Introduction
Acute kidney injury (AKI) is a serious and common
complication of sepsis (1), and
is also a risk factor for progression to chronic kidney disease
(2). Substantial evidence
indicates that patients who are diagnosed with septic AKI have a
higher mortality risk and generally have longer ICU and hospital
stays than patients with non-septic AKI (3,4).
Unfortunately, as largely supportive therapies of sepsis-induced
AKI are often ineffective, the mortality related to AKI remains
high. Therefore, efforts focused on clarifying the specific
pathogenesis are important for finding a new therapy for
sepsis-induced AKI.
In recent years, evidence has demonstrated that
autophagy has a protective role in AKI (5,6).
For instance, numerous studies have revealed that autophagy is
activated in renal tubular epithelial cells upon the occurrence of
AKI (7), and blockade of
autophagy pharmacologically or genetically leads to more severe
AKI, while induction of autophagy reduces kidney injury (8,9).
However, the regulatory mechanism of autophagy in AKI is
complicated and has not been fully elucidated.
Increasing evidence suggests that metabolic
reprogramming has a central role in the immune response of host
defense to infection and represents a novel target for inflammatory
diseases. Early metabolic reprogramming in sepsis-induced AKI not
only protects the kidney from further injury, but also determines
the fate of tissue repair and the progression to fibrosis and
chronic organ dysfunction (10,11). Smith et al demonstrated a
shift in metabolism towards aerobic glycolysis in mouse kidney
tissues after administration with lipopolysaccharide (LPS), which
is associated with a decline in renal function (12). In cecal ligation and puncture
(CLP)-induced septic mice, metabolomic analysis of kidney tissues
revealed increases in the levels of glycolytic intermediates
(13). In addition, growing
evidence indicates that autophagy regulates or controls metabolism,
which in turn affects autophagy (14). For instance, the knockdown of the
key glycolysis enzyme pyruvate kinase M2 (PKM2) decreased
glycolysis and induced autophagy in human A549 alveolar
adenocarcinoma cells (15).
Furthermore, our previous work revealed that inhibition of aerobic
glycolysis improved survival rates and protected septic mice from
kidney injury (16). However, the
mechanism of the protective effect of inhibiting aerobic glycolysis
on sepsis-induced AKI is unknown, and whether this protective
effect is achieved by regulating autophagy in sepsis-induced AKI is
also unknown.
The aim of the present study, was to study the exact
mechanism by which aerobic glycolysis regulates autophagy in
sepsis-induced AKI. It was hypothesized that aerobic glycolysis
contributes to modulating autophagy via the lactate/sirtuin 3
(SIRT3)/AMP-activated protein kinase (AMPK) pathway in CLP-induced
septic AKI. Pharmacological inhibition of glycolysis by
2-deoxy-D-glucose (2-DG) and AKI was induced by CLP in BALB/C mice
in the present study.
Materials and methods
Mice
In total 60 BALB/c mice (male; 6-8 weeks old; 20-25
g) were purchased from Hunan SJA Laboratory Animal Co., Ltd. All
mice were housed individually and were maintained at 22°C with
40-60% humidity and a 12-h light/dark cycle starting at 6:00 am.
The mice were fed with normal chow and had free access to food and
water throughout the experiment. All animal experimental protocols
were approved by the Institutional Animal Care and Use Committee of
Central South University (approval no. 2019sydw0027) and performed
according to the guidelines of the Ethics Committee of the Animal
Experimental Institute of Central South University, Changsha,
China.
Cecal ligation and puncture (CLP)-induced
sepsis
Compared to LPS-induced sepsis, the CLP-induced
septic model is closer to human sepsis since it is characterized by
a polymicrobial insult, hyper-then hypodynamic hemodynamic
transition, the generation of detectable bacteremia, and the
increase in damage-associated molecular patterns. Therefore, CLP
was selected to induce sepsis.
Sepsis was induced in BALB/c mice by CLP as
previously described (17). The
mice were anaesthetized by isoflurane and fixed on the table. A
middle incision of the belly was made and the abdomen was opened.
Then, the cecum was exposed and ligated. Two punctures were
produced with a 22-gauge needle at the end of the cecum and some
feces were extruded from the punctures. The cecum was turned back
inside the abdomen, and the abdomen was closed. Resuscitative fluid
(normal saline, 50 ml/kg) was administrated by subcutaneous
injection. All operations were the same in the sham group except
for no puncture and ligation of the cecum.
Animal treatment
From the 60 mice, 24 mice were randomly divided into
four groups: The sham group, the sham+2-DG group, the CLP group,
and the CLP+2-DG group. The glycolysis inhibitor 2-DG
(Sigma-Aldrich; Merck KGaA) was dissolved in sterile saline and
injected intraperitoneally (2 g/kg) 3 h before the CLP operation.
An equal volume of vehicle was intraperitoneally injected as a
control. The dose and time-point of 2-DG was selected based on
previous studies (16,18).
From the 60 mice, 36 mice were randomly divided into
six groups: The sham group, the sham+2-DG group, the CLP group, the
CLP+2-DG group, the CLP+3-methyladenine (3-MA) group, and the
CLP+2-DG+3-MA group. Autophagy inhibitor 3-MA (Selleck Chemicals)
was dissolved in PBS by heating and injected intraperitoneally (30
mg/kg) 30 min before the CLP operation. An equal volume of vehicle
was intraperitoneally injected as a control. The dose and
time-point of 3-MA were selected based on previous studies
(19).
At 24 h of the CLP or sham operation, serum and
kidney tissues were harvested from mice and stored at −80°C for the
following experiments.
Haematoxylin and eosin (H&E) dye and
transferase terminal UTP nick-end labeling (TUNEL) staining
Kidney sections were fixed in 10% buffered formalin
at room temperature for 24 h, embedded in paraffin, cut into
3-µm sections, stained with H&E at room temperature for
3 min, and visualized at a magnification of ×400 under an optical
microscope (Olympus Optical Co., Ltd.).
Apoptosis of kidney tissue was detected using the
TUNEL assay (Roche Diagnostics) according to the manufacturer's
instructions. The renal tissues (4 µm) were fixed in 10%
buffered formalin at room temperature for 24 h, paraffin-embedded
and labeled with TUNEL reaction mixture containing terminal
deoxynucleotidyl transferase and nucleotides including
tetramethylrhodamine labeled (TMR-labeled) dUTP at room temperature
for 1 h. DAPI was then used to stain the nucleus at room
temperature for 15 min followed by the addition of a fluorescent
mounting medium. The TUNEL-positive cells were counted in 5
randomly selected fields from each slide at a magnification of
×400, and the percentage of TUNEL-positive cells was calculated
from four kidney sections of different mice.
Cell treatment in vitro
A human renal proximal tubular epithelial cell line
(HK-2 cells) purchased from ATCC was cultured in DMEM/F12 medium
supplemented with 10% fetal bovine serum at 37°C in an incubator
supplemented with 5% CO2. HK-2 cells (1×106)
were plated into 6-well plates, and pre-treated with or without
2-DG (2 mM, before 3 h) or/and 3-MA (5 mM, before 1 h) and/or
lactate (25 µM), followed by stimulation with or without LPS
(1 µg/ml) for 12 h. Lactate was added to the culture medium
of HK-2 cells without adjustment of pH. It was determined that the
extracellular pH changed from 7.2 to 6.6 after the addition of
lactate in LPS-stimulated HK-2 cells. Then cells were harvested and
protein and total RNA were isolated for subsequent protein and RNA
studies, and the culture medium was collected and stored at
-20°C.
Measurement of lactate
The lactate concentration of serum and culture
medium was measured using a Lactate Assay kit (cat. no. A019-2-1;
Nanjing Jiancheng Bioengineering Institute), according to the
manufacturer's instructions. The soluble fraction was assayed
directly at 530 nm using ELX-800 absorbance microplate reader
(BioTek Instruments, Inc.) until completion of the reaction. Before
the sample was analyzed, several samples we tested to determine the
applicable loading volume, which ensures the readings within the
standard curve. Lactate contents in the samples were calculated
according to the corresponding formula: Lactate (mmol/l)=(sample
OD−blank OD)/(standard OD−blank OD) × standard concentration
(mmol/l) × diluent times.
Biochemical assays
The sera of mice were collected at the indicated
time-points. Commercially available blood urea nitrogen (BUN) and
serum creatinine (Scr) detection kits (cat. nos. C010 and C074;
Changchun Huili Biotech Co., Ltd.) were used to measure serum
levels of biochemical parameters including blood urea nitrogen
(BUN) and serum creatinine (Scr), according to the manufacturer's
instructions, using an Olympus AU5400 Automated Biochemical
Analyzer (Olympus Corporation). The serum concentrations of kidney
injury molecular-1 (KIM-1) were measured with an ELISA kit (cat.
no. EK0880; BOSTER Biological Technology Co., Ltd.) according to
the manufacturer's instructions.
Apoptosis detection using flow
cytometry
Cell apoptosis analysis using the Annexin V-FITC/PI
Apoptosis Detection kit (BD Biosciences) was measured by flow
cytometry according to the manufacturer's instructions. After
stimulation, 1.0×106 HK-2 cells were washed with cold
PBS. Then, Annexin V-FITC and propidium iodide (PI) staining was
performed in the dark at room temperature for 15 min. Apototic
cells were detected by flow cytometry using a BD FACSCantoII flow
cytometer (BD Biosciences) and were analyzed with FlowJo analytical
software (Version X; TreeStar, Inc.).
Reverse transcription-quantitative
(RT-q)PCR analysis
Total cell or tissue RNA was extracted with TRIzol
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
quantitated by Nanodrop spectrophotometry. Reverse transcription
was performed with a PrimeScript™ RT Reagent kit according to the
manufacturer's instructions (Takara Bio, Inc.). The cDNAs were
subjected to quantitative real-time PCR using gene-specific primers
(300 nM per reaction) according to TB Green™ Premix Ex Taq™ (Takara
Bio, Inc.) protocols. The amplification level was programmed with a
denaturation step of 3 min at 95°C, followed by 40 cycles of
denaturation at 95°C for 12 sec, and extension at 62°C for 40 sec.
Human-18S ribosomal and mouse-actin mRNA levels served as internal
controls. Data were calculated using the 2−ΔΔCq method
method (20). The primers used in
the present study are presented in Table I.
 | Table IPrimers used for qPCR in the present
study. |
Table I
Primers used for qPCR in the present
study.
| Primer names | Forward
(5′-3′) | Reverse
(5′-3') |
|---|
Human-PKM2
NM002654.6 |
ATTATTTGAGGAACTCCGCCGCCT |
ATTCCGGGTCACAGCAATGATGG |
Human-LDHA
NM005566.4 |
ATCTTGACCTACGTGGCTTGGA |
CCATACAGGCACACTGGAATCTC |
Human-PDK1
NM002610.5 |
ACCAGGACAGCCAATACAAG |
CCTCGGTCACTCATCTTCAC |
Human-18S
NM022551.3 |
ATCCTCAGTGAGTTCTCCCG |
CTTTGCCATCACTGCCATTA |
Mouse-PKM2
NM011099.3 |
TGTCTGGAGAAACAGCCAAG |
TCCTCGAATAGCTGCAAGTG |
Mouse-LDHA
NM001136069.2 |
AGAGCGGGAGGGCAGCTTTCT |
GGGCAAGCTCATCCGCCAAGT |
Mouse-PDK1
NM001360002.1 |
TTCTCGCCGTCGCCACTCTC |
TGTCGGGGAGGAGGCTGATTTC |
Mouse-actin
NM205518.1 |
AGAAAATCTGGCACCACACC |
CAGAGGCGTACAGGGATAGC |
Western blotting
After stimulation, HK-2 cells were washed twice with
ice-cold PBS and lysed by RIPA (Thermo Fisher Scientific, Inc.)
with phosphatase and protein inhibitor for 30 min on ice, followed
by sonication twice. Then whole cell lysate was centrifuged at 4°C,
and 16,000 × g for 10 min. The supernatants were collected for
proteins analysis. Protein concentration in the supernatants was
determined using the bicinchoninic acid (BCA) protein assay. Cell
proteins (10 µg) were separated by 4-12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to a
polyvinylidene fluoride membrane. The membrane was blocked in 5%
milk at room temperature for 2 h and then were incubated in primary
antibodies against LC3-I/II (product no. ABC929; Sigma-Aldrich;
Merck KGaA), SIRT3 (product no. 5490S), phosphorylated (p)-AMPKα
(Thr172) (product no. 2535S), AMPKα (product no. 2603S), p62
(product no. 5114S) and β-actin (product no. 3700S) (all 1:1,000;
from Cell Signaling Technology, Inc.) at 4°C overnight. Membranes
were washed in TBS-T (0.01% Tween) 3 times and incubated with
HRP-conjugated secondary antibody (goat anti-mouse; cat. no. BA1050
and goat anti-rabbit, cat. no. BA1054 both from BOSTER Biological
Technology co. ltd.) at room temperature for 1 h. The signals were
visualized with ECL substrate (Bio-Rad Laboratories, Inc.) and
quantified using ImageJ 1.48V software (NIH).
Immunofluorescence staining
HK-2 cells (1.0×106) were washed with
ice-cold PBS twice and fixed with 4% methanol for 15 min at room
temperature. Then the cells were washed with PBS three times and
blocked with 5% PBS-BSA at room temperature for 1 h. Coverslips
were incubated with primary antibody against LC3 (dilution 1:200;
product no. L8918; Sigma-Aldrich; Merck KGaA) overnight at 4°C.
FITC goat anti-rabbit IgG (dilution 1:200; cat. no. AS011; ABclonal
Biotech Co., Ltd.) served as the secondary antibody at room
temperature for 1 h. DAPI was then used to stain the nucleus at
room temperature for 15 min, followed by the addition of a
fluorescent mounting medium. Cells were visualized at a
magnification of ×400 using fluorescence microscopy (Olympus BX53;
Olympus Corporation.).
Statistical analysis
All statistical analyses were performed with
GraphPad Prism version 7.0 software (GraphPad Software, Inc.).
Comparisons between two groups were performed with either an
unpaired two-tailed Student's t-test (parametric) or the
Mann-Whitney test (non-parametric). Comparisons between multiple
groups were analyzed using one-way analysis of variance, followed
by Tukey's multiple comparison test. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
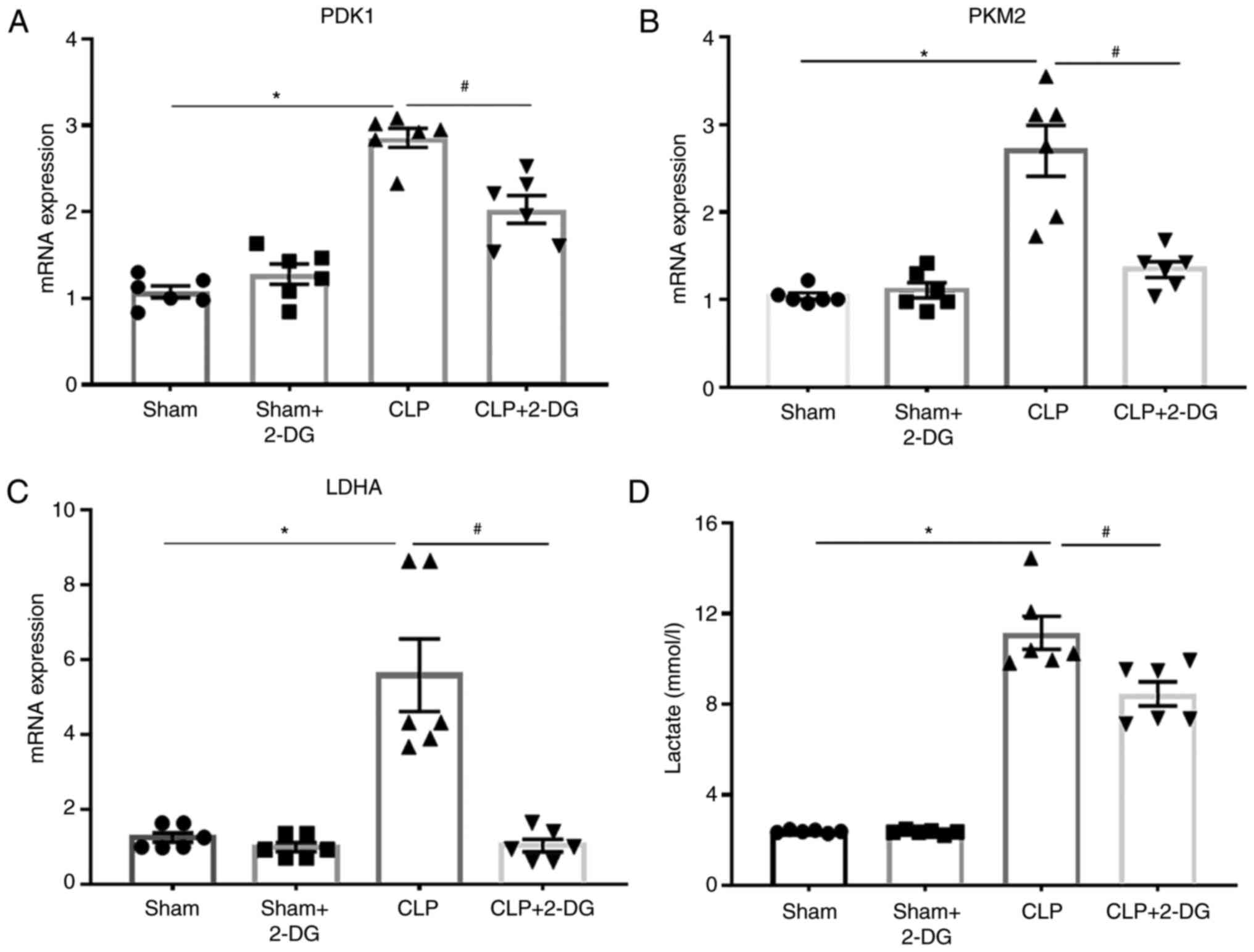
Aerobic glycolysis is upregulated in
CLP-induced AKI
First, the mRNA levels of glycolysis-related genes
and the glycolysis metabolite lactate in vivo were detected
to verify that glycolysis is upregulated in sepsis-induced AKI. In
fact, it was revealed that the mRNA expression levels of
glycolysis-related genes pyruvate dehydrogenase kinase 1 (PDK1),
lactate dehydrogenase A (LDHA), and pyruvate kinase M2 (PKM2) were
upregulated in the kidney tissue of septic mice compared to those
in the sham group. In contrast, inhibition of aerobic glycolysis by
2-DG downregulated the mRNA expression levels of PDK1, LDHA, and
PKM2 in the kidney tissue of septic mice (Fig. 1A-C). Moreover, the concentration
of serum lactate was increased in septic mice, while it was
decreased in septic mice by treatment with 2-DG (Fig. 1D). These results indicated that
aerobic glycolysis was upregulated in sepsis-induced AKI, which
could be reversed by the glycolysis inhibitor 2-DG.
Aerobic glycolysis inhibitor 2-DG
alleviates sepsis-induced AKI
To assess the effect of aerobic glycolysis on the
kidney function of septic mice, we assessed the serum levels of
BUN, creatinine and KIM-1 in CLP-induced septic mice. Mice were
treated with 2-DG (2 g/kg) or vehicle (0.9% NaCl) before CLP 3 h.
The dose of 2-DG administered was established by our previous work,
which improved the survival rates of septic mice (16). In the present study, the serum
levels of BUN, creatinine and KIM-1 were increased in septic mice
compared to those in the sham group, and this increase was
significantly decreased by 2-DG (Fig.
2A-C). In addition, the morphological tubular H&E dye
revealed that the tubular epithelial cells of septic mice were
edematous with larger cellular volume, vacuolar degeneration and
glomerular structure was disordered accompanied by narrowed lumen
of the renal tubules. These pathological lesions were alleviated by
2-DG treatment in the septic mice (Fig. 2D). These results indicated that
inhibition of aerobic glycolysis has a protective effect on
sepsis-induced AKI.
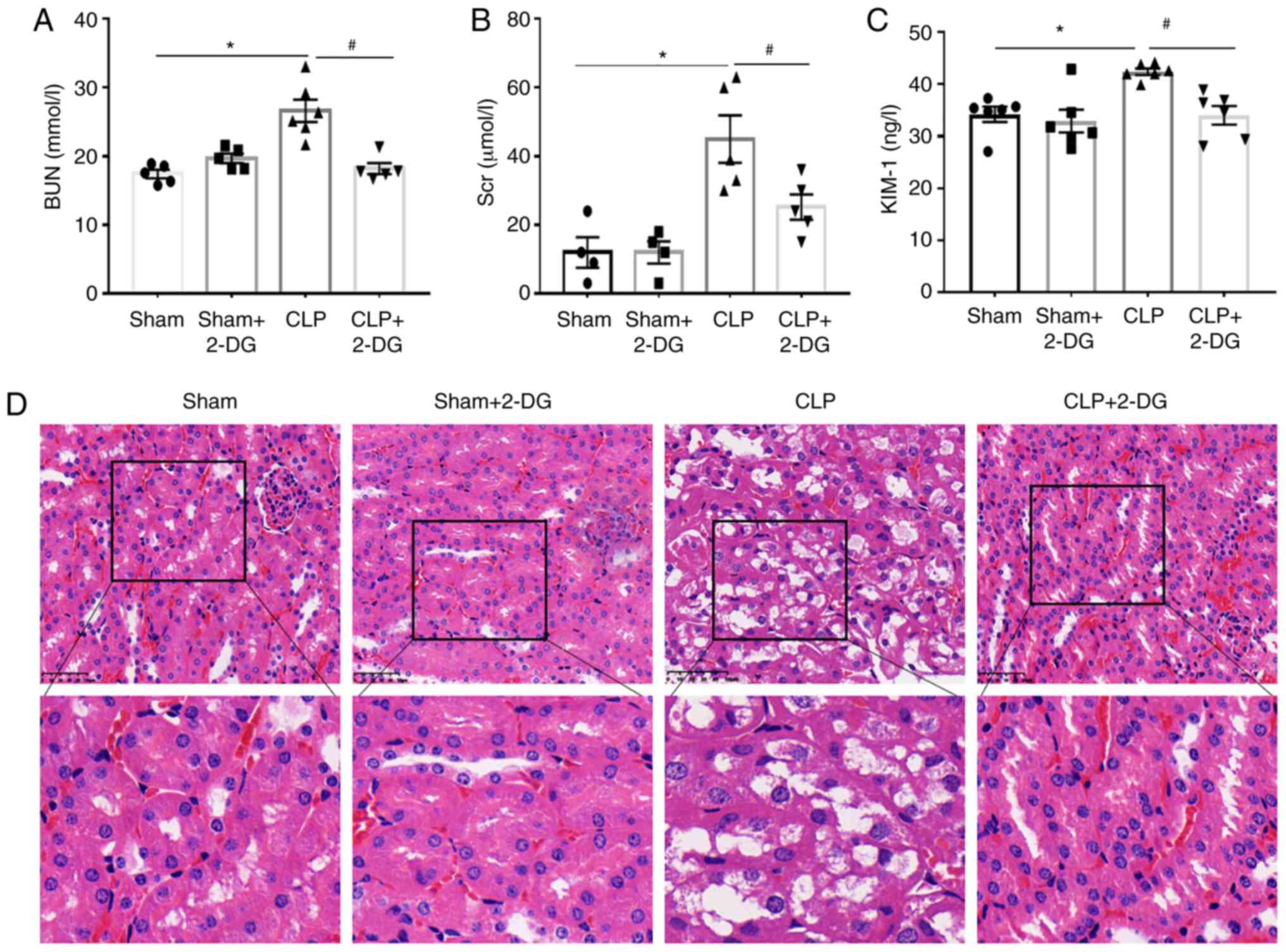 | Figure 2Glycolysis inhibitor 2-DG alleviates
sepsis-induced AKI and apoptosis. Sepsis was induced in BALB/c mice
by CLP. BALB/c mice were injected i.p. with either 2-DG (2 g/kg) or
PBS in equivalent volumes 3 h before CLP. Sham-operated animals
served as negative controls. At 24 h of CLP or sham operation,
serum and kidneys were harvested from mice. (A-C) The levels of
BUN, SCr and KIM-1 in mouse sera were analyzed using commercial
kits. n=4-6 mice per group. (D) The effect of 2-DG on the
morphological changes of kidney tissues in CLP or sham mice (×40).
Data are presented as the means ± SEM. *P<0.05 vs.
the vehicle-treated sham. #P<0.05 vs. CLP+2-DG group.
2-DG, 2-deoxy-D-glucose; AKI, acute kidney injury; CLP, cecal
ligation and puncture; i.p., intraperitoneally; PBS,
phosphate-buffered saline; BUN, blood urea nitrogen; Scr, serum
creatinine; KIM-1, kidney injury molecule-1. |
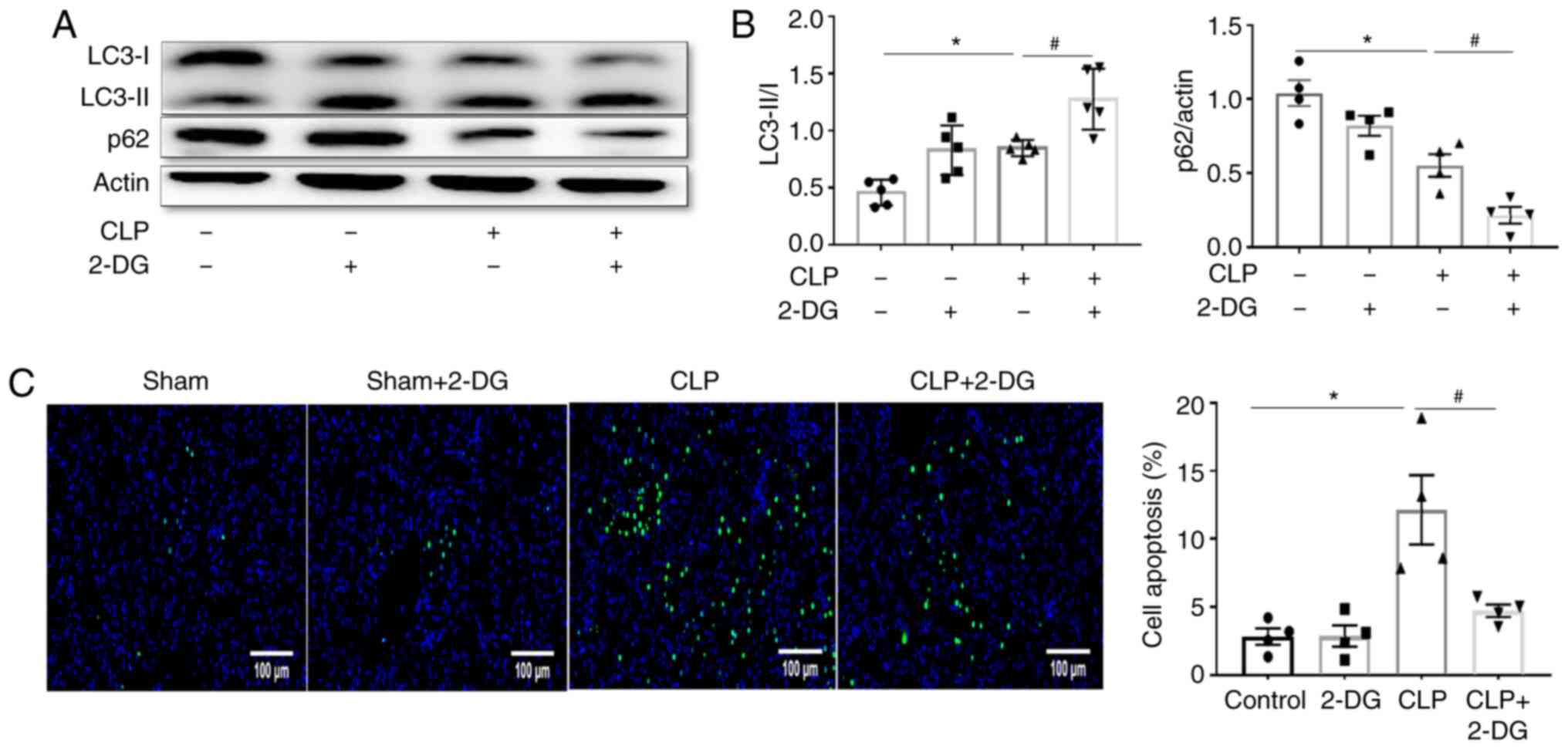
Aerobic glycolysis inhibitor 2-DG
alleviates sepsis-induced AKI by upregulating autophagy
Next, the role of autophagy in the protective effect
of the aerobic glycolysis inhibitor 2-DG against sepsis-induced AKI
was investigated. Western blotting revealed that CLP upregulated
autophagy as evidenced by the increase of the ratio of LC3-II/I and
decrease of the expression of p62 compared to the sham group, and
this effect was further improved by treatment with 2-DG in septic
mice (Fig. 3A and B). In
addition, it has been established that autophagy always interacts
with apoptosis (21). In the
present study it was revealed that CLP enhanced the apoptosis of
renal tubular epithelial cells compared to the sham group, and this
increase was significantly suppressed by 2-DG in septic mice
treated with 2-DG (Fig. 3C).
These results indicated that 2-DG-mediated protection against
sepsis-induced AKI was related to an increased regulation of
autophagy.
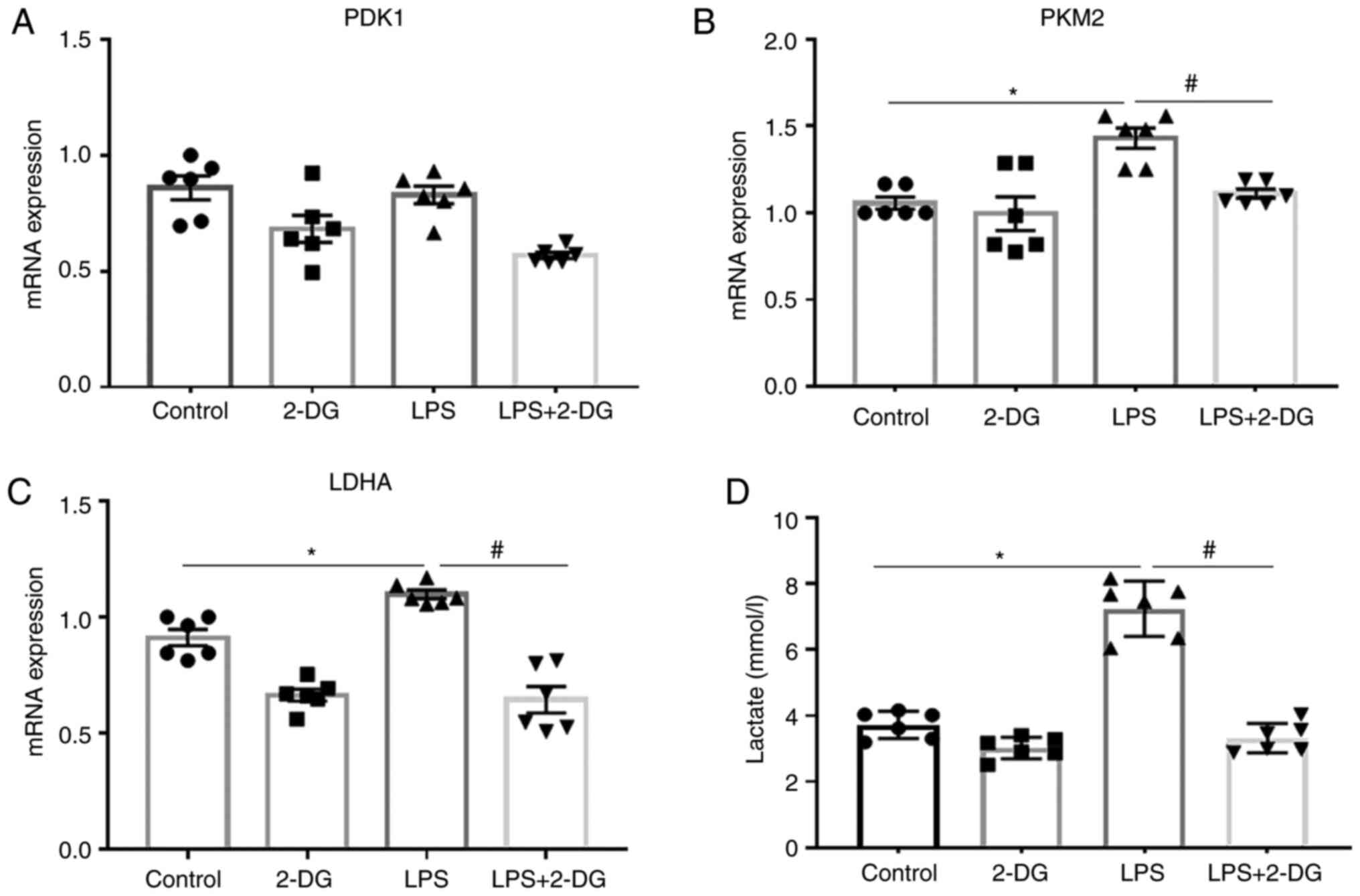
Aerobic glycolysis is upregulated in HK-2
cells stimulated with LPS
The mRNA levels of glycolysis-related genes and the
glycolysis metabolite lactate were also detected in vitro in
HK-2 cells stimulated with LPS. It was revealed that the mRNA
expression of LDHA and PKM2 was upregulated in HK-2 cells
stimulated with LPS compared to those in the control group. In
contrast, 2-DG decreased the mRNA expression of LDHA and PKM2 in
HK-2 cells stimulated with LPS (Fig.
4B and C). The mRNA expression of PDK1 was unchanged in HK-2
cells (Fig. 4A). Moreover, LPS
enhanced the lactate levels in the culture supernatants of HK-2
cells compared to the control group, while 2-DG reduced the lactate
levels in the culture supernatant of HK-2 cells stimulated with LPS
(Fig. 4D). These results
indicated that aerobic glycolysis was upregulated in HK-2 cells
stimulated with LPS, which could be reversed by the glycolysis
inhibitor 2-DG.
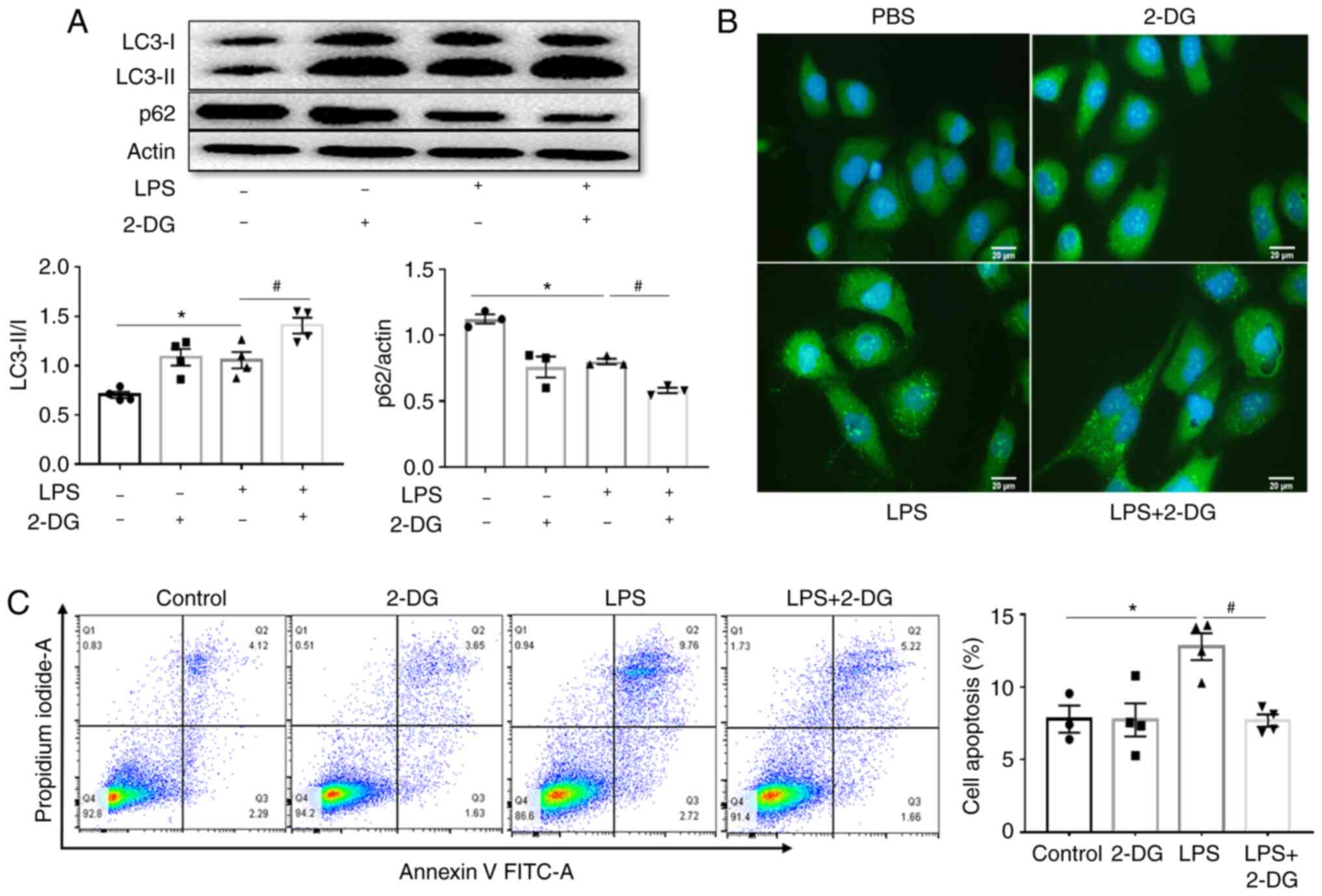
Aerobic glycolysis inhibitor 2-DG further
upregulates autophagy in HK-2 cells stimulated with LPS
The relationship between autophagy and aerobic
glycolysis in vitro in HK-2 cells stimulated with LPS was
also investigated. It was observed that LPS enhanced the ratio of
LC3-II/I and decreased p62 expression compared to those in the
control group, and this effect was further promoted by 2-DG
(Fig. 5A). Immunofluorescence
also revealed that LPS enhanced the expression of LC3-II, and this
effect was further improved by 2-DG (Fig. 5B). In the present study, LPS
increased the apoptosis of HK-2 cells compared to the control
group, and 2-DG significantly decreased this increase of apoptosis
in HK-2 cells stimulated with LPS (Fig. 5C). These results indicated that
2-DG further upregulated autophagy in HK-2 cells stimulated with
LPS.
Aerobic glycolysis inhibitor 2-DG
increases autophagy via the SIRT3/AMPK pathway in septic mice and
in HK-2 cells stimulated with LPS
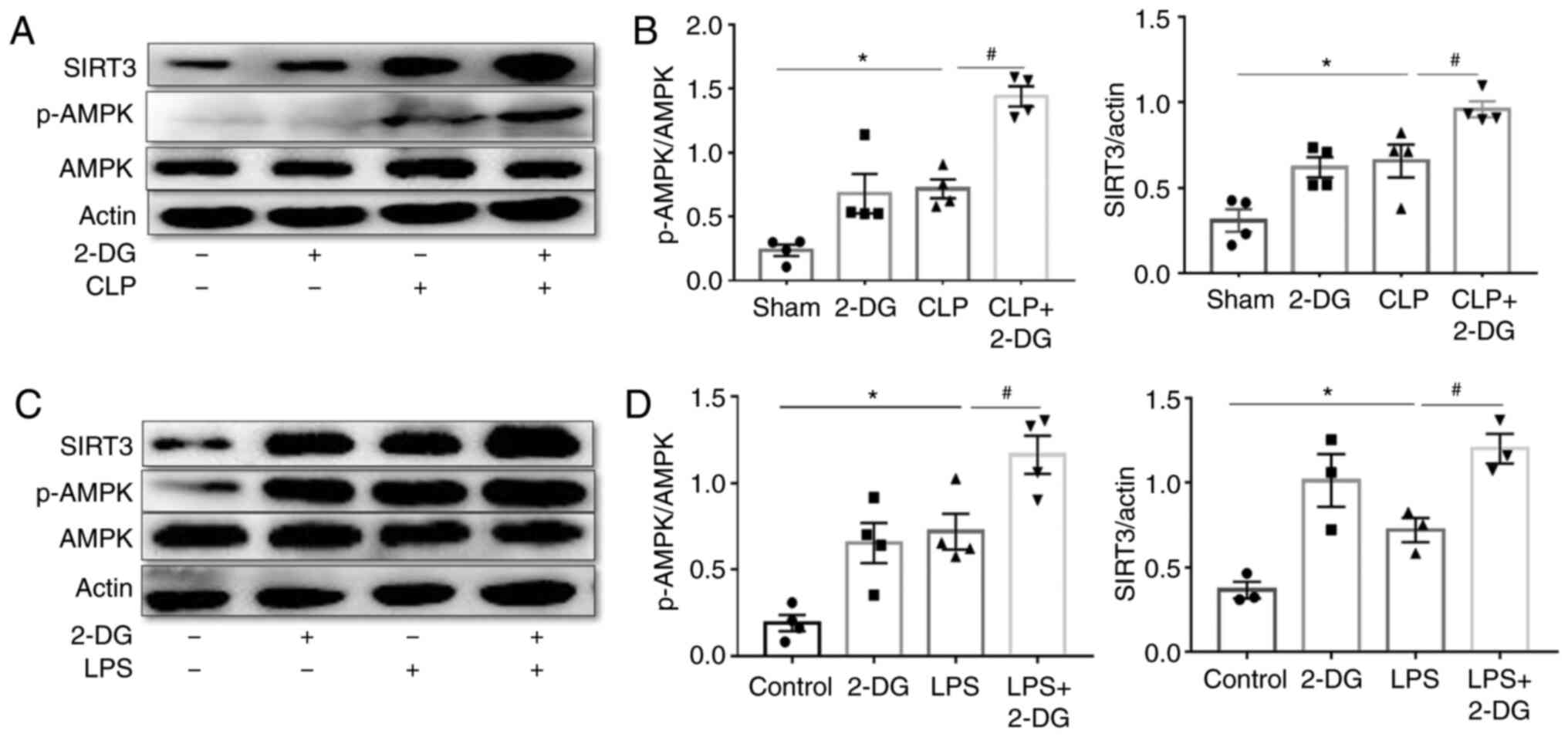
The mechanism of modulation of autophagy by 2-DG was
further investigated by assessing the levels of autophagy
regulators SIRT3 and AMPK in the kidney tissues of sepsis-induced
AKI and HK-2 cells. p-AMPK and SIRT3 were evaluated by western
blotting. The results revealed that the expression levels of SIRT3
and p-AMPK were upregulated in the kidney tissue of septic mice
compared to the sham group, and 2-DG further increased SIRT3 and
p-AMPK expression in septic mice (Fig. 6A and B). Concurrently it was
revealed that LPS promoted the expression of SIRT3 and p-AMPK in
HK-2 cells compared to the control group, and 2-DG further enhanced
the expression of SIRT3 and p-AMPK in HK-2 cells stimulated with
LPS (Fig. 6C and D). These
results indicated that 2-DG increased autophagy via the SIRT3/AMPK
pathway in sepsis-induced AKI.
Glycolysis metabolite lactate decreases
autophagy via the SIRT3/AMPK pathway in HK-2 cells stimulated with
LPS
Metabolism provides energy and intermediate
metabolites may serve as signaling molecules to regulate cellular
immune functions (22). Lactic
acid is one of the most important metabolites of the glycolysis
pathway. In the present study it was revealed that lactate
treatment reduced the ratio of LC3-II/I and increased the
expression of p62 in LPS-stimulated HK-2 cells compared with LPS
group (Fig. S1A-C).
Concurrently, lactate treatment decreased the expression of SIRT3
and p-AMPK in LPS-stimulated HK-2 cells compared with the LPS group
(Fig. S1A, D and E).
Furthermore, lactate partly abolished the increased effect on the
ratio of LC3II/I and the decreased effect of p62 by 2-DG in
LPS-treated HK-2 cells (Fig. 7A and
B). Lactate treatment also decreased the increased effect on
the expression of SIRT3 and p-AMPK by 2-DG in LPS-treated HK-2
cells (Fig. 7C and D). In
addition, lactate treatment reversed the reduction in apoptosis
induced by 2-DG in LPS-treated HK-2 cells (Fig. 7E). These results indicated that
the glycolysis metabolite lactate was involved in regulating
autophagy via the SIRT3/AMPK pathway.
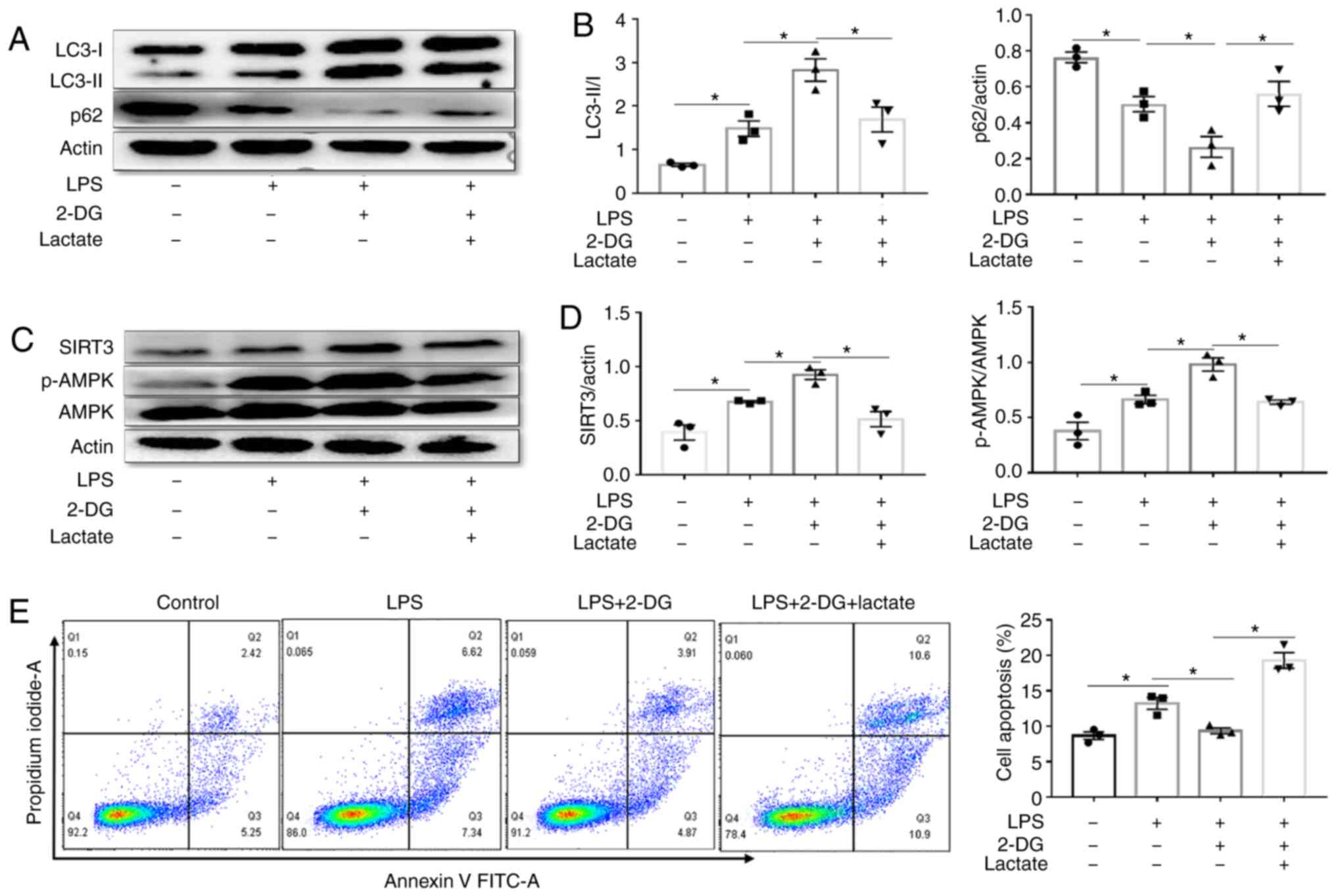 | Figure 7Lactate decreases autophagy via the
SIRT3/AMPK pathway in HK-2 cells. A total of 1×106 HK-2
cells were treated with the indicated doses of LPS (1
µg/ml), LPS+2-DG (2 mM), LPS+2-DG+lactate (25 µM) for
12 h at 37°C in 5% CO2. (A and B) Immunoblot analysis
and quantification of LC3-I/II and p62 in HK-2 cells stimulated
with LPS (1 µg/ml) in the presence/absence of lactate (25
µM) or 2-DG (2 mM, before 3 h). n=3 mice per group. (C and
D) Immunoblot analysis and quantification of SIRT3 and p-AMPK/AMPK
in HK-2 cells stimulated with LPS in the presence/absence of
lactate (25 µM) or 2-DG (2 mM, before 3 h). n=4 experiments.
(E) Apoptosis of HK-2 cells stimulated with LPS in the
presence/absence of lactate (25 µM) or 2-DG (2 mM, before 3
h). n=3 experiments. The data are presented as the means ± SEM.
*P<0.05, vs. the indicated groups. SIRT3, sirtuin 3;
AMPK, AMP-activated protein kinase; LPS, lipopolysaccharides; 2-DG,
2-deoxy-D-glucose; p-, phosphorylated. |
2-DG-induced protective effects against
sepsis-induced AKI depend on regulating autophagy
Finally, the involvement of autophagy in the
protective effect of 2-DG on AKI was further examined using the
autophagy inhibitor 3-MA. Inhibition of autophagy by 3-MA
suppressed the upregulation of LC3II and the decrease of p62 by
2-DG in the kidney tissues of septic mice (Fig. 8A and B). 3-MA partly abolished the
decreases in BUN, Scr and KIM-1 levels by 2-DG in septic mice
(Fig. 8C and D). It was
determined that 3-MA partially abrogated the decrease in apoptosis
induced by 2-DG in the kidney tissues of septic mice (Fig. 8E). Furthermore, similar results
were obtained with HK-2 cells. 3-MA suppressed the increase in
LC3II and the reduction in p62 by 2-DG in LPS-treated HK-2 cells
(Fig. S2A and B). 3-MA also
partially abrogated the decreased effect of 2-DG on apoptosis in
HK-2 cells stimulated with LPS (Fig.
S2C and D). These results indicated that 2-DG-mediated
protection against sepsis-induced AKI was related to the regulation
of autophagy.
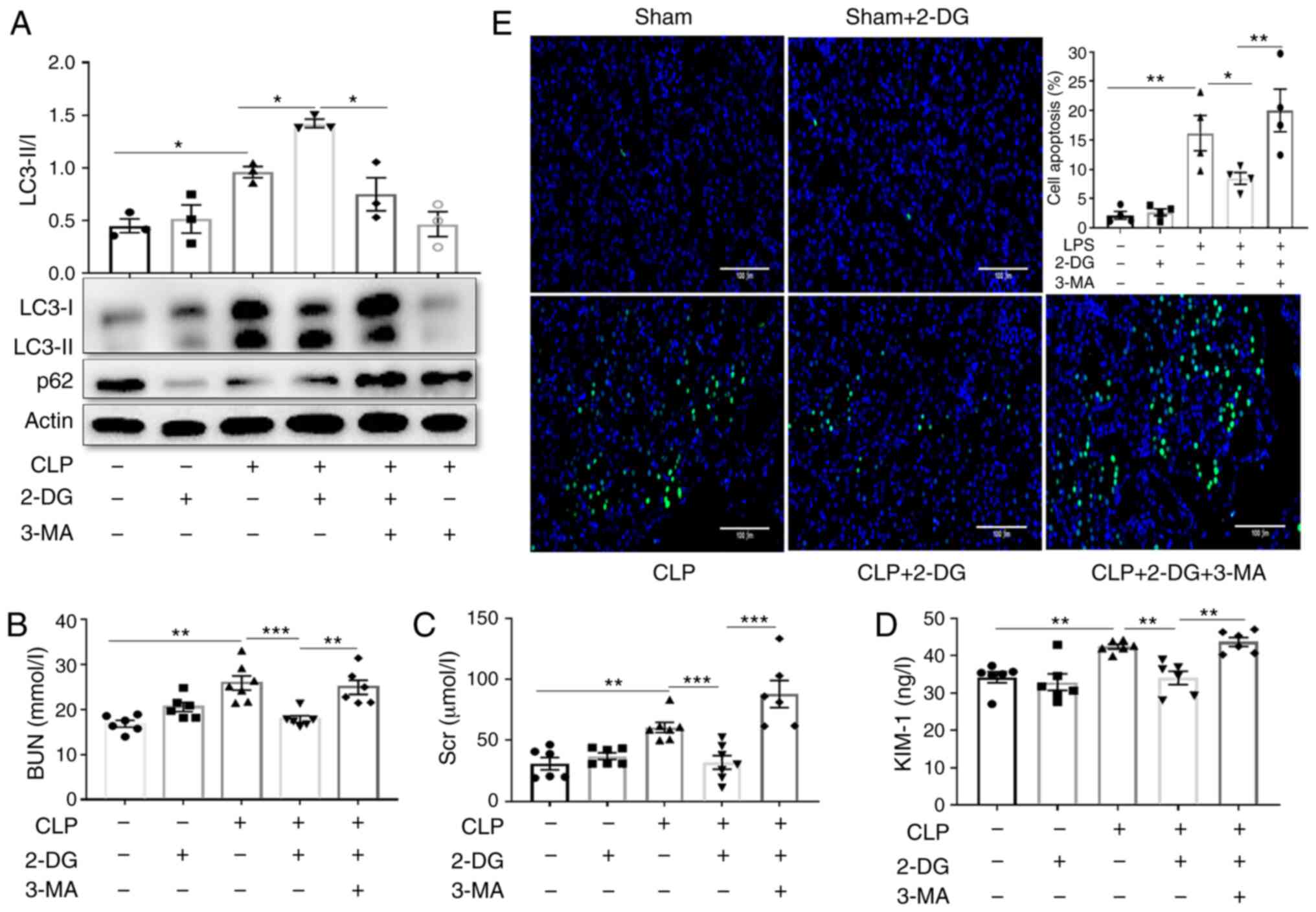 | Figure 82-DG-induced protective effects
against sepsis-induced AKI depend on regulating autophagy. Mice
were injected i.p. with either 3-MA (30 mg/kg, CLP+3-MA) or PBS in
equivalent volumes 1 h before CLP in the presence/absence of 2-DG
(2 g/kg, before 3 h). Kidney tissues and blood samples were
collected at 24 h after CLP. (A) Immunoblot analysis and
quantification of LC3-I/II and in kidney tissue of septic mice. n=3
mice per group. (B-D) BUN and Scr and KIM-1 were analyzed using
commercial kits. n=7 mice per group. (E) Apoptosis of renal tubular
cells of mice was measured by TUNEL assay and quantified. Scale
bars, 100 µm. n=4 mice per group. Data are presented as the
means ± SEM. *P<0.05, **P<0.01 and
***P<0.001 vs. the indicated groups. 2-DG,
2-deoxy-D-glucose; AKI, acute kidney injury; i.p.,
intraperitoneally; 3-MA, 3-methyladenine; CLP, cecal ligation and
puncture; PBS, phosphate-buffered saline; BUN, blood urea nitrogen;
Scr, serum creatinine; KIM-1, kidney injury molecule-1; TUNEL,
terminal UTP nick end labeling. |
Discussion
In our previous study, we revealed that inhibition
of aerobic glycolysis played a protective role in sepsis by
promoting neutrophil migration into the infectious site, and we
determined that treatment with the glycolysis inhibitor 2-DG
improved the survival rates and attenuated sepsis-induced kidney
injury (16). In the present
study, we further examined the mechanisms underlying the protective
effect of the inhibition of aerobic glycolysis on sepsis-induced
AKI by focusing on the potential involvement and modulation of
autophagy by SIRT3 and AMPK. AKI was induced by CLP in BALB/C mice,
and examination of serum and kidney tissues and assessment of
autophagy modulated by activation of SIRT3 and AMPK, revealed the
protective effects of 2-DG, on kidney function and structure.
Aerobic glycolysis (Warburg effect), first observed
in various tumor cells, is also increasingly recognized as an
essential regulator of the immune response in both immune and
nonimmune cells and represents a novel target for inflammatory
diseases (23). Previous studies
revealed that patients with AKI converted metabolism from oxidative
phosphorylation to aerobic glycolysis (24,25). Our group as well as other
researchers revealed that experimental sepsis induces a shift of
metabolism towards aerobic glycolysis in renal tissue. In a rodent
model of CLP-induced sepsis, metabolomics analysis demonstrated an
increase in the level of glycolytic intermediates concomitant with
a decrease in flux through the tricarboxylic acid cycle (13). In the present study, it was
revealed that sepsis enhanced aerobic glycolysis as evidenced by
increased production of lactate and upregulated mRNA expression of
glycolysis-related genes in kidney tissues in vivo. 2-DG
markedly reduced this effect in vivo. This is consistent
with previous studies (13,24,25). In addition, HK-2 cells stimulated
with LPS were used to establish a cell model of AKI. It was also
determined that LPS enhanced aerobic glycolysis as evidenced by
increased lactate production and upregulated mRNA expression of
glycolysis-related genes in HK-2 cells in vitro, which could
be reduced by 2-DG. These data indicated that tubular epithelial
cells (TECs) undergo similar metabolic changes in response to
inflammation and suggest that aerobic glycolysis is upregulated in
sepsis-induced AKI.
Studies have identified that aerobic glycolysis
(namely the Warburg effect) is involved in the regulation of innate
immune function and is a novel target of kidney disease (11,26). Chiaravalli et al and
Riwanto et al revealed that inhibition of aerobic glycolysis
with 2-DG greatly reduced disease progression and improved the
function of the kidney in polycystic kidney disease (27,28). Numerous studies have demonstrated
that a switch to aerobic glycolysis may be harmful to sepsis with
evidence that stimulating the promoters of OXPHOS protected organs
from damage and improved survival in experimental sepsis (11,16,27,28). Treatment with a PKM2 inhibitor or
knockdown of PKM2 significantly decreased glycolysis and reduced
mortality in CLP-induced septic mice (13). Our group as well as other
researchers verified that inhibition of aerobic glycolysis improved
the survival rate and protected against multiorgan injury in sepsis
(16,18). Early reprogramming of metabolic
pathways is not only able to protect the kidney from further
injury, but also determines the fate of tissue repair and the
progression to fibrosis and chronic organ dysfunction in
sepsis-induced AKI (10,11). In the present study, it was
revealed that inhibition of aerobic glycolysis had a protective
effect on sepsis-induced AKI with evidence of decreased BUN and Scr
levels and apoptosis of tubular epithelial cells. Therefore,
interventions designed to regulate the metabolism of the immune
system may have potential therapeutic significance for
sepsis-induced AKI.
Autophagy is a process of lysosome-mediated
degradation of intracellular organelles, proteins, and other
macromolecules (29,30), which is detected at low levels in
the normal physiological situation, but it can be significantly
activated under starvation, hypoxia, and infection (31). The dysregulation of autophagy
pathways has been implicated in the pathogenesis of numerous renal
diseases including AKI, polycystic kidney disease, and diabetic
nephropathy (32,33). Numerous studies have revealed that
autophagy is activated in renal tubular epithelial cells upon the
occurrence of AKI, and activation of autophagy has a protective
effect on the AKI model (3,5,34).
In addition, immune metabolism plays a critical role in modulating
autophagy. Some studies have verified that knockdown of PKM2
decreases glycolysis and induces autophagy (15,35). In the present study, it was
determined that CLP upregulated the autophagy marker, LC3-II and
decreased p62, while 2-DG further induced autophagy in CLP mice in
parallel with decreased levels of BUN, Scr and KIM-1, which
indicated renal dysfunction. Similar results were obtained in
LPS-treated HK-2 cells where 2-DG also upregulated autophagy.
However, inhibition of autophagy by 3-MA suppressed the
upregulation of LC3II and the decrease in p62 by 2-DG in the kidney
tissues of septic mice. Similar results were obtained in
LPS-treated HK-2 cells. Moreover, 3-MA partly abolished the
decreases in the BUN, Scr and KIM-1 levels induced by 2-DG in
septic mice. According to the present results, LPS enhanced aerobic
glycolysis and autophagy in vivo and in vitro
concurrently, while inhibition of aerobic glycolysis by 2-DG
further increased autophagy in LPS-treated HK-2 cells. It is
inferred that LPS enhanced autophagy via other signaling pathways,
and this effect was stronger than the inhibitory effect by
LPS-increased aerobic glycolysis on autophagy, and thus inhibition
of aerobic glycolysis by 2-DG further increased autophagy. All the
aforementioned results emphasized the importance of our findings in
elucidating a potential mechanism underlying the protective effect
of inhibition of aerobic glycolysis against AKI, which is related
to an augmented regulation of autophagy.
However, the mechanism by which aerobic glycolysis
regulates autophagy in sepsis-induced AKI in HK-2 cells is unclear.
Numerous studies have demonstrated that SIRT3 and AMPK regulated
autophagy in AKI (19,36). It has been reported that SIRT3
knockdown can reduce AMPK phosphorylation and significantly inhibit
the activation of AMPK kinase (37). SIRT3 deletion inhibited autophagy
in CLP mice in parallel with increased levels of BUN and Scr, which
indicated renal dysfunction. These effects were accompanied by
downregulation of p-AMPK and upregulation of p-mTOR (36). In the present study, western
blotting and quantification of the results revealed that treatment
with 2-DG induced autophagy in parallel with upregulation of SIRT3
and p-AMPK in septic mice. Based on the aforementioned results, it
was concluded that autophagy was associated with SIRT3/AMPK, in the
CLP mouse model. Concurrently, the same results were also obtained
in HK-2 cells. It was revealed that 2-DG further induced autophagy
and increased SIRT3 and p-AMPK in HK-2 cells stimulated with LPS.
In addition, glycolysis has been reported to regulate autophagy as
evidenced by 2-DG enhancement of autophagy through activation of
the AMPK/ULK1 pathway and in cancer cells (38). Modulation of sepsis-enhanced
glycolysis with 2-DG improved the survival outcome by decreasing
MKK3 phosphorylation and increasing SIRT3 expression (18). Moreover, Jin et al recently
revealed that renal AMPK activation protected from sepsis-induced
AKI by preserving mitochondrial function and metabolic fitness
likely through SIRT3 signaling (39). The aforementioned results are
consistent with the results of the present study. It was concluded
that 2-DG decreased aerobic glycolysis and increased autophagy
through the SIRT3/AMPK pathway, and protected against
sepsis-induced AKI.
Lactic acid is one of the most important metabolites
of the glycolysis pathway. Lactate is an immunosuppression factor
and high serum lactic acid levels are a strong negative prognostic
factor in severe septic patients (40). In the present study upregulated
production of lactate in the serum of septic mice was observed.
Lactate may be a potential and critical contributory factor in the
regulation of immune function in sepsis. It has been reported that
increased production of lactate suppressed autophagy in cancer
cells (41). In the present
study, it was revealed that lactate treatment decreased the
autophagy marker LC3-II and increased p62 in the HK-2 cells
stimulated with LPS. However, the molecular mechanism for the
suppression of autophagy by lactate remains unclear. It was
revealed that lactate treatment decreased p-AMPK and SIRT3 in
LPS-treated HK-2 cells. Lactate also partly abolished the increased
effect on LC3II/I and p62 by 2-DG in LPS-treated HK-2 cells.
Notably, Xu et al already demonstrated that lower pH and
higher pH conditions induced completely opposite effects on
autophagy, with its activity suppressed at lower pH whereas
stimulated at higher pH (42). It
is not certain whether lactate-suppressed autophagy is a direct
effect of lactate or an indirect effect of extracellular pH,
further research is required to confirm this hypothesis.
In conclusion, the present study revealed a
potential mechanism underlying the protective role of inhibition of
aerobic glycolysis against sepsis-induced AKI, which was involved
in the induction of autophagy through the lactate/SIRT3/AMPK
pathway. These findings suggest that the reprogramming of the
metabolism is important in renal injury and is a potential
therapeutic target of AKI.
Supplementary Data
Funding
The present study was supported by funding from the
National Natural Science Foundation of China, grant nos. 81871610,
81870071, 81471897 and 81671895.
Availability of data and materials
The datasets generated during the present study are
not currently available to the public but will be available from
the corresponding author on reasonable request.
Authors' contributions
XX and HZ oversaw the study. XX and CT designed and
conceived the study. CT, JG, TL, HC, and KL collected samples,
performed experiments, and analyzed data. CT and ML prepared the
figures and wrote the manuscript. HZ and XX reviewed the manuscript
for important intellectual content. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animal experimental protocols were approved by
the Institutional Animal Care and Use Committee of Central South
University (approval no. 2019sydw0027) and performed according to
the guidelines of the Ethics Committee of the Animal Experimental
Institute of Central South University, Changsha, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors are grateful to Dr Kewen Ma (Department
of Pathology, Xiangya Hospital, Central South University, Changsha,
China) for her help with the analysis of the morphological tubular
H&E dye in this study. We also thank the associate editor and
the reviewers for their useful feedback that improved this
study.
Abbreviations:
|
2-DG
|
2-deoxy-D-glucose
|
|
AKI
|
acute kidney injury
|
|
AMPK
|
AMP-activated protein kinase
|
|
BUN
|
blood urea nitrogen
|
|
CLP
|
cecal ligation and puncture
|
|
LDHA
|
lactate dehydrogenase A
|
|
LPS
|
lipopolysaccharides
|
|
mTOR
|
mammalian target of rapamycin
|
|
PDK1
|
pyruvate dehydrogenase kinase 1
|
|
PKM2
|
pyruvate kinase M2
|
|
Scr
|
serum creatinine
|
|
SIRT3
|
sirtuin 3
|
|
3-MA
|
3-methyladenine
|
|
PBS
|
phosphate-buffered saline
|
|
DAPI
|
4,6-diamino-2-phenyl indole
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
HK-2
|
human renal proximal tubular
epithelial cell line
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FITC
|
fluorescein isothiocyanate
|
|
RIPA
|
radio-immunoprecipitation
|
References
|
1
|
Uchino S, Kellum JA, Bellomo R, Doig GS,
Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, et al:
Acute renal failure in critically ill patients: A multinational,
multicenter study. JAMA. 294:813–818. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murugan R and Kellum JA: Acute kidney
injury: What's the prognosis? Nat Rev Nephrol. 7:209–217. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cruz MG, Dantas JG, Levi TM, Rocha Mde S,
de Souza SP, Boa-Sorte N, de Moura CG and Cruz CM: Septic versus
non-septic acute kidney injury in critically ill patients:
Characteristics and clinical outcomes. Rev Bras Ter Intensiva.
26:384–391. 2014.In En, Portuguese. View Article : Google Scholar
|
|
4
|
Mehta RL, Bouchard J, Soroko SB, Ikizler
TA, Paganini EP, Chertow GM and Himmelfarb J; Program to Improve
Care in Acute Renal Disease (PICARD) Study Group: Sepsis as a cause
and consequence of acute kidney injury: Program to improve care in
acute renal disease. Intensive Care Med. 37:241–248. 2011.
View Article : Google Scholar :
|
|
5
|
Kaushal GP and Shah SV: Autophagy in acute
kidney injury. Kidney Int. 89:779–791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Duann P, Lianos EA, Ma J and Lin PH:
Autophagy, innate immunity and tissue repair in acute kidney
injury. Int J Mol Sci. 17:6622016. View Article : Google Scholar :
|
|
7
|
Jiang M, Wei Q, Dong G, Komatsu M, Su Y
and Dong Z: Autophagy in proximal tubules protects against acute
kidney injury. Kidney Int. 82:1271–1283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li T, Liu Y, Zhao J, Miao S, Xu Y, Liu K,
Liu M, Wang G and Xiao X: Aggravation of acute kidney injury by
mPGES-2 down regulation is associated with autophagy inhibition and
enhanced apoptosis. Sci Rep. 7:102472017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu S, Hartleben B, Kretz O, Wiech T,
Igarashi P, Mizushima N, Walz G and Huber TB: Autophagy plays a
critical role in kidney tubule maintenance, aging and
ischemia-reperfusion injury. Autophagy. 8:826–837. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zager RA: 'Biologic memory' in response to
acute kidney injury: Cytoresistance, toll-like receptor
hyper-responsiveness and the onset of progressive renal disease.
Nephrol Dial Transplant. 28:1985–1993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gomez H, Kellum JA and Ronco C: Metabolic
reprogramming and tolerance during sepsis-induced AKI. Nat Rev
Nephrol. 13:143–151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith JA, Stallons LJ and Schnellmann RG:
Renal cortical hexokinase and pentose phosphate pathway activation
through the EGFR/Akt signaling pathway in endotoxin-induced acute
kidney injury. Am J Physiol Renal Physiol. 307:F435–F444. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Waltz P, Carchman E, Gomez H and
Zuckerbraun B: Sepsis results in an altered renal metabolic and
osmolyte profile. J Surg Res. 202:8–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang J, Zhou R and Ma Z: Autophagy and
energy metabolism. Adv Exp Med Biol. 1206:329–357. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chu B, Wang J, Wang Y and Yang G:
Knockdown of PKM2 induces apoptosis and autophagy in human A549
alveolar adenocarcinoma cells. Mol Med Rep. 12:4358–4363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan C, Gu J, Chen H, Li T, Deng H, Liu K,
Liu M, Tan S, Xiao Z, Zhang H and Xiao X: Inhibition of aerobic
glycolysis promotes neutrophil to influx to the infectious site via
CXCR2 in sepsis. Shock. 53:114–123. 2020. View Article : Google Scholar
|
|
17
|
Hubbard WJ, Choudhry M, Schwacha MG, Kerby
JD, Rue LW III, Bland KI and Chaudry IH: Cecal ligation and
puncture. Shock. 24(Suppl 1): S52–S57. 2005. View Article : Google Scholar
|
|
18
|
Zheng Z, Ma H, Zhang X, Tu F, Wang X, Ha
T, Fan M, Liu L, Xu J, Yu K, et al: Enhanced glycolytic metabolism
contributes to cardiac dysfunction in polymicrobial sepsis. J
Infect Dis. 215:1396–1406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao W, Zhang L, Chen R, Lu H, Sui M, Zhu
Y and Zeng L: SIRT3 protects against acute kidney injury via
AMPK/mTOR-regulated autophagy. Front Physiol. 9:15262018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ganeshan K and Chawla A: Metabolic
regulation of immune responses. Annu Rev Immunol. 32:609–634. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Palsson-McDermott EM and O'Neill LA: The
Warburg effect then and now: From cancer to inflammatory diseases.
Bioessays. 35:965–973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bar-Or D, Carrick M, Tanner A II, Lieser
MJ, Rael LT and Brody E: Overcoming the Warburg effect: Is it the
key to survival in sepsis? J Crit Care. 43:197–201. 2018.
View Article : Google Scholar
|
|
25
|
Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng
L, Sun X, Yang M, Billiar TR, Wang H, et al: PKM2-dependent
glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat
Commun. 7:132802016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rowe I, Chiaravalli M, Mannella V, Ulisse
V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, et al:
Defective glucose metabolism in polycystic kidney disease
identifies a new therapeutic strategy. Nat Med. 19:488–493. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiaravalli M, Rowe I, Mannella V, Quilici
G, Canu T, Bianchi V, Gurgone A, Antunes S, D'Adamo P, Esposito A,
et al: 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc
Nephrol. 27:1958–1969. 2016. View Article : Google Scholar :
|
|
28
|
Riwanto M, Kapoor S, Rodriguez D,
Edenhofer I, Segerer S and Wuthrich RP: Inhibition of aerobic
glycolysis attenuates disease progression in polycystic kidney
disease. PLoS One. 11:e01466542016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huber TB, Edelstein CL, Hartleben B, Inoki
K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, et
al: Emerging role of autophagy in kidney function, diseases and
aging. Autophagy. 8:1009–1031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kume S, Thomas MC and Koya D: Nutrient
sensing, autophagy, and diabetic nephropathy. Diabetes. 61:23–29.
2012. View Article : Google Scholar :
|
|
33
|
Belibi F, Zafar I, Ravichandran K, Segvic
AB, Jani A, Ljubanovic DG and Edelstein CL: Hypoxia-inducible
factor-1α (HIF-1α) and autophagy in polycystic kidney disease
(PKD). Am J Physiol Renal Physiol. 300:F1235–F1243. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Wang L, Meng L, Cao G and Wu Y:
Sirtuin 6 overexpression relieves sepsis-induced acute kidney
injury by promoting autophagy. Cell Cycle. 18:425–436. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dey P, Kundu A, Sachan R, Park JH, Ahn MY,
Yoon K, Lee J, Kim ND, Kim IS, Lee BM and Kim HS: PKM2 knockdown
induces autophagic cell death via AKT/mTOR pathway in human
prostate cancer cells. Cell Physiol Biochem. 52:1535–1552.
2019.PubMed/NCBI
|
|
36
|
Li S, Dou X, Ning H, Song Q, Wei W, Zhang
X, Shen C, Li J, Sun C and Song Z: Sirtuin 3 acts as a negative
regulator of autophagy dictating hepatocyte susceptibility to
lipotoxicity. Hepatology. 66:936–952. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi T, Fan GQ and Xiao SD: SIRT3 reduces
lipid accumulation via AMPK activation in human hepatic cells. J
Dig Dis. 11:55–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li W, Tanikawa T, Kryczek I, Xia H, Li G,
Wu K, Wei S, Zhao L, Vatan L, Wen B, et al: Aerobic glycolysis
controls myeloid-derived suppressor cells and tumor immunity via a
specific CEBPB isoform in triple-negative breast cancer. Cell
Metab. 28:87–103.e106. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin K, Ma Y, Manrique-Caballero CL, Li H,
Emlet DR, Li S, Baty CJ, Wen X, Kim-Campbell N, Frank A, et al:
Activation of AMP-activated protein kinase during
sepsis/inflammation improves survival by preserving cellular
metabolic fitness. FASEB J. 34:7036–7057. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nolt B, Tu F, Wang X, Ha T, Winter R,
Williams DL and Li C: Lactate and immunosuppression in sepsis.
Shock. 49:120–125. 2018. View Article : Google Scholar :
|
|
41
|
Im JH and Kang KW, Kim SY, Kim YG, An YJ,
Park S, Jeong BH, Choi SY, Lee JS and Kang KW: GPR119 agonist
enhances gefitinib responsiveness through lactate-mediated
inhibition of autophagy. J Exp Clin Cancer Res. 37:2952018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu T, Su H, Ganapathy S and Yuan ZM:
Modulation of autophagic activity by extracellular pH. Autophagy.
7:1316–1322. 2011. View Article : Google Scholar : PubMed/NCBI
|