Introduction
Head and neck squamous cell carcinoma (HNSCC)
originates from the mucosal surfaces of the oral cavity,
oropharynx, larynx, and hypopharynx, accounting for more than 90%
of the cancers of the head and neck (1). As the sixth most common cancer
worldwide, HNSCC is highly aggressive and characterized by complex
genetic alterations, and current treatment options for HNSCC
consist of surgical interventions, radiotherapy, and chemotherapy
(2,3). Although a number of advances have
been made in these modalities, the recurrence rate remains high
owing to the development of chemotherapy resistance, resulting in a
low overall patient survival rate (4,5).
Therefore, the identification of novel chemotherapeutic agents is
urgently needed to prevent cancer recurrence and delay cancer
progression.
Compounds derived from plants have significantly
contributed to the development of novel anticancer therapeutics.
Nelumbo nucifera (common name: Lotus) is widely used in
Indian and Chinese medicine for cardiovascular (6) and pulmonary (7) diseases as well as nervous
system-related disorders (8). In
addition, neferine, the major bisbenzylisoquinoline alkaloid
isolated from the seed embryo of the lotus, has recently been
revealed to exert antitumor effects through various pathways. For
instance, neferine reportedly inhibited the proliferation and
growth of prostate cancer cell (9), osteosarcoma (10), hepatocellular carcinoma (11), and lung cancer cells (12). Neferine also exhibited the
ability to suppress the migration of gastrointestinal stromal tumor
cells (13) and glioma cells
(14). Furthermore, recent
research suggests that neferine induces autophagy (15) and inhibits ovarian cancer cell
angiogenesis (16). However, the
effects of neferine on HNSCC have not yet been elucidated.
Macroautophagy is a major intracellular catabolic
mechanism that directs the degradation of cytoplasmic components
and organelles in the lysosome (17) that can have cytoprotective or
cytopathic roles in response to various stresses, including
therapeutic stress. Autophagic flux denotes the complete process of
autophagy, including autophagosome biogenesis, maturation, fusion
with lysosomes, and the breakdown of autophagic substrates inside
the lysosome (18). Indeed,
autophagic flux induction or inhibition using natural compounds has
shown promise in the treatment of diseases such as cancer (19). Additionally, apoptosis, another
critical catabolic pathway essential for the cellular response to
toxic agents, participates in extensive crosstalk with autophagy
(20), rendering both pathways
crucial for the development of effective cancer therapeutics.
Therefore, in the present study, HNSCC cell lines
and a xenograft mouse model were used to investigate the antitumor
mechanism of neferine between autophagy and apoptosis, with the aim
of presenting a promising alternative therapeutic agent for
HNSCC.
Materials and methods
Cell lines and culture
The HNSCC cell lines used in the present study were
HN6 (tongue squamous cell carcinoma; provided by the Shanghai Ninth
People's Hospital, Shanghai, China), HN30 (pharyngeal squamous cell
carcinoma; provided by the University of Maryland School of
Dentistry, Maryland, USA) and Cal27 (tongue squamous cell
carcinoma; purchased from ATCC). The control cell line was the
human immortalized oral epithelial cell line (HIOEC), which was
established by the Shanghai Ninth People's Hospital (21). All cell lines were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). Cells were cultured in a humidified atmosphere containing 5%
CO2 at 37°C.
Cell proliferation assay
Cell viability was determined using the Cell
Counting Kit-8 (CCK-8) assay kit (Dojindo Molecular Technologies,
inc.). Briefly, HN6, HN30 and CAL27 cells were seeded at a density
of 5×103 cells/well in a 96-well plate, with 200
µl DMEM medium (10% FBS). After incubation with indicated
concentrations of neferine (0, 7.5, 15, 22.5 and 30 µM;
MCE), 10 µl CCK-8 reagent was added to each well, and the
absorbance was measured at 450 nm after 2 h of incubation at 37°C
in the incubator. All experiments were repeated in triplicate.
Clonogenic assay
For the clonogenic assay, HN6, HN30 and CAL27
single-cell suspensions were prepared. A total of 500 cells were
seeded/well in 6-well plates at 37°C overnight. The cells were
treated with various concentrations (0, 1, 2, 5 and 10 µM)
of neferine for 2 weeks. Cell colonies were fixed with pure
methanol for 15 min and stained with 0.1% crystal violet (Beyotime
Biotechnology) for 30 min at room temperature. The images were
captured and scored with CanoScan 5600F (Canon, Inc.). Colonies of
>50 cells were counted to determine the surviving fraction. All
experiments were repeated in triplicate.
Cell cycle analysis
Single-cell suspensions were prepared, and
3×105 cells were seeded per well in 6-well plates
overnight in the cell incubator. After treatment with various
concentrations of neferine (0, 5, 10, 15 and 20 µM) for 24
h, HN6, HN30 and CAL27 cells were collected and fixed in 3 ml cold
75% ethanol at -20°C overnight. After washing with 2 ml of cold
PBS, the cells were resuspended and incubated with 0.5 ml of PBS
containing 100 µg/ml RNase (Beyotime Biotechnology) and 5
µg/ml propidium iodide (Shanghai Yeasen Biotechnology Co.,
Ltd.) at room temperature for 30 min. The cell cycle distribution
was analyzed via BD FACSCalibur flow cytometer (BD Biosciences) and
ModFIT 5.0 software (Verity Software House, Inc.).
Annexin V apoptosis assay
Apoptotic cells were identified using the FITC
Annexin V Apoptosis Detection Kit (BD Biosciences). A total of
3×105 cells were seeded per well in 6-well plates
overnight in the cell incubator. After treatment with the indicated
concentrations (0, 5, 10, 15 and 20 µM) of neferine for 48
h, the cells were resuspended in binding buffer. FITC Annexin V (5
µl) and PI (5 µl) were then added and were incubated
at room temperature for 15 min in the dark. Apoptotic cells were
analyzed via BD FACSCalibur flow cytometer and FlowJo (V10)
software (BD Biosciences).
Reactive oxygen species (ROS) assay
Intracellular ROS levels were determined using the
DCFH-DA fluorescent probe (Beyotime Biotechnology). Briefly,
3×105 cells were seeded per cell in 6-well plates
overnight in the cell incubator. After treatment with the indicated
concentrations of neferine (0, 5, 10 and 20 µM) for 12 h,
HN6, HN30 and CAL27 cells were incubated with DCFH-DA for 30 min in
the cell incubator. Dichlorofluorescein (DCF) fluorescence was then
detected via BD FACSCalibur flow cytometer (FITC-channel).
Fluorescence microscopy (magnification, ×100; Carl Zeiss AG) was
used to capture images of the cells.
Cell migration assay
The cell migration assay was performed using the
Transwell system (24-wells, 8-µm pore size with
polycarbonate membrane; Corning Costar; Corning, Inc.). HN30 and
CAL27 cells were suspended in serum-free medium
(5.0×105/ml), and 100 µl was added to the upper
chamber, whereas 600 µl of complete medium (10% FBS) was
added to the lower chamber. After 4 h, the media in the upper
chamber was replaced with media containing various concentrations
of neferine (0, 5, 10 and 20 µM, serum-free). After 36 h,
the chambers were fixed with 4% paraformaldehyde for 15 min and
stained with 0.1% crystal violet for 30 min at room temperature.
The non-migrated cells were wiped from the upper surface of the
chamber using cotton swabs. The successfully migrated cells were
counted in three random fields using a light microscope
(magnification, ×40; Olympus Corporation).
Scratch assay
Scratch assays were applied to determine cell
mobility. First, 1×106 HN6, HN30 and CAL27 cells were
seeded per well in a 6-well plate in complete medium overnight in
the cell incubator to obtain a fully confluent monolayer. After 12
h of starvation with DMEM (serum-free), a 20-µl pipette tip
was used to make a straight cell-free 'scratch' in each well. The
cells were washed with PBS, and serum-free medium was added with
various concentrations of neferine (0, 5, 10, 15 and 20 µM).
Finally, the migration of the cells was captured using a phase
contrast microscope (magnification, ×100; Olympus Corporation).
Images of cells were obtained at the matching reference points
initially and then at 12-h intervals. The wound closure of the
scratch was analyzed quantitatively.
Western blot analysis
Briefly, HN6, HN30 and CAL27 cells were collected
after specific treatments and lysed with RIPA buffer (Beyotime
Biotechnology). The protein concentration was measured using the
Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). A
total of 20 µg protein in each sample was run on 10%
SDS-PAGE gels and then electro-transferred onto a PVDF membrane
(EMD Millipore). The membrane was blocked with 5% skimmed milk for
1 h at room temperature and incubated overnight with primary
antibodies (1:1,000) at 4°C. The membrane was washed and incubated
with HRP-conjugated secondary antibodies [cat. nos. 70-GAR0072 and
70-GAM0072; 1:5,000; Multi Sciences (LIANKE) Biotech, Co., Ltd.]
for 1 h at the room temperature. Protein bands were then detected
using a chemiluminescence system (Immobilion Western
Chemiluminescent HRP Substrate; EMD Millipore). All bands were
quantified using ImageJ V1.8. β-Actin was used as an internal
control. The primary antibodies were as follows: Anti-cleaved
caspase-3 (product no. 9664; Cell Signaling Technology, Inc.; and
cat. no. sc-7272; Santa Cruz Biotechnology, Inc.), anti-cleaved
caspase-9 (cat. no. 10380-1-AP), anti-cleaved PARP1 (cat. no.
13371-1-AP), anti-Beclin-1 (cat. no. 11306-1-AP) and anti-p62 (cat.
no. 18420-1-AP) (all from ProteinTech Group, Inc.), anti-BAX (cat.
no. AF0054) and anti-β-Actin (cat. no. AF0003) (both from Beyotime
Biotechnology); anti-Bcl-2 (cat. no. sc-7382; Santa Cruz
Biotechnology, Inc.), and anti-LC3-I/II (product code 128025; Abcam
Inc.).
Transmission electron microscopy (TEM)
assay
After neferine treatment (0 and 20 µM) for 24
h in the cell incubator, HN30 and CAL27 cells were fixed with 2.5%
glutaraldehyde for 4 h and 1% OsO4 (Solarbio Life
Science) for 1 h at 4°C, dehydrated with graded ethanol, embedded
in propylene oxide, sectioned at 70 nm and stained with 2% uranyl
acetate (Sigma-Aldrich; Merck KGaA) for 20 min at room temperature.
The autophagosomes were visualized using electron microscopy, as
previously described (22,23).
Fluorescence imaging
HN30 and CAL27 cells were seeded in 6-well plates
overnight in the cell incubator and then transfected with
GFP-RFP-LC3 adenovirus (108 pfu/ml MOI=10; Beyotime
Biotechnology). After 24 h in the cell incubator, the medium was
removed and replaced with the indicated treatments [neferine 10
µM, 24 h; EBSS (cat. no. E2888; Sigma Aldrich; Merck KGaA),
6 h; chloroquine (MCE) 10 µM, 24 h]. Autophagic flux was
measured via fluorescence microscopy (magnification ×400; Carl
Zeiss AG).
RNA interference
The sequence for the small interfering (si)RNA
against p62 was 5′-GCA TTG AAG TTG ATA TCG AT-3′ (24). The non-targeting siRNA control
was obtained from Dharmacon, Inc. CAL27 cells (3×105)
were seeded per well in a 6-well plate and transfected with siRNA
(75 pmol) using Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 12 h in the cell incubator. After 72 h, the
transfected cells were used for the neferine experiments.
Xenograft mouse model
The animal experiments were approved by the Ethics
Review Board at the Shanghai Ninth People's Hospital (Shanghai,
China). CAL27 cells (2×106) suspended in 20% Matrigel
(BD Biosciences) were subcutaneously injected into the dorsa of
five-week-old male BALB/c nude mice (Vital River, Inc.; Charles
River Laboratories, Inc.) in routine living and feeding conditions
(temperature 24±2°C, humidity 40-60%, 12-h light/dark cycle,
sterile feed and filtered water ad libitum). After 1 week,
ten mice (tumors had formed) were randomly divided into two groups
(5 mice per group). The mice were administered neferine (10 mg/kg)
or PBS (control) intraperitoneally. The tumor volume was measured
every 4 days (V=LengthxWidth2×0.5). The humane endpoints
were as follows: i) maximum tumor volume 1,000 mm3; ii)
tumor ulceration or necrosis. The mice were anesthetized with
diethyl ether, then euthanized by cervical dislocation. A
combination of criteria in confirming death included lack of pulse,
breathing, corneal reflex, and response to firm toe pinch.
Immunohistochemical staining
Tissues were fixed with 4% paraformaldehyde for 24 h
at 4°C and dehydrated and embedded in paraffin. The paraffin
sections (4 µm) were deparaffinized, rehydrated and
subjected to antigen reparation (microwave oven, 6 min × 4 times),
endogenous peroxidase inactivation (0.3%
H2O2, 15 min), and nonspecific antigen
blocking (FBS; Gibco; Thermo Fisher Scientific, Inc.) for 15 min,
at room temperature. The slides were then incubated with primary
antibodies overnight (4°C) and, subsequently, with a secondary
antibody for 15 min (at room temperature). Staining was detected
using a diaminobenzidine (DAB) reagent (Zhongshan Jinqiao;
ZSGB-BIO, Inc.) for 1 min at room temperature. The intensity and
proportion of staining were scored by two pathologists with an
Olympus light microscope (magnification, ×200). The primary
antibodies were as follows: Anti-cleaved caspase-3 (product no.
9664; 1:1,000; Cell Signaling Technology, Inc.); anti-cleaved PARP1
(cat. no. 13371-1-AP; 1:200), anti-Ki67 (cat. no. 10205-2-AP;
1:500), anti-LC3 (cat. no. 14600-1-AP; 1:400) and anti-p62 (cat.
no. 18420-1-AP; 1:50) (all from ProteinTech Group, Inc.). The
secondary antibody was as follows: HRP-conjugated [cat. no.
70-GAR0072; 1:1,000; Multi Sciences(LIANKE) Biotech, Co.,
Ltd.].
Statistical analysis
Statistical comparisons between two groups were
performed using non-paired Student's t-test. Statistical
comparisons between multiple groups were performed by Tukey's post
hoc test with one-way ANOVA. SPSS version 18.0 software (SPSS,
Inc.) was used for statistical analysis. For each of the three
independent experiments, the data are presented as the mean ± SEM.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Neferine inhibits the proliferation and
viability of HNSCC cells through G1 arrest
Three human HNSCC cell lines (HN6, HN30, and CAL27)
were used to detect the inhibitory effect of neferine on cell
proliferation via CCK-8 assays. The cell viabilities of HN6, HN30,
and CAL27 were significantly decreased in a concentration- and
time-dependent manner following treatment with neferine (Fig. 1A). The IC50 values at
72 h were 13.13, 21.74, and 22.54 µM in HN6, HN30, and CAL27
cells, respectively. Similarly, the colony forming capacity was
inhibited by neferine, in a concentration-dependent manner
(Fig. 1B). Hence, the cell cycle
distribution was examined via flow cytometry, which revealed that
neferine induced an obvious G1 cell cycle arrest in three cell
lines. In fact, the percentage of cells in the G1 phase increased
from 44.86% (0 µM) to 63.78% (20 µM), whereas that in
the S phase decreased from 41.42% (0 µM) to 22.14% (20
µM) in the HN6 cells. Similar results were observed in the
other two cell lines (Figs. 1C
and S1A). Collectively, these
results indicated that neferine exerted its inhibitory effect by
inducing cell cycle arrest in HNSCC cells.
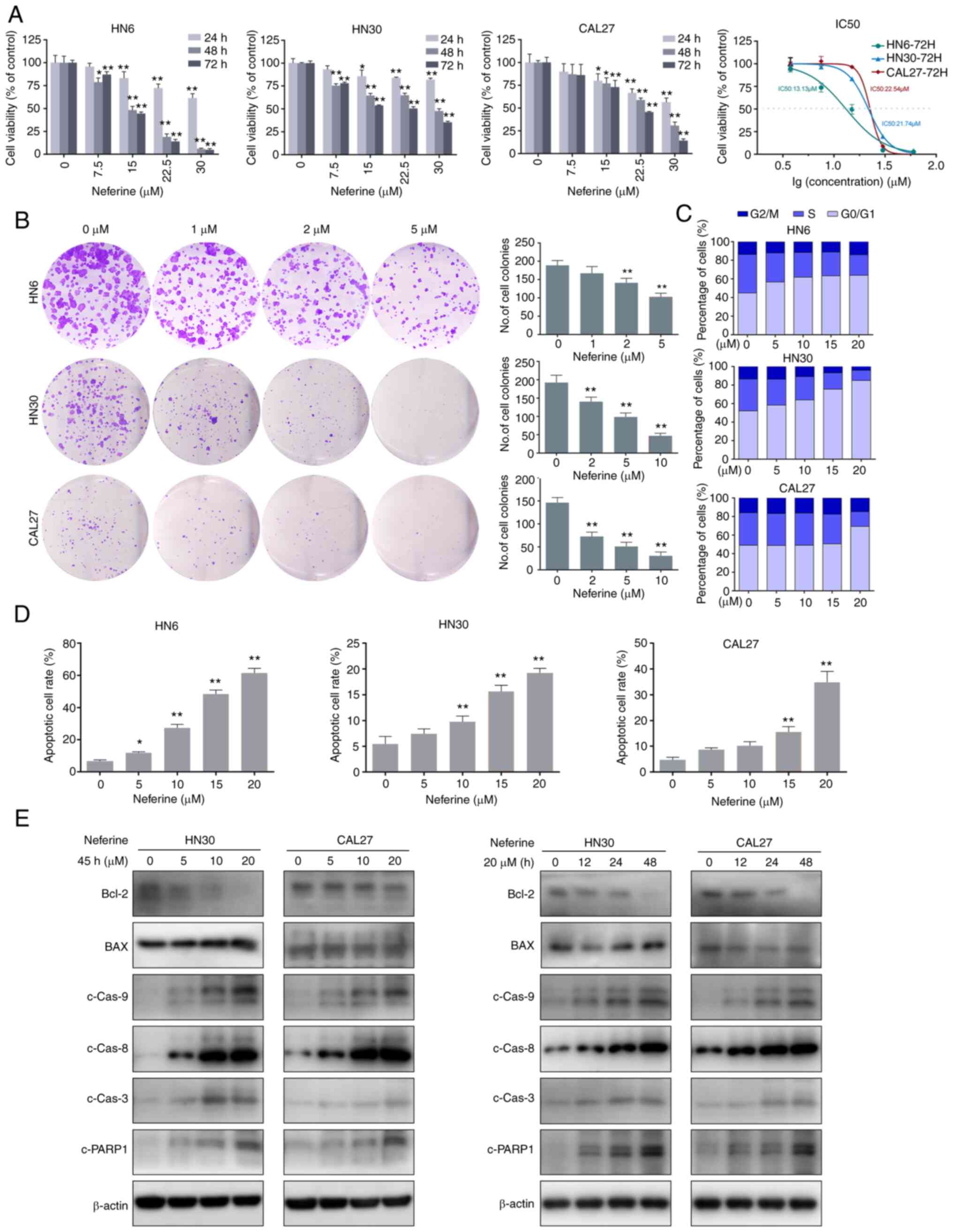 | Figure 1Neferine reduces cell viability and
induces apoptosis in HNSCC cells. (A) Histograms present the cell
viabilities of HN6, HN30, and CAL27 cells following treatment with
neferine. Line chart depicts the IC50 values at 72 h in
HN6, HN30, and CAL27 cells. (B) Plate colony formation assay
showing the inhibitory effect of neferine on HNSCC cells with the
indicated concentrations. (C) Cell cycle distribution of the cells
treated with various concentrations of neferine for 24 h, as
examined via flow cytometry. (D) Flow cytometric analysis of early
and late apoptosis in the HN6, HN30, and CAL37 cells treated with
neferine. (E) Apoptosis-related protein expression in the HN30 and
CAL37 cells treated with neferine confirmed by western blot
analysis. Left panel, HN30 and CAL37 cells treated with the
indicated concentrations of neferine for 48 h. Right panel, HN30
and CAL37 cells treated with neferine (20 µM) for the
indicated durations of time. *P<0.05 and
**P<0.01. HNSCC, head and neck squamous cell
carcinoma. |
To investigate the effects of neferine on HNSCC cell
motility, a scratch healing assay and Transwell assay were
performed. Neferine suppressed the healing and migration of HNSCC
cells in a concentration-dependent manner (Fig. S1B and C). Overall, these results
indicated that neferine exhibited potential anticancer
properties.
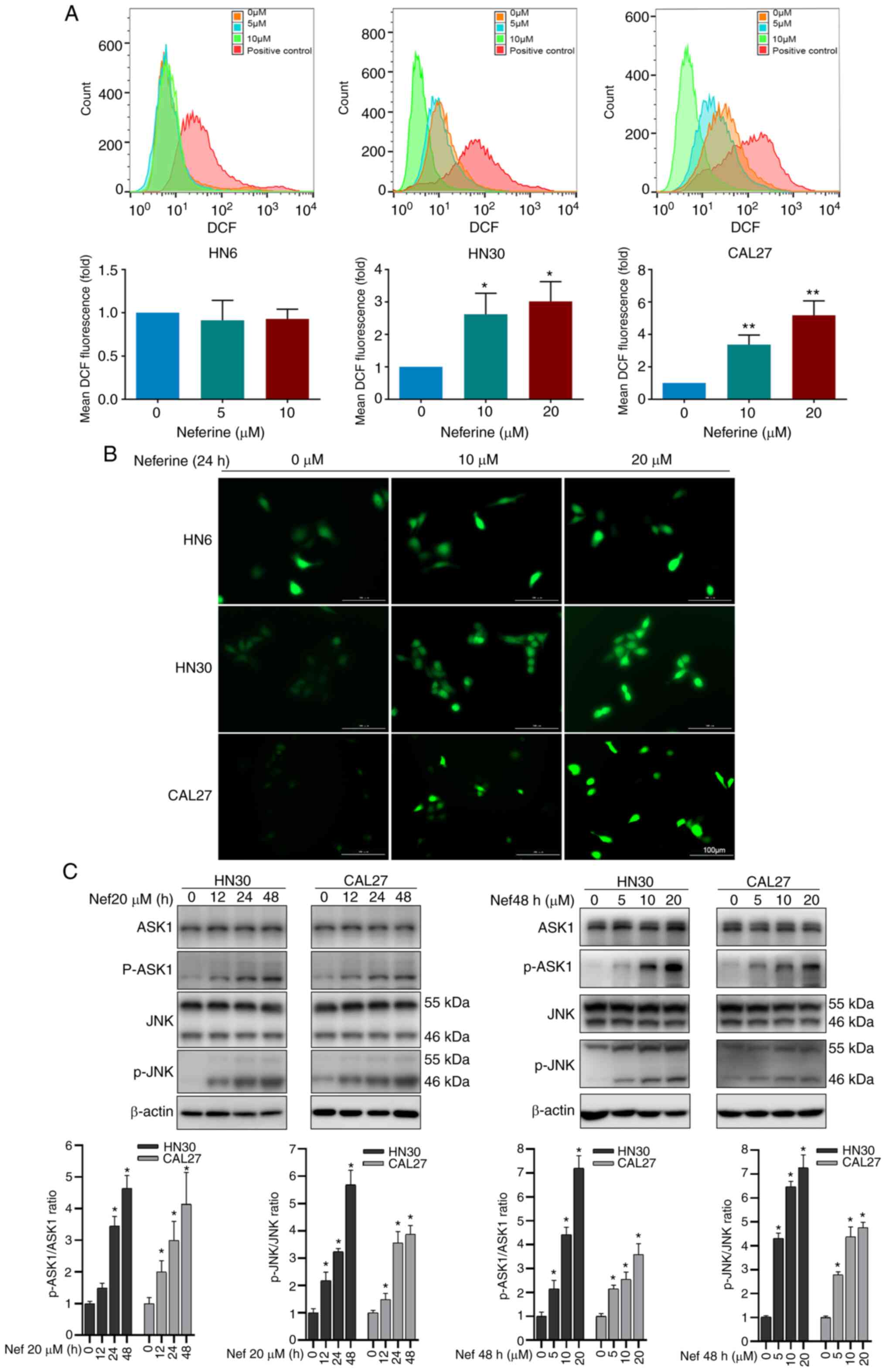
Neferine induces ROS and activates the
ASK1/JNK pathway to promote apoptosis in HNSCC cells
To investigate the underlying mechanisms of
neferine-induced cytotoxicity in HNSCC cells, the number of
apoptotic HN6, HN30, and CAL27 cells was quantified via flow
cytometry using FITC Annexin V/PI staining in the presence or
absence of neferine. The results revealed that neferine increased
apoptosis from 7.51% (0 µM) to 11.81% (5 µM), 27.90%
(10 µM), 49.10% (15 µM), and 64.70% (20 µM) in
HN6 cells (Fig. 1D). Similar
trends were observed in CAL27 and HN30 cells (Figs. 1D and S2A). In addition, neferine decreased
the Bcl-2/BAX ratio and triggered the cleavage of caspase-8,
caspase-9, caspase-3, and PARP-1 in a concentration- and
time-dependent manner (Figs. 1E
and S2B). Collectively, these
results indicated that neferine effectively induced apoptosis in
HNSCC cells.
ROS are a vital inducer of apoptosis (25,26). Therefore, to explore whether the
neferine-induced apoptosis was associated with ROS hypergeneration,
the ROS levels were measured via flow cytometry using the
oxidation-sensitive fluorescent probe DCF. The mean fluorescence
intensity of DCF in the neferine-treated HN30 cells increased by
2.62-fold (10 µM) and 3.01-fold (20 µM) compared to
that in the control cells (0 µM). Similar results were
observed in CAL27 cells, but not in HN6 (Fig. 2A). The fluorescence microscopy
images also confirmed an increase in ROS (Fig. 2B). Moreover, considering that ROS
reportedly activates the ASK1/JNK signaling pathway to induce
mitochondrial apoptosis via the caspase family and
Beclin-1-dependent autophagy (27,28), the expression of ASK1/JNK protein
in HN30 and CAL27 cells treated with neferine was also detected. It
was revealed that neferine upregulated the phosphorylation of
ASK1/JNK in a concentration- and time-dependent manner; however, it
did not alter the overall ASK1/JNK expression (Fig. 2C). These results indicated that
neferine induced ROS generation, which activated the ASK1/JNK
pathway to induce apoptosis in HNSCC cells.
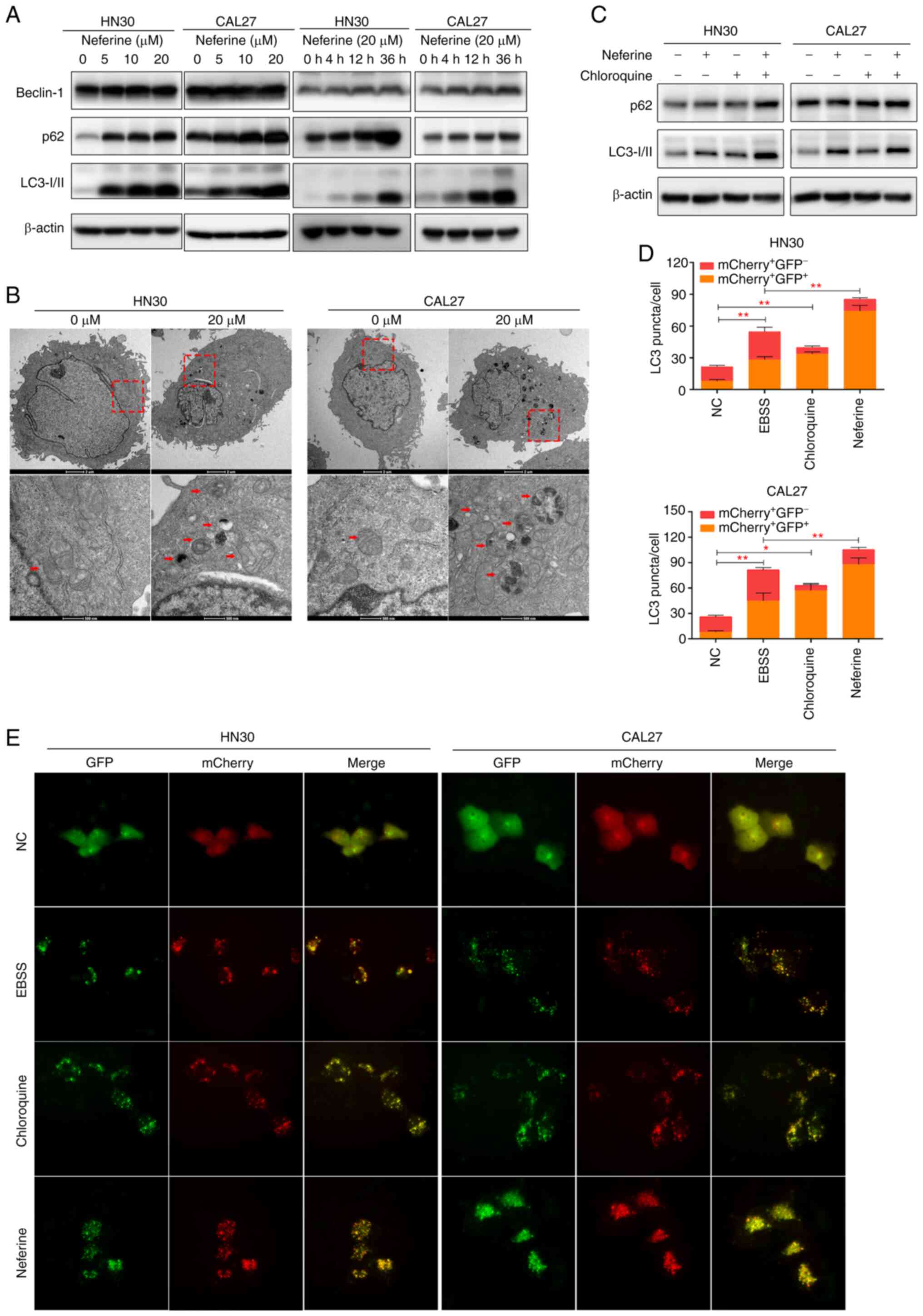
Neferine promotes the generation of
autophagosomes while inhibiting autophagy influx
A previous study revealed that autophagy can be
stimulated by ROS (26).
Considering the indistinct interaction between apoptosis and
autophagy, the effect of neferine on autophagy in the HNSCC cell
lines was also explored. Specifically, Beclin-1 expression was
analyzed owing to its important role in the initial steps of
canonical autophagy (29). The
results revealed that Beclin-1 was increased in the
neferine-treated HN30 and CAL27 cells in a concentration- and
time-dependent manner (Figs. 3A
and S3A). Moreover, LC3 and p62
represent universal markers for autophagy. Specifically, the
conversion of LC3-I to LC3-II increases as autophagosomes mature,
whereas p62 is degraded in the autolysosome, causing its abundance
to decrease with activated autophagic flux (29). Our results revealed the increased
conversion of LC3-I to the LC3-II isoform in the neferine-treated
HN30 and CAL27 cells (Figs. 3A
and S3A), indicating the
maturation of the autophagosomes. However, the p62 protein level
also increased. TEM revealed that treatment with neferine increased
the number of vacuoles containing degradative cytoplasmic materials
in the HN30 and CAL27 cells (Fig.
3B).
To further explore the influence of neferine on
autophagy, the autophagic flux was directly monitored using an
autophagosome-lysosome fusion inhibitor, chloroquine (29). First, the LC3-II levels in HN30
and CAL27 cells during neferine treatment, with or without
chloroquine were examined. Co-treatment increased the LC3-II levels
more significantly than chloroquine alone (Figs. 3C and S3B), indicating that neferine promoted
autophagosome biosynthesis. However, p62 abundance was not further
increased in the presence of chloroquine, suggesting that the
neferine-induced upregulation of p62 may be caused via the
inhibition of autophagic degradation (Figs. 3C and S3B). To further verify these results,
the autophagic flux was also examined by transfecting
double-labeled fluorescent LC3 adenovirus (mCherry-GFP-LC3)
(30,31) into HN30 and CAL27 cells.
Colocalization (yellow) of both GFP (green) and mCherry (red)
fluorescence implied the generation of autophagosomes, whereas red
puncta (mCherry+ GFP−) indicated the
generation of autolysosomes. The treatment of neferine (5 µM
and 12 h) was not presented, because the autophagosomes
(fluorescent puncta) were little or blurry. The treatment of
neferine (10 µM and 24 h) was an appropriate condition,
since autophagosomes were obvious for counting and to perform the
statistical analysis. As revealed in Fig. 3D and E, the yellow puncta were
increased without an accompanying increase in the red puncta,
indicating that the autophagic flux was blocked. These results
indicated that neferine induced autophagosome biosynthesis while
inhibiting autophagic flux at the final degradation step in HNSCC
cell lines.
Neferine-induced apoptosis is partially
mediated by autophagy influx suppression in HNSCC cells
To explore the functional relationship between
neferine-induced autophagy and apoptosis, HN30 and CAL27 cells were
treated with neferine (20 µM) in the presence or absence of
chloroquine (10 µM) and apoptosis analysis was performed.
Treatment with chloroquine alone had no obvious effect on cell
apoptosis or viability in both cell lines (Fig. 4A and B). However, chloroquine
further amplified neferine-induced cell cytotoxicity (Fig. 4A) and moderately intensified
neferine-induced apoptosis, while inducing further activation of
caspase-8, caspase-3, and PARP1 (Figs. 4B and C, and S4A and B). These results indicated
that the inhibition of autophagic flux by chloroquine promoted the
neferine-induced apoptosis of HN30 and CAL27 cells.
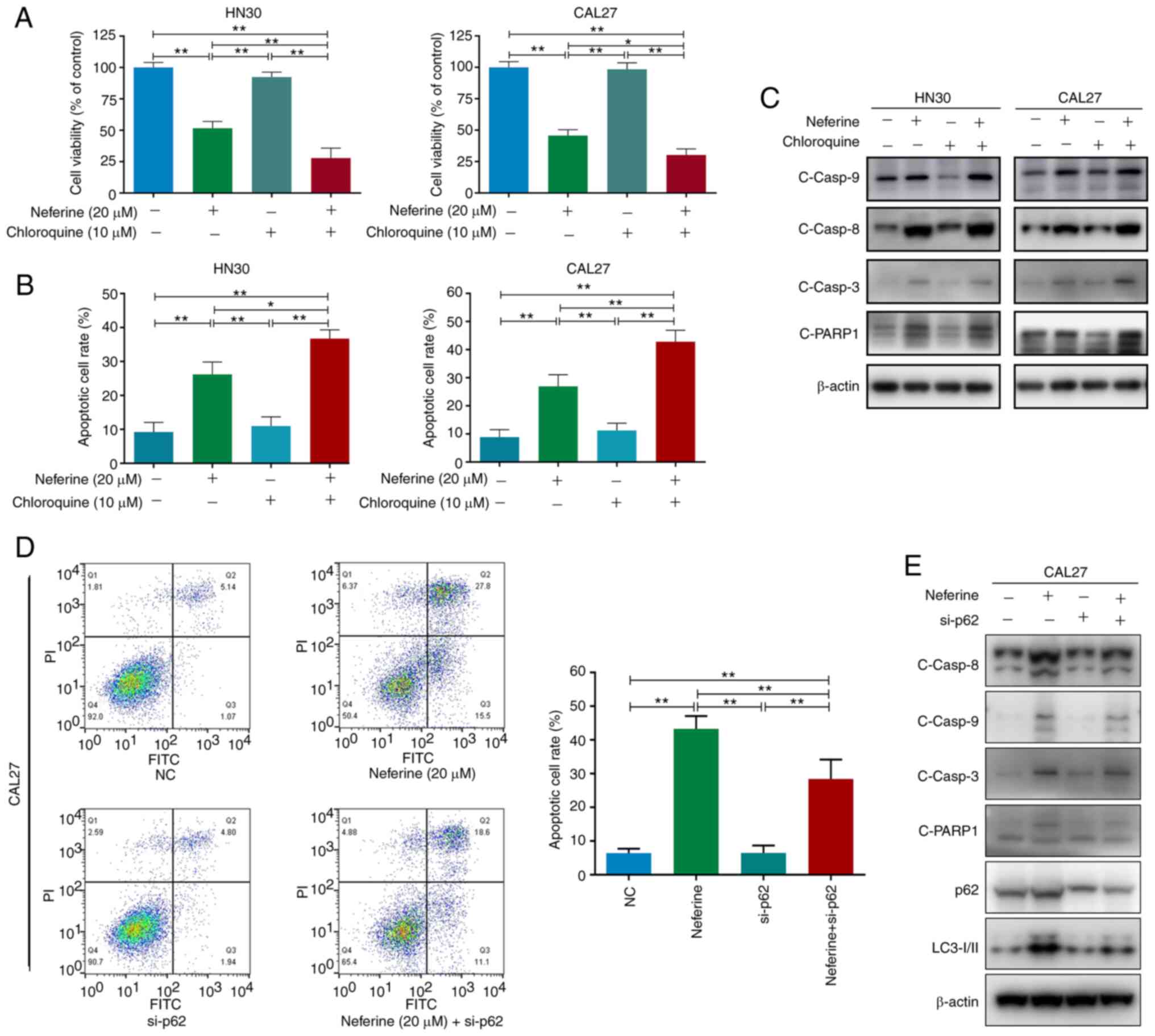 | Figure 4Neferine-induced apoptosis is
partially mediated via the accumulation of p62 due to
neferine-induced autophagic influx inhibition in head and neck
squamous cell carcinoma cells. (A) Cell viability, (B) apoptotic
cell rate, and (C) western blot analysis of cleaved caspase-8,
caspase-9, caspase-3, and PARP1 in HN30 and CAL27 cells treated
with neferine (20 µM) in the presence or absence of
chloroquine (10 µM) for 48 h. (D) Flow cytometric analysis
of early and late apoptosis and (E) western blot analysis of
cleaved caspase-8, caspase-9, caspase-3, PARP1, p62, and LC3 in
neferine-treated HN30 and CAL37 cells with or without p62
knockdown. *P<0.05 and **P<0.01. NC,
negative control; si-, small interfering. |
Since neferine was revealed to directly inhibit
autophagic flux, it could be concluded that the neferine-induced
apoptosis was partially enhanced by neferine-induced autophagic
influx inhibition. In addition, considering that the expression of
cleaved caspase-9 showed no noticeable difference between the
neferine group and the neferine + chloroquine group (Figs. 4C and S4B), the further pro-apoptotic effects
may be executed by caspase-8, but not caspase-9.
Accumulation of p62 due to
neferine-induced autophagy influx inhibition promotes apoptosis in
HNSCC cells
Previous studies have revealed that p62 acts as a
'bridge' between apoptosis and autophagy (32,33). Moreover, it was observed that the
neferine-induced autophagic flux inhibition caused an increase in
the p62 level (Fig. 3). Hence,
to investigate the role of p62 in the neferine-induced crosstalk
between apoptosis and autophagy, the expression of p62 was
downregulated in CAL27 cells, the CAL27 cells were treated with
neferine, and subsequently the level of apoptosis was quantified.
Flow cytometric analysis revealed that p62 knockdown alleviated the
neferine-induced apoptosis (from 43.3 to 29.7%) in CAL27 cells
(Fig. 4D). Moreover, the
cleavage of specific apoptosis-related proteins, caspase-8,
caspase-3, and PARP1, was decreased; however, the same effect was
not observed for cleaved caspase-9 (Figs. 4E and S4C). These results confirmed that the
neferine-induced accumulation of p62 activated caspase-8 to promote
apoptosis in HNSCC cells.
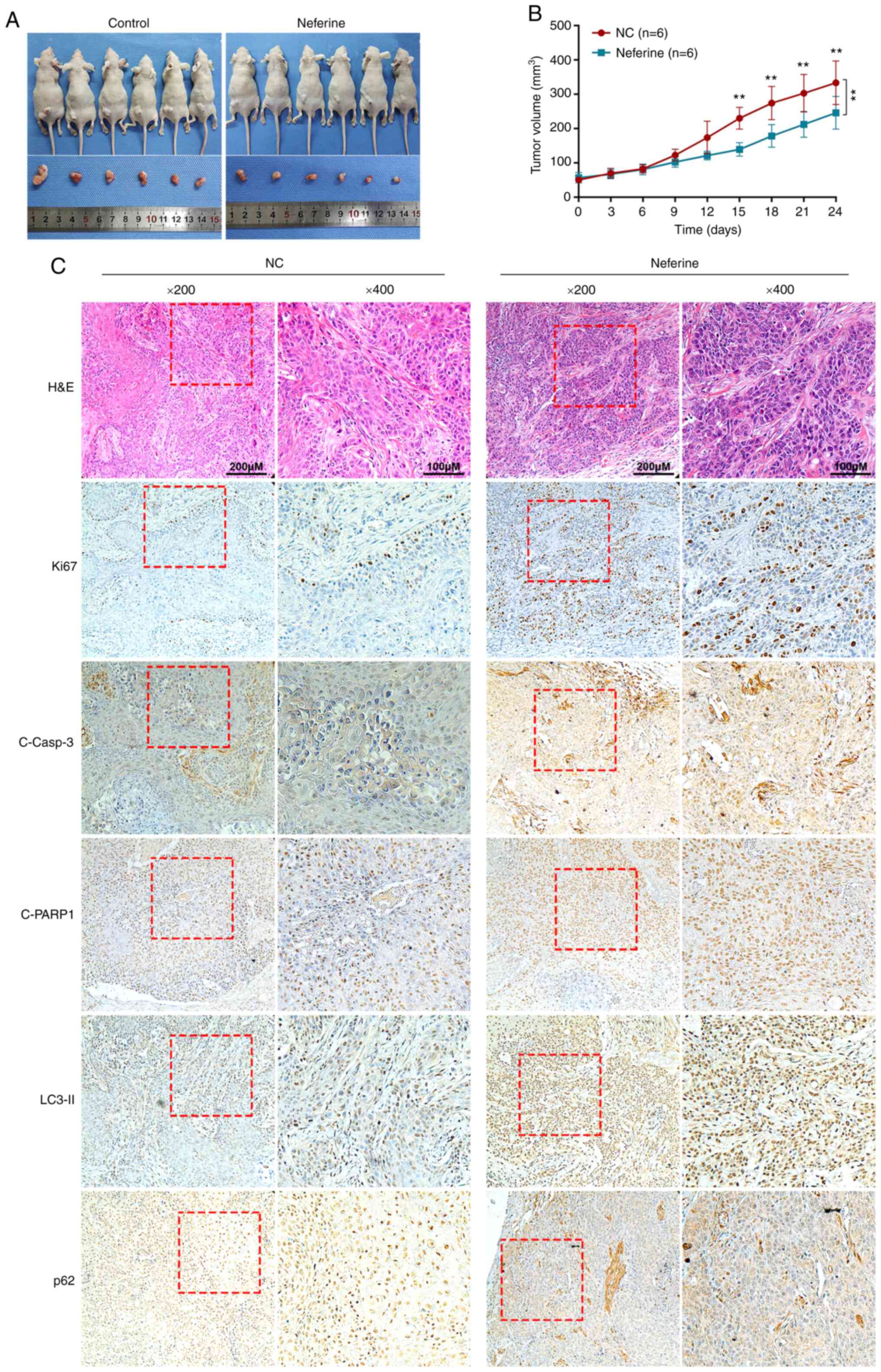
Neferine inhibits the growth of HNSCC
cells by inducing apoptosis and autophagy in vivo
Next, to examine the effects of neferine in
vivo a murine xenograft model with CAL27 cells was used. The
tumor volumes following treatment with neferine were significantly
smaller than that of the control. The growth rate of tumors treated
with neferine was also slower than that of the control (Fig. 5A and B). Furthermore, the
immunohistochemical staining of xenograft tumor tissues (Fig. 5C) revealed that treatment with
neferine decreased the expression of Ki67, a marker of cell
proliferation. In addition, neferine increased the expression of
cleaved caspase-3 and cleaved PARP1 in the nucleus, indicating that
neferine induced the apoptosis of xenograft tumors. In accordance
with the in vitro results, neferine also increased the level
of LC3-II and p62, indicating that neferine induced autophagosome
generation and inhibited autophagy influx in vivo.
Discussion
Neferine is a plant-derived reagent that induces
higher toxicity in cancer cells compared to non-transformed cells
(15). It has also been reported
to induce ovarian and hepatoma carcinoma cell apoptosis and
autophagy (16,34); however, one study reported
contradictory results indicating that neferine functions as an
autophagy inhibitor in ARPE-19 (35). In addition, neither the effects
of neferine on HNSCC nor the reciprocal interaction between
apoptosis and autophagy induced by neferine had been previously
characterized. Hence, the present study aimed to elucidate the
influence of neferine on autophagy and apoptosis in HNSCC
Numerous studies have reported the inhibitory
effects elicited by neferine on cell proliferation, growth,
migration, and angiogenesis in different tumors (10,13,16). In fact, some studies have even
focused on ROS production as well as apoptosis and autophagy
induction (15,34). However, most of these previous
studies were independent studies that did not evaluate autophagic
flux specifically. Herein, a multidimensional evaluation was
conducted and the pharmacological effect of neferine was revealed
to be undoubtedly versatile. First, it was revealed that neferine
inhibited the viability and colony formation of cancer cells while
arresting HNSCC cells in the G1/S phase. Similar results were also
reported in ovarian carcinoma via the downregulation of cyclin E1
(16). Neferine was also
revealed to suppress the healing and migration of HNSCC cells,
further highlighting its potential anticancer properties, which
were verified in an in vivo murine xenograft model.
Furthermore, it was determined that neferine induced apoptosis and
autophagy while increasing the level of ROS production and the
activation of the ASK1/JNK pathway. Indeed, ROS served as an
initiator for the activation of ASK1 (oxidative stress sensor) and
JNK. Specifically, with the activation of JNK, the mitochondrial
apoptosis induced by Bcl-2 inhibition and the canonical autophagy
induced by JNK/Beclin-1 were sequential occurrences (36,37). Hence, neferine-induced ROS likely
served as an upstream effector for apoptosis and autophagy.
Moreover, the results indicated that neferine-induced autophagosome
biogenesis was determined by the Beclin1-dependent canonical
autophagy pathway. It was also a limitation of this study that the
effect of ROS on the ASK1/JNK pathway by further interfering with
ROS was absent. The treatment of neferine increased the level of
ROS in HN30 and Cal27 cells, but not in HN6 cells. Although these 3
cell lines belong to head and neck squamous cell carcinoma, they
have different genetic backgrounds and anatomical origins. Cell
line disparity was revealed when the effect of neferine was
evaluated. In addition, this result revealed that the inhibiting
effects of neferine on HNSCC were not only through the ROS pathway,
but also other mechanisms (for example, by the autophagic flux
inhibition).
Autophagy serves as a crucial mechanism for the
degradation of harmful cellular components to mediate metabolic
adaptation and maintain energy homeostasis (38). Several studies have revealed that
autophagy exhibits a paradoxical role, having either
tumor-promoting or tumor-suppressive effects on carcinoma (39,40). Moreover, the inhibition of
autophagy is regarded as a universal target for anticancer therapy
(41). In our research, neferine
functioned as a double-edged sword for autophagy in HNSCC. It was
revealed that neferine induced autophagosome generation, while also
inhibiting autophagic flux at the autophagosome degradation step as
evidenced by the increased conversion of LC3-I to LC3-II and the
accumulation of p62.
p62/SQSTM1 is involved in the formation of
autophagosomes and is degraded by lysosomal proteases along with
autophagosomes (42). Hence, the
inhibition of autophagic flux by neferine caused the accumulation
of p62. p62 is a scaffold protein composed of five domains that
organize signal trafficking at critical points to regulate cell
death and survival (43,44). Specifically, the UBA domain of
p62 interacts with ubiquitinated proteins, thus, p62 recruits and
oligomerizes important signaling molecules (44). Moreover, p62 reportedly provides
a signal-organizing interface to recruit poly-ubiquitinated
caspase-8 and subsequently activate caspase-8 (33). The present results revealed that
neferine induced the accumulation of p62, which subsequently
enhanced neferine-induced apoptosis. This was also confirmed by
downregulation of p62, however, the absence of experiments using a
caspase-8 inhibitor is a limitation of our study. Hence,
collectively, the results indicated that p62 may function as a
bridge between the autophagy and apoptosis induced by neferine.
When treated with reagents that induce proteasome
inhibition or endoplasmic reticular stress, cells can activate
apoptosis directly through caspase-8 (45). This mechanism is strengthened by
the function of LC-3 and p62 (46,47). Therefore, a novel mechanism is
proposed for neferine in which the accumulation of p62 by
autophagic influx inhibition promotes apoptosis through the
activation of caspase-8, independent of ROS, in HNSCC cells. The
involvement of p62 in neferine-induced cell apoptosis through
caspase-8 activation broadened our understanding of the mechanism
responsible for neferine-induced cell death. Moreover, the
crosstalk between autophagy and apoptosis may have required the
participation of p62 as a conjunction point in neferine-induced
cell death. Considering that the activation of caspase-8 was
accompanied with the accumulation of p62, it is plausible that
caspase-8 may interact with p62 at the autophagosome surface during
neferine treatment.
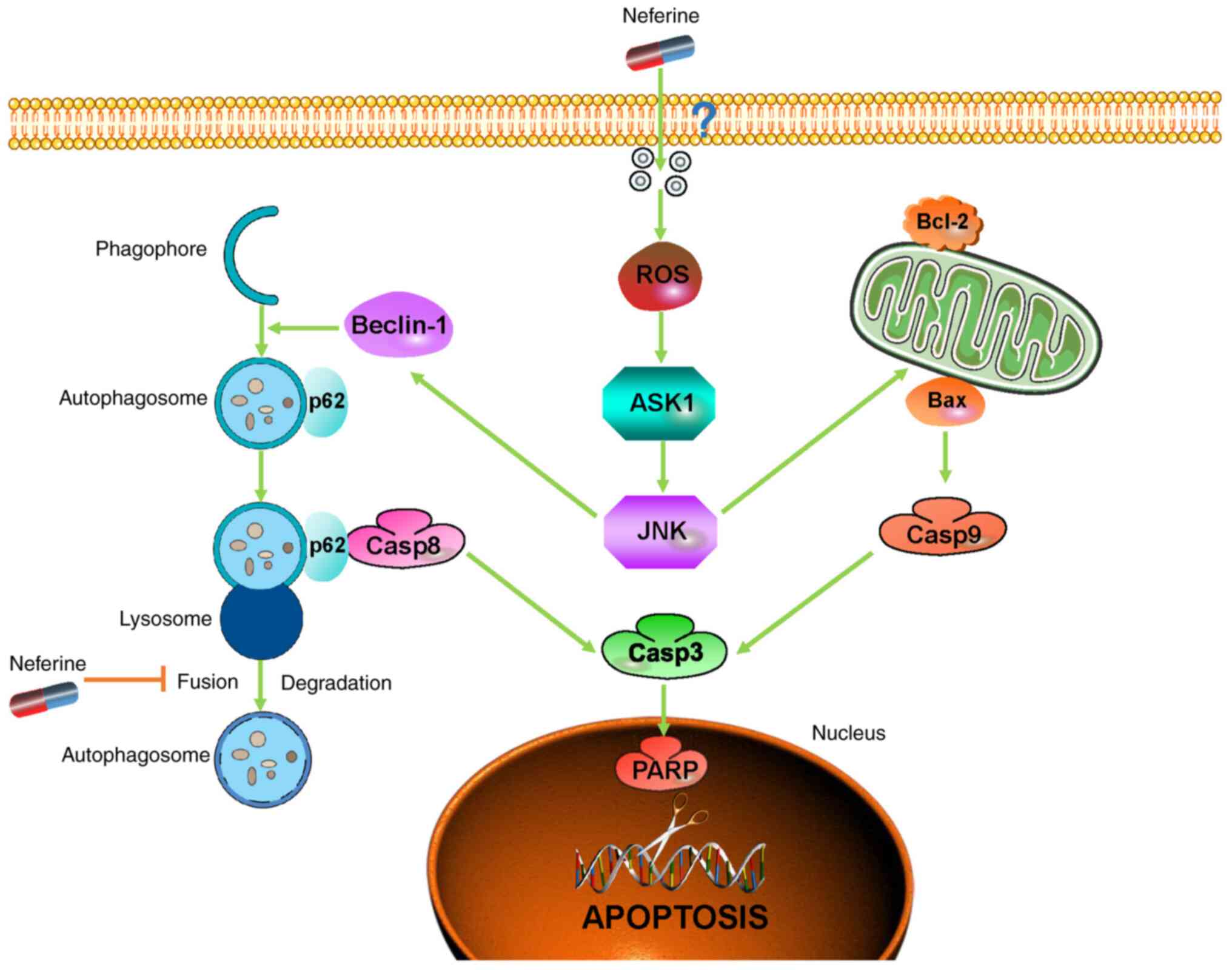
In conclusion, considering the present findings of
increased apoptosis and interrupted autophagic flux, our study
clarified the underlying pro-apoptotic mechanism of neferine in
HNSCC through the inhibition of autophagic influx mediated by p62,
thereby providing new insights into the crosstalk between apoptosis
and autophagy while highlighting neferine as a potential agent to
improve the prognosis of HNSCC patients (Fig. 6).
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FZ, XL, XT, YHa and JJ performed the research and
analyzed the results. FZ, YL, and ZL developed the methodology and
discussed the results. FZ, CM and YHe designed the research, wrote
the paper, and supervised the study. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Ethics
Review Board at the Shanghai Ninth People's Hospital (Shanghai,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 81570949).
References
|
1
|
Vigneswaran N and Williams MD:
Epidemiologic trends in head and neck cancer and aids in diagnosis.
Oral Maxillofac Surg Clin North Am. 26:123–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamano Y, Uzawa K, Saito K, Nakashima D,
Kasamatsu A, Koike H, Kouzu Y, Shinozuka K, Nakatani K, Negoro K,
et al: Identification of cisplatin-resistance related genes in head
and neck squamous cell carcinoma. Int J Cancer. 126:437–449. 2010.
View Article : Google Scholar
|
|
5
|
GBD 2015 Mortality and Causes of Death
Collaborators: Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980-2015: A systematic analysis for the Global Burden of
Disease Study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qian JQ: Cardiovascular pharmacological
effects of bisbenzylisoquinoline alkaloid derivatives. Acta
Pharmacol Sin. 23:1086–1092. 2002.PubMed/NCBI
|
|
7
|
Zhao L, Wang X, Chang Q, Xu J, Huang Y,
Guo Q, Zhang S, Wang W, Chen X and Wang J: Neferine, a
bisbenzylisoquinline alkaloid attenuates bleomycin-induced
pulmonary fibrosis. Eur J Pharmacol. 627:304–312. 2010. View Article : Google Scholar
|
|
8
|
Sugimoto Y, Furutani S, Itoh A, Tanahashi
T, Nakajima H, Oshiro H, Sun S and Yamada J: Effects of extracts
and neferine from the embryo of Nelumbo nucifera seeds on the
central nervous system. Phytomedicine. 15:1117–1124. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Erdogan S and Turkekul K: Neferine
inhibits proliferation and migration of human prostate cancer stem
cells through p38 MAPK/JNK activation. J Food Biochem.
44:e132532020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Liu Z, Xu B, Sun Z, Gong Y and
Shao C: Neferine, an alkaloid ingredient in lotus seed embryo,
inhibits proliferation of human osteosarcoma cells by promoting p38
MAPK-mediated p21 stabilization. Eur J Pharmacol. 677:47–54. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng G, Zeng S, Ma J, Zhang Y, Qu Y, Han
Y, Yin L, Cai C, Guo C and Shen H: The anti-tumor activities of
Neferine on cell invasion and oxaliplatin sensitivity regulated by
EMT via Snail signaling in hepatocellular carcinoma. Sci Rep.
7:416162017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poornima P, Weng CF and Padma VV:
Neferine, an alkaloid from lotus seed embryo, inhibits human lung
cancer cell growth by MAPK activation and cell cycle arrest.
Biofactors. 40:121–131. 2014. View Article : Google Scholar
|
|
13
|
Xue F, Liu Z, Xu J, Xu X, Chen X and Tian
F: Neferine inhibits growth and migration of gastrointestinal
stromal tumor cell line GIST-T1 by up-regulation of miR-449a.
Biomed Pharmacother. 109:1951–1959. 2019. View Article : Google Scholar
|
|
14
|
Liang HX, Sun LB and Liu NJ: Neferine
inhibits proliferation, migration and invasion of U251 glioma cells
by down-regulation of miR-10b. Biomed Pharmacother. 109:1032–1040.
2019. View Article : Google Scholar
|
|
15
|
Xu L, Zhang X, Li Y, Lu S, Lu S, Li J,
Wang Y, Tian X, Wei JJ, Shao C and Liu Z: Neferine induces
autophagy of human ovarian cancer cells via p38 MAPK/JNK
activation. Tumour Biol. 37:8721–8729. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Q, Li Y, Miao C, Wang Y, Xu Y, Dong
R, Zhang Z, Griffin BB, Yuan C, Yan S, et al: Anti-angiogenesis
effect of Neferine via regulating autophagy and polarization of
tumor-associated macrophages in high-grade serous ovarian
carcinoma. Cancer Lett. 432:144–155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vakifahmetoglu-Norberg H, Xia HG and Yuan
J: Pharmacologic agents targeting autophagy. J Clin Invest.
125:5–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng Y, Ren X, Hait WN and Yang JM:
Therapeutic targeting of autophagy in disease: Biology and
pharmacology. Pharmacol Rev. 65:1162–1197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su M, Mei Y and Sinha S: Role of the
Crosstalk between Autophagy and Apoptosis in Cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhong LP, Pan HY, Zhou XJ, Ye DX, Zhang L,
Yang X, Chen WT and Zhang ZY: Characteristics of a cancerous cell
line, HIOEC-B(a)P-96, induced by benzo(a)pyrene from human
immortalized oral epithelial cell line. Arch Oral Biol. 53:443–452.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lucocq JM and Hacker C: Cutting a fine
figure: On the use of thin sections in electron microscopy to
quantify autophagy. Autophagy. 9:1443–1448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eskelinen EL, Reggiori F, Baba M, Kovacs
AL and Seglen PO: Seeing is believing: The impact of electron
microscopy on autophagy research. Autophagy. 7:935–956. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pankiv S, Lamark T, Bruun JA, Overvatn A,
Bjorkoy G and Johansen T: Nucleocytoplasmic shuttling of p62/SQSTM1
and its role in recruitment of nuclear polyubiquitinated proteins
to promyelocytic leukemia bodies. J Biol Chem. 285:5941–5953. 2010.
View Article : Google Scholar :
|
|
25
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen Y and Gibson SB: Is mitochondrial
generation of reactive oxygen species a trigger for autophagy?
Autophagy. 4:246–248. 2008. View Article : Google Scholar
|
|
27
|
Liu J and Lin A: Role of JNK activation in
apoptosis: A double-edged sword. Cell Res. 15:36–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang D, Kang R, Zeh HJ III and Lotze MT:
High-mobility group box 1, oxidative stress, and disease. Antioxid
Redox Signal. 14:1315–1335. 2011. View Article : Google Scholar
|
|
29
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kimura S, Noda T and Yoshimori T:
Dissection of the autophagosome maturation process by a novel
reporter protein, tandem fluorescent-tagged LC3. Autophagy.
3:452–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Philip NH, DeLaney A, Peterson LW,
Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu
B, Olsen TM, et al: Activity of uncleaved caspase-8 controls
anti-bacterial immune defense and TLR-induced cytokine production
independent of cell death. PLoS Pathog. 12:e10059102016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang YB, Gong JL, Xing TY, Zheng SP and
Ding W: Autophagy protein p62/SQSTM1 is involved in HAMLET-induced
cell death by modulating apotosis in U87MG cells. Cell Death Dis.
4:e5502013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Poornima P, Quency RS and Padma VV:
Neferine induces reactive oxygen species mediated intrinsic pathway
of apoptosis in HepG2 cells. Food Chem. 136:659–667. 2013.
View Article : Google Scholar
|
|
35
|
Xu T, Singh D, Liu J, Li H, Peng S,
Rizzolo LJ and Wang SB: Neferine, is not inducer but blocker for
macroautophagic flux targeting on lysosome malfunction. Biochem
Biophys Res Commun. 495:1516–1521. 2018. View Article : Google Scholar
|
|
36
|
Zhao Q, Liu Y, Zhong J, Bi Y, Liu Y, Ren
Z, Li X, Jia J, Yu M and Yu X: Pristimerin induces apoptosis and
autophagy via activation of ROS/ASK1/JNK pathway in human breast
cancer in vitro and in vivo. Cell Death Discov. 5:1252019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma L, Wei J, Wan J, Wang W, Wang L, Yuan
Y, Yang Z, Liu X and Ming L: Low glucose and metformin-induced
apoptosis of human ovarian cancer cells is connected to ASK1 via
mitochondrial and endoplasmic reticulum stress-associated pathways.
J Exp Clin Cancer Res. 38:772019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li T, Su L, Zhong N, Hao X, Zhong D,
Singhal S and Liu X: Salinomycin induces cell death with autophagy
through activation of endoplasmic reticulum stress in human cancer
cells. Autophagy. 9:1057–1068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rosenfeldt MT and Ryan KM: The role of
autophagy in tumour development and cancer therapy. Expert Rev Mol
Med. 11:e362009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ichimura Y and Komatsu M: Selective
degradation of p62 by autophagy. Semin Immunopathol. 32:431–436.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Moscat J, Diaz-Meco MT and Wooten MW:
Signal integration and diversification through the p62 scaffold
protein. Trends Biochem Sci. 32:95–100. 2007. View Article : Google Scholar
|
|
44
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hou W, Han J, Lu C, Goldstein LA and
Rabinowich H: Autophagic degradation of active caspase-8: A
crosstalk mechanism between autophagy and apoptosis. Autophagy.
6:891–900. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Young MM, Takahashi Y, Khan O, Park S,
Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al:
Autophagosomal membrane serves as platform for intracellular
death-inducing signaling complex (iDISC)-mediated caspase-8
activation and apoptosis. J Biol Chem. 287:12455–12468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pan JA, Fan Y, Gandhirajan RK, Madesh M
and Zong WX: Hyperactivation of the mammalian degenerin MDEG
promotes caspase-8 activation and apoptosis. J Biol Chem.
288:2952–2963. 2013. View Article : Google Scholar :
|