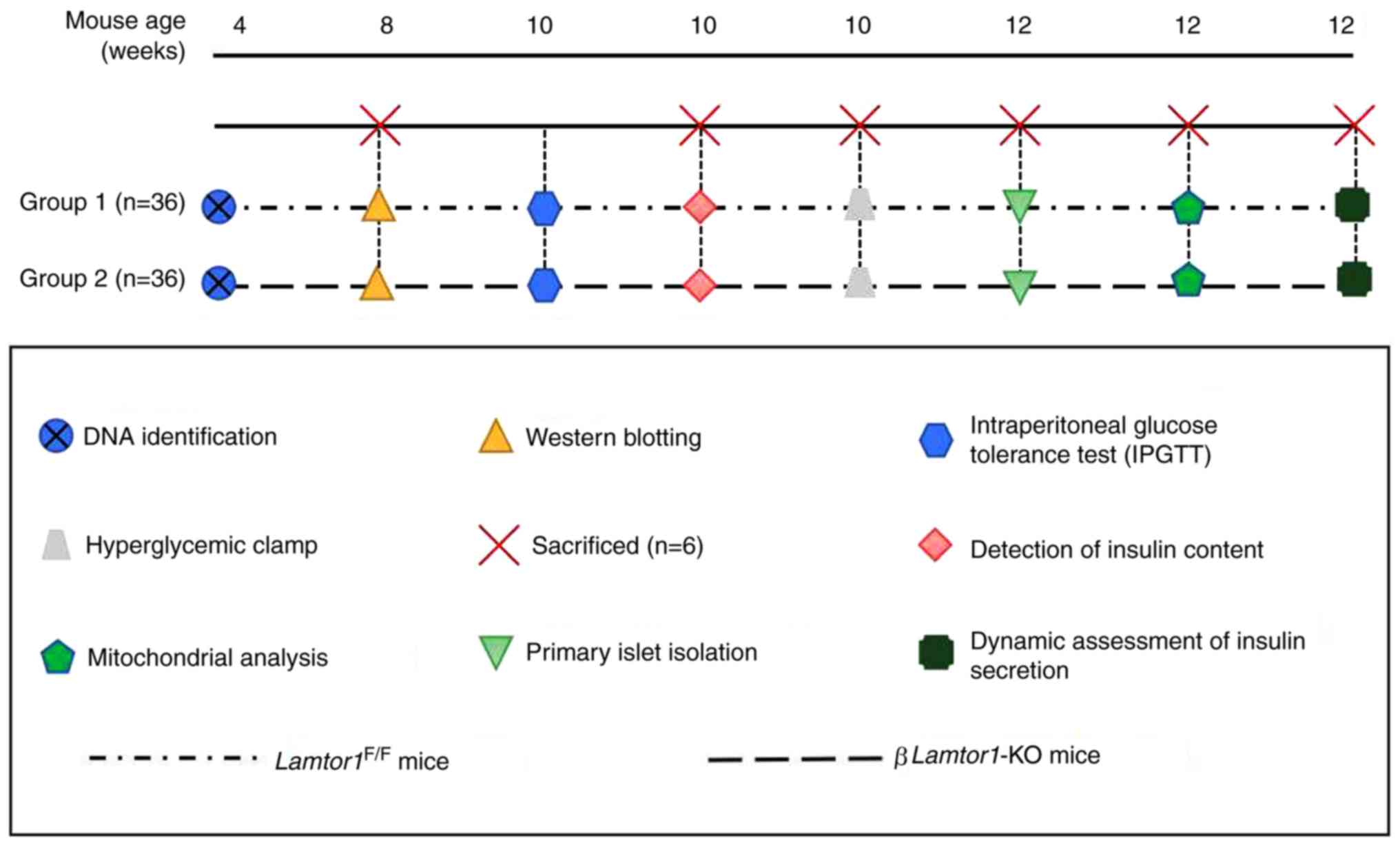
Introduction
Glucose is the most widely distributed
monosaccharide in nature. It is an important component of the human
body and the main source of energy for various tissues and organs.
The interaction between insulin and glucose plays a crucial role in
the stabilization of blood glucose, which is closely related to
various diseases including diabetes. It is well known that insulin
secreted by pancreatic β-cells plays an important role in the
development of diabetes. Glucose uptake causes a series of
responses in pancreatic β-cells ultimately leading to insulin
release (1). In pancreatic
β-cells, glucose is transported by the glucose transporter 2
(GLUT2) and phosphorylated by glucokinase to produce glucose
6-phosphate, which is then oxidatively phosphorylated by the
mitochondria, resulting in an increase in the adenosine
triphosphate/adenosine diphosphate (ATP/ADP) ratio in the cytoplasm
(2). This in turn leads to the
closure of ATP-sensitive K+ channels (KATP
channels) and results in cell membrane depolarization and opening
of voltage-dependent calcium channels (VDCCs). Ca+
influx triggers the release of insulin from β-cell insulin granules
(3-5).
Adenosine monophosphate-activated protein kinase
(AMPK) is one of the most important regulators of metabolic
homeostasis and energy metabolism, maintaining the balance of
energy supply and demand, as well as improving insulin resistance
(6). When cellular nutrition and
energy are deficient, AMPK is activated by an allosteric mechanism.
Once activated, AMPK increases catabolism and inhibits anabolism in
order to respond to stress signals induced by nutrient or energy
deficiency and increase intracellular ATP stores (7). The tumor suppressor liver kinase B1
(LKB1) complex, comprising of STRAD α/β and MO25 α/β, is the major
upstream kinase for AMPK. It activates AMPK by phosphorylating
Thr172 (8). The LKB1/AMPK pathway
plays a crucial role in regulating the polarity, size and total
mass of β-cells, thus limiting glucose-stimulated insulin secretion
(GSIS) (9). Moreover, it has been
reported that an increase of insulin secretion occurs in specific
Lkb1-knockout (KO) mice, partly as a result of improved
glutamine levels and acetyl-CoA carboxylase 1 (ACC1) activity
(10). All of these observations
point to the essential effects of LKB1 on mitochondrial homeostasis
in pancreatic β-cells and its strong negative regulatory function
with regards to insulin secretion.
Late endosomal/lysosomal adaptor MAPK and mTOR
activator 1 (LAMTOR1) is a membrane protein specifically localized
to the surface of endosomes/lysosomes. It serves as an anchor for
the 'Ragulator' complex with LAMTOR2 (P14), LAMTOR3 (MP1), LAMTOR4
(C7OR F59) and LAMTOR5 (HBXIP) (11). LAMTOR1 promotes the activation of
AMPK and inhibits mTORC1 activity, thereby turning off the anabolic
pathway and switching on the catabolic pathway when there is a need
for increased energy production (12). As mentioned previously, insulin
secretion is increased in the pancreatic β-cells of specific
Lkb1 knockout mice. However, the role of LAMTOR1 in the
regulation of β-cell function remains unknown. Herein, the role of
LAMTOR1 in pancreatic β-cell function is explored in order to
elucidate its molecular mechanisms both in vivo and in
vitro.
Materials and methods
Animals
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Nanchang University.
F1 generation mice (age, 4-6 weeks; weight, 20-25 g) were purchased
from Model Animal Research Center Of Nanjing University and kept in
an air-conditioned room (25°C; relative humidity, 50±20%; 12-h
light/dark cycle) with free access to food and water. The control
group included 36 male Loxp/Loxp mice while the experimental group
included 36 male βLamtor1-KO mice. Control Loxp/Loxp and
βLamtor1-KO male mice were obtained by mating homozygous
Lamtor1-floxed mice (Lamtor1F/F) with
RIP-Cre mice, on a C57BL/6N background. All animal experiments were
performed in accordance with the ARRIVE guidelines.
Glucose tolerance test
Mice received a 20% glucose solution (Sangon Biotech
Co., Ltd.; 1.5 g/kg) by intraperitoneal (i.p.) injection. Blood
glucose levels were measured at 0, 30, 60, 90 and 120 min after
injection.
Hyperglycemic clamp
The hyperglycemic clamp was conducted strictly
according to standardized procedures. Chronic cannulation of the
right jugular vein was performed 4-5 days prior to intravenous
glucose infusion. The mice were fasted for 6 h in a restrainer
before the experiment. The experimental mice were subsequently
infused with 20% glucose until plasma glucose levels reached
approximately 16-18 mM.
Dynamic assessment of insulin
secretion
To assess insulin secretion, a perfusion system
equipped with a peristaltic pump was used to deliver Krebs-Ringer
bicarbonate (KRB) buffer (Sigma-Aldrich; Merck KGaA) to isolated
pancreatic islets. Fifty size-matched islets were placed in columns
and perfused at a flow rate of 100 µl/min with KRB buffer at
37°C. Perfusion with 2.8 mM glucose was used to balance and measure
basal secretion before the islets were exposed to different
treatments. The medium was transferred to 96-well plates, and
insulin levels were measured by the high sensitive mouse Insulin
ELISA kit (cat. no. 32270; ImmunoDiagnostics), normalized to total
islet DNA or protein as indicated.
Mitochondrial analysis
The real-time oxygen consumption of mitochondrial
preparations was measured using a Seahorse XF analyzer (Agilent
Technologies, Inc.). Islets (50/well) were cultured in 24-well
plates in a medium consisting of unbuffered DMEM, 1% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 2.8 mM glucose at
37°C without CO2. The islets were then incubated in a
high level of glucose (16.7 mM) and continuously treated with 1M
FCCP [carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone] and 5
M rotenone plus 5 M antimycin. The oxygen consumption rate (OCR)
was calculated using an XF analyzer AKOS algorithm (Agilent
Technologies, Inc.) and normalized to basal levels or to total
protein content. Protein was extracted with a radioimmune
precipitation lysis buffer, and the total protein content was
determined using a Pierce BCA Protein Assay kit (Beyotime Institute
of Biotechnology).
Western blotting
Protein was extracted from fresh islets using the
radioimmune precipitation assay lysis buffer (Beijing Solarbio
Science & Technology Co., Ltd.) supplemented with protease and
phosphatase inhibitors (leupeptin, aprotinin and vanadate). Total
protein was determined using a Pierce BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Equal amounts of protein (30
µg) were separated by SDS-PAGE on 8 and 10% gels.
Electrophoresis was performed under identical running and
transferring conditions for all samples. Proteins were subsequently
transferred to immun-Blot PVDF membrane and blocked using 5% w/v
non-fat dry milk in TBST (Sigma-Aldrich; Merck KGaA) for 1 h at
room temperature. Membranes were incubated with the primary
antibodies for 16-20 h at 4°C. Antibody information is presented in
Table I. Subsequently, the
membranes were incubated with horseradish peroxidase-conjugated
antibodies, specifically anti-rabbit (1:2,000; cat. no. 7074) and
anti-mouse IgG (1:2,000; cat. no. 7076; both from Cell Signaling
Technology, Inc.), for 1 h at room temperature. Immunoreactive
signals were detected using enhanced chemiluminescence reagents
(EMD Millipore; Merck KGaA). Finally, densitometric analysis of the
protein strips was performed using ImageJ version 1.46 (NIH).
 | Table IAntibody information. |
Table I
Antibody information.
| Antibody | Manufacturer | Catalogue
number | Type | Species | Dilution
factor |
|---|
| Anti-LAMTOR1 | Abcam | ab229760 | Polyclonal | Rabbit | 1:1,000 |
| Anti-Tubulin | Abcam | ab6046 | Polyclonal | Rabbit | 1:500 |
| Anti-Hsp90 | Santa Cruz | sc-13119 | Monoclonal | Mouse | 1:1,000 |
| Anti-p-AMPK | Cell Signaling
Technology | 4186 | Polyclonal | Rabbit | 1:1,000 |
| Anti-AMPK | Cell Signaling
Technology | 5831 | Monoclonal | Rabbit | 1:1,000 |
| Anti-p-ACC1 | Cell Signaling
Technology | 3661 | Polyclonal | Rabbit | 1:1,000 |
| Anti-ACC1 | Cell Signaling
Technology | 4190 | Polyclonal | Rabbit | 1:1,000 |
| Anti-β-actin | Cell Signaling
Technology | 4967 | Polyclonal | Rabbit | 1:1,000 |
Quantitative PCR
Total RNA from fresh islets was extracted and
purified with Trizol (Sigma-Aldrich; Merck KGaA) and collected by
centrifugation at 10,000 g for 10 min at 4°C. Following quality
analysis of the RNA samples, cDNA was synthesized by reverse
transcription using 200 ng RNA and a High-capacity cDNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Quantitative PCR was performed using 1X SYBR-Green Universal
PCR Mastermix (Takara Bio, Inc.). The following primer sequences
were used: PDX1, forward 5′-GGT ATA GCC GGA GAG ATG C-3′, reverse
5′-CTG GTC CGT ATT GGA ACG-3′; INS2, forward 5′-TGG AGG CTC TCT ACC
TGG TG-3′, reverse 5′-TCT ACA ATG CCA CGC TTC TG-3′; GLUT2, forward
5′-CTT GGT TCA TGG TTG CTG AAT-3′, reverse 5′-GCA ATG TAC TGG AAG
CAG AGG-3′; GCK, forward 5′-ATC TTC TGT TCC ACG GAG AGG-3′, reverse
5′-GAT GTT AAG GAT CTG CCT TCG-3′. The thermocycling conditions
were as follows: 95°C for 10 min; 40 cycles at 95°C for 7 sec, 57°C
for 30 sec and extension at 72°C for 30 sec. Reactions were
performed in triplicate in 96-well plates, using the CFX96
real-time System (Bio-Rad Laboratories, Inc.). Transcript levels
were calculated using the 2−ΔΔCq method and normalized
to the expression of internal reference gene GAPDH (13).
Statistical analysis
The data were analyzed using an unpaired two-tailed
Student's t-test for experiments with only two groups and presented
as mean ± standard deviation (SD). For multiple group comparisons,
one-way ANOVA followed by Tukey's post hoc test was used. SPSS
version 20.0.0 (IBM Corp.) was used for statistical analyses.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Production of βcell-specific Lamtor1-KO
mice
To determine the physiological role of LAMTOR1 in
pancreatic β-cells, β cell-specific Lamtor1-KO mice
(βLamtor1-KO) were bred by crossing Lamtor1-floxed
mice (Lamtor11F/F) with RIP-Cre mice (Fig. 1). Immunoreactivity analysis
confirmed the deletion of a floxed sequence from the primary islet
cell genomic DNA of βLamtor1-KO mice. LAMTOR1 protein
expression in the pancreatic islet cells of βLamtor1-KO mice
was almost undetectable by western blotting compared with that in
their Lamtor1F/F littermates (Fig. 2A).
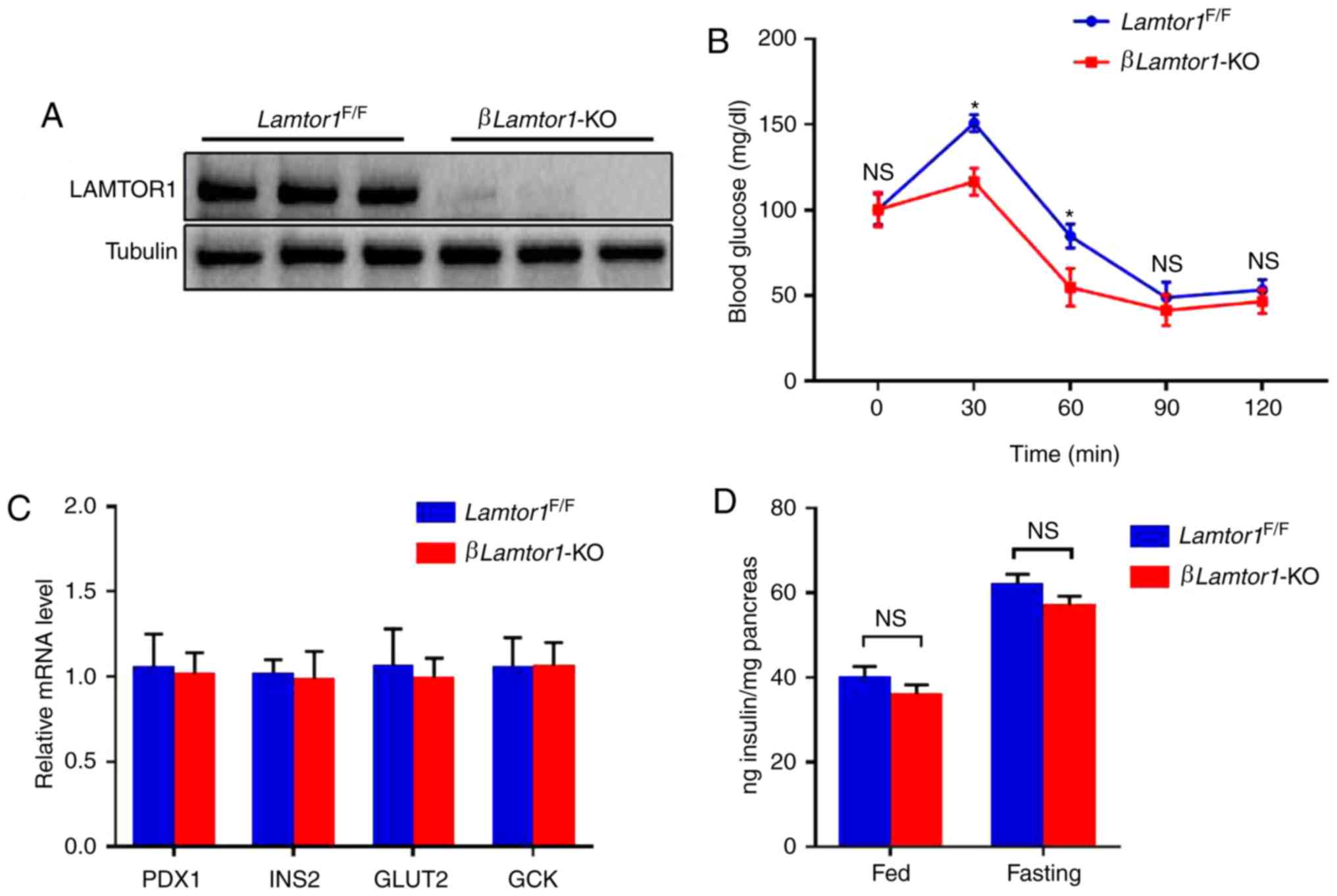 | Figure 2Effect of Lamtor1 deficiency
on insulin production. (A) Demonstration of successful
Lamtor1 knockout in βLamtor1-KO mice. Western blot
showing the depletion of Lamtor1 in islet lysates. (B) IPGTT
was conducted on 8-week-old mice after 12 h of fasting, and blood
glucose levels were measured. Data are shown as mean ± SD.
*P<0.05, n=6. (C) Expression of Pdx1, Ins2, Glut2,
and Gck was detected by real-time fluorescence quantitative-PCR,
n=6. (D) Insulin content from the islets of fed and fasted
βLamtor1-KO mice and Lamtor1F/F littermate
controls. Insulin content is presented relative to that of the
pancreas. n=6. IPGTT, intraperitoneal glucose tolerance test;
Lamtor1, late endosomal/lysosomal adaptor MAPK and mTOR activator
1; KO, knockout; NS, no significance. |
Comparison of glucose tolerance between
the two experimental animal groups
Glucose tolerance levels of 10-week-old mice in the
βLamtor1-KO and Lamtor1F/F groups were
determined by an in vivo i.p. glucose tolerance test
(IPGTT). Comparative analysis revealed that the glucose tolerance
of βLamtor1-KO mice was significantly lower than that of
Lamtor1F/F mice at 30 and 60 min after glucose
injection (Fig. 2B). As
previously described, pancreatic β-cells play an important role in
the development of diabetes, since glucose-induced insulin
secretion is the key physiological function of these cells
(14). Therefore, the potential
involvement of LAMTOR1 in the regulation of β-cell function was
evaluated. Quantitative PCR was performed to determine the mRNA
expression levels of several related genes including Pdx1,
Ins2, Glut2 and Gck. Pdx1 and
Ins2 are key regulators in insulin biosynthesis, while
Glut2 and Gck are associated with glucose uptake and
glucose metabolism, respectively (15). The results suggest that their mRNA
expression levels were similar between the two groups (Fig. 2C). In addition, there were no
significant differences in the total pancreatic insulin content
between the βLamtor1-KO and Lamtor1F/F
groups of mice, whether they were fasted or fed (Fig. 2D). These results suggest that
enhanced glucose-stimulated insulin secretion may explain the
improvement in β-cell function observed in the β Lamtor1-KO
group.
Lamtor1 knockout improves
glucose-stimulated insulin secretion
To determine whether insulin secretion is enhanced
in the absence of LAMTOR1, hyperglycemic clamping experiments were
performed. The hyperglycemic clamp is the gold standard test for
detecting pancreatic β-cells and can be used to identify both the
first and second phases of insulin secretion (16). The hyperglycemic clamp experiments
were conducted 4-5 days after the external jugular vein of
10-week-old mice had been catheterized. The two groups of mice were
then injected with a 20% glucose solution to maintain their
baseline blood glucose level at around 16-18 mM for 90 min
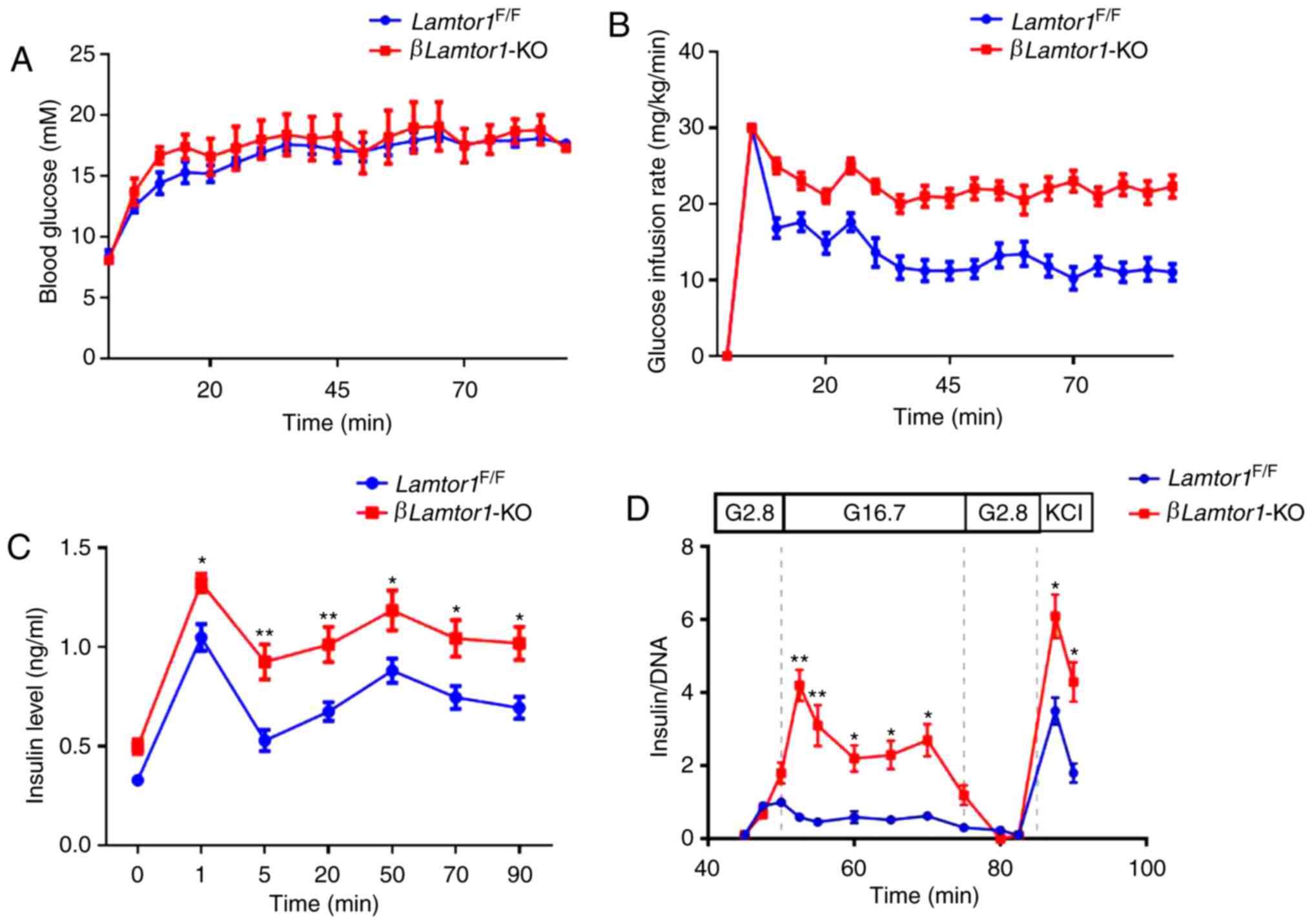
(Fig. 3A). The mean glucose
infusion levels were 21±1.1 mg/kg/min in the βLamtor1-KO
mice, and 13±1.2 mg/kg/min in the Lamtor1F/F
mice, at an overall duration of 90 min (Fig. 3B). The resulting curves showed
that the temporal pattern of insulin secretion was normal in the
two groups of mice, however the βLamtor1-KO mice released
higher insulin levels compared with the
Lamtor1F/F mice during both the first and second
phases of the experiment (Fig.
3C). These data suggest that the loss of Lamtor1
enhances insulin secretion in pancreatic β-cells.
To further confirm this conclusion, islet perfusion
experiments were performed. The GSIS response curves revealed that
insulin secretion in perfused βLamtor1-KO islets was similar
to that in the control animals at low glucose concentrations (2.8
mM). However, increased glucose concentrations (16.7 mM)
significantly enhanced insulin secretion in the islets of
βLamtor1-KO mice (Fig.
3D). In addition, this experiment produced similar results to
the hyperglycemic clamping experiment in that the mice in the
βLamtor1-KO group secreted higher levels of insulin during
both phases of the experiment.
Effect of the triggering pathway on
insulin secretion in the islets of βLamtor1-KO mice
Triggering and amplification are two action pathways
that regulate insulin secretion in response to glucose (17,18), in which cytosolic Ca2+
is a triggering signal. The potential enhancement of insulin
secretion via the triggering pathway in Lamtor1-deficient
β-cells was evaluated using islet perfusion experiments (19). The βLamtor1-KO perfused
islets were found to secrete more insulin than the control islets
during stimulation with 16.7 mM glucose (Fig. 4A). Treating mice with diazoxide, a
reagent that effectively opens KATP channels (20), completely inhibited
glucose-induced insulin secretion in both experimental groups. On
this basis, the secretion of insulin was also monitored in the two
groups following the addition of potassium chloride (KCl). In these
experiments, higher insulin levels were secreted in the
βLamtor1-KO group than in the control group, which is
consistent with previous results. Subsequently, to further analyze
the problem, the KATP channels were closed by glyburide
treatment. Higher insulin secretion was observed in the
βLamtor1-KO group than in the control group during
stimulation with either basal (2.8 mM) or high glucose
concentrations (16.7 mM) (Fig.
4B). Thus, higher insulin levels were produced in the
βLamtor1-KO group with a similar degree of membrane
depolarization, suggesting that KATP channels are not
responsible for enhancing insulin secretion in the
Lamtor1-deficient β-cells.
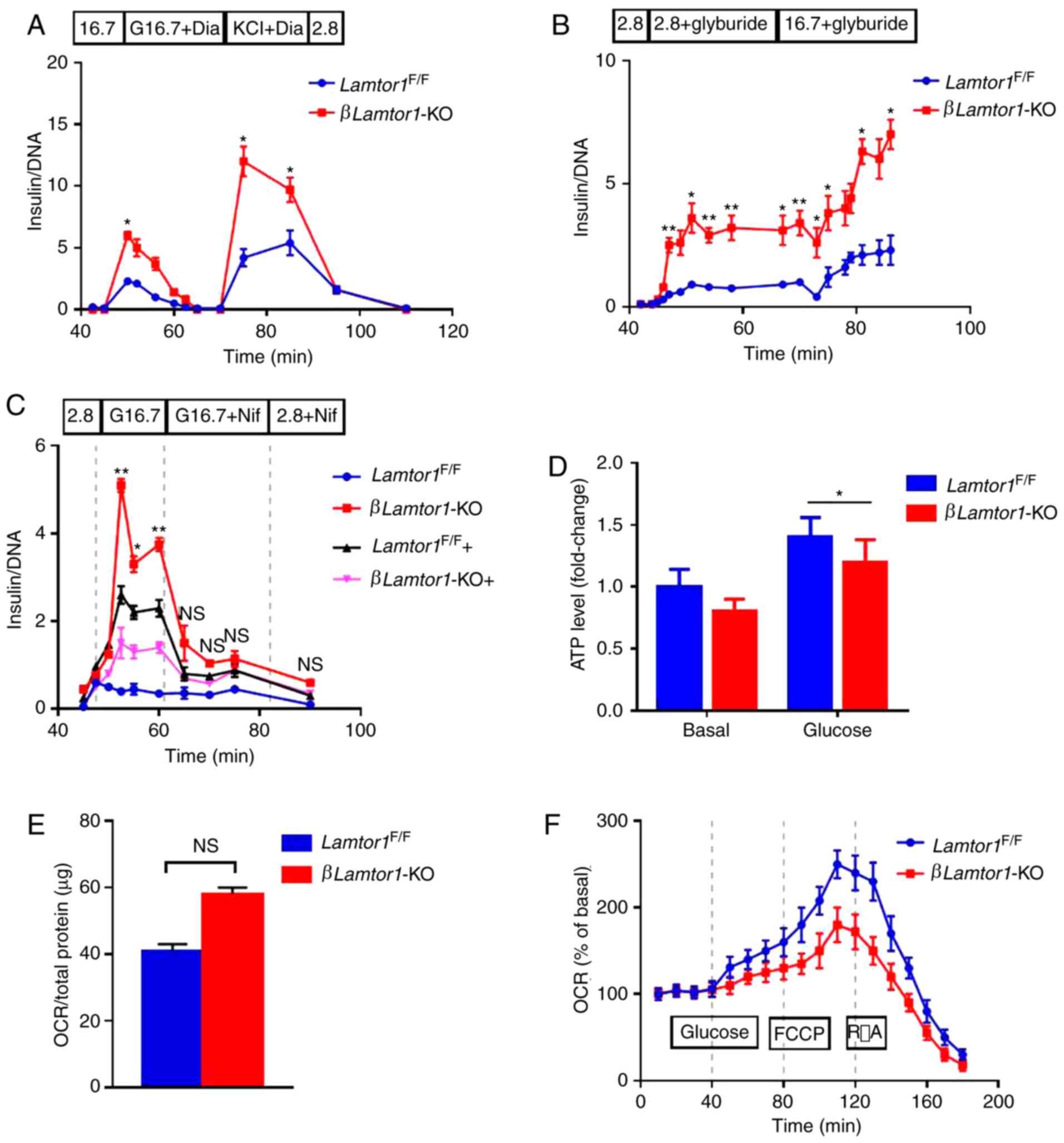 | Figure 4Lamtor1 deficiency increases insulin
secretion but impairs mitochondrial function. (A) Insulin levels
measured during the perifusion of isolated islets.
Lamtor1-deficient islets secrete higher levels of insulin
than the control under 16.7 mM glucose stimulation, and
administration of 100 µM diazoxide eliminates this
difference; however, the addition of 30 mM KCl causes a second peak
in insulin secretion. (B) Insulin secretion followed by glyburide
treatment. 1 µM glyburide induces higher levels of insulin
secretion from Lamtor1-deficient islets, under both low and
high glucose conditions. (C) Glucose-stimulated insulin secretion
from perifused islets treated with nifedipine. Black and pink
lines; nifedipine was added before high glucose. Blue and red
lines; nifedipine was added 15 min after the addition of high
glucose. Data are shown as mean ± SD. *P<0.05,
**P<0.01, n=6. (D) Adenosine triphosphate levels of
islets responsive to basal and high glucose (16.7 mM).
*P<0.05. (E) Basal OCR measured by the Seahorse XF24
analyzer in the presence of 2.8 mM glucose. n=6. (F) OCR measured
over a time period of 180 min. Glucose (20 mM), FCCP (1 µM)
and rotenone plus antimycin A (5 µM each) were added at the
indicated times. Día, diazoxide; Nif, nifedipine; ATP, adenosine
triphosphate; OCR, oxygen consumption rate; FCCP, carbonyl cyanide
4-(trifluoromethoxy) phenylhydrazone; R/A, rotenone plus antimycin
A; NS, no significance. |
Perfused islets were also treated with nifedipine,
an inhibitor of voltage-gated calcium channels. Results showed that
the addition of nifedipine led to a significant reduction in
insulin secretion in both groups (Fig. 4C). The results suggest that VDCCs
are necessary for insulin secretion in the islets of
βLamtor1-KO mice. Combined with our conclusions about
KATP channels, the triggering pathway does not
sufficiently explain the effect of glucose on insulin secretion in
βLamtor1-KO islets.
Deletion of Lamtor1 leads to
mitochondrial dysfunction
It was hypothesized that the deletion of
Lamtor1 would enhance mitochondrial function, and thus
experiments were designed to verify this possibility. However, the
results of the present study could not confirm our initial
hypothesis. While the ATP content increased in both the
βLamtor1-KO and control groups, after stimulation with high
glucose concentrations, it was markedly lower in the
βLamtor1-KO group than in the control group, indicating that
insulin secretion did not enhance mitochondrial function in the
β-cells of βLamtor1-KO mice (Fig. 4D).
In addition, the oxygen consumption rate (OCR), an
indicator of the extent of aerobic glucose metabolism was assessed.
OCR at the basal glucose level was slightly increased in the
βLamtor1-KO islets but did not reach a level of statistical
significance (Fig. 4E). However,
Lamtor1 deletion eliminated the high glucose-induced OCR
(Fig. 4F). Moreover, the OCR was
still lower after the addition of the mitochondrial un-coupler FCCP
than in the control islets. Taken together, these results indicate
that LAMTOR1 deficiency leads to mitochondrial dysfunction in
pancreatic β-cells, further supporting the conclusion that the
enhanced insulin secretion from βLamtor1-KO islets is not
caused by the triggering pathway of insulin secretion.
Deletion of Lamtor1 amplifies insulin
secretion by increasing glutamate
Previous results have demonstrated that enhanced
insulin secretion in the absence of LAMTOR1 is not associated with
the triggering pathway. Thus, the next step in the present study
was to examine the role of the amplifying pathway. Glutamate is
derived from the malate-aspartate shuttle upon glucose stimulation
and has an enhanced effect on calcium function (21-23). To determine whether
glucose-stimulated insulin secretion in βLamtor1-KO mice was
enhanced by increasing glutamate production, the 13C
enrichment of glutamate was measured in whole cells and the cytosol
of islet cells with or without aminooxyacetate [AOA, an inhibitor
of the malate-aspartate shuttle (24)] treatment, while stimulating with
[U-13C]-glucose (Fig.
5A). The glutamate levels in the whole cells and cytosol were
compared between the βLamtor1-KO and control groups after
treatment with high glucose, AOA and glucagon-like peptide 1
(GLP-1). We found that the glutamate content in both the whole
cells and the islet cytosol of βLamtor1-KO mice was
significantly higher than that of the controls, when exposed to
high glucose (16.7 mM). In addition, treatment with AOA
significantly inhibited glutamate production in both the whole
cells and the cytosol. The results indicated that Lamtor1
deletion led to increased glutamate production. Moreover, the
glutamate content of the insulin granules and the insulin secretion
in the βLamtor1-KO group were further increased by the
addition of GLP-1 (Fig. 5B). This
indicated that Lamtor1 deletion increased GLP-1-stimulated
insulin secretion.
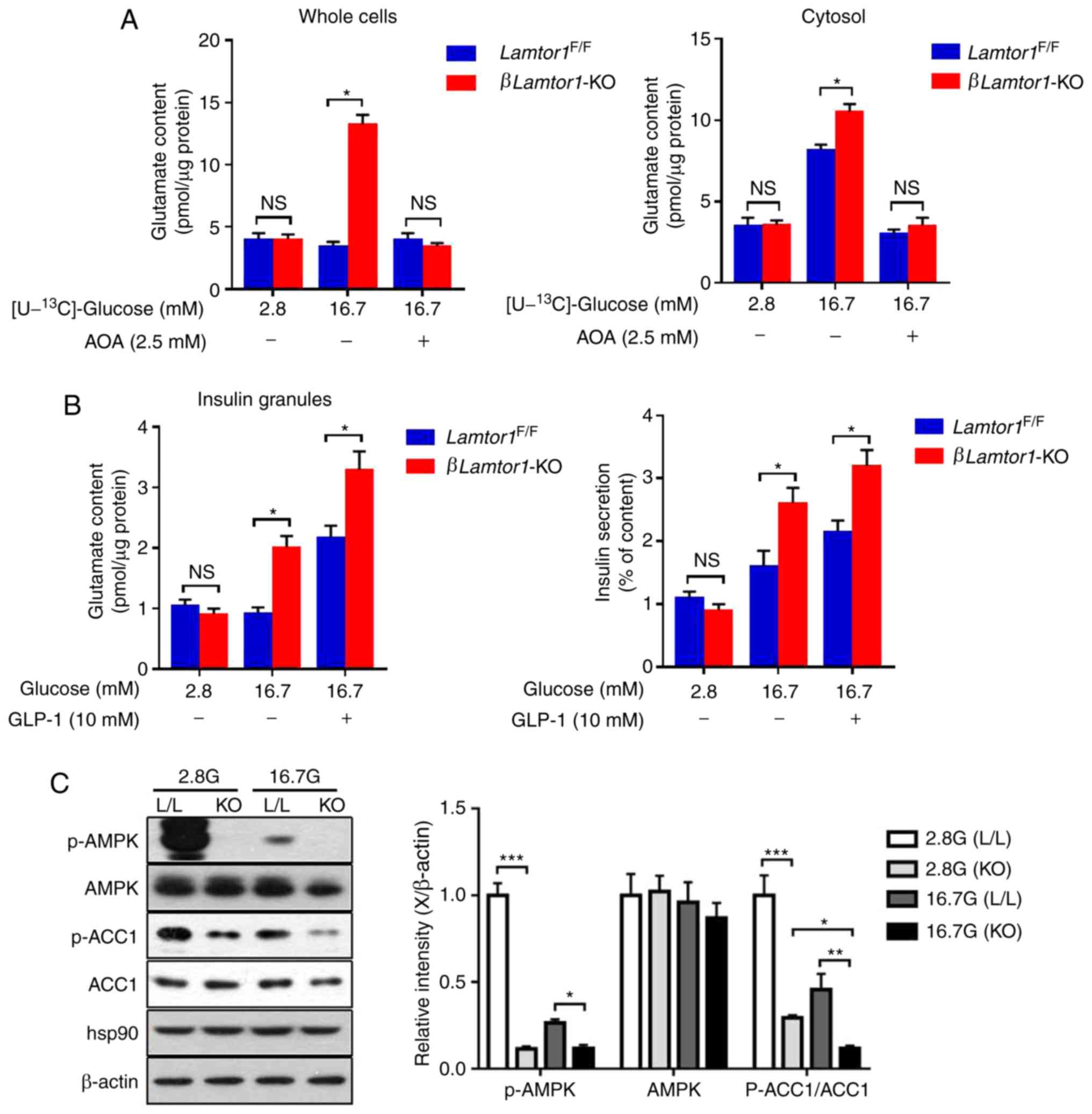 | Figure 5Lamtor1 deficiency in
pancreatic β-cells amplifies insulin secretion by increasing
glutamate production and ACC1 activity. (A) Effects of
amino-oxyacetate (2.5 mM) on the content of glutamate isotopomers
in whole cells and the cytosol in isolated Lamtor1-deficient
and control pancreatic β-cells. n=6. (B) Changes in glutamate
content in the insulin granules and insulin secretion under glucose
stimulation in the absence or presence of glucagon-like peptide 1
(10 nM). (C) Western blots showing phosphorylated adenosine
5′-monophosphate-activated protein kinase, total adenosine
5′-monophosphate-activated protein kinase, phosphorylated ACC1,
total ACC1, heat-shock protein 90 and β-actin protein levels in
Lamtor1F/F and βLamtor11-KO mouse islets
under low- and high-glucose stimulation. *P<0.05,
**P<0.01, ***P<0.001, n=6. ACC1,
acetyl-CoA carboxylase 1; p-ACC1, phosphorylated ACC1; AOA,
aminooxyac-etate; GLP-1, glucagon-like peptide 1; AMPK, adenosine
5′-monophosphate-activated protein kinase; p-AMPK, phosphorylated
AMPK; hsp90, heat-shock protein 90; NS, no significance. |
Lamtor1 knockout affects ACC1 activity in
pancreatic β-cells
AMPK phosphorylation negatively regulates ACC1 and
ACC2 enzyme activity in vivo, inhibiting the activity of
ACC1 and ACC2 (25). It has been
reported that Lkb1 deletion inhibits the phosphorylation of
AMPK, which in turn inhibits ACC1 phosphorylation, leading to the
increase of ACC1 activity and promotion of insulin secretion
(10). Therefore, we speculated
that LAMTOR1 affects the activity of ACC1. Western blotting
analysis showed that the level of AMPK phosphorylation in the
βLamtor1-KO group was lower than that in the control group
under both low and high glucose conditions, resulting in a decrease
in the ratio of p-ACC1/ACC1 in the βLamtor1-KO group,
thereby increasing the ACC1 activity. The level of phosphorylation
of ACC1 decreased with increasing glucose concentration, leading to
an increase in ACC1 activity (Fig.
5C). In agreement with this hypothesis, as demonstrated in
Fig. 5C, knocking out
Lamtor1 in β-cells inhibited phosphorylation of AMPK, and
resulted in increased ACC1 activity.
Discussion
Diabetes is a systemic disease characterized by
abnormal glucose metabolism, which is mainly caused by insufficient
insulin secretion and insulin resistance (26). In the present study, the effect of
LAMTOR1 on insulin secretion, in response to stimulation by glucose
and glucose metabolic pathways, was analyzed. Results suggest that
Lamtor1 deletion increases glucose tolerance and insulin
secretion, which is associated with certain side effects, leading
to mitochondrial dysfunction. In addition, this study explored the
possible mechanisms of action employed by LAMTOR1 in the context of
glucose-stimulated insulin secretion. It was found that LAMTOR1
deficiency stimulated an alternative pathway that amplified insulin
secretion, thus compensating for the effects of the classical
triggering pathway.
Mitochondria are the processing plants for cellular
energy metabolism. Their main function is to remove hydrogen from
glucose, fat and protein molecules that are being metabolized as
foodstuffs by oxidation-phosphorylation to form ATP, thus fueling
the body. Mitochondria combine glucose metabolism with
extracellular insulin secretion through the tricarboxylic acid
(TCA) cycle (27). Mitochondrial
dysfunction decreases ATP production and reduces the ATP/ADP ratio
in pancreatic β-cells, opening the ATP-sensitive K+
channels in the cell membrane. This enhances the efflux of
intracellular K+ and the hyperpolarization of the cell
membrane, inhibiting insulin secretion. In the present study,
opposite results were identified. The deletion of Lamtor1 in
pancreatic β-cells caused a decrease in mitochondrial function but
an increase in insulin secretion. Moreover, the glucose
oxidation-dependent triggering pathway was found to be defective in
βLamtor1-KO mice, implying that other more effective insulin
secretion mechanisms may be involved, such as the amplifying
pathway in Lamtor1-deficient β-cells.
In order to further characterize the pathways by
which glucose stimulates insulin secretion, an amplifying pathway
was analyzed by increasing glutamate production, which could in
turn promote the action of calcium. It was found that the glutamate
content in the whole cells and the islet cytosol of
βLamtor1-KO mice was higher than that in the control groups,
indicating that the amplifying pathway may play an important role
in enhanced glucose-stimulated insulin secretion in the
Lamtor1-deficient β-cells. Therefore, it appears that the
amplifying effect is not mediated by increased ATP production but
instead by glutamate. It is well known that incretin/cAMP signaling
stimulates insulin secretion in a glucose-dependent manner.
Cytosolic glutamate is transported into the insulin granules via
incretin/cAMP signaling and amplifies insulin release (23). Our results indicate that
Lamtor1 deletion enhanced insulin release by increasing
glutamate content in insulin granules via incretin-induced cAMP/PKA
signaling.
Increasing ACC1 activity represents an additional
pathway for the enhancement of insulin secretion in the absence of
LAMTOR1. ACC1 catalyzes the carboxylation of acetyl-CoA to form
malonyl-CoA, which inhibits mitochondrial fatty acid oxidation and
results in insulin secretion (28). Inhibition of ACC1 activity leads
to a decrease in pyruvate cycle activity, resulting in a
significant drop in glucose-stimulated insulin secretion,
suggesting that ACC1 may play a role in GSIS through pyruvate
metabolism (29).
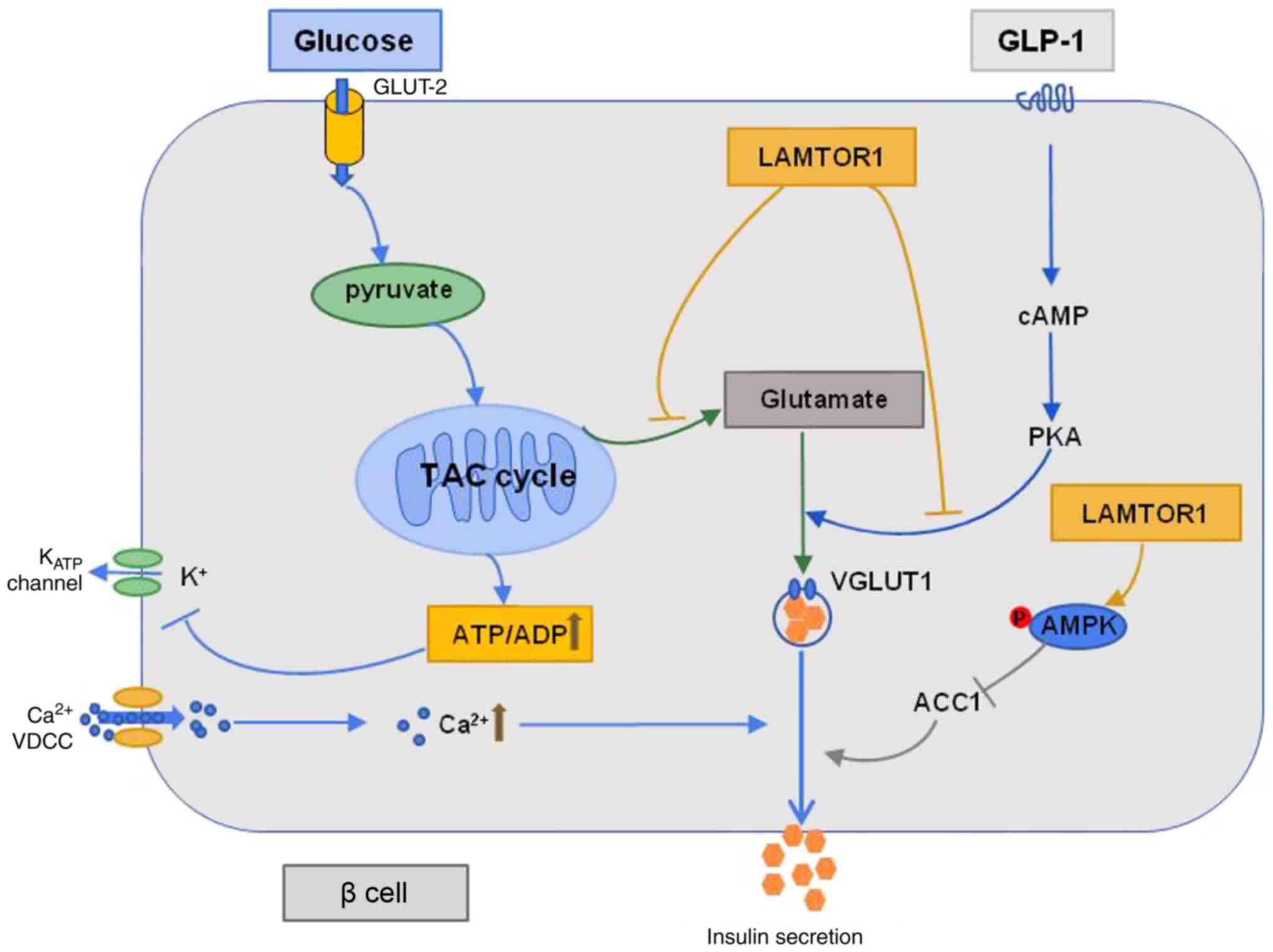
Taken together, LAMTOR1 is important for the
maintenance of mitochondrial function in pancreatic β-cells.
Although Lamtor1 deletion impairs insulin secretion via the
triggering pathway, it improves insulin secretion via the
amplifying pathways associated with glutamate and ACC1 metabolism.
These amplifying pathways compensate for the defective triggering
pathway, and ultimately lead to an increase in glucose-stimulated
insulin secretion (Fig. 6). These
findings emphasize the importance of LAMTOR1 in modulating insulin
secretion and propose the deletion of Lamtor1 as a viable
therapeutic strategy for diabetes and the improvement of pancreatic
β-cell function.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (81860153), the Youth Science
Foundation of China (81700513), the Natural Science Foundation of
Jangxi Province (2016ZRMSB1355) and the China Postdoctoral Initial
Foundation (2018M643013).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QH, QG and TW performed the experiments and wrote
the manuscript. SF, JX and JL assisted in the conduct of the
experiments. XH, QL and JH performed the literature search and
designed the study. LZ contributed to project design, data
acquisition and edited the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Nanchang University
(Nanchang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maechler P, Li N, Casimir M, Vetterli L,
Frigerio F and Brun T: Role of mitochondria in beta-cell function
and dysfunction. Adv Exp Med Biol. 654:193–216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thorens B: GLUT2, glucose sensing and
glucose homeostasis. Diabetologia. 58:221–232. 2015. View Article : Google Scholar
|
|
3
|
MacDonald PE, Joseph JW and Rorsman P:
Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R
SocLond B Biol Sci. 360:2211–2225. 2005. View Article : Google Scholar
|
|
4
|
Tarasov A, Dusonchet J and Ashcroft F:
Metabolic regulation of the pancreatic beta-cell ATP-sensitive K+
channel: A pas de deux. Diabetes. 53(Suppl 3): S113–S122. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fridlyand LE and Phillipson LH: Mechanisms
of glucose sensing in the pancreatic β-cell: A computational
systems-based analysis. Islets. 3:224–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lei Y, Gong L, Tan F, Liu Y, Li S, Shen H,
Zhu M, Cai W, Xu F, Hou B, et al: Vaccarin ameliorates insulin
resistance and steatosis by activating the AMPK signaling pathway.
Eur J Pharmacol. 851:13–24. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hawley SA, Boudeau J, Reid JL, Mustard KJ,
Udd L, Mäkelä TP, Alessi DR and Hardie DG: Complexes between the
LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are
upstream kinases in the AMP-activated protein kinase cascade. J
Biol. 2:282003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Granot Z, Swisa A, Magenheim J,
Stolovich-Rain M, Fujimoto W, Manduchi E, Miki T, Lennerz JK,
Stoeckert CJ Jr, Meyuhas O, et al: LKB1 regulates pancreatic beta
cell size, polarity, and function. Cell Metab. 10:296–308. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu A, Robitaille K, Faubert B, Reeks C,
Dai XQ, Hardy AB, Sankar KS, Ogrel S, Al-Dirbashi OY, Rocheleau JV,
et al: LKB1 couples glucose metabolism to insulin secretion in
mice. Diabetologia. 58:1513–1522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bar-Peled L, Schweitzer LD, Zoncu R and
Sabatini DM: Ragulator is a GEF for the rag GTPases that signal
amino acid levels to mTORC1. Cell. 150:1196–1208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang CS, Jiang B, Li M, Zhu M, Peng Y,
Zhang YL, Wu YQ, Li TY, Liang Y, Lu Z, et al: The lysosomal
v-ATPase-Ragulator complex is a common activator for AMPK and
mTORC1, acting as a switch between catabolism and anabolism. Cell
Metab. 20:526–540. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu Z, Gilbert ER and Liu D: Regulation of
insulin synthesis and secretion and pancreatic beta-cell
dysfunction in diabetes. Curr Diabetes Rev. 9:25–53. 2013.
View Article : Google Scholar
|
|
15
|
Li X, Cheng KKY, Liu Z, Yang JK, Wang B,
Jiang X, Zhou Y, Hallenborg P, Hoo RL, Lam KSL, et al: The
MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial
metabolism to glucose-stimulated insulin secretion in pancreatic
β-cells. Nat Commun. 7:117402016. View Article : Google Scholar
|
|
16
|
Henquin JC, Nenquin M, Stiernet P and
Ahren B: In vivo and in vitro glucose-induced biphasic insulin
secretion in the mouse: Pattern and role of cytoplasmic Ca2+ and
amplification signals in beta-cells. Diabetes. 55:441–451. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Henquin JC, Ravier MA, Nenquin M, Jonas JC
and Gilon P: Hierarchy of the beta-cell signals controlling insulin
secretion. Eur J Clin Invest. 33:742–750. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henquin JC, Dufrane D and Nenquin M:
Nutrient control of insulin secretion in isolated normal human
islets. Diabetes. 55:3470–3477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Swisa A, Granot Z, Tamarina N, Sayers S,
Bardeesy N, Philipson L, Hodson DJ, Wikstrom JD, Rutter GA,
Leibowitz G, et al: Loss of liver kinase B1 (LKB1) in beta cells
enhances glucose-stimulated insulin secretion despite profound
mitochondrial defects. J BiolChem. 290:20934–20946. 2015.
|
|
20
|
Misler S, Gee WM, Gillis KD, Scharp DW and
Falke LC: Metabolite-regulated ATP-sensitive K+ channel in human
pancreatic islet cells. Diabetes. 38:422–427. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maechler P: Glutamate pathways of the
beta-cell and the control of insulin secretion. Diabetes Res Clin
Pract. 131:149–153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murao N, Yokoi N, Honda K, Han G, Hayami
T, Gheni G, Takahashi H, Minami K and Seino S: Essential roles of
aspartate aminotransferase 1 and vesicular glutamate transporters
in β-cell glutamate signaling for incretin-induced insulin
secretion. PLoS One. 12:e01872132017. View Article : Google Scholar
|
|
23
|
Gheni G, Ogura M, Iwasaki M, Yokoi N,
Minami K, Nakayama Y, Harada K, Hastoy B, Wu X, Takahashi H, et al:
Glutamate acts as a key signal linking glucose metabolism to
incretin/cAMP action to amplify insulin secretion. Cell Rep.
9:661–673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eto K, Tsubamoto Y, Terauchi Y, Sugiyama
T, Kishimoto T, Takahashi N, Yamauchi N, Kubota N, Murayama S,
Aizawa T, et al: Role of NADH shuttle system in glucose-induced
activation of mitochondrial metabolism and insulin secretion.
Science. 283:981–985. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fullerton MD, Galic S, Marcinko K, Sikkema
S, Pulinilkunnil T, Chen ZP, O'Neill HM, Ford RJ, Palanivel R,
O'Brien M, et al: Single phosphorylation sites in Acc1 and Acc2
regulate lipid homeostasis and the insulin-sensitizing effects of
metformin. Nat Med. 19:1649–1654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tripathy D and Chavez AO: Defects in
insulin secretion and action in the pathogenesis of type 2 diabetes
mellitus. Curr Diab Rep. 10:184–191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Supale S, Li N, Brun T and Maechler P:
Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol
Metab. 23:477–487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang S and Kim KH: Essential role of
acetyl-CoA carboxylase in the glucose-induced insulin secretion in
a pancreatic beta-cell line. Cell Signal. 10:35–42. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ronnebaum SM, Joseph JW, Ilkayeva O,
Burgess SC, Lu D, Becker TC, Sherry AD and Newgard CB: Chronic
suppression of acetyl-CoA carboxylase 1 in beta-cells impairs
insulin secretion via inhibition of glucose rather than lipid
metabolism. J Biol Chem. 283:14248–14256. 2008. View Article : Google Scholar : PubMed/NCBI
|