Introduction
Acute lung injury and acute respiratory distress
syndrome (ALI/ARDS) is one of the leading causes of morbidity and
mortality in critically ill patients. ALI/ARDS causes ~75,000
deaths every year in the USA alone (1,2).
The mortality associated with ALI/ARDS continues to be 25-40% and
few pharmacological interventions are able to improve this
mortality rate (1,3).
c-Jun-N-terminal kinase (JNK), a member of the
mitogen-activated protein kinase (MAPK) family, is a
stress-activated protein kinase that is modulated by the MAPK
signaling cascade (4). JNK is
also known as stress-activated kinase (SAPK) and it is a key factor
that regulates the physiological and pathological reactions of the
body (5,6). After sensing cellular or
extracellular stress, upstream MAP kinases, such as MAPK kinase
kinase (MAP3K) and MAPK kinase (MAP2K), can activate JNK by
threonine-tyrosine phosphorylation (7,8).
Betigeri et al (9) showed
that JNK can be activated by some stimuli, such as inflammatory
cytokines, bacterial endotoxins, osmotic shock, ultraviolet (UV)
radiation and hypoxia. JNK activation regulates the cellular
response to stress, such as adaptation to stress or programmed cell
death, including apoptosis and necrosis (8). These processes are carried out by
regulating the phosphorylation and subcellular localization of the
substrates and downstream factors of JNK (10). For example, JNK phosphorylates
activator protein-1 (AP-1) and increases the AP-1-dependent
transcription of genes involved in cell proliferation, inflammatory
cytokine production and cell death (10). Li et al (11) showed that JNK is involved in the
occurrence of ALI/ARDS by regulating the apoptosis of lung cells,
activating the nuclear factor kappa-B (NF-κB) pathway and
disrupting intercellular tight junctions (12).
Endoplasmic reticulum (ER) stress is a disorder of
ER protein-folding homeostasis caused by the accumulation of
unfolded and misfolded proteins. During the biosynthesis and
assembly of proteins, secretory complex and transmembrane proteins
enter the ER as unfolded polypeptides and emerge as folded and
processed proteins (13).
However, unfolding and misfolding occasionally occur and cause an
unfolded protein response (UPR) to repair improperly folded
proteins or degrade the proteins that cannot be repaired (14). When these mistakes accumulate and
exceed the recovery capacity, ER stress occurs. BiP (also known as
Grp78) enters the lumen of the ER after dissociating from protein
kinase RNA-activated-like ER kinase (PERK) (15), inositol-requiring transmembrane
kinase (IRE-1) (16) and
activating transcription factor 6 (AFT6) (17). These changes lead to the
dimerization and activation of PERK and IRE-1 as well as the
activation of ATF6 (15-17). Irreversible ER stress and ER
homeostasis disturbance are related to the deterioration of
cellular function and even cell death (13). According to previous studies
(13,16,18,19), a number of cellular insults may
lead to UPR and ER stress, such as infection, hypoxia, reactive
oxygen species (ROS), nutrient deprivation and lack of ATP.
Previous studies (13,16,18) have suggested that ER stress is
critical to various diseases, such as atherosclerosis, diabetes,
non-alcoholic fatty liver disease/non-alcohol related
steatohepatitis, obesity and cancer.
Mitochondria are the major producers of ATP in
mammalian cells and serve critical roles in a variety of events
associated with the initiation of apoptosis (20). These roles include receiving
stress signals from the cytosol and other organelles, disrupting
electron transport and energy generation, altering the cellular
oxidation-reduction state, generating excessive levels of ROS,
inducing mitochondrial transmembrane potential (ΔΨ) loss and
releasing cytochrome c to the cytosol (21). Moreover, mitochondria are
involved in the upregulation of proapoptotic proteins and
downregulation of antiapoptotic proteins and these functions
include interacting with apoptosis protease-activating factor 1,
triggering the activation of caspase-9 and leading to subsequent
apoptotic processes (20,21).
During these actions, some proteins are translated in the cytosol
and imported into the mitochondrion. These proteins carry messages
and initiate the changes. JNK is one such protein. Activated JNK
can interact with Sab, which is expressed on the mitochondrial
outer membrane and translocate to the mitochondria (8). The location of JNK on mitochondria
can lead to a sequence of events, such as sustained activation of
JNK, loss of ΔΨ, overgeneration of ROS and even cell death
(22). Sab is composed of an
N-terminal SH3 domain binding site in the intermembrane space, one
membrane spanning domain and a JNK kinase interaction motif (KIM)
on the C-terminus that faces the cytoplasm (23). Sab is the only docking site for
JNK on mitochondria. The knockdown of Sab or inhibition of the
JNK-Sab interaction using KIM1 peptides can block the translocation
of JNK to mitochondria and inhibit JNK-induced sequence events in
mitochondria (24-26). According to a study conducted by
Li et al (27), the
pathological changes in ALI/ARDS are partly associated with the
abnormal regulation of mitochondria and maintaining the stability
of mitochondrial function is vital to ameliorating ALI/ARDS as
mitochondria serve crucial roles in energy generation, ROS
production and cell survival, autophagy and apoptosis
modulation.
ER stress can activate JNK through the IRE1α
pathway. Studies (28,29) have also confirmed that JNK
activation induced by ER stress can also interact with Sab and then
lead to the disruption of mitochondrial homeostasis and function.
Tunicamycin or brefeldin A (BFA), specific inducers of ER stress,
can induce cell apoptosis by triggering ER-induced sustained JNK
activation and subsequent JNK-mitochondria localization and
silencing of Sab can reverse BFA-induced sustained JNK activation
(28).
Accumulating evidence suggests that ER stress and
JNK mitochondrial localization serve important roles in
mitochondrial dysfunction and cell death and the key component Sab
might be a potentially attractive target for ALI/ARDS treatment.
However, abnormalities in ER stress and the localization of JNK to
the mitochondria are rarely reported with respect to the occurrence
and progression of ALI/ARDS and their underlying role in ALI/ARDS
remains unknown and requires further study. Therefore, the present
study hypothesized that abnormal activation of the
JNK-mitochondrial pathway could significantly disrupt the normal
physiological function of lung cells, resulting in the occurrence
of ALI/ARDS and also suggested that selective inhibition of JNK
mitochondrial localization by Tat-SabKIM1 had a
protective effect against the mitochondrial dysfunction and cell
death caused by ER stress in mice with lipopolysaccharide
(LPS)-induced ALI/ARDS.
Materials and methods
Reagents
Antibodies against Bip, p-PERK, PERK, p-IRE1, IRE1,
ATF6, Chop and Caspase-3 were obtained from Cell Signaling
Technology, Inc. Antibodies against cytochrome c, cytochrome c
oxidase IV (COX IV) and GAPDH were purchased from Abcam.
Phosphorylated (p)-Bcl-2 (Ser70) rabbit mAb, p-JNK (Thr183/Tyr185)
rabbit mAb and JNK rabbit mAb were obtained from Cell Signaling
Technology, Inc. LC3 rabbit mAb was purchased from Santa Cruz
Biotechnology, Inc. The MDA Assay kit (TBA method) and Hydrogen
Peroxide Assay kit were obtained from Nanjing Jiancheng
Bioengineering Institute. An In Situ Cell Death Detection
kit was obtained from Roche Applied Science. Dexamethasone (DEX)
was purchased from MilliporeSigma. Tat-scramble (LPS VFG DVG APS
RLP EVS LSP PRR RQR RKK RG-NH2) and Tat-SabKIM1 (GFE SLS
VPS PLD LSG PRV VAP PRR RQR RKK RG-NH2) were purchased from
NeoPeptide. TRIzol was purchased from Thermo Fisher Scientific,
Inc.
Animal procedures
The animal procedures conducted in the present study
were approved by the Animal Care and Use Committee of the Fourth
Military Medical University (approval no. TDLL20160194) and were
carried out in accordance with the National Institutes of Health
Guide for Care and Use of Laboratory Animals (30). A total of 90 male BALB/c mice (8
weeks old and weighing 20-24 g) were purchased from the Animal
Center of the Fourth Military Medical University and maintained on
a 12-h light/dark cycle with free access to food and water; the
ambient temperature was 18-26°C and the relative humidity was
40-70%. The BALB/c mice were randomly divided into the following
experimental groups (n=15): the control group, LPS 12 h group
(LPS-induced ALI/ARDS was established by intraperitoneal injection
of 5 mg/kg LPS), LPS 24 h group, DEX pretreatment group (in which
the mice were pretreated intraperitoneally with 2.5 mg/kg body
weight DEX 30 min before modelling), Tat-SabKIM1
pretreatment group (in which the mice were pretreated with tracheal
injection of 2 mg/kg body weight Tat-SabKIM1 30 min
before modelling) and SP600125 pretreatment group (in which the
mice were pretreated with intravenous injection of 20 mg/kg body
weight SP600125 30 min before modelling). At the preset time (24 h
if not otherwise specified), the mice were euthanized by
pentobarbital overdose (200 mg/kg, intraperitoneal injection), the
lungs were harvested and samples were collected.
Lung wet (W)/dry (D) weight ratio
To evaluate the severity of pulmonary oedema, the
W/D ratio of the lung tissue was calculated. Briefly, the left
lungs of the mice were harvested and weighed to determine the wet
weight. Then, the lungs were placed in an oven and incubated at
75°C for 72 h to obtain a constant weight, which was denoted as the
dry weight. The W/D ratio was calculated by dividing the wet weight
by the dry weight.
Assessment of lung cell apoptosis
To quantify cell apoptosis in the injured mouse
lungs, a TUNEL assay was conducted using the In-Situ Cell
Death Detection kit according to the protocol provided by the
manufacturer. Lung tissues were fixed with 4% paraformaldehyde for
24 h at room temperature. Then the lung tissues were paraffin
embedded and sectioned at 5 µm). After dewaxing, TUNEL
working solution was added to the tissue sections for 1 h at 37°C
to label apoptotic cells, which were then stained with the nuclear
stain DAPI (5 µg/ml; Merck KGaA) for 5 min at room
temperature. The slides were mounted with 50% glycerol (Merck
KGaA). The result was analyzed by a digital imaging system
(Pannoramic Viewer 1.15.3; Silicon Graphic, Inc.). Images from
three slides/groups were randomly captured and the cells exhibiting
positive staining for apoptosis were counted manually.
Histopathological evaluation
Histopathological evaluation was conducted by
staining the lung tissue with hematoxylin and eosin (H&E).
First, the lung tissue of the mice was harvested at the preset
time, fixed with 4% paraformaldehyde at room temperature for 24 h,
embedded in paraffin and cut into 5-µm sections before
staining with H&E (at room temperature for 3 min). The
differences among the groups were examined by optical
microscopy.
XBP1 mRNA splicing
XBP1 mRNA splicing was detected by reverse
transcription PCR. The tissue samples were homogenized using TRIzol
and mRNA was collected. Then, reverse transcription was conducted
using the PrimeScript RT reagent kit (Takara Bio, Inc.). The
primers used to detect XBP1 (spliced form) and XBP1 mRNA were
5′-GAA CCA GGA GTT AAG AAC ACG-3′ (forward) and 5′-AGG CAA CAG TGT
CAG AGT CC-3′ (reverse). Finally, the PCR products were analyzed by
agarose gel electrophoresis. The DNA ladder we used was purchased
from Beyotime Institute of Biotechnology (cat. no. D0107) and the
EtBr was purchased from MilliporeSigma.
Neutrophil numbers in bronchoalveolar
lavage fluid (BALF)
At the preset time, the mice were euthanized and the
lungs were surgically removed. Then, the lungs were lavaged with 1
ml ice-cold PBS three times. Then, the cells in the BALF were
harvested by centrifugation (2,500 × g at 4°C for 5 min). The
number of neutrophils was calculated after staining with Wright's
stain (at room temperature for 3 min) according to the
manufacturer's instructions and pale purple neutrophils were
counted using a cell counting plate.
Evans blue extravasation assessment
Evans blue extravasation analysis was used to
measure the barrier permeability of the lungs. Evans blue dye
(MilliporeSigma; 20 mg/kg) was injected into the mice through the
tail vein 30 min before the mice were euthanized and the lungs were
surgically removed. Then, Evans blue dye was extracted from the
lung tissue by incubation in formamide (3 ml/100 mg) at room
temperature for 24 h. Finally, the total Evans blue (µg/g)
in each sample was calculated using spectrophotometry (620 nm).
Survival analysis
To observe the mortality rates, the mice were
administered LPS (50 mg/kg) intraperitoneally 30 min after
pretreatment with DEX (DEX pretreatment group) or
Tat-SabKIM1 (Tat-SabKIM1 pretreatment group).
Then, the mortality of the mice in each group was recorded every 6
h for 3 days.
Cell culture and treatment
As blood-air barrier disruption is the vital
pathophysiological changes during ALI/ARDS and alveolar epithelial
cell and vascular endothelium are the important element of
alveolar-capillary membrane. So immortalized human umbilical vein
endothelial cells (HUVECs) and A549 cells were used to represent
endothelial cells and epithelial cells respectively in this study.
HUVECs and A549 cells were purchased from Jenniobio Biotechnology
Co. HUVECs were cultured in F-12K medium (0.1 mg/ml heparin and
0.03 mg/ml endothelial cell growth supplement; Gibco; Thermo Fisher
Scientific, Inc.) with 10% fetal bovine serum (Cytiva) at 37°C and
in a 5% CO2 atmosphere and A549 cells were cultured in
RPMI 1640 medium with 10% fetal bovine serum at 37°C and in a 5%
CO2 atmosphere. LPS challenge was performed by exposing
HUVECs to 1 µg/ml LPS at 37°C for 12 h before harvesting or
testing.
Preparation of mitochondrial and
cytosolic/nuclear proteins
Mitochondrial and cytosolic/nuclear proteins were
prepared by isolating mitochondria from cells or tissue as
described by Xu et al (22). Briefly, the mouse lungs were
washed and homogenized using isolation buffer (210 mM mannitol, 70
mM sucrose, 5 mM HEPES, 1 mM EGTA and 0.5 mg/ml BSA, pH=7.4). Next,
the homogenate was centrifuged at 1,000 × g for 10 min at 4°C and
the supernatant was collected and centrifuged at 10,000 × g for 10
min at 4°C. This second supernatant was used as the soluble
cytosolic/nuclear fraction with excluded mitochondria and the
sedimentation pellet was resuspended in lysis buffer for western
blot analysis of the mitochondrial proteins. COX IV was used as an
internal mitochondrial control and GAPDH served as the control for
other organelles.
Western blot analysis
Protein was extracted by RIPA lysis buffer with
protease inhibitor cocktail and the protein concentration was
determined by a BCA kit (Beyotime Institute of Biotechnology)
according to the protocol provided by the manufacturer.
Subsequently, equivalent amounts of proteins (20 µg) were
separated by 12% SDS-PAGE and transferred to nitrocellulose
membranes. Then, the membranes were blocked with 10% nonfat dry
milk in Tris-buffered saline (TBS) at room temperature for 30 min
and probed overnight with primary antibodies at 4°C. Subsequently,
the membranes were washed with TBST and then incubated with an
appropriate horseradish peroxidase-conjugated secondary antibody
(1:10,000; cat. no. ab6721; Abcam) at room temperature for 2 h. The
immunoreactive target proteins were detected by an enhanced
chemiluminescent detection system (Thermo Fisher Scientific, Inc.).
Band intensities were quantified using Image Lab 4.1 (Bio-Rad
Laboratories, Inc.). The following primary antibodies were used:
anti-cytochrome c (1:10,000, cat. no. ab133504, Abcam), anti-p-JNK
(1:1,000, cat. no. 4668, Cell Signaling Technology, Inc.),
anti-GAPDH (1:2500, cat. no. ab181602, Abcam), anti-JNK (1:1,000,
cat. no. 9258, Cell Signaling Technology, Inc.), anti-COX IV
(1:1,000, cat. no. ab202554, Abcam), anti-LC3 (1:1,000, cat. no.
sc-398822, Santa Cruz Biotechnology, Inc.), anti-phospho-Bcl-2
(Ser70) rabbit mAb (1:1,000, cat. no. 2827, Cell Signaling
Technology, Inc.) and anti-cleaved caspase-3 (1:1,000, cat. no.
9664, Cell Signaling Technology, Inc.).
Determination of cell apoptosis
Cell apoptosis was detected by double labelling with
annexin-V-FITC (Molecular Probes; Thermo Fisher Scientific, Inc.)
and PI (Molecular Probes; Thermo Fisher Scientific, Inc.). Briefly,
cells from different groups were harvested with trypsin and washed
with PBS. Annexin V-FITC and PI were added to the cells as the
manual described (15 min at room temperature in the dark). After
labelling, the cell apoptosis ratios were detected and analyzed
with flow cytometer (FACScan; BD Biosciences) and the software
supplied with the machine. The percentage of early and late
apoptotic cells were calculated.
Mitochondrial homeostasis
As the mito-JNK pathway participates in LPS-induced
ALI/ARDS, the changes in ROS production and mitochondrial functions
following LPS exposure were explored. Briefly, to detect the
changes in superoxide anion content, the cells from different
groups were stained with dihydroethidium (10 µM) and
cultured at 37°C for 30 min to load the fluorescent indicator.
Then, the medium was replaced and the fluorescence intensity, which
reflected the superoxide anion concentration, was recorded by
fluorescence microscopy.
Mitochondrial membrane permeability (ΔΨ) was also
measured using a JC-1 fluorescence ratio of 590:538 nm. Briefly, to
detect changes in mitochondrial membrane permeability, the cells
from the different groups were stained with JC-1 (10 µM) and
cultured at 37°C for 20 min to allow the fluorescent indicator to
enter the cells. Then, the medium was replaced. The fluorescence
intensity (590 and 538 nm) was recorded by fluorescence microscopy
and a fluorescence ratio of 590:538 nm reflected ΔΨ.
Statistical analysis
Statistical analyses were conducted using GraphPad
Prism v8 software (GraphPad Software, Inc.). The data are reported
as the mean ± standard error of the mean. Comparisons between
experimental groups were performed by one-way ANOVA and the
Bonferroni test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ER stress is enhanced during LPS-induced
ALI/ARDS
To characterize the role and status of ER stress in
LPS-induced ALI/ARDS, the expression of the ER stress sensors
Bip/GRP78 and ATF6 and the phosphorylation of PERK and IRE-1 were
determined by western blotting. The data (as shown in Fig. 1A) showed that the expression of
both Bip and ATF6 was significantly increased after LPS challenge
compared with those in the control group and this was especially
true in the LPS 24 h group; these results indicated the activation
of ER stress. In contrast, the expression of these proteins in the
DEX pretreatment group was decreased compared with that in the LPS
24 h group. In addition, LPS also increased the phosphorylation of
PERK, which was slightly but significantly attenuated by DEX
pretreatment (Fig. 1A). A
similar change was also detected in IRE-1 (Fig. 1A). The results suggested that
anti-inflammatory therapy with dexamethasone could alleviate
LPS-induced ER stress during ALI/ARDS.
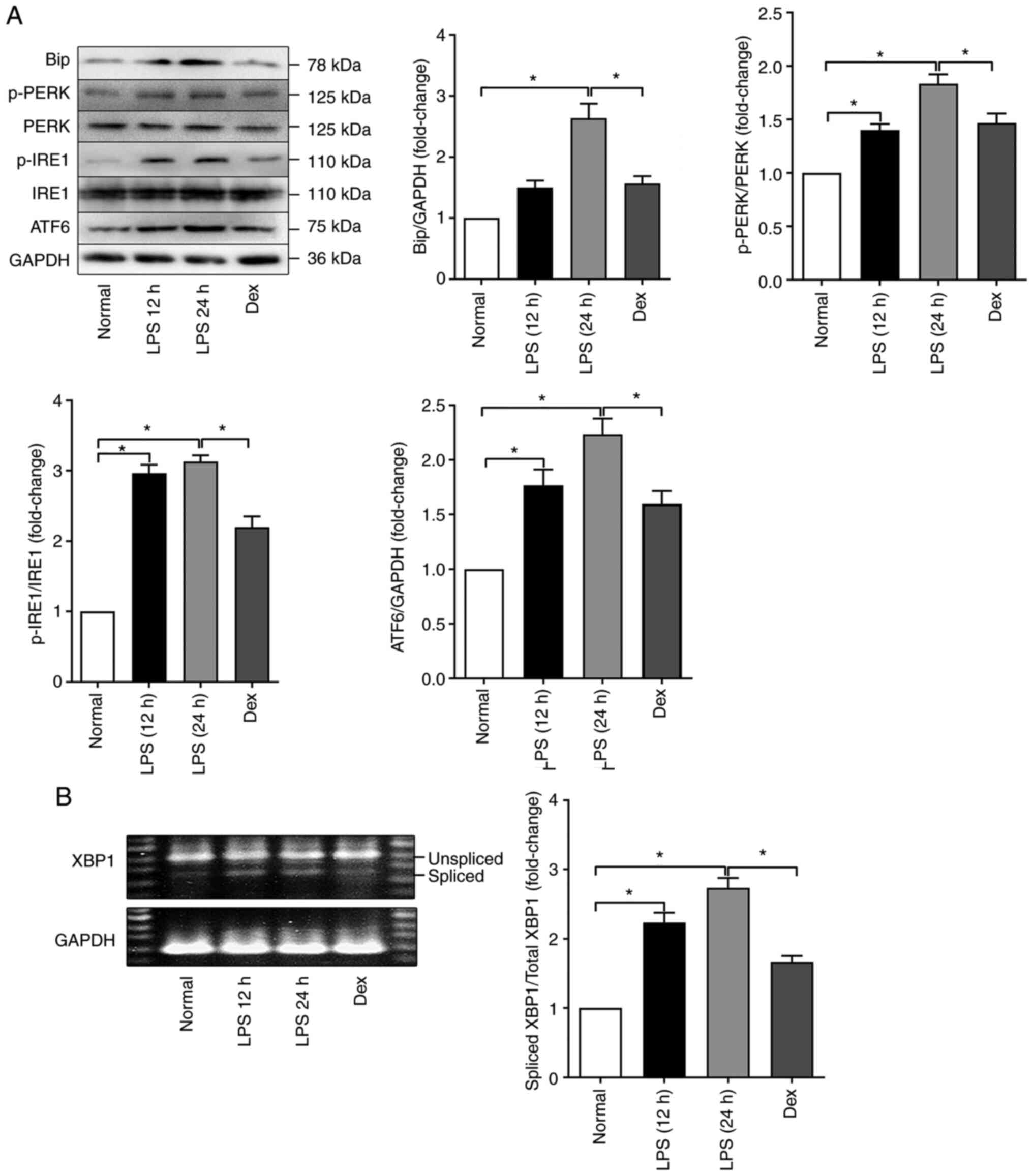 | Figure 1LPS treatment induces ER stress in
the lungs of mice. (A) Representative western blots of ER
stress-related genes (Bip, p-PERK, PERK, p-IRE1, IRE1 and ATF6) in
the Normal, LPS 12 h, LPS 24 h and Dex groups. (B) Splicing of
XBP-1 in the Normal, LPS 12 h, LPS 24 h and Dex groups. The data
are expressed as the mean ± standard error of the mean, n=4/group,
*P<0.05. LPS, lipopolysaccharide; ER, endoplasmic
reticulum; p-, phosphorylated; PERK, protein kinase
RNA-activated-like ER kinase; IRE1, inositol-requiring
transmembrane kinase; ATF6, activating transcription factor 6. |
To further confirm the occurrence of ER stress, the
splicing of XBP-1 was also detected; this is the active form of the
transcription factor XBP-1. XBP-1 is one of the downstream
signaling molecules of IRE-1 and it can increase protein folding,
reduce protein translation and eventually restore protein folding
homeostasis. As shown in Fig.
1B, the amount of spliced XBP-1 was elevated in the lung after
intraperitoneal LPS injection. Collectively, these results clearly
indicated that ER stress in the lung was enhanced during
LPS-induced ALI/ARDS.
ER stress can induce mitochondrial
dysfunction and cell death in A549 cells and HUVECs
Next, the effect of ER stress pathway activation on
lung cells, which were represented by A549 cells and HUVECs was
explored. First the expression of apoptosis-related genes, such as
p-Bcl-2, cleaved caspase-3 and Bax and the leakage of cytochrome c
into the cytosol were detected (Fig.
2A). After treatment with the ER stress activator tunicamycin
(20 µg/ml), more cytochrome c was released from mitochondria
to the cytosol in both A549 cells and HUVECs and less p-Bcl-2 and
more cleaved caspase-3 and Bax were detected in the activated
groups. These results indicated that ER stress activated by
tunicamycin could lead to the excessive activation of apoptosis
pathways.
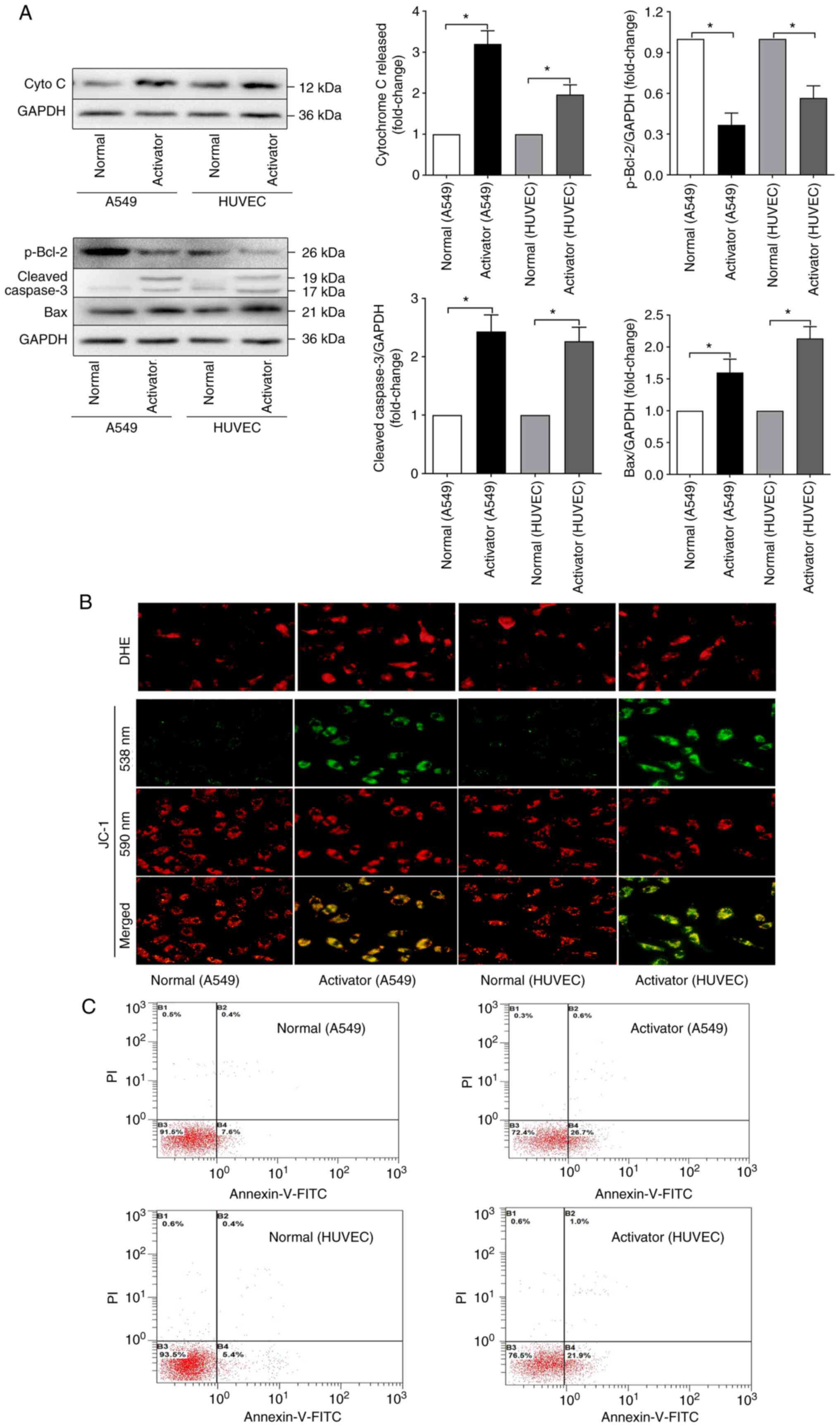 | Figure 2ER stress can induce mitochondrial
dysfunction and cell death. (A) Representative western blots of
cyto c leakage to the cytosol, p-Bcl-2, cleaved caspase3 and Bax in
HUVECs and A549 cells treated with the ER stress activator
tunicamycin. (B) Changes in superoxide anion content and
mitochondrial membrane permeability (ΔΨ) after HUVECs and A549
cells were treated with the ER stress activator tunicamycin;
magnification, ×200. (C) Cell apoptosis detected by double
labelling of HUVECs and A549 cells treated with the ER stress
activator tunicamycin with annexin-V-FITC and PI. The data are
expressed as the mean ± standard error of the mean, n=4/group,
*P<0.05. ER, endoplasmic reticulum; cyto c,
cytochrome c; HUVECs, human umbilical vein endothelial cells; p-,
phosphorylated; DHE, dihydroethidium. |
The leakage of cytochrome c indicated that an
oxidant-antioxidant imbalance might occur. For confirmation, the
changes in ROS production and mitochondrial function upon ER stress
were further explored. The results (as shown in Fig. 2B) showed that tunicamycin
exposure could significantly elevate the production of ROS in both
A549 cells and HUVECs. The mitochondrial membrane permeability (ΔΨ)
and ΔΨ dissipation was detected using a JC-1 fluorescence ratio of
590:538 nm. These results suggest that overactivation of ER stress
could induce mitochondrial dysfunction and ROS accumulation and
even lead to apoptosis.
Cell apoptosis were quantitated by flow cytometry
with annexin-V-FITC and PI double labelling. The results (Fig. 2C) showed that tunicamycin
increased the percentage of apoptotic cells to >20%. All of
these results indicated that ER stress could induce mitochondrial
dysfunction and cell death in A549 cells and HUVECs.
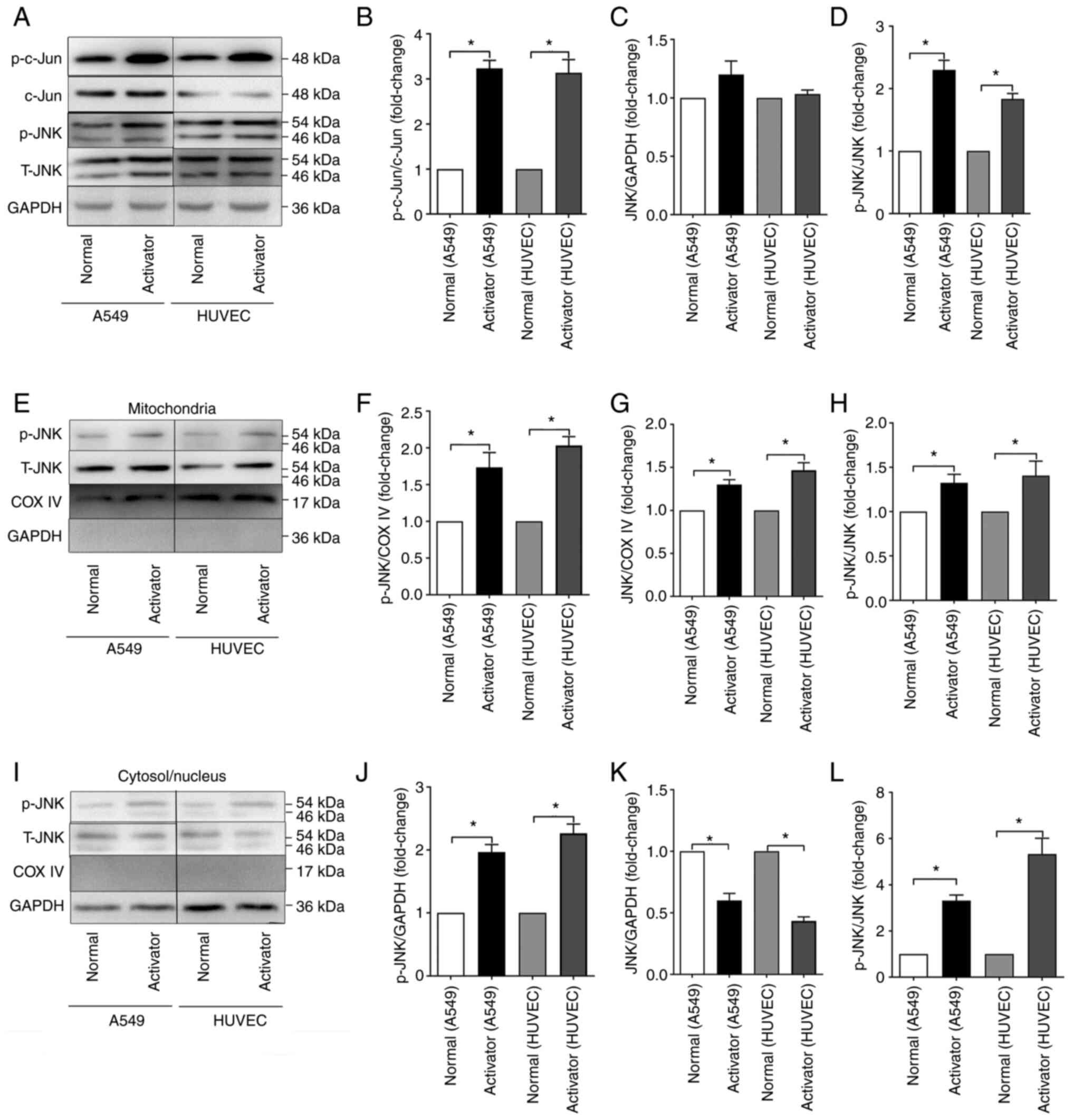
ER stress can induce activation of JNK
and JNK mitochondrial localization in A549 cells and HUVECs
To elucidate the role of JNK, especially the
mitochondria-JNK interaction, during ER stress, the expression of
p-JNK and JNK was assessed in total cellular proteins,
mitochondrial proteins and cytosolic/nuclear proteins by western
blotting. According to the results (Fig. 3), ER stress induced by
tunicamycin could activate the JNK pathway by phosphorylating JNK
and c-jun. Furthermore, it was found that the phosphorylation level
of JNK was significantly elevated in both the mitochondria and
cytosol/nucleus after tunicamycin challenge. Although the total JNK
in the mitochondria was also elevated, that in the cytosol/nucleus
was decreased, indicating that ER stress could induce JNK
translocation from the cytosol/nucleus to mitochondria in A549
cells and HUVECs.
LPS induces JNK activation and JNK
mitochondrial localization in mice
To further observe the role of the mitochondria-JNK
interaction in LPS-induced ALI/ARDS, the activation of JNK pathways
in the lungs was detected following LPS challenge. According to the
results (Fig. 4), p-JNK and
c-jun in the lung tissues were elevated following intraperitoneal
LPS injection, especially in the 24 h group. The amount of JNK
mitochondrial localization during LPS-induced ALI/ARDS was also
detected by western blotting. Similar to tunicamycin exposure, LPS
exposure could induce the phosphorylation of JNK in both
mitochondria and the cytosol/nucleus. The total JNK in the
mitochondria was also elevated in the LPS groups, whereas that in
the cytosol/nucleus was decreased, indicating that in LPS-induced
ALI/ARDS, JNK could translocate from the cytosol/nucleus to
mitochondria.
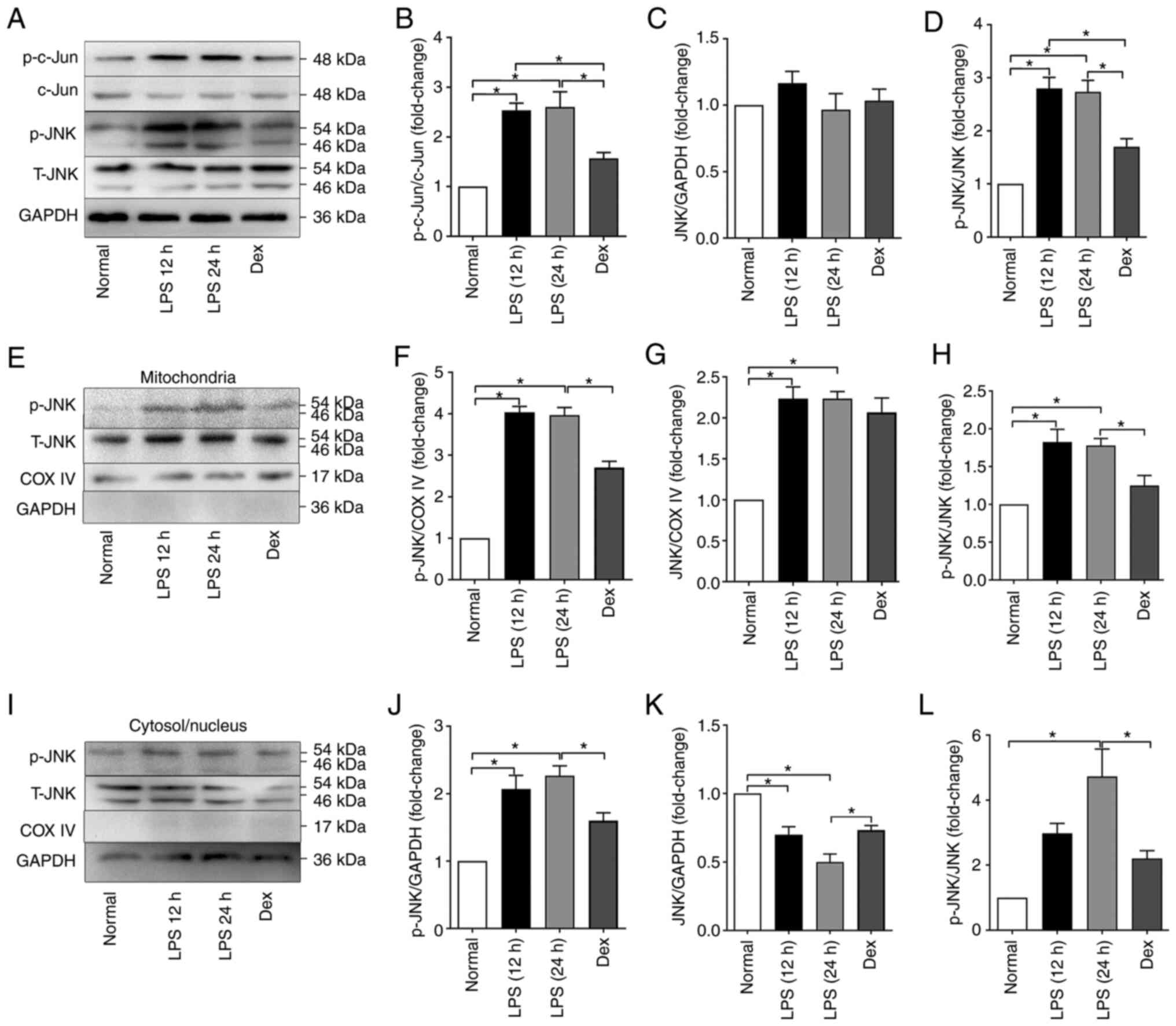 | Figure 4LPS induced JNK activation and JNK
mitochondrial localization in mice. (A) Representative western
blots of p-c-Jun, c-Jun, p-JNK and JNK in the total proteins of the
Normal, LPS 12 h, LPS 24 h and Dex groups. (B) Normalized ratio of
p-c-Jun/c-Jun in the total proteins. (C) Relative content of JNK in
the total proteins. (D) Normalized ratio of p-JNK/JNK in the total
proteins. (E) Representative western blots of p-JNK and JNK in the
mitochondrial proteins. (F) Relative content of p-JNK in the
mitochondrial proteins. (G) Relative content of JNK in the
mitochondrial proteins. (H) Normalized ratio of p-JNK/JNK in the
mitochondrial proteins. (I) Representative western blots of p-JNK
and JNK in the cytosolic/nuclear proteins. (J) Relative content of
p-JNK in the cytosolic/nuclear proteins. (K) Relative content of
JNK in the cytosolic/nuclear proteins. (L) Normalized ratio of
p-JNK/JNK in the cytosolic/nuclear proteins. The data are expressed
as the mean ± standard error of the mean, n=4/group,
*P<0.05. LPS, lipopolysaccharide; p-, phosphorylated;
t-, total; Dex, dexamethasone; COX IV, cytochrome c oxidase IV. |
The protective role of DEX was also examined. As
shown in Fig. 4, DEX
pretreatment significantly alleviated the LPS-induced increase in
phosphorylated JNK in both the mitochondria and cytosol/nucleus,
whereas it did not affect the total JNK content.
Efficiency of Tat-SabKIM1 and
SP600125 on JNK activation
To further explore the effect of JNK mitochondrial
localization during LPS-induced ALI/ARDS, the protective effects of
SP600125 and Tat-SabKIM1 were first verified. SP600125 is a
specific inhibitor of the JNK pathway. Tat-SabKIM1 is a
peptide that can act on the SabKIM1 domain expressed in
mitochondria and selectively block JNK translocation to
mitochondria both in vitro and in vivo by inhibiting
the binding of JNK to SabKIM1. Tat-SabKIM1
does not exert any effect on JNK translocation to the nucleus. As
shown in Fig. 5, tracheal
Tat-SabKIM1 injection significantly decreased the level
of phosphorylated JNK and the total JNK level in mitochondria.
Tat-SabKIM1 also inhibited the LPS-induced decrease in
the total JNK level in the cytosol/nucleus. These results indicated
that Tat-SabKIM1 could specifically inhibit JNK
localization to mitochondria and mito-JNK signal activation without
affecting cytosolic/nuclear JNK activation. SP600125 also inhibited
the phosphorylation of c-jun and JNK. However, it had no selective
effect on the distribution of JNK.
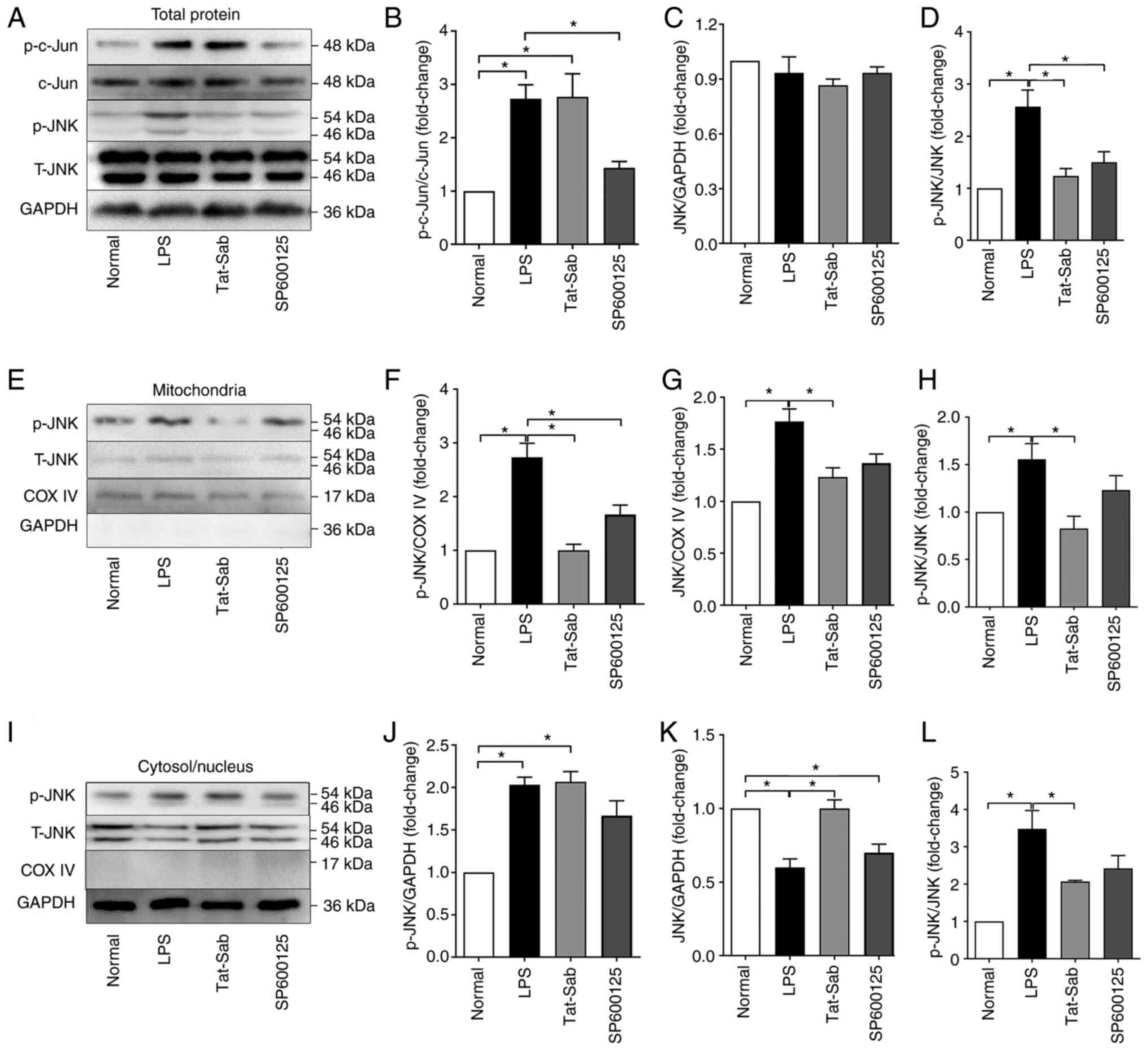 | Figure 5Efficiency of Tat-SabKIM1
and SP600125 on the activation of JNK. (A) Representative western
blots of p-c-Jun, c-Jun, p-JNK and JNK in the total proteins of the
Normal, LPS, Tat-SabKIM1 and SP600125 groups. (B)
Normalized ratio of p-c-Jun/c-Jun in the total proteins. (C)
Relative content of JNK in the total proteins. (D) Normalized ratio
of p-JNK/JNK in the total proteins. (E) Representative western
blots of p-JNK and JNK in the mitochondrial proteins. (F) Relative
content of p-JNK in the mitochondrial proteins. (G) Relative
content of JNK in the mitochondrial proteins. (H) Normalized ratio
of p-JNK/JNK in the mitochondrial proteins. (I) Representative
western blots of p-JNK and JNK in the cytosolic/nuclear proteins.
(J) Relative content of p-JNK in the cytosolic/nuclear proteins.
(K) Relative content of JNK in the cytosolic/nuclear proteins. (L)
Normalized ratio of p-JNK/JNK in the cytosolic/nuclear proteins.
The data are expressed as the mean ± standard error of the mean,
n=4/group, *P<0.05. KIM, kinase interaction motif;
p-, phosphorylated; t-, total; COX IV, cytochrome c oxidase IV. |
Treatment effect of
Tat-SabKIM1 on LPS-induced ALI/ARDS
As Tat-SabKIM1 can selectively inhibit
JNK mitochondrial localization, it was further utilized to explore
the role of mitochondrial JNK pathway activation in the progression
of ALI/ARDS. As shown in Fig.
6A, the mice in the control group had clear and normal alveolar
structures. Tat-SabKIM1 alleviated LPS injection-induced
lung tissue structure destruction, alveolar wall thickening,
interstitial lung inflammatory cells and liquid exudation.
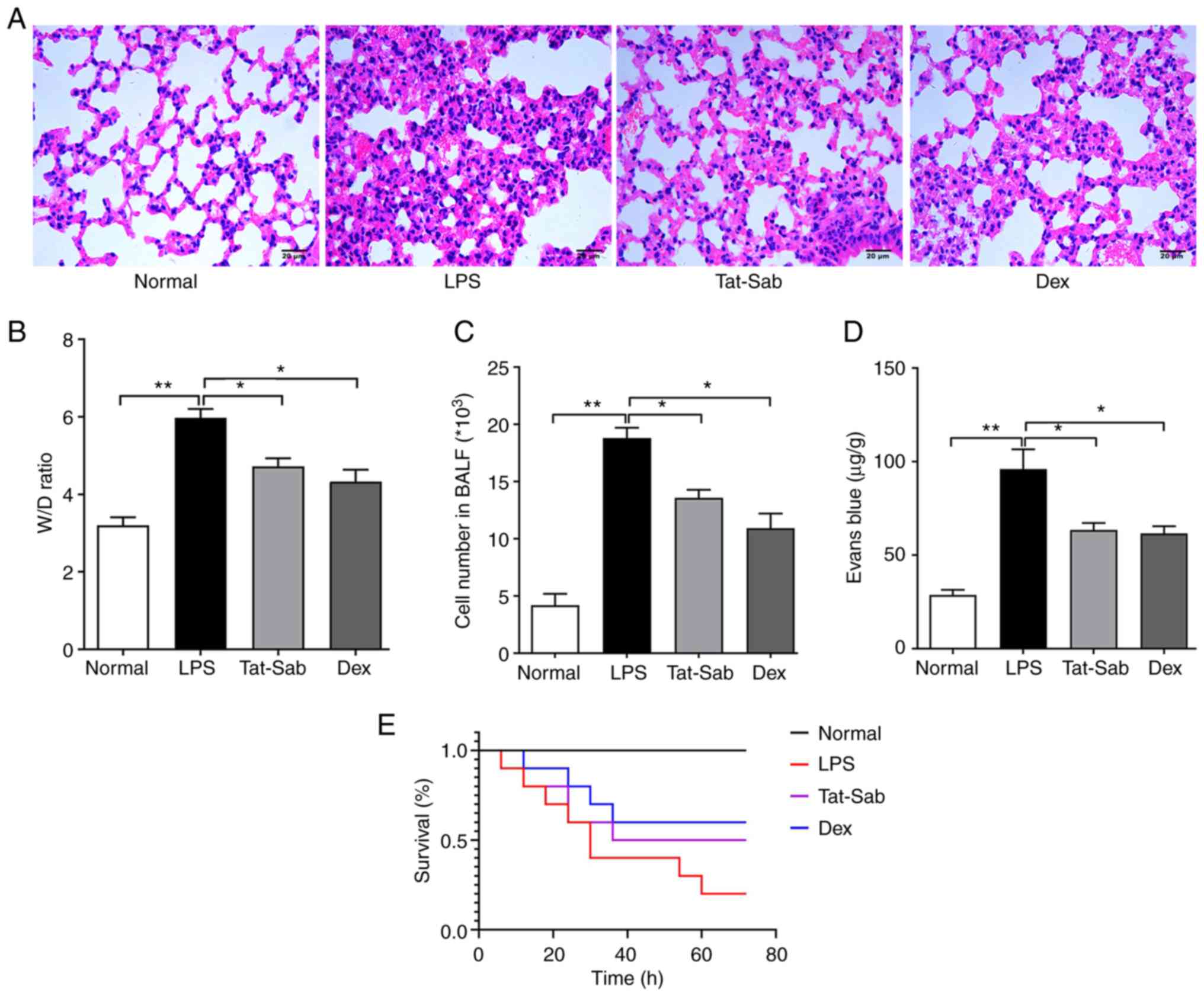 | Figure 6Treatment effect of
Tat-SabKIM1 on LPS-induced ALI/ARDS. (A) Histological
changes in mouse lung tissue of the Normal, LPS,
Tat-SabKIM1 and Dex groups; magnification, ×200. (B)
Change in the lung W/D ratio of the Normal, LPS,
Tat-SabKIM1 and Dex groups. (C) Cell number in BALF of
the Normal, LPS, Tat-SabKIM1 and Dex groups. (D) Evans
blue extravasation assessment of the Normal, LPS,
Tat-SabKIM1 and Dex groups. The data are expressed as
the mean ± standard error of the mean, n=4/group,
*P<0.05, **P<0.01. (E) Survival
analysis of the Normal, LPS, Tat-SabKIM1 and Dex groups.
n=10/group, *P<0.05. LPS, lipopolysaccharide;
ALI/ARDS, acute lung injury and acute respiratory distress
syndrome; p-, phosphorylated; t-, total; Dex, dexamethasone; W/D,
wet/dry; BALF. |
To observe pulmonary oedema, the W/D rate was also
measured (Fig. 6B). LPS caused
pulmonary oedema and the W/D ratio of the LPS group was nearly
twice that of the control group. Pretreatment with
Tat-SabKIM1 significantly alleviated tissue oedema.
In addition, the cell number in the BALF and Evans
blue extravasation were measured to evaluate the barrier
permeability of the lungs. The results (Fig. 6C and D) suggested that
Tat-SabKIM1 could maintain the barrier function of the
lung and exert a protective effect against ALI/ARDS.
These results were consistent with those of the
survival analysis (Fig. 6E). The
number of deaths 72 h after LPS in the LPS group was significantly
higher than that in the control groups. Tat-SabKIM1
treatment significantly improved the mouse survival rates compared
with LPS treatment alone.
Effect of Tat-SabKIM1 on cell
death in the mouse lung
Apoptosis was also examined during LPS-induced
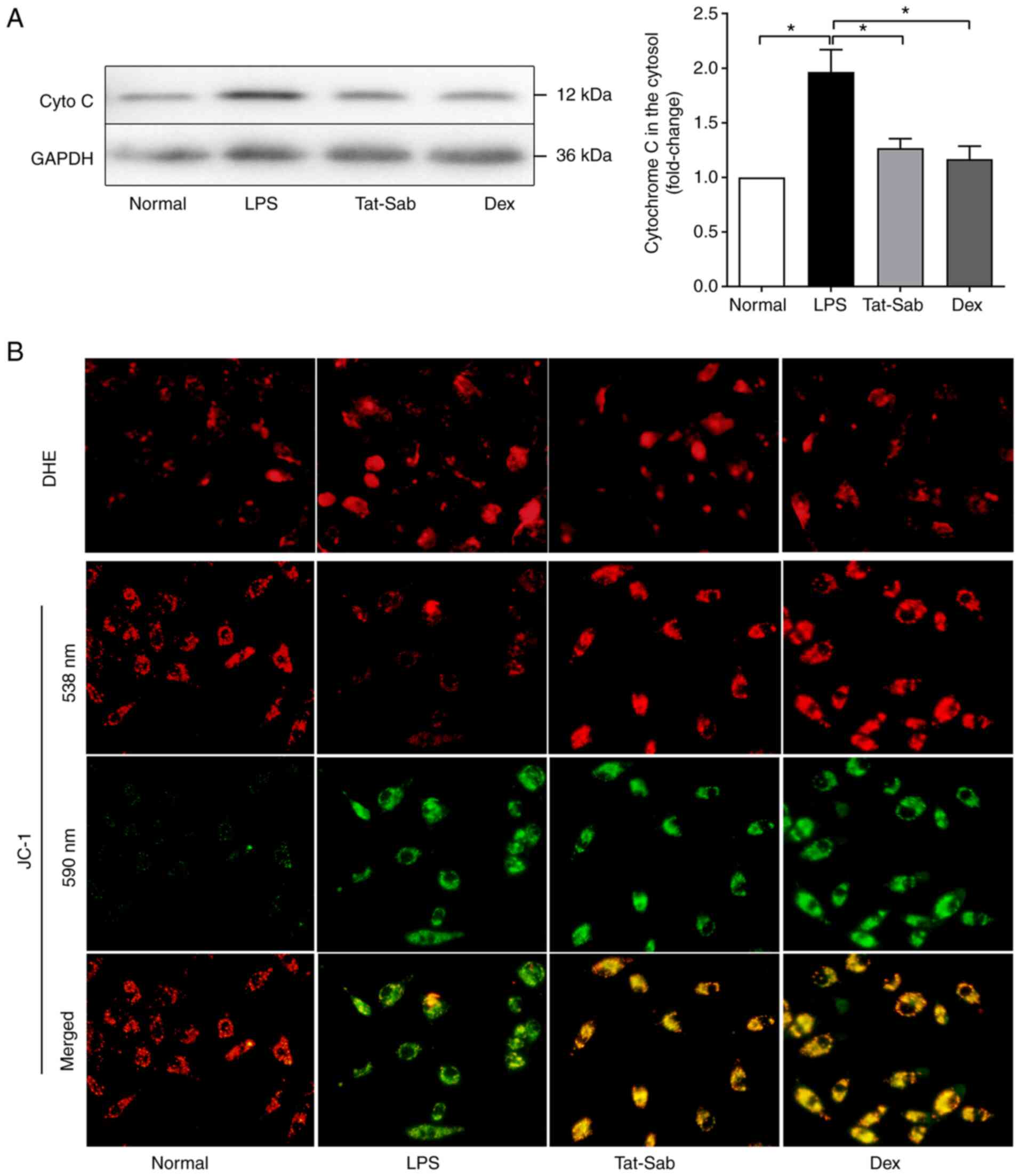
ALI/ARDS and the protective effect of Tat-SabKIM1. As
shown in Fig. 7A, in the LPS
group, the release of cytochrome c from the mitochondria into the
cytosol was considerably increased compared to that in the control
group. Treatment with Tat-SabKIM1 or DEX reduced cytochrome c
release from the mitochondria. The expression of p-Bcl-2, cleaved
caspase-3 and Bax was also detected. More p-Bcl-2 and less cleaved
caspase-3 and Bax were detected in the Tat-SabKIM1
groups than in the LPS group. These results indicated that blocking
JNK mitochondrial localization could inhibit the excessive
activation of apoptosis pathways.
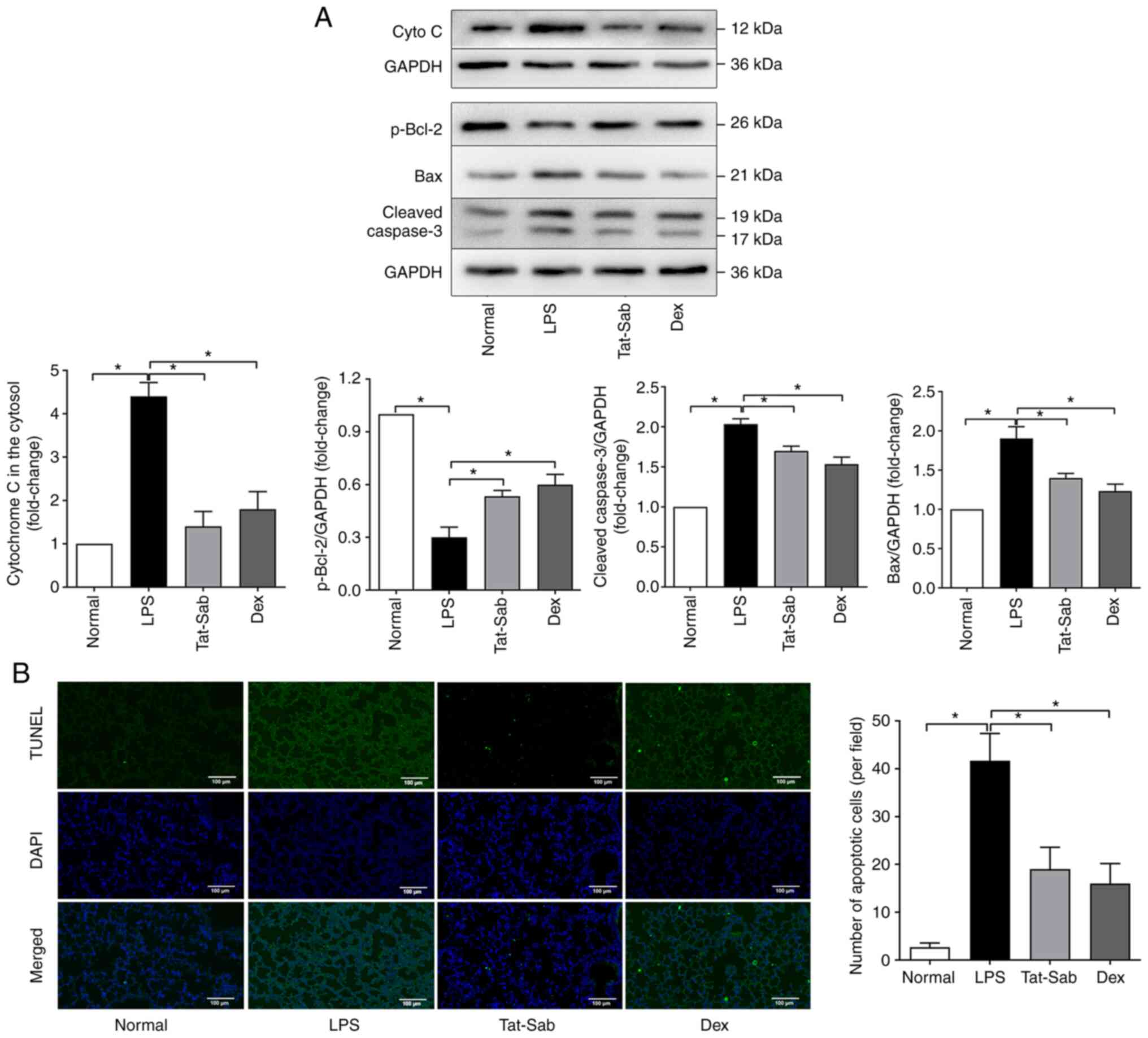 | Figure 7Effect of Tat-SabKIM1 on
cell death in the mouse lung. (A) Representative western blots of
cyto c in the cytosol, p-Bcl-2, cleaved caspase3 and Bax of the
Normal, LPS, Tat-SabKIM1 and Dex groups. (B) Changes in
the cell apoptosis ratio in the lungs detected by TUNEL staining of
the Normal, LPS, Tat-SabKIM1 and Dex groups;
magnification, ×100. The data are expressed as the mean ± standard
error of the mean, n=4/group, *P<0.05. cyto c,
cytochrome c; p-, phosphorylated; t-, total; Dex, dexamethasone;
LPS, lipopolysaccharide. |
The cell apoptosis ratio in the lungs by was also
detected TUNEL staining. The results (Fig. 7B) indicated that the inhibition
of mitochondrial JNK signaling exerted an antiapoptotic effect.
Tat-SabKIM1 treatment significantly decreased the number
of TUNEL-positive cells compared with LPS treatment alone. These
results indicated that mitochondrial JNK signaling participated in
mitochondria-mediated apoptosis during ALI/ARDS.
Effect of Tat-SabKIM1 on
mitochondrial function in HUVECs
The results from Xu et al (22) indicate that JNK mitochondrial
localization might disrupt the function of mitochondria and even
lead to cell death. To confirm the disruption of mitochondrial
functions, the present study explored the changes in cytochrome c
leakage, ROS production and ΔΨ in HUVECs during LPS challenge. As
epithelial cells, A549 cells cannot be affected and injured by LPS
directly. LPS is generate by G-bacteria and released into blood and
the endothelial cells rather than epithelial cells can contact with
LPS directly. Thus, LPS was used to intervene HUVECs in the in
vitro experiments. The results (Fig. 8) showed that LPS exposure could
significantly elevate the leakage of cytochrome c in HUVECs and
Tat-SabKIM1 could eliminate this. The changes in ROS production and
ΔΨ dissipation induced by LPS could also be alleviated by
Tat-SabKIM1. These results suggested that LPS could
induce mitochondrial dysfunction and ROS accumulation and these
changes could be alleviated when JNK mitochondrial localization was
blocked by Tat-SabKIM1.
 | Figure 8Effect of Tat-SabKIM1 on
mitochondrial function in HUVECs. (A) Representative western blots
of cyto c in the cytosol of HUVECs. (B) Changes in superoxide anion
content and mitochondrial membrane permeability (ΔΨ) after HUVECs
were challenged with LPS and pre-treated with
Tat-SabKIM1 or Dex; magnification, ×200. The data are
expressed as the mean ± standard error of the mean, n=4/group,
*P<0.05. HUVECs, human umbilical vein endothelial
cells; cyto c, cytochrome c; p-, phosphorylated; t-, total; cyto c,
cytochrome c; LPS, lipopolysaccharide; Dex, dexamethasone; DHE,
dihydroethidium. |
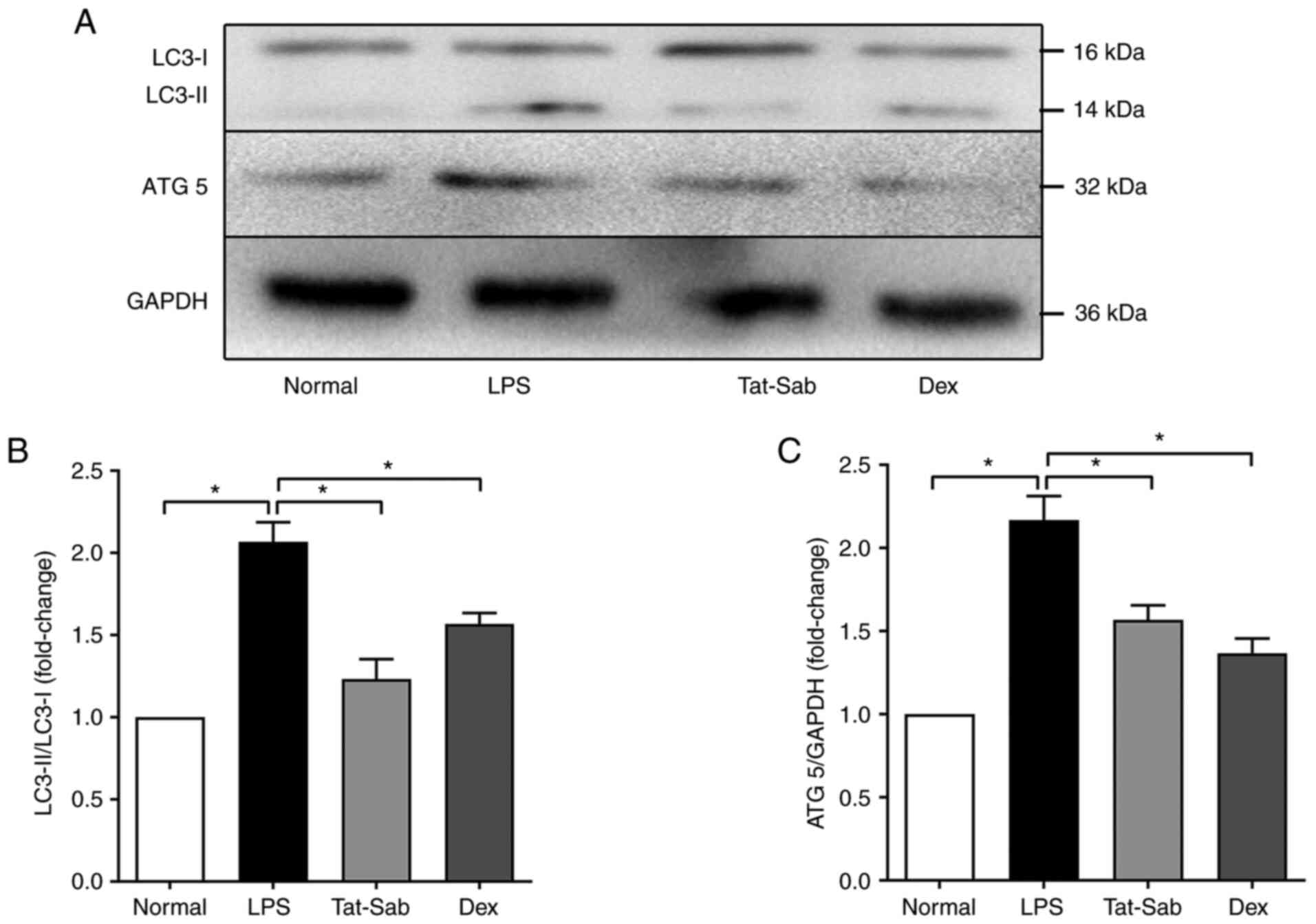
Effect of Tat-SabKIM1 on
autophagy in the mouse lung
Autophagy has been demonstrated to participate in
the development of ALI/ARDS, as per the conversion of LC3, i.e.,
LC3-I to LC3-II (31). ATG5
might also modulate autophagy. As shown in Fig. 9, LPS exposure increased the
conversion of LC3 to LC3-II, which is the active form and increased
the expression of ATG5. The inhibition of mitochondrial JNK by
Tat-SabKIM1 restored the levels of ATG5 and LC3,
indicating that the inhibition blocked the autophagy induced by
ALI/ARDS.
Discussion
The mortality rate associated with ALI/ARDS varies
between 25-40%. To date, there is no evidence showing that any
pharmacological interventions are associated with an ameliorative
mortality rate (1,3). ALI/ARDS can be caused by diseases
such as pneumonia, trauma and sepsis, which means that the
heterogeneity of its etiology is huge. Therefore, it is difficult
to elucidate the pathogenesis of ALI/ARDS and identify drugs with
definite effects.
The present study mainly explored the role of ER
stress and JNK-mitochondria pathways in ALI/ARDS. The major goal
was to explore ER stress-JNK-mitochondria abnormalities during
ALI/ARDS and confirm the hypothesis that abnormal activation of the
JNK-mitochondrial pathway could significantly disrupt the normal
physiological function of lung cells, resulting in the occurrence
of ALI/ARDS. Furthermore, selective inhibition of JNK mitochondrial
localization by Tat-SabKIM1 had a protective effect
against mitochondrial dysfunction and cell death caused by ER
stress in mice with LPS-induced ALI/ARDS.
Studies have implicated ER stress-related cellular
dysfunction and cell death in the occurrence and progression of a
number of diseases and these changes may be potential therapeutic
targets (13,16,18). Under normal circumstances,
BiP/GRP78 binds to the ER stress sensor proteins PERK, IRE1 and
ATF6, which prevents their dimerization and UPR activation.
However, during the stress response, the accumulation of unfolded
proteins leads to the release of BiP/GRP78 from IRE1α, ATF6 and
PERK and then to the activation of downstream signaling components,
such as XBP-1, eIF2α, ATF4 and cleaved ATF6 (15-17). These reactions are helpful for
protein folding and degradation and ER expansion. After releasing
BiP/GRP78, activated PERK dimerizes and phosphorylates eIF2α, which
can suppress 5′-capped mRNA translation (14,15). The cleaved ATF6 fragment can
modulate apoptosis and protein folding by regulating the expression
of some ER chaperones. XBP-1 can lead to ER-associated degradation
by upregulating the expression of ER chaperones and genes. XBP-1
activation is initiated by the splicing of XBP-1 mRNA by activated
IRE1α and then, the spliced XBP-1 mRNA binds to open reading frames
and promotes translation (32).
The experimental results of the present study
demonstrated that ER stress was enhanced during LPS-induced
ALI/ARDS. Bip/GRP78 and p-PERK and ATF6 were significantly
increased after LPS challenge, indicating the activation of ER
stress. The percentage of spliced XBP-1 was elevated in the lung
after intraperitoneal LPS injection.
Li et al (33) show that IRE1α can also activate
JNK by recruiting TRAF2 and ASK1. JNK regulates cellular adaptation
to stress and causes cell death. Vannuvel et al (29) show that JNK can translocate to
the mitochondrial outer membrane following activation and then lead
to Bim phosphorylation and activation. Then, Bim induces the
oligomerization of Bax and Bak, finally resulting in the release of
cytochrome c and activation of the caspase-dependent apoptotic
pathway.
In the present study, tunicamycin, a specific and
widely used chemical inducer of lethal ER stress, inhibited protein
glycosylation in the ER and led to ER stress due to protein
misfolding. ER stress could induce mitochondrial dysfunction and
cell death in lung cells, which were represented by A549 cells and
HUVECs. Tunicamycin exposure significantly increased ROS production
and ΔΨ dissipation. Tunicamycin increased the percentage of
apoptotic cells to >20% and more cytochrome c was released from
mitochondria into the cytosol. Less p-Bcl-2 and more cleaved
caspase-3 and Bax were detected in the activated groups. These
results suggested that overactivation of ER stress could induce
mitochondrial dysfunction and ROS accumulation and even lead to
apoptosis.
Studies (23,25,26,34) show that JNK can transfer from
cytoplasm to mitochondria in some conditions by interacting with
Sab, which expresses in the outer membrane of mitochondria.
Following knockdown of Sab, the change of JNK in cytoplasm and
mitochondria is prevented. N-terminal KIM in Sab is essential for
JNK binding and confocal immunocytochemistry and cell fractionation
studies indicate that Sab is associated with mitochondria, where it
co-localizes with a fraction of JNK. These reported properties of
Sab suggest its role in targeting JNK to this subcellular
compartment (mitochondria). The present study used western blot
analysis to detect the transfer of JNK as previous studies
(22,35) report and proved that ER stress
could induce JNK activation and mitochondrial localization in A549
cells and HUVECs. The total JNK in the mitochondria was also
elevated, whereas that in the cytosol/nucleus was decreased,
indicating that ER stress could induce JNK translocation from the
cytosol/nucleus to mitochondria in A549 cells and HUVECs.
Therefore, blocking the interaction of JNK with mitochondria and
inhibiting secondary apoptosis might be potential therapeutic
targets.
Previous studies (8,22,28) show that JNK can interact with
mitochondria by binding to Sab (SH3BP5). Sab is a mitochondrial
outer membrane protein with one SH3 domain binding site at the
N-terminus, one membrane spanning domain and two D-motifs (KIMs) at
the C-terminus. KIM is similar to c-Jun and can link JNK with Sab.
Studies (22,26,35) also prove that the activation of
JNK and interaction with mitochondria by the docking protein Sab
are involved in the regulation of mitochondrial functions,
impairment of electron transport and mitochondrial bioenergetics
and participation in ROS generation and apoptosis.
As Sab is the only JNK docking site in mitochondria,
depleting Sab could completely prevent JNK translocation to
mitochondria. Previous studies (26,36) show that knockdown of Sab blocks
JNK translocation to mitochondria in in vivo or in
vitro models of JNK-dependent toxicity (APAP, TNF/GalN, ER
stress and palmitic acid lipotoxicity) and inhibit JNK
activation-induced mitochondrial dysfunction and cell death. Study
(28) showed that silencing Sab
in PMH and HeLa cells can prevent BFA-induced JNK-mitochondria
pathway activation and subsequent cell death. The synthesis of the
KIM1-specific binding peptide Tat-SabKIM1 can also
selectively block the binding of JNK to Sab without blocking the
kinase activity of JNK, the ratio of p-JNK/JNK or the activation of
cytosolic/nuclear JNK (22). The
results of the present study showed that Tat-SabKIM1
could successfully reach the cytoplasm through the cell membrane,
its concentration was stable and the concentration in the cells
after 24 h could still reach the initial concentration of up to 90%
(34). Therefore,
Tat-SabKIM1 might be optimal for blocking the binding of
Sab and JNK. Studies (22,24) have shown that inhibition of p-JNK
binding to Sab using Tat-SabKIM1 prevents ischemic
necrosis in the heart and brain.
ER stress could trigger the interaction of JNK with
mitochondrial Sab, followed by impaired respiration and increased
mitochondrial ROS and cell death. ROS accumulation was
significantly blocked by Tat-SabKIM1 but not by
scrambled peptide. Therefore, it was hypothesized that in ALI/ARDS,
abnormal activation of the JNK-mitochondrial pathway could
significantly disrupt the normal physiological function of lung
cells and the initial activation of JNK in the ER is followed by
its interaction with Sab, leading to impaired mitochondrial
function and amplification of mitochondrial ROS release and serving
a key role in the occurrence of ALI/ARDS. Selective inhibition of
the mitochondrial localization of JNK by Tat-SabKIM1
protected against the mitochondrial dysfunction and cell death
caused by endoplasmic reticulum stress in LPS-induced ALI/ARDS
mice.
The present study verified this hypothesis and
showed that LPS induced JNK activation and JNK mitochondrial
localization in mice. In LPS-induced ALI/ARDS, LPS exposure induced
the phosphorylation of JNK in both mitochondria and the
cytosol/nucleus. The total JNK expression in the mitochondria was
also increased in the LPS groups, whereas that in the
cytosol/nucleus was decreased, indicating that in LPS-induced
ALI/ARDS, JNK could translocate from the cytosol/nucleus to
mitochondria. Tat-SabKIM1 specifically inhibited JNK
localization to mitochondria and the activation of mito-JNK
signaling without affecting cytosolic/nuclear JNK activation.
Tat-SabKIM1 does not exhibit any effect on the
translocation of JNK to the nucleus. The experimental results of
the present study also showed that Tat-SabKIM1 could
alleviate LPS injection-induced lung tissue structure destruction,
alveolar wall thickening, interstitial lung inflammatory cells and
liquid exudation. Tat-SabKIM1 treatment significantly
improved the mouse survival rates compared with LPS treatment
alone. Blocking JNK mitochondrial localization also inhibited the
excessive activation of apoptosis pathways. Tat-SabKIM1
treatment significantly decreased the number of TUNEL-positive
cells compared with LPS treatment alone. These results indicated
that mitochondrial JNK signaling participated in
mitochondria-mediated apoptosis during ALI/ARDS. LPS could induce
mitochondrial dysfunction and ROS accumulation and these changes
could be alleviated when JNK mitochondrial localization was blocked
by Tat-SabKIM1. The inhibition of mitochondrial JNK by
Tat-SabKIM1 restored the levels of ATG5 and LC3, indicating that
mitochondrial JNK inhibition blocked the autophagy induced by
ALI/ARDS. Therefore, inhibiting the translocation of JNK to
mitochondria can be used to repair damage by protecting the normal
physiological function of organelles.
There are also some limitations in this study that
should be mentioned. Alveolar epithelial cells and pulmonary
vascular endothelial cells extracted from LPS-interfered animals
might be the best choice to explore ALI/ARDS in vitro.
However, because extracting and cultivate primary cells needed time
and the effect of LPS on the extracted cells from LPS-interfered
animals might already lose efficacy, so HUVECs and A549 cells were
used instead.
Collectively, the results presented clearly
indicated that during ALI/ARDS, abnormal activation of ER stress
and JNK-mitochondrial pathways could significantly disrupt the
normal physiological function of lung cells, resulting in the
occurrence of ALI/ARDS. Through selective inhibition, JNK
mitochondrial localization by Tat-SabKIM1 exerted a
protective effect against the mitochondrial dysfunction and cell
death caused by ER stress in mice with LPS-induced ALI/ARDS.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL and LB contributed to the study design. LB, DM
and YC also contributed to the conduct of the study. WL and FJ
contributed to the data analysis. CL and LB confirm the
authenticity of all the raw data. All authors contributed to
drafting the manuscript and have read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal procedures conducted in the present study
were approved by the Animal Care and Use Committee of the Fourth
Military Medical University (approval no. TDLL20160194) and were
carried out in accordance with the National Institutes of Health
Guide for Care and Use of Laboratory Animals (30).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81900083 and 81800076) and China
Postdoctoral Science Foundation (grant no. 2019M653911).
References
|
1
|
ARDS Definition Task Force; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The berlin definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
2
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schattauer SS, Bedini A, Summers F,
Reilly-Treat A, Andrews MM, Land BB and Chavkin C: Reactive oxygen
species (ROS) generation is stimulated by kappa opioid receptor
activation through phosphorylated c-Jun N-terminal kinase and
inhibited by p38 mitogen-activated protein kinase (MAPK)
activation. J Biol Chem. 294:16884–16896. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Negishi T, Matsumoto M, Kobayashi Y,
Kojima M, Sakaguchi F, Takahata K, Kanehira T, Arakaki R, Aoyama Y,
Yoshida H, et al: Dysregulation of MAP kinase signaling pathways
including p38MAPK, SAPK/JNK and ERK1/2 in cultured rat cerebellar
astrocytes exposed to diphenylarsinic acid. Toxicol Sci.
156:509–519. 2017.PubMed/NCBI
|
|
6
|
Nguyen HT, Hsieh MH, Gaborro A, Tinloy B,
Phillips C and Adam RM: JNK/SAPK and p38 SAPK-2 mediate mechanical
stretch-induced apoptosis via caspase-3 and -9 in NRK-52E renal
epithelial cells. Nephron Exp Nephrol. 102:e49–e61. 2006.
View Article : Google Scholar
|
|
7
|
Chuang HC, Wang X and Tan TH: MAP4K family
kinases in immunity and inflammation. Adv Immunol. 129:277–314.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Win S, Than TA and Kaplowitz N: The
regulation of JNK signaling pathways in cell death through the
interplay with mitochondrial sab and upstream post-translational
effects. Int J Mol Sci. 19:36572018. View Article : Google Scholar :
|
|
9
|
Betigeri S, Pakunlu RI, Wang Y, Khandare
JJ and Minko T: JNK1 as a molecular target to limit cellular
mortality under hypoxia. Mol Pharm. 3:424–430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Komatsu W, Kishi H, Yagasaki K and Ohhira
S: Urolithin A attenuates pro-inflammatory mediator production by
suppressing PI3-K/Akt/NF-kappaB and JNK/AP-1 signaling pathways in
lipopolysaccharide-stimulated RAW264 macrophages: Possible
involvement of NADPH oxidase-derived reactive oxygen species. Eur J
Pharmacol. 833:411–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li B, Zeng M, He W, Huang X, Luo L, Zhang
H and Deng DY: Ghrelin protects alveolar macrophages against
lipopolysaccharide-induced apoptosis through growth hormone
secretagogue receptor 1a-dependent c-Jun N-terminal kinase and
Wnt/beta-catenin signaling and suppresses lung inflammation.
Endocrinology. 156:203–217. 2015. View Article : Google Scholar
|
|
12
|
Zheng Y, Zhang M, Zhao Y, Chen J, Li B and
Cai W: JNK inhibitor SP600125 protects against
lipopolysaccharide-induced acute lung injury via upregulation of
claudin-4. Exp Ther Med. 8:153–158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Di Conza G and Ho PC: ER stress responses:
An emerging modulator for innate immunity. Cells. 9:6952020.
View Article : Google Scholar :
|
|
14
|
Amin-Wetzel N, Saunders RA, Kamphuis MJ,
Rato C, Preissler S, Harding HP and Ron D: A J-protein Co-chaperone
recruits BiP to monomerize IRE1 and repress the unfolded protein
response. Cell. 171:1625–1637. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kopp MC, Larburu N, Durairaj V, Adams CJ
and Ali M: UPR proteins IRE1 and PERK switch BiP from chaperone to
ER stress sensor. Nat Struct Mol Biol. 26:1053–1062. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu
H, Zhang XP and Jiang H: Endoplasmic reticulum stress of
neutrophils is required for ischemia/reperfusion-induced acute lung
injury. J Immunol. 195:4802–4809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sommer T and Jarosch E: BiP binding keeps
ATF6 at bay. Dev Cell. 3:1–2. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu SK, Chiu CC, Dahms HU, Chou CK, Cheng
CM, Chang WT, Cheng KC, Wang HD and Lin IL: Unfolded cprotein
response (UPR) in survival, dormancy, immunosuppression, metastasis
and treatments of cancer cells. Int J Mol Sci. 20:25182019.
View Article : Google Scholar
|
|
19
|
Wang M, Law ME, Castellano RK and Law BK:
The unfolded protein response as a target for anticancer
therapeutics. Crit Rev Oncol Hematol. 127:66–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HC and Wei YH: Mitochondrial role in
life and death of the cell. J Biomed Sci. 7:2–15. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J,
Zhang L, Tang Y, Liu J, Zhang X and Gao F: Mitochondrial JNK
activation triggers autophagy and apoptosis and aggravates
myocardial injury following ischemia/reperfusion. Biochim Biophys
Acta. 1852:262–270. 2015. View Article : Google Scholar
|
|
23
|
Wiltshire C, Matsushita M, Tsukada S,
Gillespie DA and May GH: A new c-Jun N-terminal kinase
(JNK)-interacting protein, Sab (SH3BP5), associates with
mitochondria. Biochem J. 367:577–585. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chambers JW, Pachori A, Howard S, Iqbal S
and LoGrasso PV: Inhibition of JNK mitochondrial localization and
signaling is protective against ischemia/reperfusion injury in
rats. J Biol Chem. 288:4000–4011. 2013. View Article : Google Scholar :
|
|
25
|
Ngoei KR, Catimel B, Church N, Lio DS,
Dogovski C and Perugini MA: Characterization of a novel JNK (c-Jun
N-terminal kinase) inhibitory peptide. Biochem J. 434:399–413.
2011. View Article : Google Scholar
|
|
26
|
Win S, Than TA, Le BH, Garcia-Ruiz C,
Fernandez-Checa JC and Kaplowitz N: Sab (Sh3bp5) dependence of JNK
mediated inhibition of mitochondrial respiration in palmitic acid
induced hepatocyte lipotoxicity. J Hepatol. 62:1367–1374. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li T, Liu Y, Xu W, Dai X, Liu R, Gao Y,
Chen Z and Li Y: Polydatin mediates Parkin-dependent mitophagy and
protects against mitochondria-dependent apoptosis in acute
respiratory distress syndrome. Lab Invest. 99:819–829. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Win S, Than TA, Fernandez-Checa JC and
Kaplowitz N: JNK interaction with Sab mediates ER stress induced
inhibition of mitochondrial respiration and cell death. Cell Death
Dis. 5:e9892014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vannuvel K, Renard P, Raes M and Arnould
T: Functional and morphological impact of ER stress on
mitochondria. J Cell Physiol. 228:1802–1818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
National Research Council (US): Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
31
|
Chichger H, Rounds S and Harrington EO:
Endosomes and autophagy: Regulators of pulmonary endothelial cell
homeostasis in health and disease. Antioxid Redox Signal.
31:994–1008. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Glimcher LH, Lee AH and Iwakoshi NN: XBP-1
and the unfolded protein response (UPR). Nat Immunol. 21:963–965.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Jiang W, Niu Q, Sun Y, Meng C, Tan
L, Song C, Qiu X, Liao Y and Ding C: eIF2alpha-CHOP-BCl-2/JNK and
IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and
support the proliferation of newcastle disease virus. Cell Death
Dis. 10:8912019. View Article : Google Scholar
|
|
34
|
Chambers JW, Cherry L, Laughlin JD,
Figuera-Losada M and Lograsso PV: Selective inhibition of
mitochondrial JNK signaling achieved using peptide mimicry of the
Sab kinase interacting motif-1 (KIM1). ACS Chem Biol. 6:808–818.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bo L, Li Y, Liu W, Jin F and Li C:
Selective inhibition of JNK mitochondrial location is protective
against seawater inhalation-induced ALI/ARDS. Mol Med Rep.
24:5152021. View Article : Google Scholar :
|
|
36
|
Win S, Than TA, Han D, Petrovic LM and
Kaplowitz N: c-Jun N-terminal kinase (JNK)-dependent acute liver
injury from acetaminophen or tumor necrosis factor (TNF) requires
mitochondrial Sab protein expression in mice. J Biol Chem.
286:35071–35078. 2011. View Article : Google Scholar : PubMed/NCBI
|