Introduction
Renal ischemia-reperfusion (IR) injury (IRI) is one
of the leading causes of acute kidney injury (AKI), as it causes
renal cell dysfunction to varying degrees, which ultimately leads
to death (1,2). The increased demand for kidney
allografts worldwide suggests that there is an increased risk of
developing AKI (3,4). However, there is currently no cure
available for renal IRI, at least to the best of our knowledge;
therefore, the further exploration of the underlying pathological
mechanisms may lead to the development of more effective treatment
options and a more successful transplantation rate.
Heme oxygenase (HO) is a rate-limiting enzyme that
degrades heme into iron, carbon monoxide and biliverdin (5). In total, three distinct subtypes of
the enzyme have been identified: Inducible HO-1, also known as heat
shock protein 32, and the constitutively expressed HO-2 and HO-3
(6). The increased expression of
HO-1 has been suggested to play a key cytoprotective role in
maintaining the redox homeostasis and activating the oxidative
stress defense mechanism when cells are subjected to certain types
of stress, such as inflammation, ischemia, hypothermia or radiation
exposure (7). HO-1 was
discovered to be induced in response to renal IRI (8-10), and to prolong renal allograft
survival, reduce renal tubular damage induced by IRI and improve
renal function (11,12).
However, to the best of our knowledge, the molecular
mechanisms through which HO-1 exerts its cytoprotective effects
remain poorly understood. Leukocytes are key players in the immune
and inflammatory responses activated following transplantation, and
must anchor to vascular endothelial cells (ECs) first to exert
their functions (13). ECs are
activated in response to inflammatory stimuli through the
upregulation of a number of adhesion molecules, such as P-selectin,
intracellular adhesion molecular (ICAM)-1 (also known as CD54) and
vascular cell adhesion molecule (VCAM)-1 (also known as CD106),
which promotes the adhesion, activation and migration of
circulating leukocytes (14-17). HO-1 has been found to be one of
the principal proteins regulating the expression of adhesion
molecules associated with EC activation (18). Previous studies have demonstrated
that hemoglobin exposure is able to increase vascular permeability
and thereby improve leukocyte infiltration into the spleen,
intestine and liver, which is associated with the upregulation of
ICAM-1, P-selectin and fibronectin expression (19,20).
VCAM-1 is an important adhesion molecule associated
with EC activation (21,22). The induced expression of VCAM-1
has been reported to increase the adhesion of leukocytes expressing
the counter receptor, integrin α4β1 (23,24). However, to the best of our
knowledge, whether HO-1 affects the recruitment of neutrophils by
regulating VCAM-1 expression during renal IRI has not yet been
investigated.
The present study thus aimed to investigate the
mechanisms through which HO-1 regulates the recruitment of
neutrophils in renal IRI. The present study used HO-1 knockdown
(HO-1+/−) mice, rather than HO knockout
(HO-1−/−) mice, as the animal model since a partial
reduction in HO-1 levels is more common and clinically relevant
than the complete elimination of HO-1, and HO-1−/− mice
also have a high mortality rate (25). The present study reveals a
protective role of HO-1 in renal IRI by regulating VCAM-1
expression. It is possible to explore some treatment methods
through this target in the future.
Materials and methods
Genotype identification of mice
HO-1+/− and wild-type
(HO-1+/+) mice (n=7 in each group) were provided by
Jackson Laboratory. DNA from mouse tail tips were extracted using a
Genomic DNA Extraction kit from Accurate Biotechnology Co., Ltd.
(cat. no. AG21009). The DNA was used as a template to perform the
PCR assay. PCR was performed with 10 µl Taq 2X PCR master
mix (K0171, Thermo Fisher Scientific, Inc.), 2 µl mixed
primers (10 µM of primer each), 1 µl genomic DNA and
7 µl ddH2O. Target DNA was amplified in a
thermocycler machine (ABI Veriti), with initial denaturation at
95°C for 5 min, followed by 35 cycles of denaturation at 95°C for
20 sec, annealing at 58°C for 30 sec, and extension at 72°C for 30
sec. The PCR products were electrophoresed on a 2% TBE Agarose gel
and imaged in a Gel Imaging System (Tanon 2500, Tanon Science &
Technology Co., Ltd.). Primers P1 (GTA CAT GCT GGC TGG GTT CT), P2
(CCA TTT CTC AGG CAA GAA GG) and P3 (GCC AGA GGC CAC TTG TGT AG)
were used to identify HO-1+/+ [size, 280 base pair (bp)]
and HO-1+/− (size, 280 and 225 bp) mice.
Renal ischemia reperfusion model
All mice were housed under specific pathogen-free
conditions in strict compliance with the facility standards
approved by the China Laboratory Animal Management Accreditation
Association. All experiments performed in the study were approved
by the Institutional Animal Care and Use Committee of Binzhou
Medical University (PJ2018-10-16). HO-1+/+ and
HO-1+/− mice were anesthetized by an inhalation of
isoflurane (5% for induction and 2% for maintenance). The hair on
the backs of each mouse was partially removed using hair removal
cream and the exposed skin was disinfected with 75% alcohol; the
mice were then fixed on a 37°C thermostat (heating pad) in the
prone position. A midline incision was made, the vessels of the
bilateral renal pedicles were exposed, and the right renal artery
was clamped using a non-traumatic vascular clamp (size B-1 V;
S&T AG-Microsurgical Instruments) for 60 min. After the midline
incision was made, the vessels of the bilateral renal pedicles were
exposed and the right renal artery was clamped using a temporary
clip (Serrefine Vascular Clamps, MDG0795861; Medline) for 60 min.
During the clamping process, the color of the kidney changed from
bright red to purple/black. After 60 min, the renal artery clip was
released to initiate blood flow reperfusion. Furthermore, to
prevent functional compensation by the left kidney, it was removed
following left renal pedicle ligation. After the wound was sutured,
the mice were woken and returned to a clean cage to recover from
the anesthetic.
In vivo blocking experiment
A total of 30 male mice were randomly divided into
the following 3 groups (n=10/group): i) The HO-1+/−
group; ii) HO-1+/+ group; and iii) the
HO-1+/− + anti-VCAM-1 antibody group. Prior to renal IR
surgery, the animals were administered mouse anti-VCAM-1 monoclonal
antibody (cat. no. AF643; R&D Systems, Inc.) at 1 mg/kg through
the tail vein, which was set as the trial dose (26).
The control animals were injected with equal amounts
of isotype-matched antibodies (1 mg/kg, cat. no. MAB002; R&D
Systems, Inc.). Each animal received the same dose of monoclonal
antibody injection at 24, 48 and 72 h after surgery. At the end of
the monoclonal antibody treatment, the animals were anesthetized
with 5% inhaled isoflurane for induction and 2% for maintenance,
the kidneys were harvested, and the mice were immediately
euthanized by cervical dislocation.
Western blot analysis
The mouse kidney tissue was lysed with Protein
Extraction Reagent (cat. no. 78505; Thermo Fisher Scientific, Inc.)
containing protease-inhibitor (cat. no. P8340, Sigma-Aldrich; Merck
KGaA) and the protein concentration was quantified using a BCA
protein assay kit (Beijing Solarbio Science & Technology Co.,
Ltd.) and 30 µg protein from each group was separated via
10% SDS-PAGE. The separated proteins were subsequently transferred
onto a polyvinylidene fluoride membrane and blocked with 5% milk
for 1 h. The membranes were then incubated with the following
primary antibodies overnight at 4°C: Anti-VCAM-1 (0.25
µg/ml, cat. no. AF643; R&D systems), anti-HO-1 (1:1,000,
cat. no. 27282-1-AP; ProteinTech Group, Inc.), anti-cleaved
caspase-3 (1:1,000, cat. no. 9664; Cell Signaling Technology, Inc.)
and anti-β-actin (1:1,000, cat. no. 58169; Cell Signaling
Technology, Inc.), followed by the horseradish
peroxidase-conjugated anti-goat (1:2,000, cat. no. ab97110; Abcam),
anti-rabbit (1:2,000, cat. no. 7074; Cell Signaling Technology,
Inc.) or anti-mouse secondary antibodies (1:2,000, cat. no. 7076;
Cell Signaling Technology, Inc.) at room temperature for 1 h.
Protein bands were visualized using an ECL reagent (Beyotime
Institute of Biotechnology) and analyzed using densitometry with
ImageJ software (version 1.48; National Institutes of Health).
Determination of histological scores
After the mice were sacrificed, the kidneys were
immediately fixed with 4% paraformaldehyde for 24 h and embedded in
paraffin according to the standard protocol as previously described
(27). The kidneys were then
sectioned into 5-µm-thick slices using a paraffin microtome
(Leica RM CoolClamp; Leica Microsystems, Inc.). Hematoxylin and
eosin (H&E) staining was performed according to the standard
protocol as previously described (28,29). To quantify the extent of tubular
injury, the H&E-stained sections were visualized under a light
microscope (Olympus IX83; Olympus Corporation) and the degree of
tubular injury was scored using a five-point scale, based on the
following criteria: 0, normal tubule; 1, slight blistering and loss
of brush borders; 2, severe blistering and mild vacuolization; 3,
significant vacuolization and shrunken nuclei; 4, presence of
necrotic or apoptotic cells and denudation of the basement
membrane; and 5, complete tubular necrosis (30).
Determination of renal function
To evaluate the changes in renal function following
renal I/R surgery, the HO+/+ and HO-1+/− mice
were sacrificed at 8, 24 and 72 h post-reperfusion, and 200
µl of blood were collected from the dorsal aorta into
heparinized Eppendorf tubes as previously described (31). The blood was left to clot in an
upright position for 30 min and then centrifuged for 15 min at
1,480 × g at 4°C, then serum was obtained for the measurement of
blood urea nitrogen (BUN) and serum creatinine (SCr) levels
(28). The concentrations of SCr
(mg/dl) and BUN (mg/dl) were measured using a Cobas Fara
spectrophotometer system (UV-7504; Roche Diagnostics).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from the mouse kidney s was extracted
using TRIzol® reagent and reverse transcribed into cDNA
using a SuperScript III First-Strand synthesis kit (18080051,
Invitrogen; Thermo Fisher Scientific, Inc.). qPCR to determine the
mRNA expression levels of IFN-γ, IL-6, TNF-α, IL-10 and MMP-13 was
subsequently performed using a SYBR-Green PCR master mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The relative mRNA
expression levels were calculated using the 2−ΔΔCq
method and GAPDH was used as the endogenous control for
normalization (32). The primer
sequences used for qPCR are listed in Table I.
 | Table ICytokine-specific primer pair
sequences used for RT-qPCR. |
Table I
Cytokine-specific primer pair
sequences used for RT-qPCR.
| Gene name | Primer
sequences |
|---|
| IFN-γ | F:
AGCGGCTGACTGAACTCAGATTGTAG |
| IFN-γ | R:
GTCACAGTTTTCAGCTGTATAGGG |
| IL-6 | F:
CTGGTGACAACCACGGCCTTCCCTA |
| IL-6 | R:
ATGCTTAGGCATAACGCACTAGGTT |
| TNF-α | F:
GGCAGGTCTACTTTGGAGTCATTGC |
| TNF-α | R:
ACATTCGAGGCTCCAGTGAATTCGG |
| IL-10 | F:
ACCTGGTAGAAGTGATGCCCCAGGCA |
| IL-10 | R:
CTATGCAGTTGATGAAGATGTCAAA |
| MMP-13 | F:
GATGATCCCACCTTAGACATCATGAGAAAA |
| MMP-13 | R:
AAAGTGGTCTTAGATACTACCGTGACG |
Mouse glomerular EC (mGEC) culture
The mice were anesthetized with 5% inhaled
isoflurane for induction and 2% for maintenance and disinfected
with 75% alcohol, and placed on a clean hood. The chest was opened
for bloodletting from the heart, followed by the opening of the
abdominal cavity and the removal of the kidneys. The kidneys were
placed in PBS at 4°C and the renal capsule and medulla were
removed; the kidney cortex was then collected and cut into small (1
mm3) sections. After washing three times with cold PBS,
the sections were further homogenized into a slurry and digested
with 0.1% type IV collagenase (HBSS dilution) at 37°C for 25-30
min, which was terminated when the glomerulus was slightly broken
and loosened. The renal cortex tissue mass was poured into a
stainless steel 200 wire mesh (1985-00200, Bellco), gently grinded
and washed repeatedly with PBS at 4°C to collect the suspension.
The latter was poured onto a 400 mesh screen (1985-00400; Bellco)
and thoroughly rinsed with PBS at 4°C so that the kidney tubules
and cell debris were filtered out and the glomerulus remained on
the 400 mesh screen for collection. The glomerular suspension was
centrifuged at 120 × g for 5 min at room temperature, resuspended
in endothelial cell culture medium (DMEM plus 20% FBS, L-glutamine
30 µg/ml, bovine insulin 0.6 U/ml, heparin 5 U/ml,
penicillin 100 U/ml), and placed in an incubator at 37°C for 3 days
without disturbance. The ECs were allowed to grow out from the
adherent glomeruli until 80-90% confluency and passaged with 0.25%
trypsin. Cells of two to three generations were used in the
subsequent experiments.
Immunohistochemistry
Immunohistochemical staining of the
paraffin-embedded tissues was performed as follows: Following
deparaffinization and antigen retrieval, the kidney tissue sections
were blocked with 10% goat serum for 1 h and then incubated
overnight at 4°C with the following antibodies: Anti-CD68 antibody
(1:100, cat. no. ab283654; Abcam) and anti-lymphocyte antigen 6
complex, locus G (Ly-6G) (1:200, ab25377; Abcam). The following
day, following incubation with HRP-conjugated secondary antibodies
(1:500, cat. no. ab97057; Abcam; 1:500, cat. no. 7074, Cell
Signaling Technology, Inc.) for 1 h at room temperature, the slides
were developed with DAB until the signal clearly appeared, and the
nuclei were stained with hematoxylin for 5 min at room temperature,
and images were obtained using a microscope (Olympus IX83, Olympus
Corporation).
TUNEL staining
TUNEL staining was performed using an In Situ
Cell Death Detection kit (cat. no. 12156792910, Sigma-Aldrich Merck
KGaA) according to the manufacturer's instructions on cryosections.
Briefly, the sections were washed with PBS and permeabilized with
0.1% Triton X-100 + 0.1% sodium citrate on ice for 5 min. The TUNEL
reaction mixture was then added to the tissue followed by
incubation at 37°C for 1 h. Following rinsing three times with PBS,
the tissues were mounted with mounting media containing DAPI (cat.
no. H-1200; Vector Laboratories, Inc., part of Maravai
LifeSciences) and visualized using a fluorescence microscope
(Olympus IX83; Olympus Corporation).
Neutrophil purification
Mouse neutrophils were extracted using a peripheral
blood Neutrophil isolation kit (cat. no. LZS1100, TBD Hao Yang
Biological Manufacture Co., Ltd.). The blood samples were carefully
sucked through a straw and added to the surface of the separation
solution. Following centrifugation for 30 min at 400 × g and 4°C,
the neutrophil layer was carefully absorbed and the red blood cells
were lysed with lysis buffer. After washing, neutrophils were
resuspended in DMEM at a concentration of 1×106
cells/ml. Neutrophils were labeled with PKH26 (cat. no. PKH26GL,
Sigma-Aldrich; Merck KGaA) to prepare for the adhesion and
Transwell migration assays.
Neutrophil adhesion assay
mGECs were grown to confluency in a 96-well plate.
Confluent cells were stimulated with 100 U/ml TNF-α (Millipore
Sigma) for 4 h. The medium was then replaced with fresh medium and
supplemented with 10 µg/ml VCAM-1 (cat. no. AF643; R&D
systems) or isotype Mb (cat. no. AB-108-C; R&D systems) for 1
h. Neutrophils labeled with PKH26 were then added to the 96-well
plate for 6 h. The cells were then washed with PBS three times and
photographed using a fluorescence microscope (Olympus IX83; Olympus
Corporation). ImageJ software (version 1.48; National Institutes of
Health) was used to quantify the fluorescence area.
Neutrophil Transwell migration assay
A transwell assay was used to detect neutrophil
migration through the mGECs. mGECs were grown to confluency on
transwells with 12-µm pores. Confluent cells were stimulated
with 100 U/ml TNF-α (Millipore Sigma) for 4 h. The medium was then
replaced with fresh medium and supplemented with 10 µg/ml
VCAM-1 or isotype Mb for 1 h. A neutrophil migration assay was
performed as previously described (33). Fluorescently-stained neutrophils
(3×104) were added to the upper chamber for 6 h. The
culture medium was aspirated, the upper chamber was removed and the
membrane was gently wiped with a paper towel. The membrane was then
fixed with 4% PFA for 10 min, washed twice with PBST and images
were captured using an Olympus IX83 fluorescence microscope
(Olympus Corporation).
Statistical analysis
The IBM SPSS 22 version program was used for
analysis. An unpaired t-test and one-way ANOVA were used to analyze
the data. The results are expressed as the mean ± SD. P<0.05 was
considered to indicate a statistically significant difference.
Results
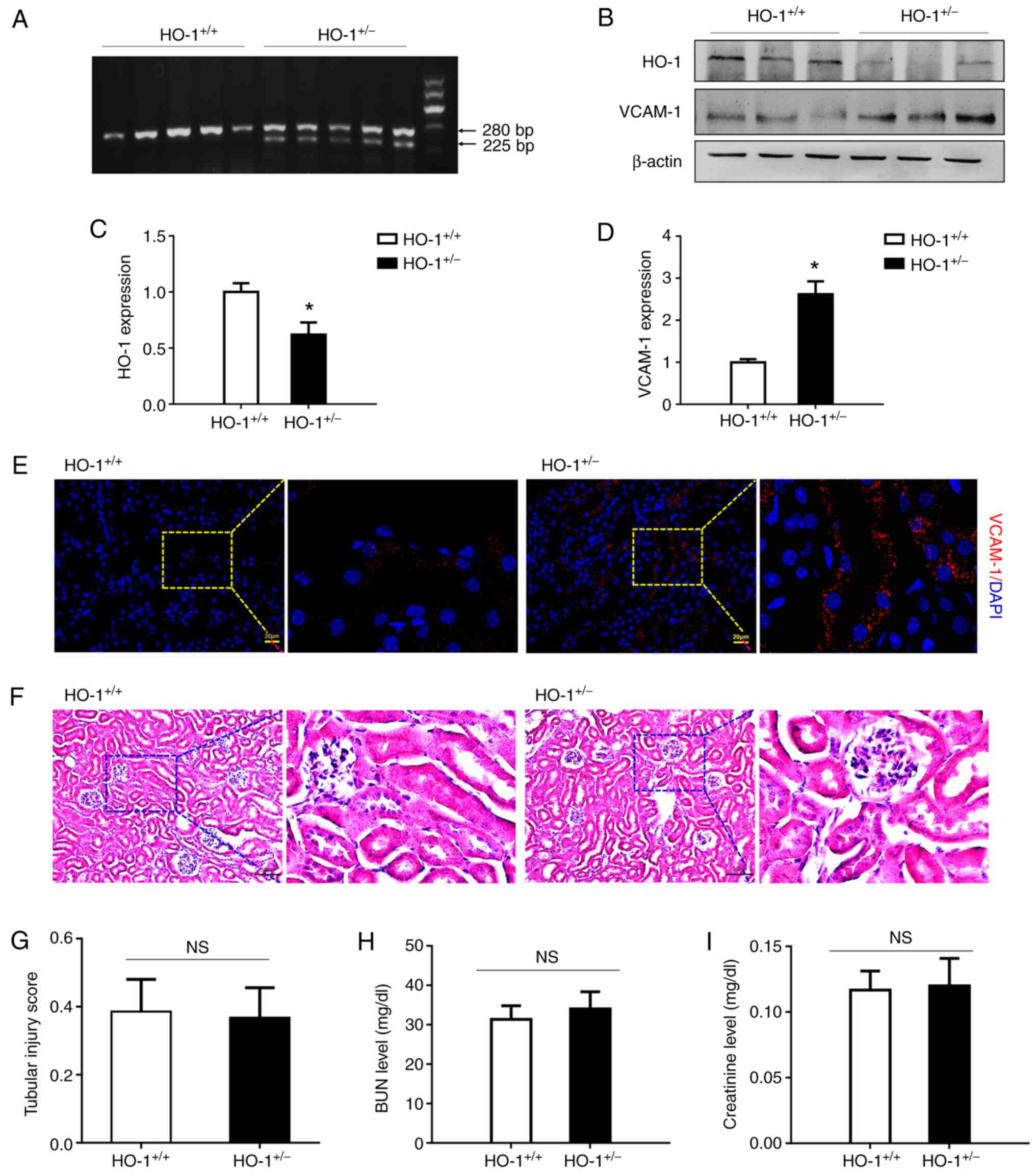
HO-1+/− knockdown mice exhibit
elevated expression levels of VCAM-1 without changes in renal
structure and function
Genotypes of HO+/+ and HO+/−
mice were first identified. As illustrated in Fig. 1A, by agarose gel electrophoresis,
the PCR products in the HO+/+ group produced a band of
280 bp, and those in the HO+/− group produced two bands
of 225 and 280 bp, which suggested that the genotypes of the
HO+/+ and HO+/− mice were correct. Western
blot analysis was used to compare VCAM-1 protein expression in the
HO-1+/− knockdown and wild-type mice. As shown in
Fig. 1B-D, HO-1 expression in
the HO-1+/− mice was decreased by 38% relative to that
in the wild-type mice, while the expression of VCAM-1 was increased
2.62-fold (P<0.05). Furthermore, immunohistochemical staining
revealed that VCAM-1 was expressed at a high level in the
HO-1+/− group, while its expression was relatively low
in the HO-1+/+ group (Fig. 1E). These results revealed a
significant increase in VCAM-1 expression and a decrease in that of
HO-1 in the HO-1+/− mice. In addition, H&E staining
was used to detect renal tissue morphology in the
HO-1+/+ and HO+/− mice. As shown in Fig. 1F and G, no marked differences
were observed in renal tissue morphology and the tubular injury
score between the HO-1+/+ and HO+/− group.
The BUN and SCr levels were then analyzed to assess renal function.
As shown in Fig. 1H and I, no
significant differences were observed in the BUN and SCr levels
between the HO-1+/+ and HO+/− groups.
Exacerbation of renal IRI in
HO-1+/− mice
To examine the role of HO-1 in the development of
AKI, a model of renal IRI was established by clamping the right
renal artery for 60 min and blocking left kidney function in the
HO-1+/− and wild-type mice. Right renal tissues and
blood were then harvested for analysis at 8, 24 and 72 h following
surgery (Fig. 2A). As shown in
Fig. 2B and C, HO-1 expression
in the HO-1+/− mice was decreased significantly at 24 h
following surgery (P<0.05). However, the expression of VCAM-1
and cleaved caspase-3 was significantly increased (P<0.05).
Immunofluorescence staining was further used to assess the
expression of VCAM-1. Higher expression levels of VCAM-1 were
observed in the HO-1+/− mice than in the
HO-1+/+ control group (Fig. 2D). H&E staining of the kidney
tissue also revealed more tissue damage in the HO-1+/−
mice than in the wild-type mice. In either the cortex or medulla,
cell necrosis, brush border loss, cast formation and tubular
dilation were more evident in the HO-1+/− group
(Fig. 2E). A substantial
increase in the tubular injury score was observed in the
HO-1+/− group. Over time, the tubular injury score
changed, increasing during the early time period, peaking at 24 h,
and gradually decreasing thereafter (Fig. 2F). The BUN and SCr concentrations
were also analyzed to assess renal function. Similar to the tubular
injury score, both the BUN and SCr levels were higher in the
HO-1+/− knockdown group, exhibiting maximal values at 24
h (Fig. 2G and H). As shown in
Fig. 2G, the BUN level in the
HO-1+/+ group was 53 mg/dl at 8 h post-IRI, increased to
110 mg/dl at 24 h and then decreased to 82 mg/dl at 72 h. By
contrast, the BUN levels in the HO-1+/− experimental
group were 82 mg/dl at 8 h post-IRI, increased to 191 mg/dl at 24 h
and then decreased to 139 mg/dl at 72 h. Compared with the
HO-1+/+ control group, the BUN levels in the
HO-1+/− group were elevated at various time points
(P<0.05). As shown in Fig.
2H, in the HO-1+/+ group, the SCr level was 0.6
mg/dl at 8 h post-IRI, increased to 1.1 mg/dl at 24 h and then
decreased to 0.9 mg/dl at 72 h. However, in the HO-1+/−
group, the SCr level was 0.9 mg/dl at 8 h post-IRI, increased to
1.5 mg/dl at 24 h and decreased to 1.2 mg/dl at 72 h. Compared with
the HO-1+/+ control group, the SCr levels in the
HO-1+/− group were elevated at different time points
(P<0.05).
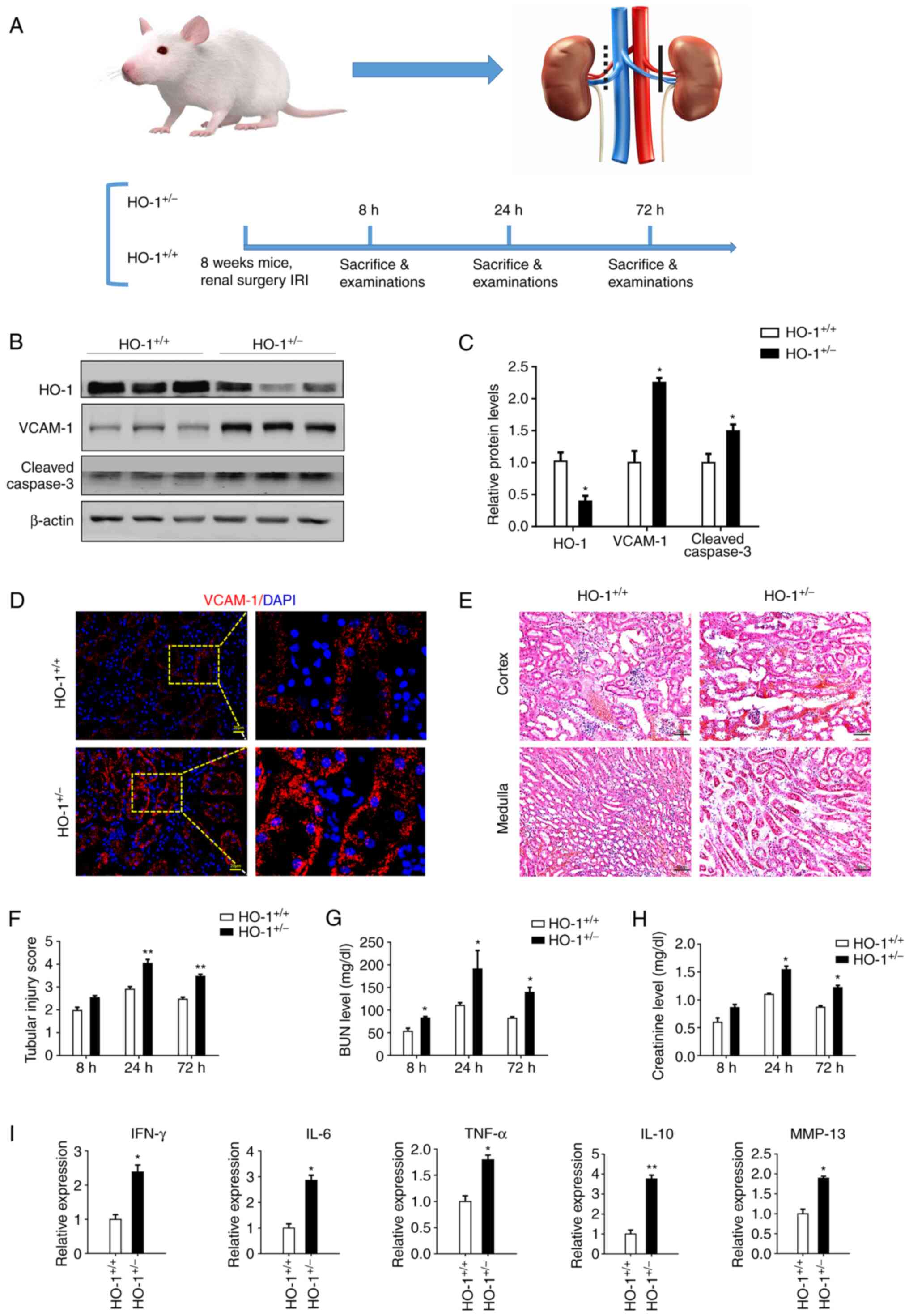 | Figure 2Exacerbation of renal IRI in the
HO-1+/− mice. (A) Renal ischemia reperfusion model were
established by clamping the right renal artery for 60 min and
blocking left kidney function in the HO-1+/−, as well as
the wild-type mice and tissue harvesting at 8, 24 and 72 h
following surgery. (B) Western blot analysis of the expression
levels of HO-1, VCAM-1, cleaved caspase-3 and β-actin proteins at
24 h post-IRI in the HO-1+/− vs. those in the wild-type
mice. (C) Densitometric-based quantification of the western blot
analysis results shown in panel B using ImageJ software.
Densitometry values are expressed as the mean ± SD (n=3).
*P<0.05 vs. HO-1+/+. (D)
Immunohistochemical staining of VCAM-1-expressing cells at 24 h
post-IRI in the HO-1+/+ and HO-1+/− mice. (E)
Representative images of H&E-stained sections of the cortical
and medullary renal tissue showing the structure of the renal
tissue. (F) Tissue injury was assessed by using the scoring scale
from 0 to 5 points (n=3, **P<0.01 vs.
HO-1+/+). (G) Serum BUN concentration at 8, 24 and 72 h
post-IRI in the HO-1+/+ and HO-1+/− mice
(n=3, *P<0.05 vs. HO-1+/+). (H) Serum
creatinine concentration at 8, 24 and 72 post-IRI in the
HO-1+/+ and HO-1+/− mice (n=3,
*P<0.05 vs. HO-1+/+). (I) Expression
levels of IFN-γ, IL-6, TNF-α, IL-10 and MMP-13 in the
HO-1+/+ vs. the HO-1+/− group assessed by
RT-qPCR (n=3, *P<0.05, **P<0.01 vs.
HO-1+/+). IRI, ischemia-reperfusion injury; HO-1, heme
oxygenase-1; VCAM-1, vascular cell adhesion molecule-1; BUN, blood
urea nitrogen. |
IRI can cause an inflammatory
response
In order to determine the effects of HO-1 knockdown
on the inflammatory response, the expression of inflammatory
factors in the injured kidney tissue were analyzed at 24 h
post-IRI. The expression levels of IFN-γ, IL-6, TNF-α, IL-10 and
MMP-13 in the HO-1+/− group were 2.4-, 2.9-, 1.8-, 3.8-
and 1.9-fold higher relative to those in the HO-1+/+
group, respectively (Fig. 2I).
All these factors exhibited elevated expression levels in the
HO-1+/− group, as compared with the HO-1+/+
group.
VCAM-1 blocking alleviates renal IRI
VCAM-1 antibody was infused into the
HO-1+/− mice to block VCAM-1 expression on the inner
vascular endothelium. Tubular injury was found to be visibly
reduced in the VCAM-1 antibody infusion group (Fig. 3A). The estimated
histopathological scoring also suggested alleviated tubular injury
in the HO-1+/− knockdown mice upon VCAM-1 blocking
(Fig. 3B). In addition, the BUN
and SCr levels were measured to assess kidney functionality. As
shown in Fig. 3C, at 24 h
post-IRI, the BUN concentration value was 156.3±18.6 mg/dl in the
HO-1+/− group, which decreased to 119.3±11.7 mg/dl in
the HO-1+/− + VCAM-1 anti-body group (P<0.05,
compared with the HO-1+/− group); at 72 h post-IRI, the
BUN concentration value in the HO-1+/− group was
137.3±5.5 mg/dl, which decreased to 104.7±13.6 mg/dl in the
HO-1+/− + VCAM-1 antibody animal group (P<0.05,
compared with the HO-1+/− group). In addition, as shown
in Fig. 3D, the SCr level in the
HO-1+/− group at 24 h post-IRI was 1.56±0.13 mg/dl,
which was decreased to 1.31±0.05 mg/dl in the HO-1+/− +
VCAM-1 antibody group (P<0.05, compared with the
HO-1+/− group). The SCr level in the HO-1+/−
group at 72 h post-IRI was 1.32±0.04 mg/dl, which decreased to
1.13±0.04 mg/dl in the HO-1+/− + VCAM-1 antibody group
(P<0.05, compared with the HO-1+/− group).
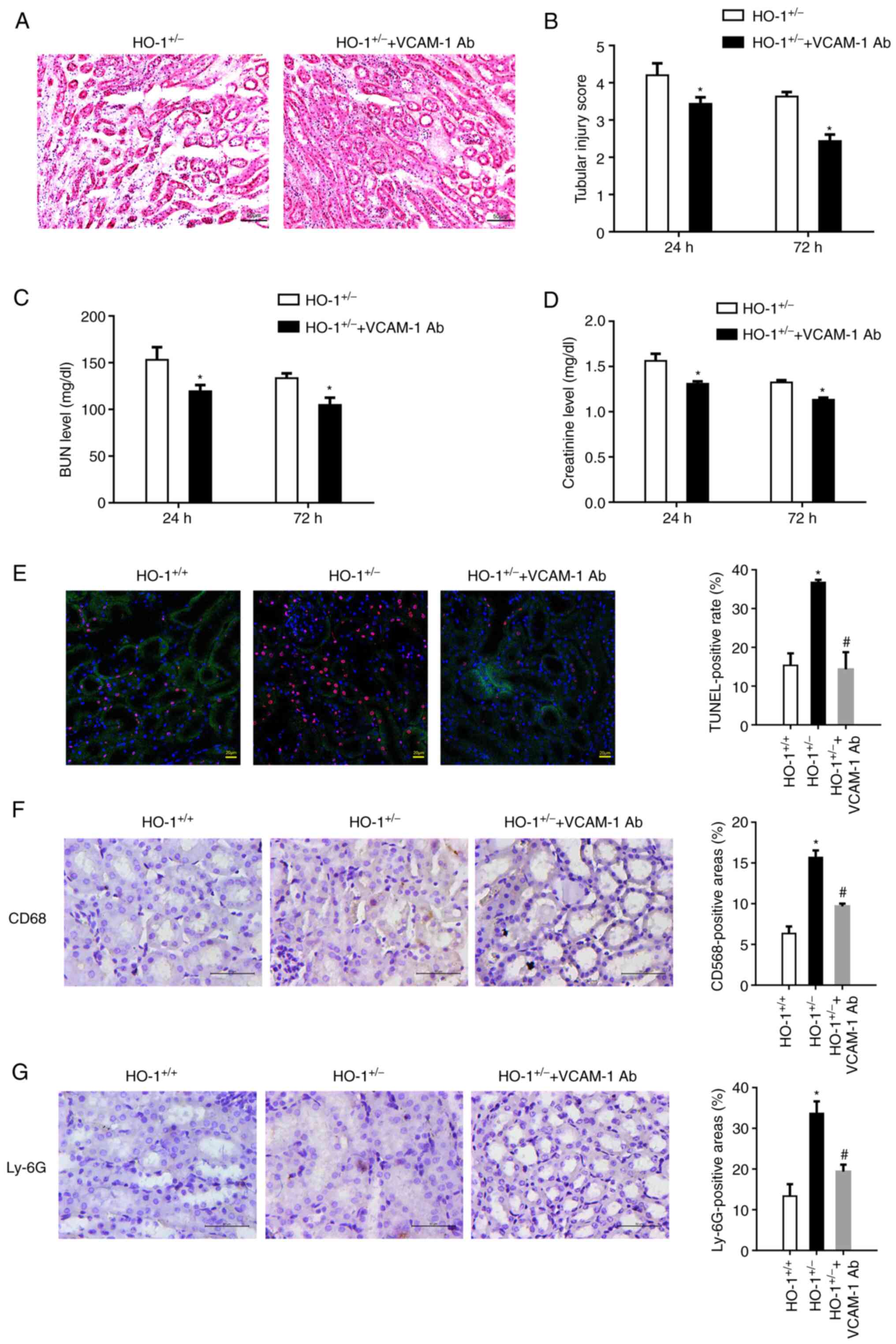 | Figure 3VCAM-1 blocking alleviates renal IRI.
VCAM-1 antibody was infused into the HO-1+/− knockdown
mice through the tail vein to block VCAM-1 expression on the
vascular endothelium. (A) Representative images of H&E-stained
sections of renal tissue showing its morphology. (B) Extent of the
kidney tissue injury was assessed using the 0 to 5-point scoring
system (n=3, *P<0.05 vs. HO-1+/−). (C)
Serum BUN concentration at 24 and 72 h post-IRI in the
HO-1+/+ and HO-1+/− mice (n=3,
*P<0.05 vs. HO-1+/−). (D) Serum creatinine
concentration at 24 and 72 h post-IRI in the HO-1+/+ and
HO-1+/− mice (n=3, *P<0.05 vs.
HO-1+/−). (E) Cell death upon IRI was measured using
TUNEL assay. The TUNEL-positive rate was analyzed using ImageJ
software in the HO-1+/+, HO-1+/− and
HO-1+/− + VCAM-1 Ab groups (*P<0.05 vs.
HO-1+/+; #P<0.05 vs. HO-1+/−).
(F) Immunohistochemical staining and quantification analysis of
CD68-expressing cells in mouse kidneys in the HO-1+/+,
HO-1+/− and HO-1+/− + VCAM-1 Ab groups
(*P<0.05 vs. HO-1+/+;
#P<0.05 vs. HO-1+/−). (G)
Immunohistochemical staining and quantification of Ly-6G-expressing
cells in mouse kidneys of the HO-1+/+,
HO-1+/− and HO-1+/− + VCAM-1 Ab groups
(*P<0.05 vs. HO-1+/+;
#P<0.05 vs. HO-1+/−). IRI,
ischemia-reperfusion injury; HO-1, heme oxygenase-1; VCAM-1,
vascular cell adhesion molecule-1; BUN, blood urea nitrogen; Ab,
antibody. |
Cell death upon IRI measured by TUNEL
assay
The numbers TUNEL-positive cells in the
HO-1+/− group were evidently higher than those in the
HO-1+/+ group. However, in the HO-1+/− +
VCAM-1 antibody group, the TUNEL-positive rate was significantly
attenuated (Fig. 3E).
Furthermore, the accumulation of macrophages (Fig. 3F) and neutrophils (Fig. 3G) was observed in the renal
tubular area upon IRI, and this was more evident in the
HO-1+/− group, as compared with the HO-1+/+
group. However, in the HO-1+/− + VCAM-1 antibody group,
leukocyte accumulation was significantly alleviated, indicating
that HO-1 affects leukocyte recruitment and inflammatory injury at
least partly through the VCAM-1 pathway.
In vitro blocking of VCAM-1 suppresses
neutrophil adhesion and migration through Transwells
mGECs from the HO-1+/− knockdown and the
wild-type mice were isolated, and following TNF-α stimulation,
western blot analysis was used to identify the expression of HO-1
(Fig. 4A). A lower expression of
HO-1 was observed in the HO-1+/− mGECs. However, higher
expression levels of VCAM-1 were observed in the HO-1+/−
mGECs than in the HO-1+/+ mGECs by means of
immunofluorescence staining. The fluorescence intensity of VCAM-1
expressed on the mGEC of the HO-1+/− mice was 2.04 times
higher than that in the HO-1+/+ control group (Fig. 4B). Neutrophil recruitment is an
indispensable process of the immune response (34). Neutrophils were then isolated and
labeled with PKH26 to perform adhesion assay and Transwell
migration assay (Fig. 4C). As
shown in Fig. 4D, the number of
neutrophils adhered to the mGECs of the HO-1+/− mice was
higher than that in the HO-1+/+ group. However, when the
VCAM-1 Ab were added to the culture medium, the number of
neutrophils adhered to the mGECs was significantly reduced as
compared with the HO-1+/− knockdown only group. In the
Transwell migration assay, more neutrophils were observed migrating
through the perforated membrane in the HO-1+/− group
than in the HO-1+/+ control group. However, when the
VCAM-1 antibody was added to the culture medium, the number of
migrating neutrophils was significantly reduced as compared with
the HO-1+/− knockdown only group (Fig. 4E). These data suggest that HO-1
knockdown attenuated the adherence of neutrophils and their
migration at least partly through the vascular basement membrane
via VCAM-1.
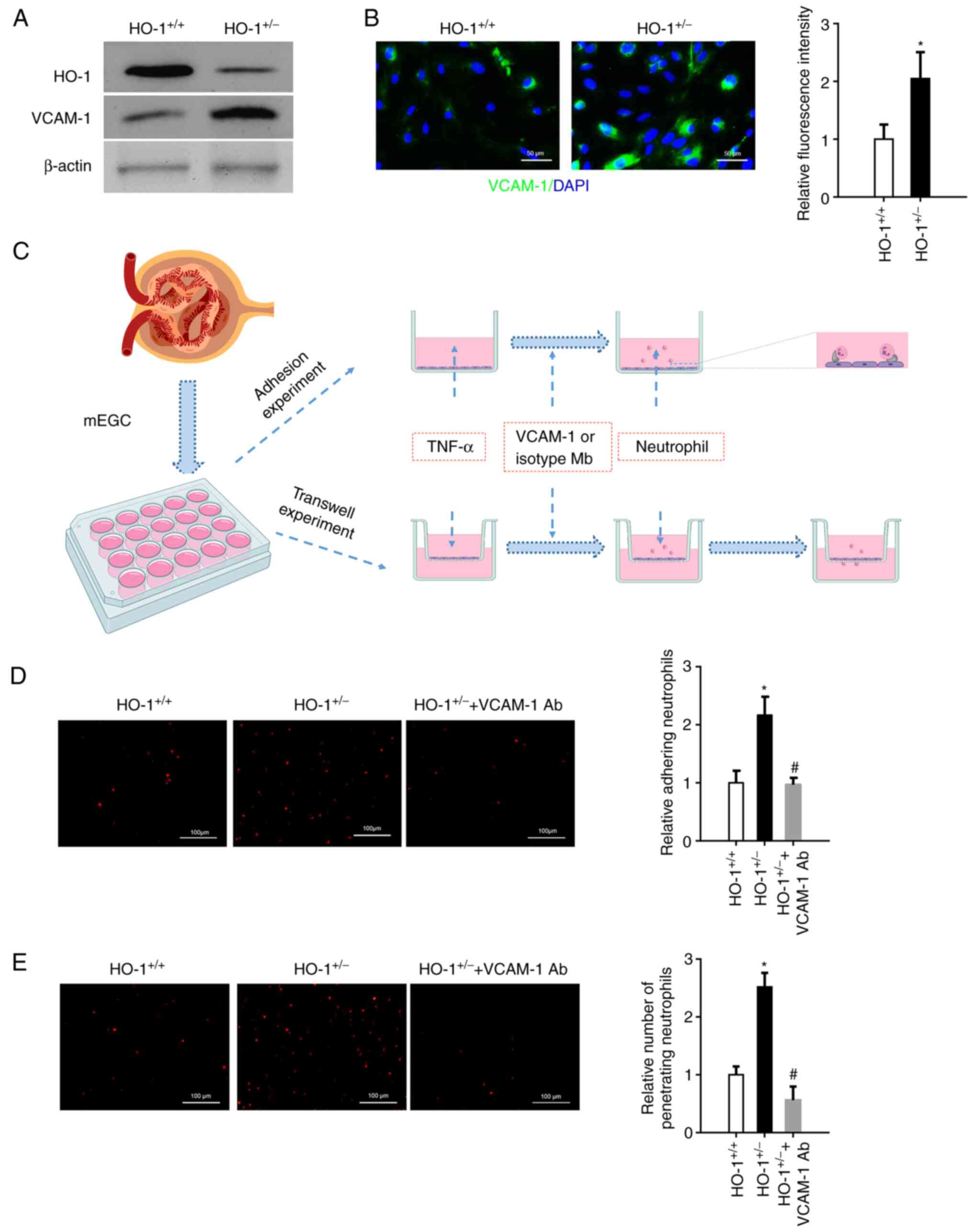 | Figure 4In vitro blocking of VCAM-1
suppresses neutrophil adhesion and migration through Transwells.
mGECs from the HO-1+/− and wild-type mice were isolated.
(A) Western blot analysis of the expression levels of HO-1, VCAM-1
and β-actin proteins in the mGECs extracted from the
HO-1+/+ and HO-1+/− mice. (B)
Immunofluorescence staining of VCAM-1 in the mGECs extracted from
the HO-1+/+ and HO-1+/− mice. Relative
fluorescent intensity was quantified using ImageJ software (n=3,
*P<0.05 vs. HO-1+/+). (C) mGECs were grown
in a 96-well plate or Transwell chamber and stimulated with 100
U/ml TNF-α for 4 h. Neutrophils were isolated and labeled with
PKH26 to perform adhesion assay or Transwell migration assay. (D)
Neutrophils adhered to mGECs were photographed using a fluorescence
microscope, and the fluorescence area was quantified using ImageJ
software (n=3, *P<0.05 vs. HO-1+/+;
#P<0.05 vs. HO-1+/−). (E) Neutrophil
migration through mGECs was photographed using a fluorescence
microscope, and the fluorescence area was quantified using ImageJ
software (n=3, *P<0.05 vs. HO-1+/+;
#P<0.05 vs. HO-1+/−). mGECs, mouse
glomerular endothelial cells; HO-1, heme oxygenase-1; VCAM-1,
vascular cell adhesion molecule-1; Ab, antibody. |
Discussion
The present study demonstrates a protective role of
HO-1 in renal IRI, leading to the following conclusions: i) HO-1
knockdown in HO-1+/− mice exacerbates renal IRI; ii)
HO-1 knockdown aggravates renal IRI through the upregulation of
VCAM-1 with a concomitant augmentation of leukocyte recruitment and
inflammatory damage; iii) HO-1 knockdown increases neutrophil
adherence and the migration of the vascular basement membrane in
vitro, mediated by the upregulation of VCAM-1.
The protective effect of HO-1 on renal IRI has been
widely recognized (35,36). Furthermore, HO-1 has been
utilized as a therapeutic target for renal IRI. For example,
Pannexin 1 silencing has been shown to attenuate renal IRI by
inducing HO-1 expression (37).
Hydrogen sulfide has also been shown to attenuates renal IRI by the
upregulation of HO-1 (37). The
results of the present study also demonstrated that HO-1 knockdown
in HO-1+/− mice significantly exacerbated renal IRI with
a concomitant increase in the serum levels of SCr and BUN markers,
as well as the renal tubule injury score, which is consistent with
the findings of previous studies (36,38,39).
HO-1 deficiency increases the infiltration of
myeloid cells following IRI (37). The results of the present study
demonstrated leukocyte accumulation in the renal tubular area
following IRI, which included the accumulation of macrophages and
neutrophils, that were more evident obvious in the
HO-1+/− group than in the HO-1+/+ control
group. IRI is caused by the accumulation of neutrophils at the site
of tissue injury and the release of a large number of inflammatory
mediators, such as reactive oxygen species and cytokines.
Neutrophil recruitment is a hallmark of an immune response, which
is regulated by a cascade process, including cell roll, activation,
adhesion and migration through the endothelium. During this
process, an essential role is played by very late antigen 4 (VLA-4)
expressed on neutrophils binding to ICAM-1 and VCAM-1 expressed on
the vascular endothelial cell surface (40,41). Therefore, the levels of VCAM-1
and ICAM-1 determine whether neutrophils can migrate through the
endothelium. In the present study, VCAM-1 expression was
significantly upregulated in the HO-1+/− mice as
compared with the wild-type mice, which confirms that HO-1 inhibits
the expression of VCAM-1.
To verify the effects of HO-1 on VCAM-1, VCAM-1
antibody was infused into HO-1+/− mice to block the
endogenous pool of kidney VCAM-1. The results revealed that tubular
injury was significantly reduced in the VCAM-1 antibody infusion
group, exhibiting a decreased renal tubule injury score, BUN and
SCr levels, and leukocyte recruitment, including neutrophils. This
indicates that the effect of HO-1 knockdown requires the
involvement of VCAM-1.
Leukocyte adhesion and migration to inflammatory
sites through vascular endothelial cells are important features of
the inflammatory processes accompanying IRI. The molecular basis of
this process lies in the interaction between leukocytes and
vascular endothelial cell surface adhesion factors, as well as in
the regulation of the expression of adhesion molecules by cytokines
and other factors. The effect of TNF-α on endothelial cells can
lead to the upregulation of VCAM-1, leading to the increased
adhesion of neutrophils to endothelial cells (40,42). The present study revealed
markedly higher numbers of adhering and migrating neutrophils in
the TNF-α treated HO-1+/− group than in the
HO-1+/+ control group, suggesting that the mGECs in the
HO-1+/− group exhibited a higher VCAM-1 expression
level, which was consistent with the results of western blot
analysis. The anti-VCAM-1 antibody incubation experiments revealed
that if the VCAM-1 on the mGEC surface in the HO+/−
group was blocked, the number of adhering and migrating neutrophils
significantly decreased, further indicating that the enhanced
adhesive and migratory ability of neutrophils through mGECs in the
HO-1+/− group was achieved by increasing VCAM-1
expression.
However, the present study has some limitations.
Although it was demonstrated that HO-1 knockdown upregulated VCAM-1
expression to mediate neutrophil infiltration, it remains critical
to further validate the detailed mechanisms through which HO-1
regulates VCAM-1 expression. Moreover, conditional HO-1 knockout
models are also necessary for further validation.
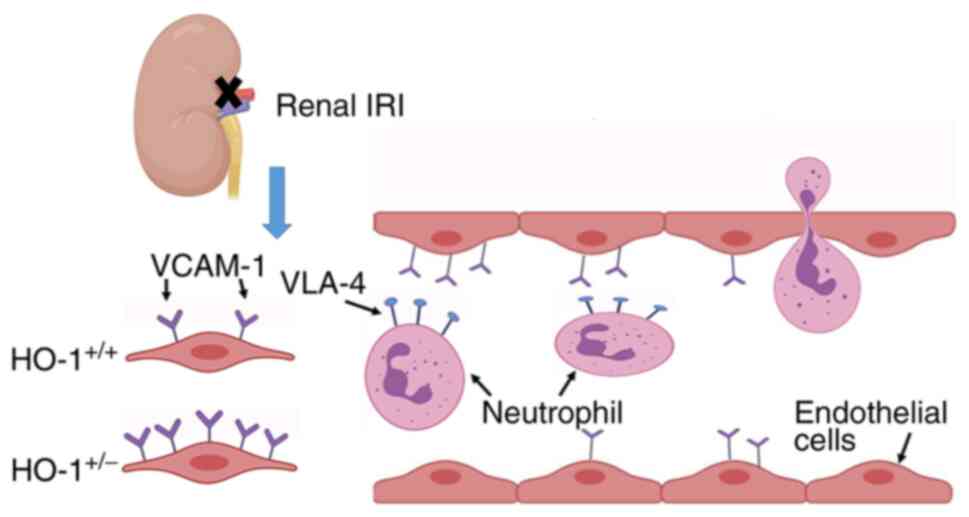
In conclusion, the present study demonstrated that
HO-1 knockdown in HO-1+/− mice exacerbated renal IRI via
the upregulation of VCAM-1 with a concomitant augmentation of
leukocyte recruitment and inflammatory damage. This, was further
verified by neutrophil adherence and migration on a vascular
basement membrane in vitro. The schematic diagram in
presented Fig. 5 illustrates the
mechanisms through which HO-1 protects against renal IRI injury
proposed herein. These data thus suggest novel potential strategies
for the clinical treatment of IR injury that typically follows
renal transplantation.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WB and YH conceived the study and established the
initial design of the study. YH, HL, JY, HZ, YK and XL performed
the experiments and analyzed the data. YH prepared the manuscript.
WB and YH confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments performed in the present study were
approved by the Institutional Animal Care and Use Committee of
Binzhou Medical University (PJ2018-10-16).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was funded by the National Natural Science
Foundation of China (grant no. 81801192) and by the Higher
Educational Science and Technology Program of Shandong Province
(grant no. J18KA141).
References
|
1
|
Dong Y, Zhang Q, Wen J, Chen T, He L, Wang
Y, Yin J, Wu R, Xue R, Li S, et al: Ischemic duration and frequency
determines AKI-to-CKD progression monitored by dynamic changes of
tubular biomarkers in IRI mice. Front Physiol. 10:1532019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sheashaa H, Lotfy A, Elhusseini F, Aziz
AA, Baiomy A, Awad S, Alsayed A, El-Gilany AH, Saad MA, Mahmoud K,
et al: Protective effect of adipose-derived mesenchymal stem cells
against acute kidney injury induced by ischemia-reperfusion in
Sprague-Dawley rats. Exp Ther Med. 11:1573–1580. 2016. View Article : Google Scholar
|
|
3
|
Cheng Q and Wang L: LncRNA XIST serves as
a ceRNA to regulate the expression of ASF1A, BRWD1M, and PFKFB2 in
kidney transplant acute kidney injury via sponging hsa-miR-212-3p
and hsa-miR-122-5p. Cell Cycle. 19:290–299. 2020. View Article : Google Scholar :
|
|
4
|
Yang C, Qi R and Yang B: Pathogenesis of
chronic allograft dysfunction progress to renal fibrosis. Adv Exp
Med Biol. 1165:101–116. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang A, Wan B, Jiang D, Wu Y, Ji P, Du Y
and Zhang G: The cytoprotective enzyme heme oxygenase-1 suppresses
pseudorabies virus replication in vitro. Front Microbiol.
11:4122020. View Article : Google Scholar
|
|
6
|
Haines DD and Tosaki A: Role of heme
oxygenases in cardiovascular syndromes and co-morbidities. Curr
Pharm Des. 24:2322–2325. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun XQ, Wu C, Qiu YB, Wu YX, Chen JL,
Huang JF, Chen D and Pang QF: Heme oxygenase-1 attenuates seawater
drowning-induced acute lung injury through a reduction in
inflammation and oxidative stress. Int Immunopharmacol.
74:1056342019. View Article : Google Scholar
|
|
8
|
Xu X, He X, Liu J, Qin J, Ye J and Fan M:
Protective effects of hydrogen-rich saline against renal
ischemia-reperfusion injury by increased expression of heme
oxygenase-1 in aged rats. Int J Clin Exp Pathol. 12:1488–1496.
2019.
|
|
9
|
Seo MS, Kim HJ, Kim H and Park SW: Ethyl
pyruvate directly attenuates active secretion of HMGB1 in proximal
tubular cells via induction of heme oxygenase-1. J Clin Med.
8:6292019. View Article : Google Scholar
|
|
10
|
Barakat M, Gabr MM, Zhran F, El-Adl M,
Hussein AM, Barakat N and Eldemerdash R: Upregulation of heme
oxygenase 1 (HO-1) attenuates kidney damage, oxidative stress and
inflammatory reaction during renal ischemia/reperfusion injury. Gen
Physiol Biophys. 37:193–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baan C, Peeters A, Lemos F, Uitterlinden
A, Doxiadis I, Claas F, Ijzermans J, Roodnat J and Weimar W:
Fundamental role for HO-1 in the self-protection of renal
allografts. Am J Transplant. 4:811–818. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rund KM, Peng S, Greite R, Claassen C,
Nolte F, Oger C, Galano JM, Balas L, Durand T, Chen R, et al:
Dietary omega-3 PUFA improved tubular function after ischemia
induced acute kidney injury in mice but did not attenuate
impairment of renal function. Prostaglandins Other Lipid Mediat.
146:1063862020. View Article : Google Scholar
|
|
13
|
Wadey RM, Connolly KD, Mathew D, Walters
G, Rees DA and James PE: Inflammatory adipocyte-derived
extracellular vesicles promote leukocyte attachment to vascular
endothelial cells. Atherosclerosis. 283:19–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coenen DM, Mastenbroek TG and Cosemans J:
Platelet interaction with activated endothelium: Mechanistic
insights from microfluidics. Blood. 130:2819–2828. 2017. View Article : Google Scholar
|
|
15
|
Cheng Q, McKeown SJ, Santos L, Santiago
FS, Khachigian LM, Morand EF and Hickey MJ: Macrophage migration
inhibitory factor increases leukocyte-endothelial interactions in
human endothelial cells via promotion of expression of adhesion
molecules. J Immunol. 185:1238–1247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma YR and Ma YH: MIP-1α enhances jurkat
cell transendothelial migration by up-regulating endothelial
adhesion molecules VCAM-1 and ICAM-1. Leuk Res. 38:1327–1331. 2014.
View Article : Google Scholar
|
|
17
|
Sumagin R, Lomakina E and Sarelius IH:
Leukocyte-endothelial cell interactions are linked to vascular
permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circ
Physiol. 295:H969–H977. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jung TW, Park HS, Jeong JH and Lee T:
Salsalate ameliorates the atherosclerotic response through HO-1-
and SIRT1-mediated suppression of ER stress and inflammation.
Inflamm Res. 68:655–663. 2019. View Article : Google Scholar
|
|
19
|
Pérez S, Pereda J, Sabater L and Sastre J:
Pancreatic ascites hemoglobin contributes to the systemic response
in acute pancreatitis. Free Radic Biol Med. 81:145–155. 2015.
View Article : Google Scholar
|
|
20
|
Vinchi F, Gastaldi S, Silengo L, Altruda F
and Tolosano E: Hemopexin prevents endothelial damage and liver
congestion in a mouse model of heme overload. Am J Pathol.
173:289–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hassan M, Bakar NS, Aziz MA, Basah NK and
Singh HJ: Leptin-induced increase in blood pressure and markers of
endothelial activation during pregnancy in Sprague Dawley rats is
prevented by resibufogenin, a marinobufagenin antagonist. Reprod
Biol. 20:184–190. 2020. View Article : Google Scholar
|
|
22
|
Li H, Peng W, Jian W, Li Y, Li Q, Li W and
Xu Y: ROCK inhibitor fasudil attenuated high glucose-induced MCP-1
and VCAM-1 expression and monocyte-endothelial cell adhesion.
Cardiovasc Diabetol. 11:652012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chakraborty S, Hu SY, Wu SH, Karmenyan A
and Chiou A: The interaction affinity between vascular cell
adhesion molecule-1 (VCAM-1) and very late antigen-4 (VLA-4)
analyzed by quantitative FRET. PLoS One. 10:e1213992015. View Article : Google Scholar
|
|
24
|
Hart R and Greaves DR: Chemerin
contributes to inflammation by promoting macrophage adhesion to
VCAM-1 and fibronectin through clustering of VLA-4 and VLA-5. J
Immunol. 185:3728–3739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao YA, Wagers AJ, Karsunky H, Zhao H,
Reeves R, Wong RJ, Stevenson DK, Weissman IL and Contag CH: Heme
oxygenase-1 deficiency leads to disrupted response to acute stress
in stem cells and progenitors. Blood. 112:4494–4502. 2008.
View Article : Google Scholar
|
|
26
|
Park JG, Ryu SY, Jung IH, Lee YH, Kang KJ,
Lee MR, Lee MN, Sonn SK, Lee JH, Lee H, et al: Evaluation of VCAM-1
anti-bodies as therapeutic agent for atherosclerosis in
apolipoprotein E-deficient mice. Atherosclerosis. 226:356–363.
2013. View Article : Google Scholar
|
|
27
|
Kucherenko MM, Marrone AK, Rishko VM,
Yatsenko AS, Klepzig A and Shcherbata HR: Paraffin-embedded and
frozen sections of Drosophila adult muscles. J Vis Exp.
27:24382010.
|
|
28
|
Chen CB, Liu LS, Zhou J, Wang XP, Han M,
Jiao XY, He XS and Yuan XP: Up-regulation of HMGB1 exacerbates
renal ischemia-reperfusion injury by stimulating inflammatory and
immune responses through the TLR4 signaling pathway in mice. Cell
Physiol Biochem. 41:2447–2460. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ellis JL and Yin C: Histological analyses
of acute alcoholic liver injury in zebrafish. J Vis Exp.
25:556302017.
|
|
30
|
Tanaka S, Tanaka T, Kawakami T, Takano H,
Sugahara M, Saito H, Higashijima Y, Yamaguchi J, Inagi R and
Nangaku M: Vascular adhesion protein-1 enhances neutrophil
infiltration by generation of hydrogen peroxide in renal
ischemia/reperfusion injury. Kidney Int. 92:154–164. 2017.
View Article : Google Scholar
|
|
31
|
Garcia-Cenador MB, Lorenzo-Gomez MF,
Herrero-Payo JJ, Ruiz J, de Obanos MP, Pascual J, Lopez-Novoa JM
and Garcia-Criado FJ: Cardiotrophin-1 administration protects from
ischemia-reperfusion renal injury and inflammation.
Transplantation. 96:1034–1042. 2013. View Article : Google Scholar
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Cui A, Xiang M, Xu M, Lu P, Wang S, Zou Y,
Qiao K, Jin C, Li Y, Lu M, et al: VCAM-1-mediated neutrophil
infiltration exacerbates ambient fine particle-induced lung injury.
Toxicol Lett. 302:60–74. 2019. View Article : Google Scholar
|
|
34
|
Block H, Herter JM, Rossaint J, Stadtmann
A, Kliche S, Lowell CA and Zarbock A: Crucial role of SLP-76 and
ADAP for neutrophil recruitment in mouse kidney
ischemia-reperfusion injury. J Exp Med. 209:407–421. 2012.
View Article : Google Scholar :
|
|
35
|
Rossi M, Delbauve S, Roumeguère T, Wespes
E, Leo O, Flamand V, Moine AL and Hougardy JM: HO-1 mitigates acute
kidney injury and subsequent kidney-lung cross-talk. Free Radic
Res. 53:1035–1043. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rossi M, Thierry A, Delbauve S, Preyat N,
Soares MP, Roumeguère T, Leo O, Flamand V, Moine AL and Hougardy
JM: Specific expression of heme oxygenase-1 by myeloid cells
modulates renal ischemia-reperfusion injury. Sci Rep. 7:1972017.
View Article : Google Scholar :
|
|
37
|
Su L, Jiang X, Yang C, Zhang J, Chen B, Li
Y, Yao S, Xie Q, Gomez H, Murugan R and Peng Z: Pannexin 1 mediates
ferroptosis that contributes to renal ischemia/reperfusion injury.
J Biol Chem. 294:19395–19404. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferenbach DA, Ramdas V, Spencer N, Marson
L, Anegon I, Hughes J and Kluth DC: Macrophages expressing heme
oxygenase-1 improve renal function in ischemia/reperfusion injury.
Mol Ther. 18:1706–1713. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding M, Tolbert E, Birkenbach M, Akhlaghi
F, Gohh R and Ghonem NS: Treprostinil, a prostacyclin analog,
ameliorates renal ischemia-reperfusion injury: Preclinical studies
in a rat model of acute kidney injury. Nephrol Dial Transplant.
36:257–266. 2021. View Article : Google Scholar
|
|
40
|
Buffone AJ, Anderson NR and Hammer DA:
Human neutrophils will crawl upstream on ICAM-1 If Mac-1 is
blocked. Biophys J. 117:1393–1404. 2019. View Article : Google Scholar
|
|
41
|
Chen WC, Chen NJ, Chen HP, Yu WK, Su VYF,
Chen H, Wu HH and Yang KY: Nintedanib reduces neutrophil chemotaxis
via activating GRK2 in bleomycin-induced pulmonary fibrosis. Int J
Mol Sci. 21:47352020. View Article : Google Scholar :
|
|
42
|
Taubitz A, Schwarz M, Eltrich N,
Lindenmeyer MT and Vielhauer V: Distinct contributions of TNF
receptor 1 and 2 to TNF-induced glomerular inflammation in mice.
PLoS One. 8:e681672013. View Article : Google Scholar : PubMed/NCBI
|