲P<0.05 vs. C + LPS group. PHC/P, penehyclidine
hydrochloride; LPS, lipopolysaccharide; Cav-1, caveolin-1; TLR4,
Toll-like receptor 4; NF-κB p65, nuclear factor
kappa-light-chain-enhancer of activated B cells transcription
factor p65 subunit; p, phosphorylated; p38 MAPKs, p38
mitogen-activated protein kinases; C, Cav-1-siRNA; siRNA, short
interfering RNA; S, scramble-siRNA. |
Discussion
ALI/acute respiratory distress syndrome (ARDS) is a
complex respiratory disorder associated with high morbidity and
mortality rates worldwide (25).
In the present study, it was suggested that Cav-1 may be involved
in the protective role of PHC against LPS-induced ALI. In addition,
the potential underlying mechanism associated with the inhibition
of p38 MAPKs phosphorylation and TLR4/NF-κB signaling pathway
activation was examined.
LPS is recognized as an important participant in the
pathogenesis of sepsis (26),
which may induce ALI (27). In
the present study, it was indicated that intratracheal injection of
LPS caused histological changes of lung tissues and dysfunction of
gas exchange, indicating that ALI was successfully induced by LPS
in SD rats.
NF-κB and MAPKs signal transduction pathways have
been demonstrated to mediate inflammatory responses in the lung.
Following recognition by TLR4, LPS causes inhibitor of κB
phosphorylation and degradation, which subsequently induces NF-κB
p65 translocation to the nucleus (28). The activated NF-κB in the nucleus
promotes the transcription of specific targeting genes, including
the pro-inflammatory cytokines TNF-α, IL-6 and IL-1β (29-31). In patients with ALI, NF-κB
activation significantly increases pro-inflammatory cytokine
production, which in turn promotes neutrophil recruitment into the
lung and inflammatory responses (32). The MAPKs are a group of signaling
molecules (33), among which p38
MAPKs have been demonstrated to be involved in the development of
ALI/ARDS caused by different stimuli (10,11,34). Following LPS challenge, p38 MAPKs
are activated, which subsequently induces the release of
pro-inflammatory cytokines (35,36), neutrophil recruitment into the
lung and bronchoconstriction (37). Inhibition of p38 MAPKs efficiently
decreases the effects of LPS-induced ALI, including protein leakage
and cell apoptosis in BALF, and neutrophil recruitment into the
lung (12). In the present study,
LPS challenge induced p38 MAPKs phosphorylation and NF-κB p65
translocation to the nucleus in vivo and in
vitro.
PHC is a novel anti-cholinergic drug, which may
selectively block M1 and M3 receptors, and nicotinic acetylcholine
receptors, but has almost no demonstrated cardiovascular side
effects associated with M2 receptors. A number of studies have
suggested that PHC exhibits a protective effect in pulmonary
diseases and sepsis, potentially by inhibiting pro-inflammatory
cytokine production (38).
According to Shen et al (14), a single injection of PHC
significantly decreased inflammation and lung vascular leakage in a
rat model of LPS-induced ALI, as the protective effect may involve
the inhibition of NF-κB and p38 MAPKs signaling pathways. In the
present study indicated that PHC post-treatment in vivo
decreased the degree of pulmonary edema in the rat model of
LPS-induced ALI, which was additionally verified by a significant
decrease in the lung W/D ratio, PMNs/total cells and total protein
concentration in BALF, and was associated with decreases in
histological lung damage and improvements in gas exchange
dysfunction induced by LPS. In addition, PHC significantly
decreased MPO activity in the lung tissues of ALI rats and
LPS-stimulated J774A.1 macrophages. PHC significantly inhibited
LPS-induced pro-inflammatory cytokine production in vivo and
in vitro, which indicated that PHC possesses
anti-inflammatory properties to inhibit the production of cytokines
in LPS-induced ALI. To examine the underlying anti-inflammatory
mechanism of PHC on LPS-induced ALI, the effects of PHC on NF-κB
and p38 MAPKs pathway activation were determined in vivo and
in vitro. The results indicated that LPS-induced TLR4/NF-κB
and p38 MAPKs activation were significantly inhibited by PHC
administration.
Cav-1 has been demonstrated to serve a crucial role
in acute inflammation and capillary leakage during ALI: Yuan et
al (39) observed that
Cav-1−/− mice exhibited an elevated lung inflammatory
response, an elevated pro-inflammatory cytokine production and an
increased mortality rate following LPS challenge, which may be
associated with NF-κB activation. Wang et al (19) revealed that Cav-1 regulated LPS
induced pro-inflammatory cytokines TNF-α and IL-6 production in
murine peritoneal and alveolar macrophages via the p38 MAPKs
pathway. In the present study, the results indicated that Cav-1
knockdown by siRNA in J774A.1 macrophages increased
pro-inflammatory cytokine production and MPO activity induced by
LPS, concomitant with the activation of the TLR4/NF-κB and p38
MAPKs pathways. It was also indicated that PHC upregulated Cav-1
expression following LPS stimulation in vivo and in
vitro.
There are a number of limitations in the present
study. Whether LPS-induced lung injury is aggravated in Cav-1 gene
deficiency requires additional investigation through the use of
Cav-1 gene-deficient mice. Whether other pathways have the ability
to mediate Cav-1 involvement in lung injury following LPS-induced
ALI also requires additional studies. Furthermore, a single
macrophage cell line was selected in the present study; lung
epithelial cells or other alveolar macrophage cell lines should be
included in future studies.
In conclusion, the present study indicated that PHC
possesses anti-inflammatory activity in rats with ALI and
LPS-stimulated J774A.1 cells, which may due to the inhibition of
p38 MAPKs phosphorylation and TLR4/NF-κB signaling pathway by
upregulating Cav-1 expression.
Abbreviations:
|
ALI
|
acute lung injury
|
|
PHC
|
penehyclidine hydrochloride
|
|
LPS
|
lipopolysaccharide
|
|
Cav-1
|
caveolin-1
|
|
NF-κB
|
nuclear factor
kappa-light-chain-enhancer of activated B cells
|
|
MAPK
|
mitogen activated protein kinase
|
|
siRNA
|
short-interfering RNA
|
|
TLR4
|
Toll-like receptor 4
|
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81571941).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QK and XW contributed equally to the present study.
XW, ZX and XS conceived and designed the study. QK, XW, LZ and WD
performed the experiments, analyzed the data, interpreted the
experimental results, prepared the figures and drafted the
manuscript. ZX, LZ and XS were involved in revising the manuscript.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Ethical approval was provided by the Medical Ethics
Committee of Renmin Hospital of Wuhan University. All surgical
procedures were performed in accordance with Wuhan University
Animal Care and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Strauss RH, Palmer KC and Hayes JA: Acute
lung injury induced by cadmium aerosol. I. Evolution of alveolar
cell damage. Am J Pathol. 84:561–578. 1976.PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fein AM and Calalang-Colucci MG: Acute
lung injury and acute respiratory distress syndrome in sepsis and
septic shock. Crit Care Clin. 16:289–317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matute-Bello G, Frevert CW and Martin TR:
Animal models of acute lung injury. Am J Physiol Lung Cell Mol
Physiol. 295:L379–L399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tolle LB and Standiford TJ:
Danger-associated molecular patterns (DAMPs) in acute lung injury.
J Pathol. 229:145–156. 2013. View Article : Google Scholar
|
|
8
|
Ghosh S, May MJ and Kopp EB: NF-kappa B
and Rel proteins: Evolutionarily conserved mediators of immune
responses. Annu Rev Immunol. 16:225–260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kolb M, Margetts PJ, Anthony DC, Pitossi F
and Gauldie J: Transient expression of IL-1beta induces acute lung
injury and chronic repair leading to pulmonary fibrosis. J Clin
Invest. 107:1529–1536. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haddad EB, Birrell M, McCluskie K, Ling A,
Webber SE, Foster ML and Belvisi MG: Role of p38 MAP kinase in
LPS-induced airway inflammation in the rat. Br J Pharmacol.
132:1715–1724. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nash SP and Heuertz RM: Blockade of p38
map kinase inhibits complement- induced acute lung injury in a
murine model. Int Immunopharmacol. 5:1870–1880. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HJ, Lee HS, Chong YH and Kang JL: p38
Mitogen-activated protein kinase upregulates LPS-induced NF-kappaB
activation in the development of lung injury and RAW 264.7
macrophages. Toxicology. 225:36–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han XY, Liu H, Liu CH, Wu B, Chen LF,
Zhong BH and Liu KL: Synthesis of the optical isomers of a new
anticholinergic drug, penehyclidine hydrochloride (8018). Bioorg
Med Chem Lett. 15:1979–1982. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen W, Gan J, Xu S, Jiang G and Wu H:
Penehyclidine hydrochloride attenuates LPS-induced acute lung
injury involvement of NF-kappaB pathway. Pharmacol Res. 60:296–302.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benlimame N, Le PU and Nabi IR:
Localization of autocrine motility factor receptor to caveolae and
clathrin-independent internalization of its ligand to smooth
endoplasmic reticulum. Mol Biol Cell. 9:1773–1786. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gumbleton M, Abulrob AG and Campbell L:
Caveolae: An alternative membrane transport compartment. Pharm Res.
17:1035–1048. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rothberg KG, Heuser JE, Donzell WC, Ying
YS, Glenney JR and Anderson RG: Caveolin, a protein component of
caveolae membrane coats. Cell. 68:673–682. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun Y, Hu G, Zhang X and Minshall RD:
Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary
vascular permeability via paracellular and transcellular pathways.
Circ Res. 105:676–685, 15 p following 685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang XM, Kim HP, Song R and Choi AM:
Caveolin-1 confers antiinflammatory effects in murine macrophages
via the MKK3/p38 MAPK pathway. Am J Respir Cell Mol Biol.
34:434–442. 2006. View Article : Google Scholar
|
|
20
|
Garrean S, Gao XP, Brovkovych V, Shimizu
J, Zhao YY, Vogel SM and Malik AB: Caveolin-1 regulates NF-kappaB
activation and lung inflammatory response to sepsis induced by
lipopolysaccharide. J Immunol. 177:4853–4860. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
National Research Council (US) Committee
on Recognition and Alleviation of Pain in Laboratory Animals:
Recognition and Alleviation of Pain in Laboratory Animals. National
Academies Press; Washington, DC: 2009
|
|
22
|
Wu XJ, Xia ZY, Wang LL, Luo T, Zhan LY,
Meng QT and Song XM: Effects of penehyclidine hydrochloride on
pulmonary contusion from blunt chest trauma in rats. Injury.
43:232–236. 2012. View Article : Google Scholar
|
|
23
|
Wu XJ, Liu HM, Song XM, Zhao B, Leng Y,
Wang EY, Zhan LY, Meng QT and Xia ZY: Penehyclidine hydrochloride
inhibits TLR4 signaling and inflammation, and attenuates blunt
chest trauma and hemorrhagic shock-induced acute lung injury in
rats. Mol Med Rep. 17:6327–6336. 2018.PubMed/NCBI
|
|
24
|
Su X, Wang L, Song Y and Bai C: Inhibition
of inflammatory responses by ambroxol, a mucolytic agent, in a
murine model of acute lung injury induced by lipopolysaccharide. I.
ntensive Care Med. 30:133–140. 2004. View Article : Google Scholar
|
|
25
|
Matthay MA, McAuley DF and Ware LB:
Clinical trials in acute respiratory distress syndrome: Challenges
and opportunities. Lancet Respir Med. 5:524–534. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brigham KL and Meyrick B: Endotoxin and
lung injury. Am Rev Respir Dis. 133:913–927. 1986.PubMed/NCBI
|
|
27
|
Kitamura Y, Hashimoto S, Mizuta N,
Kobayashi A, Kooguchi K, Fujiwara I and Nakajima H:
Fas/FasL-dependent apoptosis of alveolar cells after
lipopolysaccharide- induced lung injury in mice. Am J Respir Crit
Care Med. 163:762–769. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chiang EY, Yu X and Grogan JL: Immune
complex-mediated cell activation from systemic lupus erythematosus
and rheumatoid arthritis patients elaborate different requirements
for IRAK1/4 kinase activity across human cell types. J Immunol.
186:1279–1288. 2011. View Article : Google Scholar
|
|
29
|
Kagan JC and Medzhitov R:
Phosphoinositide-mediated adaptor recruitment controls toll-like
receptor signaling. Cell. 125:943–955. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wilson SJ, Leone BA, Anderson D, Manning A
and Holgate ST: Immunohistochemical analysis of the activation of
NF-kappaB and expression of associated cytokines and adhesion
molecules in human models of allergic inflammation. J Pathol.
189:265–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar
|
|
32
|
Kuo MY, Liao MF, Chen FL, Li YC, Yang ML,
Lin RH and Kuan YH: Luteolin attenuates the pulmonary inflammatory
response involves abilities of antioxidation and inhibition of MAPK
and NFκB pathways in mice with endotoxin-induced acute lung injury.
Food Chem Toxicol. 49:2660–2666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yeh CH, Yang JJ, Yang ML, Li YC and Kuan
YH: Rutin decreases lipopolysaccharide-induced acute lung injury
via inhibition of oxidative stress and the MAPK-NF-κB pathway. Free
Radic Biol Med. 69:249–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang J, Murphy C, Denham W, Botchkina G,
Tracey KJ and Norman J: Evidence of a central role for p38 map
kinase induction of tumor necrosis factor alpha in
pancreatitis-associated pulmonary injury. Surgery. 126:216–222.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JC and Young PR: Role of CSB/p38/RK
stress response kinase in LPS and cytokine signaling mechanisms. J
Leukoc Biol. 59:152–157. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carter AB, Knudtson KL, Monick MM and
Hunninghake GW: The p38 mitogen-activated protein kinase is
required for NF-kappaB-dependent gene expression. The role of
TATA-binding protein (TBP). J Biol Chem. 274:30858–30863. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schnyder-Candrian S, Quesniaux VF, Di
Padova F, Maillet I, Noulin N, Couillin I, Moser R, Erard F,
Vargaftig BB, Ryffel B and Schnyder B: Dual effects of p38 MAPK on
TNF-dependent bronchoconstriction and TNF-independent neutrophil
recruitment in lipopolysaccharide-induced acute respiratory
distress syndrome. J Immunol. 175:262–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhan J, Wang Y, Wang C, Li J, Zhang Z and
Jia B: Protective effects of penehyclidine hydrochloride on septic
mice and its mechanism. Shock. 28:727–732. 2007.PubMed/NCBI
|
|
39
|
Yuan K, Huang C, Fox J, Gaid M, Weaver A,
Li G, Singh BB, Gao H and Wu M: Elevated inflammatory response in
caveolin-1-deficient mice with Pseudomonas aeruginosa infection is
mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB). J
Biol Chem. 286:21814–21825. 2011. View Article : Google Scholar : PubMed/NCBI
|