Introduction
Posterior capsule opacification (PCO) is a common
postoperative complication of phacoemulsification or extracapsular
cataract extraction (1), which
are mainly caused by the proliferation and migration of residual
lens epithelial cells (LECs) in the anterior and equator lens
capsule after cataract surgery (2,3).
In total, 20-40% of adult and 100% of pediatric patients suffer
from visual loss due to PCO after cataract surgery (1). At present, epithelial-mesenchymal
transition (EMT) of the postoperative residual LECs is believed to
be the primary cause underlying PCO pathogenesis (4,5).
During the process of EMT, epithelial cells lose cell polarity and
cell-cell adhesion, gain migratory and invasive properties, and
acquired characteristics of mesenchymal stem cells (1). Due to hyperplasia, migration and
transdifferentiation of LECs, the lens posterior capsule may
exhibit thickening, opacification and clouding (1). Several signaling pathways may be
involved in EMT (6). The residual
LECs after cataract surgery could release autocrine molecules such
as transforming growth factor-β2 (TGF-β2), which is secreted into
the aqueous humor in the capsular bag (7,8).
Additionally, in our previous studies, TGF-β2 was found to be able
to induce EMT in human LECs (9,10).
EMT is characterized by the loss of epithelial
markers, such as E-cadherin, and the acquisition of mesenchymal
markers, such as α-Smooth Muscle Actin (α-SMA) and fibronectin.
Several transcription factors, such as snail family transcriptional
repressor 1 (SNAI1), Twist and zinc finger E-box binding homeobox
1, are involved in EMT by repressing E-cadherin transcription in
vitro and in vivo (11). Previous studies suggested that the
transcription factor SNAI1 is able to regulate the EMT process of
some cells such as alveolar epithelial cells (12). In addition, SNAI1 regulated EMT in
a variety of other physiological and pathological processes, such
as embryonic development, tissue fibrosis and metastasis of
malignant tumors (13). SNAI1
recruits multiple chromatin enzymes including lysine-specific
demethylase 1A (LSD1), histone deacetylase (HDAC) 1/2, polycomb
repressive complex 2 (PRC2), euchromatic histone lysine
methyltransferase 2 (EHMT2) and suppressor of variegation 3-9
homolog 1 (SUV39H1) to the E-cadherin promoter, thereby suppressing
E-cadherin expression in cancers cells (14). HDACs are enzymes that remove the
acetyl groups from histones and increase the affinity between DNA
and histones (2). HDAC activity
can be inhibited by inhibitors such as trichostatin A (TSA) and
suberoylanilide hydroxamic acid (SAHA) (15). HDAC1 has a zinc-dependent active
site and can be recruited by various transcription factors, such as
SNAI1, which belongs to the zinc finger transcription factor family
(3). However, to the best of our
knowledge, the possible functional relationship between SNAI1 and
HDAC1 in the induction of EMT in HLECs has not been yet
reported.
Therefore, the aim of this study was to determine
whether the transcription factor SNAI1 is involved in the EMT
process of HLECs induced by TGF-β2, and to investigate the
potential functional relationship between SNAI1 and HDAC1 during
that process.
Materials and methods
Cell culture
Immortalized HLEB-3 cells were obtained from The
American Type Culture Collection (ATCC) and cultured in Eagle's
Minimal Essential Medium (EMEM; ATCC) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
containing 5% CO2 at 37°C. HLEB-3 cells were treated
with various doses of TGF-β2 (1 µg/ml dissolved in PBS;
PeproTech, Inc.) for the indicated time with or without TSA
(Sigma-Aldrich; Merck KGaA) or SAHA (Sigma-Aldrich; Merck KGaA),
which are class I and II HDAC inhibitors. The specific small
interfering RNA (siRNA) for SNAI1 (cat. no. sc-38398) and control
siRNA (cat. no. sc-37007) were purchased from Santa Cruz
Biotechnology, Inc. The HLEB-3 cells, seeded on six-well plate,
were transfected with 5 µl SNAI1 siRNA or control siRNA at a
concentration of 10 µM using Lipofectamine RNAiMax (Thermo
Fisher Scientific, Inc.) for 48 h prior to further experimentation,
according to the manufacturer's protocol.
Immunocytochemistry
HLEB-3 cells were cultured and treated with 1 ng/ml
TGF-β2. Cells were fixed using 4% formaldehyde at room temperature
for 10 min and then permeabilized with 0.5% Triton X-100 in PBS at
room temperature for 5 min. After blocking with 1% BSA in PBS at
room temperature for 1 h, HLEB-3 cells were incubated with mouse
anti-SNAI1 antibodies (cat. no. sc-271977; 1:200; Santa Cruz
Biotechnology, Inc.) at 4°C overnight. Subsequently, samples were
incubated with Alexa Fluor 488-conjugated donkey anti-mouse
antibody (cat. no. A21202; 1:1,000; Thermo Fisher Scientific, Inc.)
at room temperature for 1 h. The nuclei were counterstained with
VECTASHIELD mounting medium containing DAPI (Vector Laboratories).
The slides were observed using a fluorescent microscope (Carl Zeiss
AG; magnification, ×400) and representative images were
obtained.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cultured HLEB-3 cells
using the RNeasy Mini kit (Qiagen, Inc.) following the
manufacturer's protocol. mRNA was reverse transcribed to cDNA using
the Maxima First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.). The reverse transcription reaction was incubated
for 10 min at 25°C, for 15 min at 50°C and for 5 min at 85°C.
RT-qPCR was conducted using the Power SYBR Green (Takara
Biotechnology, Co., Ltd.) according to the manufacturer's protocol
using the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo
Fisher Scientific, Inc.) under the following condition: Initial
denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for
30 sec, 60°C for 1 min, 95°C for 1 min and 60°C for 1 min. Data
were analyzed using the 2−ΔΔCq method (16). The primer sequence for RT-qPCR
were as follows: E-cadherin forward, 5′-AGT GAC TGA TGC TGA TGC
CC-3′ and reverse, 5′-CTG CAT CTT GCC AGG TCC TT-3′; fibronectin
forward, 5′-TAT TGA AGG CTT GCA GCC CA-3 and reverse, 5′-CAC CAT
CAG GTG CAG GGA AT-3′; α-SMA forward, 5′-CAG GCT CAA GTC TGT CTT
TGC-3′ and reverse, 5′-CCG CCT GGA TAG CCA CAT AC-3′; β-actin
forward, 5′-AAA CTG GAA CGG TGA AGG TG-3′ and reverse, 5′-GTG GCT
TTT AGG ATG GCA AG-3′. β-actin was used as the reference gene.
Western-blot analysis
HLEB-3 cells were lysed in RIPA buffer (50 mM Tris,
pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS;
Beyotime Institute of Biotechnology) containing 1% protease
inhibitors (PhosStop phosphatase inhibitor cocktail tablet; Roche
Diagnostics) at 4°C for 30 min. The cell lysates were harvested and
centrifuged at 14,000 × g at 4°C for 10 min. Subsequently, the
supernatants were collected. The protein concentration was
quantified using a Pierce bicinchoninic acid Protein Assay kit
(Thermo Fisher Scientific, Inc.). After denaturing the samples with
Laemmli buffer (Bio-Rad Laboratories, Inc.) at 95°C for 5 min, 20
µg of total protein was separated by 10% SDS-PAGE (Beyotime
Institute of Biotechnology), and then transferred onto a PVDF
membrane (EMD Millipore). After blocking with 5% non-fat dry milk
in TBST (TBS + 0.1% Tween-20) for 1 h at room temperature, the
membranes were washed with TBST three times. The membranes were
subsequently incubated overnight at 4°C with the following primary
antibodies: Rabbit anti-fibronectin (cat. no. sc-9068, Santa-Cruz
Biotechnology, Inc.), mouse anti-α-SMA (cat. no. A2547,
Sigma-Aldrich; Merck KGaA), rabbit anti-E-cadherin (cat. no. 3195;
Cell Signaling Technology, Inc.), mouse anti-SNAI1 (cat. no.
sc-271977; Cell Signaling Technology, Inc.) and mouse anti-GAPDH
(cat. no. ab9482; Abcam, Inc.) or mouse anti-β-actin (cat. no.
A5441; Sigma-Aldrich; Merck KGaA). The membranes were washed in
TBST and then incubated for 1 h at room temperature with
horseradish peroxidase-conjugated goat anti-mouse (1:2,000; cat.
no. BA1051; Wuhan Boster Biological Technology Co., Ltd.) and goat
anti-rabbit secondary antibodies (1:2,000; cat. no. BA1054; Boster
Biological Technology Co., Ltd.). The specific bands were detected
using an ECL reagent (Applygen Technologies, Inc.) and BioMax film
(Kodak). The bands were analyzed using GEL-PRO Analyzer software
(version 4.0; Media Cybernetics, Inc.).
Immunoprecipitation
HLEB-3 cells were lysed for 60 min at 4°C in
extraction buffer containing 50 mmol/l Tris-Cl (pH 7.5), 100 mmol/l
NaCl, 1% Triton X-100, 1 mmol/l DTT, 1 mmol/l EDTA, 1 mmol/l EGTA,
2 mmol/l Na3VO4, 50 mmol/l β-glycerophosphate
and a protease inhibitor (Roche Diagnostics). The cell lysates were
pre-washed with protein A/G-agarose beads (Cell Signaling
Technology, Inc.) and pelleted by centrifugation at 1,000 × g for 5
min at 4°C. The supernatant was collected and further incubated
with control IgG or mouse anti-SNAI1 (1:100; cat. no. ab78105;
Abcam, Inc.) or rabbit anti-HDAC1 (1:100; cat. no. ab7028; Abcam,
Inc.) antibody conjugated with protein A/G-agarose beads at 4°C
overnight. The beads were washed twice with extraction buffer and
twice with extraction buffer with 0.5 mol/l LiCl. Proteins were
eluted directly in non-reducing buffer for western-blot
analysis.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed using a ChIP kit (cat.
no. ab500; Abcam, Inc.) according to the manufacturer's protocol.
Briefly, 1×107 HLEB-3 cells were crosslinked with 1%
formaldehyde at room temperature for 10 min, lysed with ChIP lysis
buffer on ice for 10 min and sonicated to produce chromatin
fragments. The precipitation was performed with polyclonal
antibodies against HDAC1 (1:100; cat. no. ab7028; Abcam, Inc.) at
4°C for 1 h. The rabbit immunoglobumin G (1:100; cat. no. ab172730;
Abcam Inc.) was used as negative control. The precipitation of the
E-cadherin promoter was determined by PCR with Taq DNA polymerase
(Takara Biotechnology, Co., Ltd.) using primer spanning from
nucleotide -680 to -541 of the E-cadherin sequence under the
following conditions: Initial denaturation at 95°C for 5 min,
followed by 35 cycles of 95°C for 30 sec, 62°C for 30 sec, and 72°C
for 30 sec, with a final extension at 72°C for 5 min. The
amplification primers of E-cadherin promoter were as follows:
Forward: 5′-GTC ACC GCG TCT ATG CGA GGC CG-3′ and reverse: 5′-GCG
TGG CTG CAG CCA GGT G-3′. The PCR product was detected using a 1.5%
agarose gel and visualized with ethidium bromide.
Statistical analysis
Data were presented as the mean ± SEM and
statistical analysis were performed with SPSS 17.0 software (SPSS,
Inc.). Each experiment was independently repeated at least three
times. One way ANOVA was used to compare three or more groups,
followed by Bonferroni's post hoc test. Student's t-test was used
to analyze the differences between two independent groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TGF-β2 promotes EMT markers in HLECs
To examine whether TGF-β2 induced EMT in HLECs, the
mRNA and protein expression levels of various molecules, including
E-cadherin, fibronectin and α-SMA, were examined. After treatment
with TGF-β2 (10 ng/ml), there was a slight reduction of E-cadherin
expression at 12 h, followed by a remarkable decrease in its
expression at 24 and 48 h (Fig.
1A). Furthermore, TGF-β2 (10 ng/ml) induced a significant
upregulation of fibronectin (Fig.
1B) and α-SMA (Fig. 1C) in
HLEB-3 cells in a time-dependent manner. Consistently, the protein
level of E-cadherin was significantly downregulated by TGF-β2 after
24 h (Fig. 1D and E).
Furthermore, the expression levels of fibronectin and α-SMA, two
major fibrotic markers, were upregulated by TGF-β2 treatment
(Fig. 1D and E). Treatment with
different doses of TGF-β2 resulted in a concentration-dependent
decrease in E-cadherin expression but a concentration-dependent
increase in fibronectin and α-SMA expression in HLEB-3 cells at
both mRNA (Fig. 1F-H) and protein
level (Fig. 1I and J).
Collectively, the present results suggested that TGF-β2 upregulated
EMT markers in HLEB3 cells by reducing E-cadherin expression and
increasing fibro-nectin and α-SMA expression.
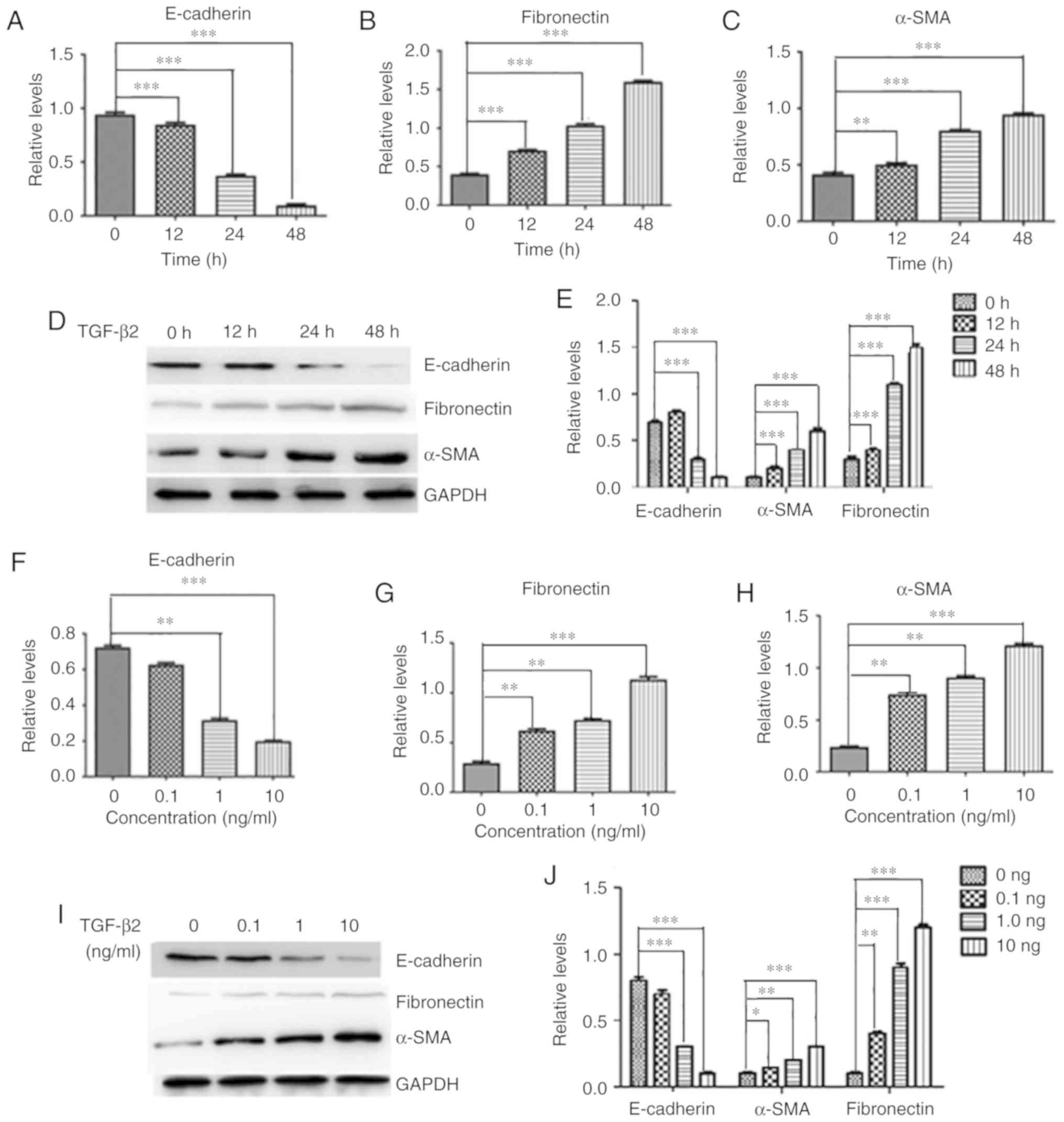 | Figure 1TGF-β2 induces the expression of EMT
markers in HLEB-3 cells. HLEB-3 cells were treated with TGF-β2 at
10 ng/ml for 12, 24 and 48 h, and the mRNA levels of (A)
E-cadherin, (B) fibronectin and (C) α-SMA were detected by RT-qPCR.
(D) Western blot analysis and (E) quantification of the protein
expression levels of E-cadherin, fibronectin and α-SMA. HLEB-3
cells were treated with TGF-β2 at various concentrations (0.1, 1
and 10 ng/ml) for 48 h, and the mRNA levels of (F) E-cadherin, (G)
fibronectin and (H) α-SMA were detected by RT-qPCR. (I) Western
blot analysis and (J) quantification of the protein expression
levels of E-cadherin, fibronectin and α-SMA. n=3.
*P<0.05, **P<0.01,
***P<0.001. RT-qPCR, reverse
transcription-quantitative PCR; SMA, smooth muscle actin; TGF,
transforming growth factor. |
Involvement of SNAI1 in TGF-β2-induced
E-cadherin downregulation in HLEB-3
SNAI1 is an important transcription factor in EMT
(17). Therefore, the role of
SNAI1 in TGF-β2-induced EMT was examined by immunostaining and
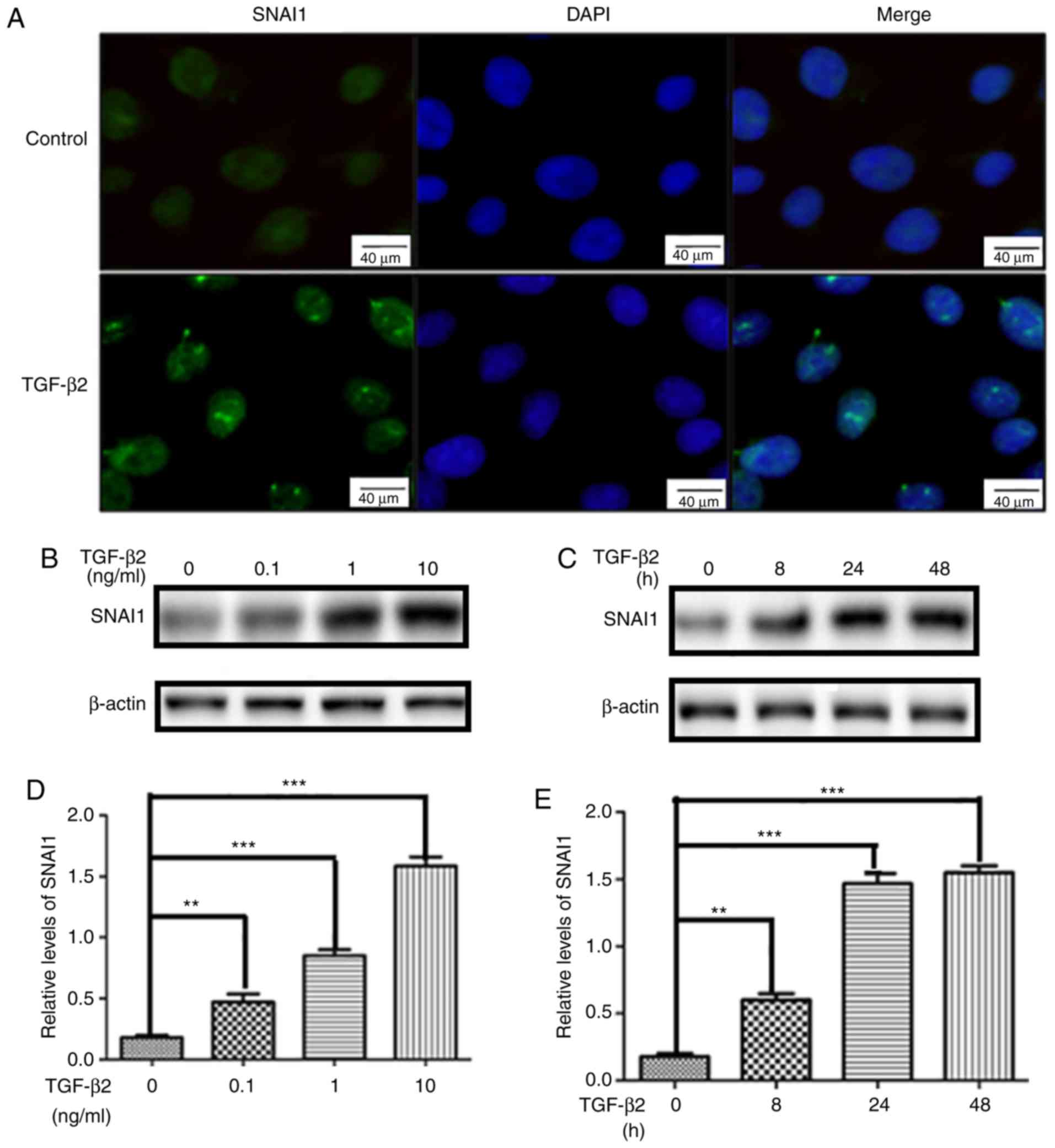
western-blot analysis. As shown in Fig. 2A, SNAI1 was weakly expressed and
mainly distributed in the nucleus of quiescent HLEB3 cells.
Consistently, nuclear SNAI1 expression was markedly increased in
HLEB-3 cells after TGF-β2-treatment. Moreover, SNAI1 expression was
dramatically increased in TGF-β2-treated HLEB-3 cells in both dose-
and time-dependent manner (Fig.
2B-E). Next, HLEB-3 cells were transfected with specific
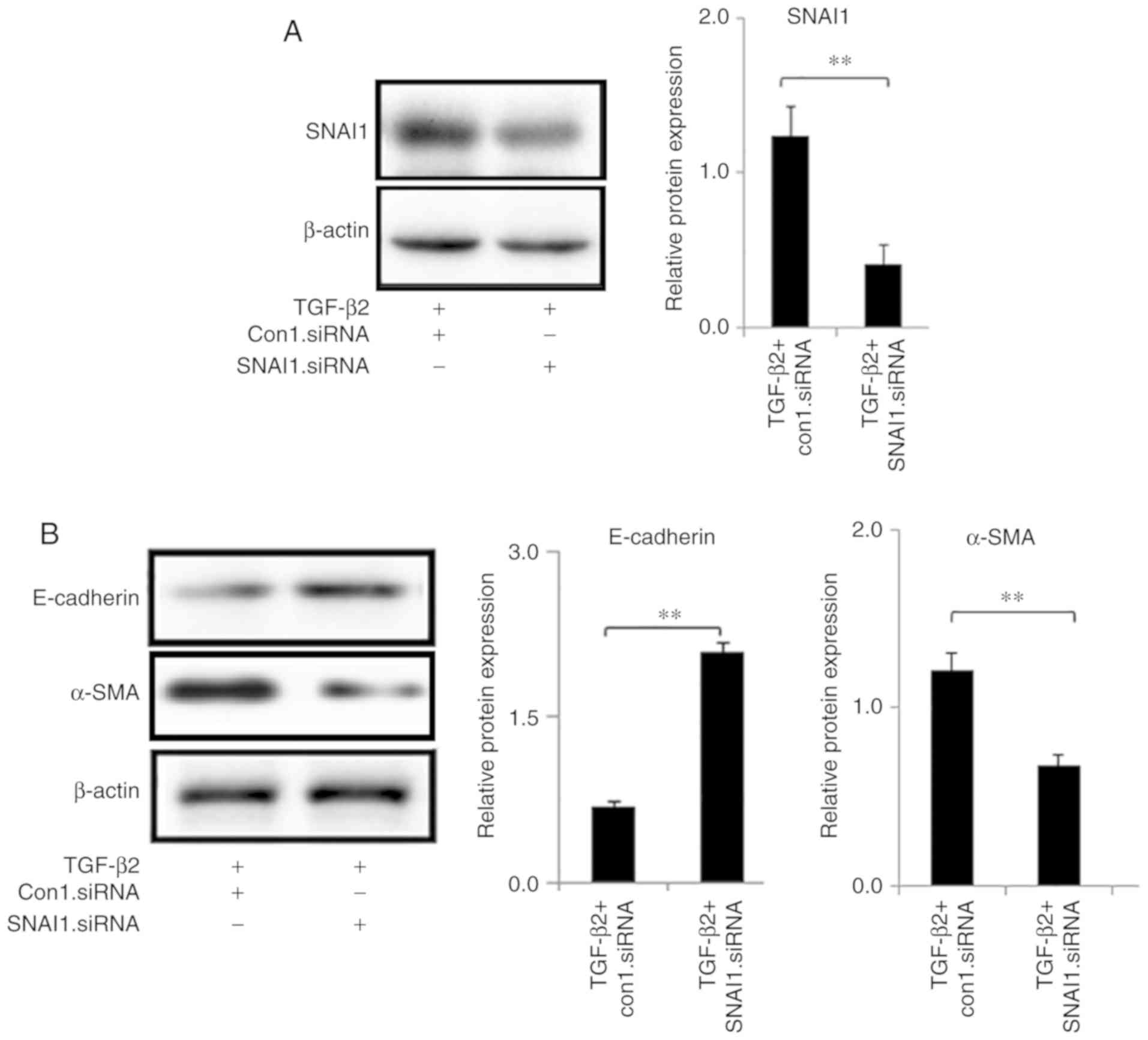
SNAI1-siRNA and control siRNA. The expression level of SNAI1 was
decreased following SNAI1-siRNA transfection (Fig. S1). The western-blot analysis
demonstrated that SNAI1-siRNA significantly reduced TGF-β2-induced
SNAI1 expression (Fig. 3A).
Moreover, knockdown of SNAI1 by siRNA dramatically reversed
TGF-β2-associated downregulation of E-cadherin and upregulation of
α-SMA (Fig. 3B), which suggested
a causal role of SNAI1 in regulating EMT markers.
SNAI1 is associated with HDAC1 in the
TGF-β2-mediated E-cadherin repression
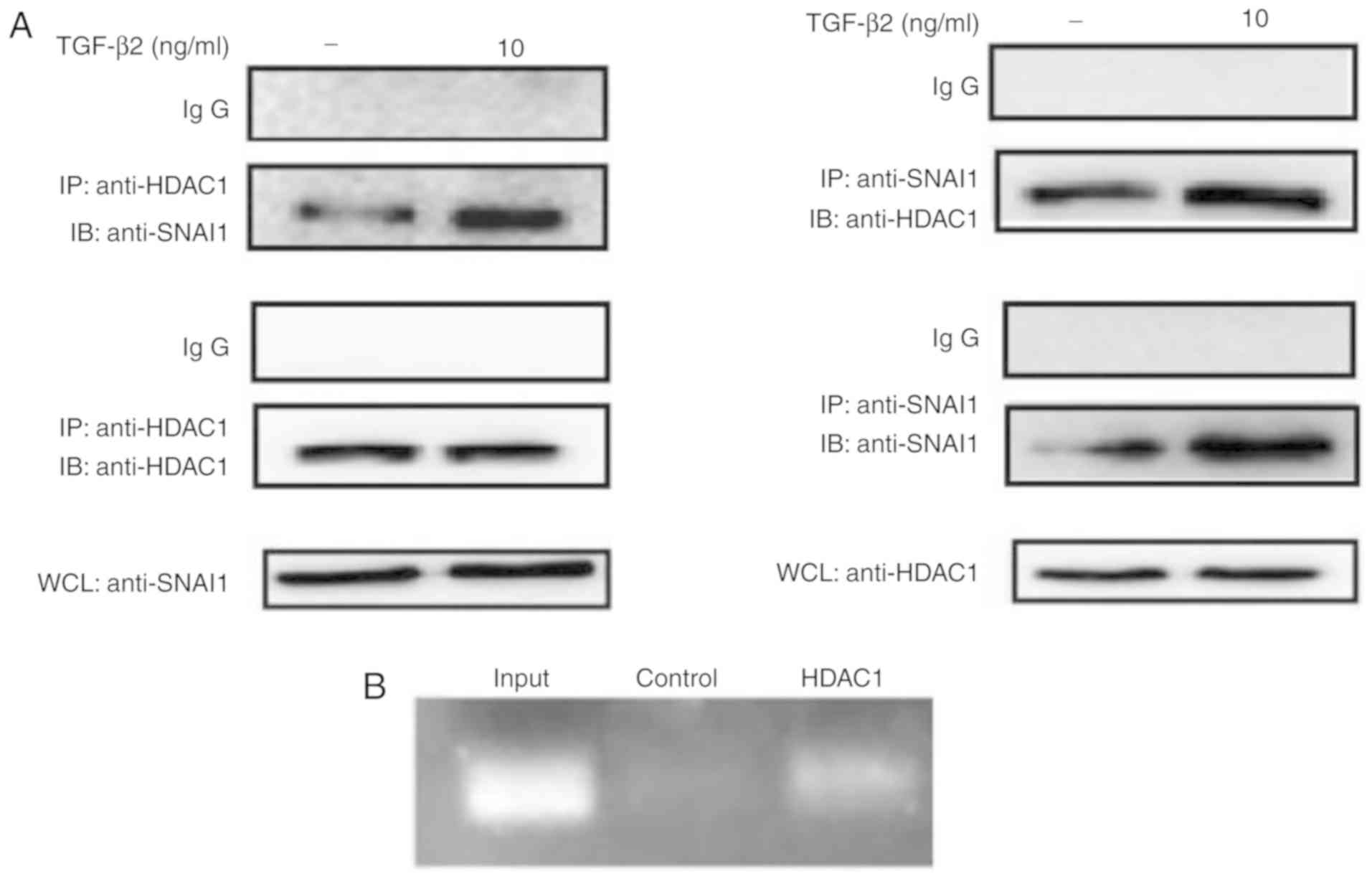
In the present study, it was investigated whether
SNAI1 was directly associated with HDAC1 at the E-cadherin promoter
by immunoprecipitation assay and ChIP. After immunoprecipitation
with SNAI1 antibody, SNAI1 was found to be associated with HDAC1
(Fig. 4A). Consistently, after
immunoprecipitation with HDAC1 antibody, HDAC1 was found to
interact with SNAI1. Intriguingly, by using ChIP assay, a direct
interaction between HDAC1 and E-cadherin promoter was confirmed
(Fig. 4B). The present data
suggested that the association of SNAI1 and HDAC1 is involved in
transcriptional inhibition E-cadherin induced by TGF-β2.
HDAC inhibition prevents TGF-β2-induced
E-cadherin down- regulation in HLEB-3 cells
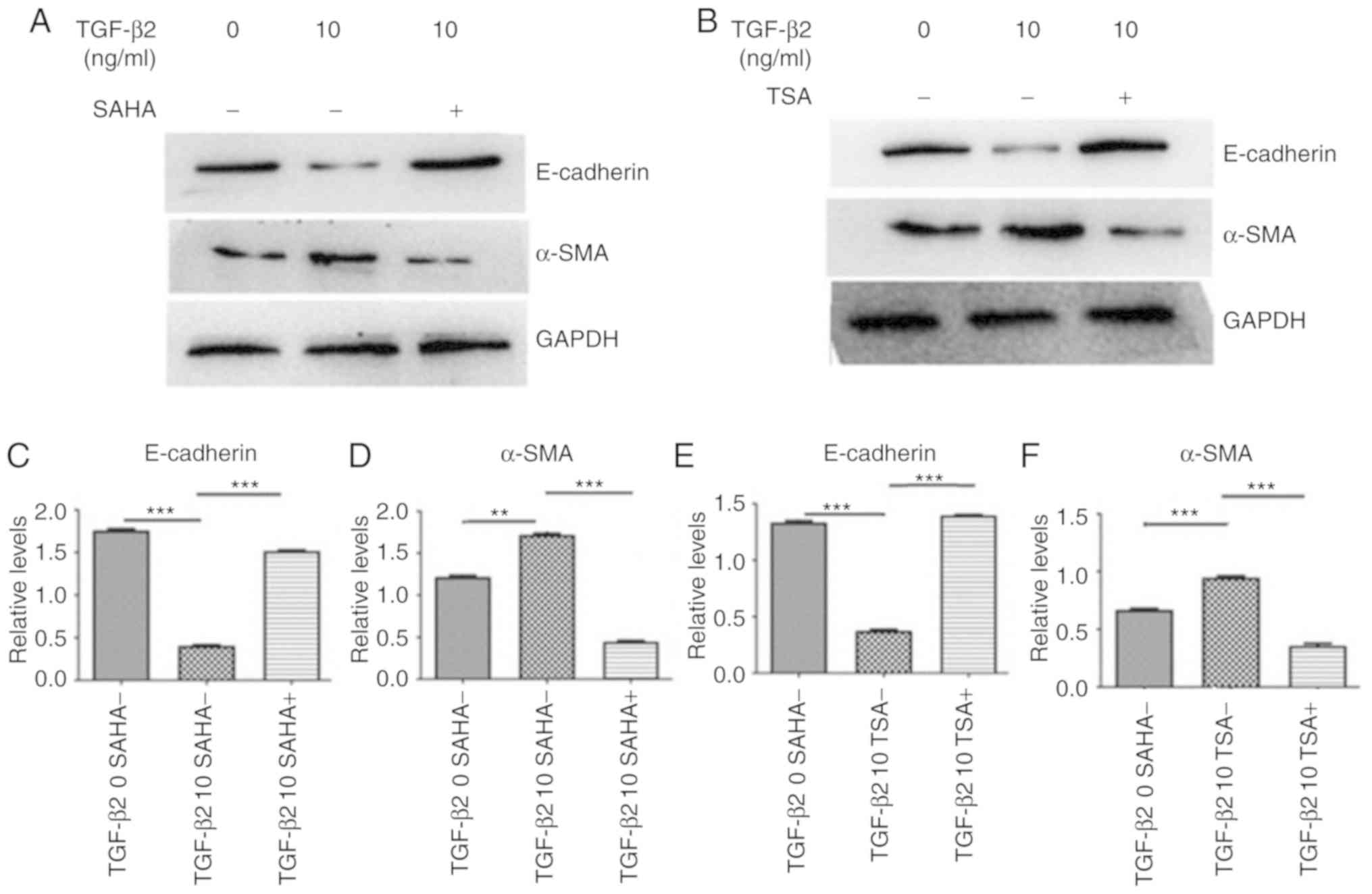
The effect of HDAC inhibition on the TGF-β2-induced
decrease of E-cadherin and increase of α-SMA was then examined. TSA
and SAHA were used as HDAC inhibitors. The present results
suggested that SAHA prevented TGF-β2 induced α-SMA upregulation and
reversed E-cadherin downregulation (Fig. 5A, C and D). Furthermore,
TGF-β2-induced α-SMA upregulation and E-cadherin downregulation
were also attenuated by TSA, another HDAC inhibitor (Fig. 5B, E and F). Collectively,
inhibition of HDAC activity could attenuate the TGF-β2-induced
increase of EMT markers in HLEB3 cells.
Discussion
Accumulating evidence demonstrated that the
underlying mechanism of PCO is a wound-healing process in response
to postoperative residual LECs initiated by surgical stress
(4,18). Some of these cells undergo EMT,
which includes cell proliferation and migration. These cells
migrate along the lens capsule into the visual axis with ectopic
proliferation and loss of epithelial phenotype (1). Therefore, they gain a
myofibroblast-like phenotype (1).
On one hand, downregulation of E-cadherin expression can reduce
intercellular adhesion (19). On
the other hand, upregulation of α-SMA expression contributes to
tissue fibrosis (19).
Additionally, cells produce more extracellular matrix containing
glycoprotein, fibrin and various types of collagen fibers (10). Cellular fibronectin is
incorporated into the fibrillar matrix of the cell surface, and its
level can be measured by examining the protein expression and mRNA
levels in cellular extracts (20,21). Therefore, the deposition of these
molecules on the surface of the posterior capsule may lead to
impaired vision (1).
During the process of EMT, epithelial cells lose
cell polarity and adhesion, gain migratory and invasive properties,
and acquire a mesenchymal stem cell-like phenotype (22). Loss of the adhesion factor
E-cadherin is one of the most important events underlying EMT
(22). Several signaling
pathways, including TGF-β2, fibroblast growth factor, epidermal
growth factor, hepatocyte growth factor, Wnt/β-catenin and Notch
pathways, may be involved in EMT. Tamiya et al (23) found that TGF-β2 can induce EMT in
retinal pigment epithelial cells, participating in proliferative
vitreous retinal disease. Dwivedi et al (24) used TGF-β2 to induce EMT in lens
epithelial cells of rats, resulting in the upregulation of matrix
metalloproteinases and the formation of cloudy lens. The residual
epithelial cells after cataract surgery can sense signal via
autocrine TGF-β2, which is secreted into the aqueous humor in the
capsular bag (25). Our previous
studies have also shown that TGF-β2 plays an important role in
inducing EMT, migration and proliferation of LECs (9).
TGF-β2 could induce EMT in HLECs (26). The signaling pathways that are
activated in response to TGF-β2 and lead to EMT have primarily been
conducted in vitro using NMuMG, MDCK and HaCaT cell lines
(27-30). In the present study, EMT in HLEB-3
cells was activated when cells were incubated with TGF-β2,
providing new insight for further exploring the potential
functional relationship between SNAI1 and HDAC1 in the pathogenesis
of PCO and its underlying mechanisms.
A recent study suggested that proteins of the SNAI1
family are important regulatory factors in the process of EMT in a
variety of cell types (17). The
transcription factors belonging to the SNAI family are highly
homologous at the N-terminal region, but variable at the C-terminal
region, which is formed by a variable number of zinc finger
structures (31). SNAI proteins
are involved in multiple steps in the process of EMT induced by
TGF-β2, causing tissue or organ fibrosis (32). Furthermore, TGF-β2 induces SNAI1
expression during skin (33),
palate (34) and heart
development (35), as well as in
mesothelial cells during pathological fibrosis (36). SNAI1 can bind to the E-boxes of
human E-cadherin promoter and repress its transcription, thus
reducing cell adhesion (37). The
present results identified that TGF-β2 induces the increased
expression of α-SMA, fibro-nectin and decreased the expression of
E-cadherin in HLECs in a time- and dose-dependent manner. Moreover,
the expression of SNAI1 was increased as a result of the TGF-β2
treatment. HDAC is found primarily in the nucleus (38). HDAC removes the acetyl groups from
histones, promoting a high-affinity interaction between histones
and DNA. The interaction between DNA and histones can modify the
DNA structure, which makes DNA less accessible and thus prevents
its transcription (38). The
abnormal expression of HDAC1 is involved in many pathological
processes. Zupkovitz et al (39) found that a specific subset of
genes was dysregulated in the absence of HDAC1 in mice. SNAI1 is a
transcriptional repressor that recruits multiple chromatin enzymes
including HDAC1/2, LSD1, PRC2, EHMT2 and SUV39H1 to the E-cadherin
promoter, thereby suppressing E-cadherin expression during cancers
cell metastasis (14,40-43). In the present study, SNAI1 was
found to directly interact with HDAC1 at E-cadherin promoter by
immunoprecipitation assay and ChIP. The present results suggested
that SNAI1 and HDAC1 may be involved in E-cadherin transcriptional
regulation.
HDAC inhibitors such as SAHA and TSA have been used
for ~10 years to treat neurological disorders (44). These inhibitors were being studied
as a novel therapy for neurodegenerative disorders (45). A previous study aimed to develop
novel HDAC inhibitors to treat cancer, since old HDAC inhibitors
have been shown to be ineffective (46). Vorinostat and romidepsin, which
belongs to the SAHA family, were approved for the treatment of
cutaneous manifestations in patients with cutaneous T cell lymphoma
(47,48). Isoform-selective HDAC inhibitors,
which can facilitate the investigation of the specific roles of
individual HDAC isoforms, have been developed (49). SNAI1 requires HDAC1 to repress
E-cadherin promoter, and treatment with TSA can block the
repressive effect of SNAI1. The present study suggested that
treatment with HDAC inhibitors, such as SAHA and TSA, completely
abrogated upregulation of α-SMA and downregulation of E-cadherin,
indicating that the HDAC inhibitors can significantly attenuate
TGF-β2-induced EMT in HLEB-3 cells.
Collectively, the present study suggested that SNAI1
interacted with HDAC1 to control TGF-β2-induced EMT in cultured
HLECs. Therefore, HDAC inhibitors may be a potential method for the
treatment of PCO.
Supplementary Data
Funding
The present study was supported by the NSFC (grant
no. 81470614), Shaanxi Young Talent Scholar Grant (grant no.
2016KJXX-12) and a research grant from First Affiliated Hospital of
Xi'an Jiaotong University (grant no. 2015YK25).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
NG, JL, YW, QK and CP designed experiments. NG, YQ
and YW performed the experiments. NG, JL, YW, QK and CP analyzed
and interpreted the data. NG, JL, YW, QK and CP wrote and revised
the manuscript. All the authors approved the final version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Yi Lv, the
head of the National Local Joint Engineering Research Center for
Precision Surgery & Regenerative Medicine and Shaanxi Province
Center for Regenerative Medicine and Surgery Engineering Research
for sharing experimental facilities.
References
|
1
|
Awasthi N, Guo S and Wagner BJ: Posterior
capsular opacification: A problem reduced but not yet eradicated.
Arch Ophthalmol. 127:555–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Apple DJ, Escobar-Gomez M, Zaugg B,
Kleinmann G and Borkenstein AF: Modern cataract surgery: Unfinished
business and unanswered questions. Surv Ophthalmol. 56(6 Suppl):
S3–S53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang Y, Ye Y, Lin X, Wu K and Yu M:
Inhibition of pirfenidone on TGF-beta2 induced proliferation,
migration and epithlial-mesen-chymal transition of human lens
epithelial cells line SRA01/04. PLoS One. 8:e568372013. View Article : Google Scholar
|
|
4
|
de Iongh RU, Wederell E, Lovicu FJ and
McAvoy JW: Transforming growth factor-beta-induced
epithelial-mesenchymal transition in the lens: A model for cataract
formation. Cells Tissues Organs. 179:43–55. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saika S, Yamanaka O, Flanders KC, Okada Y,
Miyamoto T, Sumioka T, Shirai K, Kitano A, Miyazaki K, Tanaka S and
Ikeda K: Epithelial-mesenchymal transition as a therapeutic target
for prevention of ocular tissue fibrosis. Endocr Metab Immune
Disord Drug Targets. 8:69–76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garweg JG, Zandi S, Gerhardt C and Pfister
IB: Isoforms of TGF-β in the aqueous humor of patients with
pseudoexfoliation syndrome and a possible association with the
long-term stability of the capsular bag after cataract surgery.
Graefes Arch Clin Exp Ophthalmol. 255:1763–1769. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu B, Gao J, Lyu BC, Du SS, Pei C, Zhu ZQ
and Ma B: Expressions of TGF-β2, bFGF and ICAM-1 in lens epithelial
cells of complicated cataract with silicone oil tamponade. Int J
Ophthalmol. 10:1034–1039. 2017.
|
|
9
|
Pei C, Ma B, Kang QY, Qin L and Cui LJ:
Effects of transforming growth factor β2 and connective tissue
growth factor on induction of epithelial mesenchymal transition and
extracellular matrix synthesis in human lens epithelial cells. Int
J Ophthalmol. 6:752–757. 2013.
|
|
10
|
Ma B, Kang Q, Qin L, Cui L and Pei C:
TGF-β2 induces transdifferentiation and fibrosis in human lens
epithelial cells via regulating gremlin and CTGF. Biochem Biophys
Res Commun. 447:689–695. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamada N, Sugai T, Eizuka M, Tsuchida K,
Sugimoto R, Mue Y, Suzuki M, Osakabe M, Uesugi N, Ishida K, et al:
Tumor budding at the invasive front of colorectal cancer may not be
associated with the epithelial-mesenchymal transition. Hum Pathol.
60:151–159. 2017. View Article : Google Scholar
|
|
12
|
Matsuno Y, Coelho AL, Jarai G, Westwick J
and Hogaboam CM: Notch signaling mediates TGF-β1-induced
epithelial-mesenchymal transition through the induction of Snai1.
Int J Biochem Cell Biol. 44:776–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Dong C and Zhou BP: Epigenetic
regulation of EMT: The Snail story. Curr Pharm Des. 20:1698–1705.
2014. View Article : Google Scholar :
|
|
15
|
Imai S, Armstrong CM, Kaeberlein M and
Guarente L: Transcriptional silencing and longevity protein Sir2 is
an NAD-dependent histone deacetylase. Nature. 403:795–800. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dewey S: Posterior capsule opacification.
Curr Opin Ophthalmol. 17:45–53. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang YN, Qin L, Li JM, Chen L and Pei C:
Expression of transcription factors Slug in the lens epithelial
cells undergoing epithelial-mesenchymal transition induced by
connective tissue growth factor. Int J Ophthalmol. 8:872–876.
2015.PubMed/NCBI
|
|
20
|
Liu J, Xu D, Li J, Gao N, Liao C, Jing R,
Wu B, Ma B, Shao Y and Pei C: The role of focal adhesion kinase in
transforming growth factor-β2 induced migration of human lens
epithelial cells. Int J Mol Med. 42:3591–3601. 2018.PubMed/NCBI
|
|
21
|
Li J, Tripathi BJ and Tripathi RC:
Modulation of pre-mRNA splicing and protein production of
fibronectin by TGF-beta2 in porcine trabecular cells. Invest
Ophthalmol Vis Sci. 41:3437–3443. 2000.PubMed/NCBI
|
|
22
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tamiya S, Liu L and Kaplan HJ:
Epithelial-mesenchymal transition and proliferation of retinal
pigment epithelial cells initiated upon loss of cell-cell contact.
Invest Ophthalmol Vis Sci. 51:2755–2763. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dwivedi DJ, Pino G, Banh A, Nathu Z,
Howchin D, Margetts P, Sivak JG and West-Mays JA: Matrix
metalloproteinase inhibitors suppress transforming growth
factor-beta-induced subcapsular cataract formation. Am J Pathol.
168:69–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hales AM, Chamberlain CG, Dreher B and
McAvoy JW: Intravitreal injection of TGFbeta induces cataract in
rats. Invest Ophthalmol Vis Sci. 40:3231–3236. 1999.PubMed/NCBI
|
|
26
|
Maring JA, van Meeteren LA, Goumans MJ and
Ten Dijke P: Interrogating TGF-β function and regulation in
endothelial cells. Methods Mol Biol. 1344:193–203. 2016. View Article : Google Scholar
|
|
27
|
Conway J, Al-Zahrani KN, Pryce BR,
Abou-Hamad J and Sabourin LA: Transforming growth factor β-induced
epithelial to mesenchymal transition requires the Ste20-like kinase
SLK independently of its catalytic activity. Oncotarget.
8:98745–98756. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matheus LHG, Simao GM, Amaral TA, Brito
RBO, Malta CS, Matos YST, Santana AC, Rodrigues GGC, Albejante MC,
Bach EE, et al: Indoleamine 2,3-dioxygenase (IDO) increases during
renal fibrogenesis and its inhibition potentiates TGF-β 1-induced
epithelial to mesenchymal transition. BMC Nephrol. 18:2872017.
View Article : Google Scholar
|
|
29
|
Yang L, Xing S, Wang K, Yi H and Du B:
Paeonol attenuates aging MRC-5 cells and inhibits
epithelial-mesenchymal transition of premalignant HaCaT cells
induced by aging MRC-5 cell-conditioned medium. Mol Cell Biochem.
439:117–129. 2018. View Article : Google Scholar
|
|
30
|
Guo R, Meng Q, Guo H, Xiao L, Yang X, Cui
Y and Huang Y: TGF-β2 induces epithelial-mesenchymal transition in
cultured human lens epithelial cells through activation of the
PI3K/Akt/mTOR signaling pathway. Mol Med Rep. 13:1105–1110. 2016.
View Article : Google Scholar
|
|
31
|
Vinnakota JM and Gummadi SN: Snail
represses the expression of human phospholipid scramblase 4 gene.
Gene. 591:433–441. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Medici D, Potenta S and Kalluri R:
Transforming growth factor-β2 promotes Snail-mediated
endothelial-mesenchymal transition through convergence of
Smad-dependent and Smad-independent signalling. Biochem J.
437:515–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takahashi M, Akamatsu H, Yagami A,
Hasegawa S, Ohgo S, Abe M, Iwata Y, Arima M, Mizutani H, Nakata S
and Matsunaga K: Epithelial-mesenchymal transition of the eccrine
glands is involved in skin fibrosis in morphea. J Dermatol.
40:720–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu W, Ruest LB and Svoboda KK: Regulation
of epithelial-mesenchymal transition in palatal fusion. Exp Biol
Med (Maywood). 234:483–491. 2009. View Article : Google Scholar
|
|
35
|
Biswas H and Longmore GD: Action of SNAIL1
in cardiac myofibroblasts is important for cardiac fibrosis
following hypoxic injury. PLoS One. 11:e01626362016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang L, Li Z, He W, Xu L, Wang J, Shi J
and Sheng M: Effects of astragaloside IV against the TGF-β1-induced
epithelial-to-mesenchymal transition in peritoneal mesothelial
cells by promoting Smad 7 expression. Cell Physiol Biochem.
37:43–54. 2015. View Article : Google Scholar
|
|
37
|
Batlle E, Sancho E, Franci C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
Characterization of the classical HDAC family. Biochem J.
370:737–749. 2003. View Article : Google Scholar
|
|
39
|
Zupkovitz G, Tischler J, Posch M, Sadzak
I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S,
Decker T and Seiser C: Negative and positive regulation of gene
expression by mouse histone deacetylase 1. Mol Cell Biol.
26:7913–7928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ferrari-Amorotti G, Chiodoni C, Shen F,
Cattelani S, Soliera AR, Manzotti G, Grisendi G, Dominici M, Rivasi
F, Colombo MP, et al: Suppression of invasion and metastasis of
triple-negative breast cancer lines by pharmacological or genetic
inhibition of slug activity. Neoplasia. 16:1047–1058. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
von Burstin J, Eser S, Paul MC, Seidler B,
Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, et
al: E-cadherin regulates metastasis of pancreatic cancer in vivo
and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex.
Gastroenterology. 137:361–371. 371.e1–e5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu YW, Sun M, Xia R, Zhang EB, Liu XH,
Zhang ZH, Xu TP, De W, Liu BR and Wang ZX: LincHOTAIR
epigenetically silences miR34a by binding to PRC2 to promote the
epithelial-to-mesenchymal transition in human gastric cancer. Cell
Death Dis. 6:e18022015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu S, Ye D, Guo W, Yu W, He Y, Hu J, Wang
Y, Zhang L, Liao Y, Song H, et al: G9a is essential for
EMT-mediated metastasis and maintenance of cancer stem cell-like
characters in head and neck squamous cell carcinoma. Oncotarget.
6:6887–6901. 2015.PubMed/NCBI
|
|
44
|
Ziemka-Nalecz M, Jaworska J, Sypecka J and
Zalewska T: Histone deacetylase inhibitors: A therapeutic key in
neurological disorders? J Neuropathol Exp Neurol. 77:855–870. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hahnen E, Hauke J, Trankle C, Eyupoglu IY,
Wirth B and Blumcke I: Histone deacetylase inhibitors: Possible
implications for neurodegenerative disorders. Expert Opin Investig
Drugs. 17:169–184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mwakwari SC, Patil V, Guerrant W and
Oyelere AK: Macrocyclic histone deacetylase inhibitors. Curr Top
Med Chem. 10:1423–1440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Duvic M, Talpur R, Ni X, Zhang C, Hazarika
P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel
SR: Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic
acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood.
109:31–39. 2007. View Article : Google Scholar
|
|
48
|
Piekarz RL, Frye R, Turner M, Wright JJ,
Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ,
et al: Phase II multi-institutional trial of the histone
deacetylase inhibitor romidepsin as monotherapy for patients with
cutaneous T-cell lymphoma. J Clin Oncol. 27:5410–5417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Patil V, Sodji QH, Kornacki JR, Mrksich M
and Oyelere AK: 3-Hydroxypyridin-2-thione as novel zinc binding
group for selective histone deacetylase inhibition. J Med Chem.
56:3492–3506. 2013. View Article : Google Scholar : PubMed/NCBI
|