Introduction
Chronic kidney disease (CKD) results from a variety
of disorders that affect the structure and function of the kidneys
and represents a rising global health concern due to its increasing
prevalence (8–16%) (1). There is
no cure for CKD and the available therapies only aim to slow
disease progression. The progressive nature of CKD, as well as its
associated cardiovascular morbidity and mortality and eventual
end-stage renal disease place a substantial burden on global
healthcare resources (2,3). An improved understanding of the
nature of CKD is required for the development of novel therapeutic
strategies. All types of progressive CKD inevitably induce renal
fibrosis (4). The induction of
renal fibrosis due to mitochondrial dysfunction has received
considerable research attention since the early 2000s (5). The kidney is an organ with high
energy demand. Mitochondria provide the energy required to maintain
kidney function via a number of signaling pathways, such as the
mechanistic target of rapamycin and AMP-activated protein kinase
(AMPK) signaling pathways (6).
Aberrations in energy metabolism can lead to cellular dysfunction
and death. Mitochondria not only produce cellular energy but also
modulate several cellular processes, including proliferation and
intracellular calcium homeostasis (7). Mitochondrial dysfunction induces
apoptosis and the generation of reactive oxygen species (ROS), both
of which contribute to the development and progression of various
kidney diseases, including acute kidney injury, diabetic
nephropathy and CKD (6,8–11). Reversing mitochondrial
dysfunction has the potential to halt disease progression,
emphasizing the need for mitochondria-targeting agents that can
restore mitochondrial and renal function (7).
Stanniocalcin-1 (STC1) was originally identified as
a calcium/phosphate-regulating hormone in bony fishes and is
released from the corpuscles of Stannius (organs associated with
kidneys) (12). In mammals, STC1
is expressed in various tissues (12) and is considered to function in an
autocrine and/or paracrine manner (13); however, the localization of STC1
to mitochondria suggests a possible intracellular role (14). STC1 is one of the few hormones
that target the mitochondrion (15) and demonstrates antioxidant
effects via activation of mitochondrial antioxidant pathways
(16–18). STC1 regulates AMPK activity in
the kidneys (19) and suppresses
ROS generation via the induction of the mitochondrial uncoupling
protein (UCP) in macrophages (16,17). The aims of the present study were
to ascertain whether STC1 reduces TGF-β-induced cellular fibrosis
in the kidney tubular epithelial cells by reducing mitochondrial
oxidative stress and mitochondrial damage and to explore the
associated molecular mechanisms.
Materials and methods
Reagents and antibodies
Recombinant human TGF-β was purchased from PeproTech
EC Ltd. (cat. no. 100-21) and recombinant STC1 (rSTC1; MBS1265316)
was purchased from MyBioSource, Inc. AMPK inhibitor (ab120843) was
purchased from Abcam. UCP2-targeted small interfering RNA (siRNA)
(siUCP2) was purchased from GE Healthcare Dharmacon, Inc. (cat. no.
L-005114-00-0020). Anti-fibronectin (610077) and anti-UCP2
antibodies (sc-390189) were obtained from BD Biosciences and Santa
Cruz Biotechnology, Inc. Antibodies against α-smooth muscle actin
(α-SMA; a2547) and β-actin (a3854) were obtained from
MilliporeSigma. Anti-AMPK (cat. no. 2793) and anti-phosphorylated
AMPK (recognizing phosphorylated Thr172; cat. no. 2535) antibodies
were purchased from Cell Signaling Technology, Inc.
Cell culture and TGF-β treatment
Human renal proximal tubular epithelial cells (HK2,
American Type Culture Collection) were cultured in complete
DMEM-F12 (Welgene, Inc.) supplemented with 10% FBS (Thermo Fisher
Scientific, Inc.), 50 U/ml penicillin and 50 µg/ml streptomycin
(MilliporeSigma) at 37°C in a humidified 5% CO2
atmosphere. Cells were passaged every 3–4 days and starved in
serum-free medium for 24 h before experiments. TGF-β is the central
mediator that drives fibrosis in most, if not all, forms of CKD
(20). To investigate the role
of STC1 in the AMPK pathway, the expression levels of AMPK and UCP2
were analyzed at different time points (0 min, 15 min, 30 min, 1 h,
3 h and 6 h) following TGF-β and rSTC1 treatment. To identify
whether STC1 suppressed TGF-β-induced mitochondrial dysfunction and
fibrosis, HK2 cells were pre-incubated at 37°C with 200 ng/ml STC1
for 1 h and then incubated at 37°C with fresh medium containing 10
ng/ml TGF-β for 16 h. To determine whether the effect of STC1 was
mediated by AMPK activation, pharmacological inhibition of AMPK by
compound C (Calbiochem; Merck KGaA) was performed. Compound C (5
µM/ml) was added to HK2 cells at 37°C for 1 h and then cells were
pre-incubated at 37°C with 200 ng/ml STC1 for 1 h and subsequently
incubated at 37°C with fresh medium containing 10 ng/ml TGF-β for
16 h.
siRNA transfection
To explore the molecular mechanisms underlying the
effects of STC1, RNA interference was performed using a pool of
UCP2-specific siRNAs from On-TargetPlus Human SmartPools containing
4 distinct siRNA species (cat. no. L-005114-00-0020). The negative
control siRNA was purchased from Santa Cruz Biotechnology, Inc.
(cat. no. sc 37007). siRNA sequences of UCP2 are described in
Table SI. Cells were
transfected with the indicated concentration of siRNA (40 nM) using
DharmaFECT 1 transfection reagent according to the manufacturer's
protocol. For knockdown of UCP2, siRNA and control siRNA were
transfected into HK-2 cells at 60% confluence at 37°C in a 5%
CO2 incubator for 24 h using DharmaFECT 1 transfection
reagent (T-2001-02; GE Healthcare Dharmacon, Inc.) at a final
concentration of 40 nM. After transfection, cells were incubated in
serum-free medium for 1 day in a 37°C incubator under a humidified
5% CO2 atmosphere, pretreated at 37°C with 200 ng/ml
STC1 for 1 h and stimulated at 37°C with 10 ng/ml TGF-β for 16 h.
Confirmation of Successful transfection of siRNA in HK-2 cells was
determined using semi-quantitative PCR and western blotting as
described subsequently in this section (Fig. S1).
Semi-quantitative PCR
Total RNA was extracted from HK-2 cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
First-standard complementary DNA synthesis was performed using a
SuperScript™ First-Strand Synthesis System kit (cat. no. 11904-018;
Invitrogen; Thermo Fisher Scientific, Inc.). Total RNA (1 µg) was
used for first-strand complementary DNA synthesis and First-Strand
Synthesis reaction (10 mM dNTP mix, 50 ng/µl random hexamers, 10 X
RT buffer, 25 mM MgCl2, 0.1 M DTT, 40 U/µl RNaseOUT™,
SuperScript™ II RT, DEPC-treated water) was used. After incubation
at 42°C for 50 min, the reaction was completed by incubation at
70°C for 15 min to prepare complementary DNA. The specific steps
followed the SuperScript™ First-Strand Synthesis System kit
manufacturer's protocol. UCP2 expression was normalized to that of
β-actin. The specific primers sequences were as follows: UCP2
forward, 5′-TCTACAATGGGCTGGTTGC-3′ and reverse,
5′-TGTATCTCGTCTTGACCAC-3′ (494 bp); and β-actin forward,
5′-GCTCTTTTCCAGCCTTCCTT-3′ and reverse, 5′-AGTACTTGCGCTCAGGAGGA-3′
(234 bp). After an initial denaturation step of 5 min at 94°C,
semi-quantitative PCR amplification was carried out as follows:
95°C for 30 sec, 56°C for 30 sec and 72°C for 30 sec for 35 cycle,
followed by one cycle at 72°C for 5 min. A 1% agarose gel was used
for DNA loading, and SYBR DNA gel stain (cat. no. S33102;
Invitrogen; Thermo Fisher Scientific, Inc.) was used for
visualization. The relative intensities of DNA were measured by
densitometry using Scion Image for Windows (version Alpha 4.0.3.2;
Scion Corporation).
Western blotting
The cells were harvested, washed twice with ice-cold
PBS, resuspended in lysis buffer and sonicated briefly. Protein
extraction buffer consisted of 50 mM Tris-HCl (pH 7.2), 5 mM EDTA,
150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, protease inhibitor cocktail
(P3100-001; GenDEPOT, LLC) and phosphatase inhibitor cocktail
(P3200-001; GenDEPOT, LLC). After centrifugation at 4°C at 13,500 ×
g for 5 min, supernatants containing the protein extracts were
collected and the protein concentrations were measured using a
Pierce® BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Proteins were separated on 10 and 12% sodium
dodecyl sulfate polyacrylamide gels, and transferred onto
nitrocellulose membranes. The protein concentration loaded per lane
was 30 µg, and the blot was blocked at room temperature for 1 h
with 5% skim milk in PBS containing 0.1% Tween-20. Then, the blots
were incubated overnight with the primary antibody (1:2,000) at
4°C, washed four times at 10 min intervals in 1X TBS with 0.1%
Tween-20 (TBST) at room temperature, and incubated with appropriate
anti-rabbit IgG, HRP-linked secondary antibody (1:2,500; 7074S;
Cell Signaling Technology, Inc.) and anti-mouse IgG, HRP-linked
secondary antibody (1:2,500; 7076S; Cell Signaling Technology,
Inc.) at room temperature for 2 h. After that, the blots were
washed four times in 1X TBST at 10 min intervals. Specific protein
bands were visualized using an enhanced chemiluminescence system
(cat. no. WBKLS0500; MilliporeSigma). The relative intensities of
immunoblot signals were measured by densitometry using Scion Image
for Windows (version Alpha 4.0.3.2; Scion Corporation) and were
expressed as fold-change values relative to the intensities of the
controls.
Quantification of mitochondrial
membrane potential (MMP)
MMP was assessed using a Leica TCS SP8 confocal
laser-scanning microscope (Leica Microsystems GmbH) with
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole–carbocyanide
iodine (10009172; JC-1 MMP Assay kit; Cayman Chemical Company). HK2
cells were seeded at a density of 3×105 cells/well in a
35-mm confocal special dish, followed by a 24-h incubation at 37°C.
Cells were pretreated with or without 10 ng/ml TGF-β and 200 ng/ml
rSTC1 and, then incubated for 16 h at 37°C with 5% CO2.
Cell nuclei were stained with 2 µg/ml Hoechst 33258 for 20 min in a
CO2 incubator at 37°C. Slides were analyzed with a
confocal laser scanning microscope (Leica TCS SP8; Leica
Microsystems GmbH) and LAS X software (version 4.0.2; Leica
Microsystems GmbH) was used. Fluorescence was read at an excitation
wavelength of 488 nm (green) or 530 nm (red) and an emission
wavelength of 530 nm (green) or 590 nm (red). JC-1 emits green
fluorescence in the cytoplasm and exhibits membrane
potential-dependent accumulation in mitochondria, resulting in a
shift in the emission wavelength from green to red. A reduction in
MMP is indicated by a decrease in the red/green fluorescence
intensity ratio (21).
Mitochondrial ROS production
Mitochondrial ROS levels were measured using
MitoSOX, which selectively reacts with superoxide in the
mitochondria. HK2 cells were seeded at a density of
3×105 cells/well on a cover-glass bottom dish and grown
to 60% confluence. After incubation at 37°C for 24 h, cells were
pretreated with or without 10 ng/ml TGF-β and 200 ng/ml rSTC1 and
incubated for 16 h at 37°C with 5% CO2. The cells were
then incubated with 5 µM MitoSOX for 15 min at 37°C and fixed with
4% paraformaldehyde for 10 min at 37°C. After fixation, cells were
permeabilized with 0.1% Triton X-100 for 10 min and blocked in 1X
PBS containing 5% BSA (cat. no. ALB001; BioShop Canada Inc.) for 2
h at room temperature. The cells were then washed twice with
phosphate-buffered saline and incubated with 1 µg/ml DAPI solution
at 4°C for 15 min. Images were immediately acquired by confocal
microscopy on a laser-scanning microscope (LSM 510; Carl Zeiss AG)
and analyzed using ImageJ (version 1.53; National Institutes of
Health).
Statistical analysis
All experiments were independently replicated at
least three times. Data are presented as the mean ± standard error
of the mean. Multiple comparisons among the groups were performed
by one-way analysis of variance followed by Tukey's post hoc tests.
All statistical analyses were performed using SPSS version 24.0
(IBM Corp.). P<0.05 was considered to indicate a statistically
significant difference.
Results
rSTC1 inhibits TGF-β-induced fibrosis
in HK2 cells
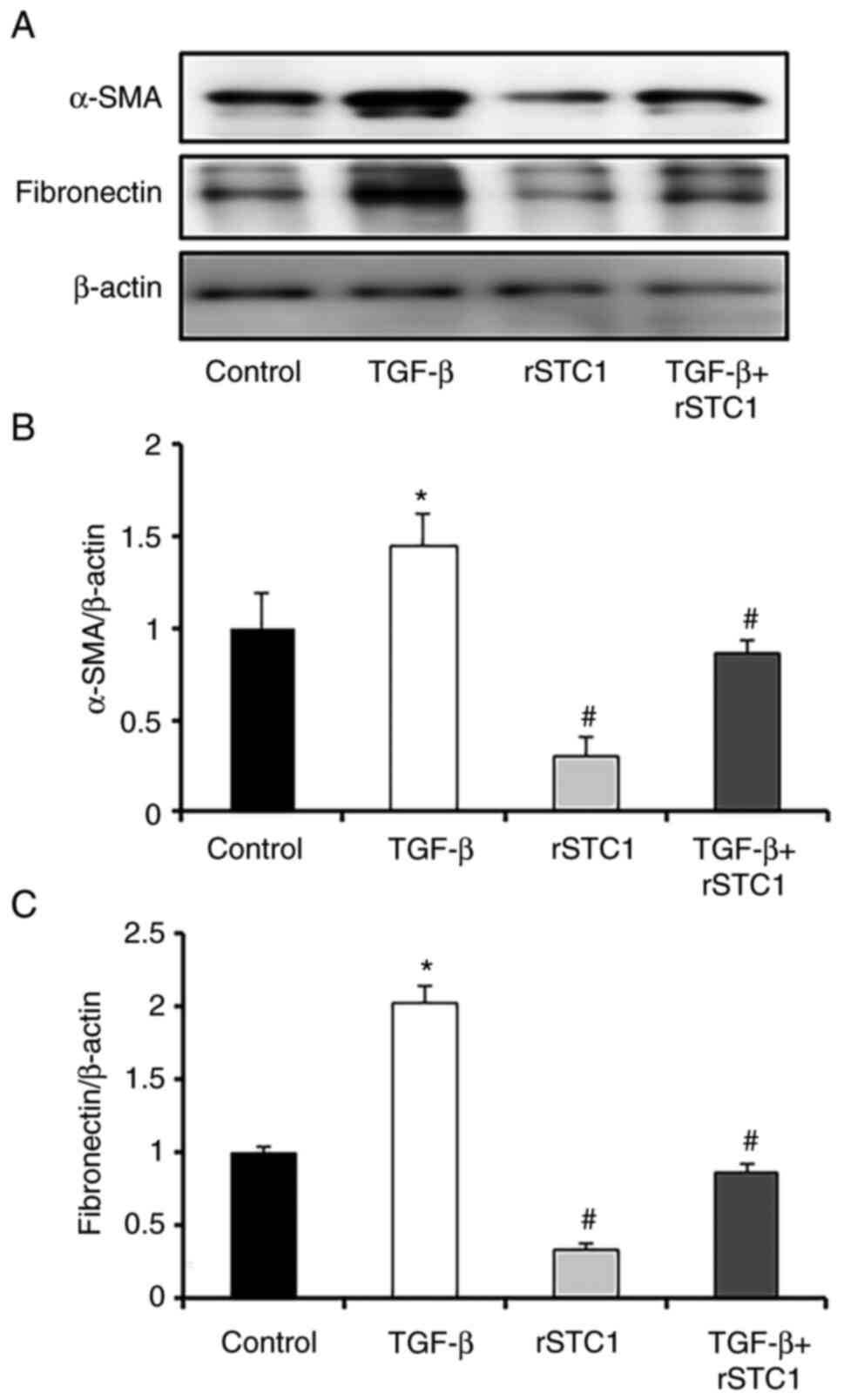
To determine whether rSTC1 inhibited fibrotic
progression in HK-2 cells, the effects of rSTC1 on TGF-β-induced
α-SMA and fibronectin expression in HK2 cells were assessed. The
protein expression levels of α-SMA and fibronectin were higher in
the TGF-β-only-treated group than in the control group (Fig. 1). Treatment with rSTC1 alone did
not affect the protein levels of α-SMA and fibronectin (Fig. 1); however, treatment with rSTC1
and TGF-β together restored the levels of α-SMA and fibronectin to
those in the control group.
rSTC1 attenuates the TGF-β-induced
suppression of AMPK-UCP2-dependent signaling
Cultured HK2 cells were treated with rSTC1 for
different durations (0 min, 15 min, 30 min, 1 h, 3 h and 6 h).
While TGF-β repressed AMPK activity for 6 h (Fig. 2A), rSTC1 significantly
upregulated AMPK activity by nearly 3-fold after treatment for 30
min to 1 h (Fig. 2B). UCP2
expression was also decreased after TGF-β treatment and upregulated
after rSTC1 treatment at certain time points (30 min to 1 h).
Similar to AMPK activity, the peak level of UCP2 was reached 30 min
after rSTC1 treatment (Fig. 2C and
D). Treatment with rSTC1 and TGF-β together prevented the
TGF-β-induced reduction in AMPK activity in HK2 cells (Fig. 2E). AMPK has been reported to
upregulate UCP2 in the kidneys (19). To determine whether the
activation of AMPK mediated the upregulation of UCP2, HK2 cells
were treated with the AMPK inhibitor, STC1, and TGF-β. Treatment
with the AMPK inhibitor diminished the upregulation of UCP2
compared with that observed after treatment with rSTC1 and TGF-β in
combination (Fig. 2F). These
results indicate that rSTC1 induces AMPK activation and the
AMPK-mediated induction of UCP2 expression.
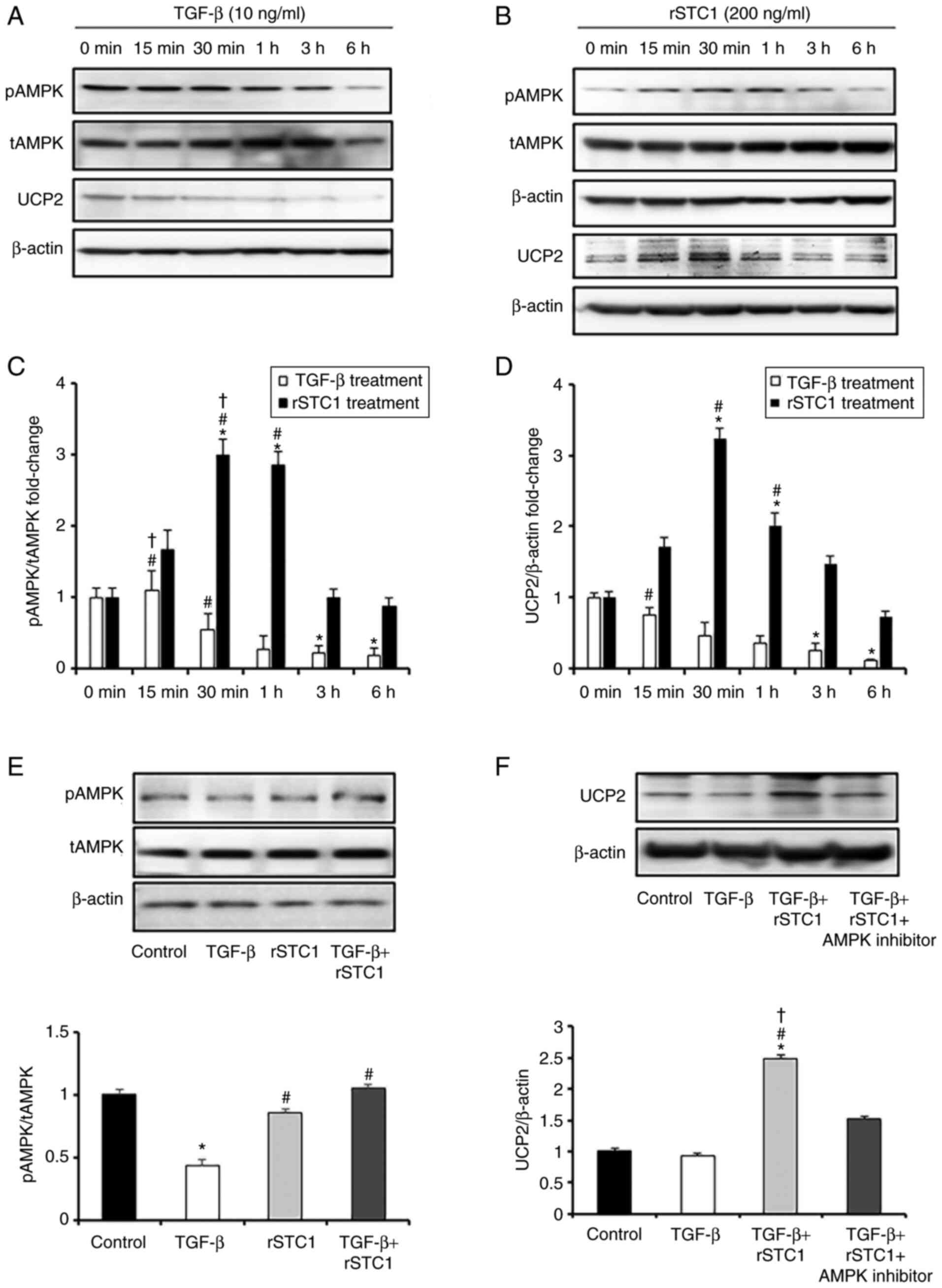 | Figure 2.Effects of rSTC1 on the AMPK/UCP2
signaling pathway in HK2 cells. (A) Protein expression levels of
AMPK and UCP2 in TGF-β-only-treated HK2 cells at the indicated time
points, as determined using western blotting. (B) Protein
expresison level of AMPK and UCP2 in rSTC1-treated HK2 cells at the
indicated time points, as determined using western blotting. (C)
Bar graph of ratio of pAMPK/tAMPK in TGF-β-only-treated and
rSTC1-treated HK2 cells at the indicated time points. (D) Bar graph
of protein expression levels of UCP2 in TGF-β-only-treated and
rSTC1-treated HK2 cells at the indicated time points. *P<0.05
compared with 0 min. #P<0.05 compared with 6 h.
†P<0.05 compared with 3 h. (E) Ratio of pAMPK/tAMPK
in response to TGF-β and rSTC1 treatment after 6 h, as determined
using western blotting. Relative protein expression levels are
shown, with the densitometric values normalized to the respective
β-actin values. *P<0.05 compared with the control;
#P<0.05 compared with TGF-β-only-treated cells. (F)
Protein expression levels of UCP2 in response to TGF-β, rSTC1, and
AMPK inhibitor treatment after 6 h, as determined using western
blotting. Relative protein expression levels are shown, with the
densitometric values normalized to the respective β-actin values.
*P<0.05 compared with the control; #P<0.05
compared with TGF-β-only-treated cells; †P<0.05
compared with TGF-β, rSTC1 and AMPK inhibitor-treated cells. AMPK,
AMP-activated protein kinase; rSTC1, recombinant stanniocalcin-1;
pAMPK, phosphorylated AMPK; tAMPK, total AMPK; UCP2, uncoupling
protein 2. |
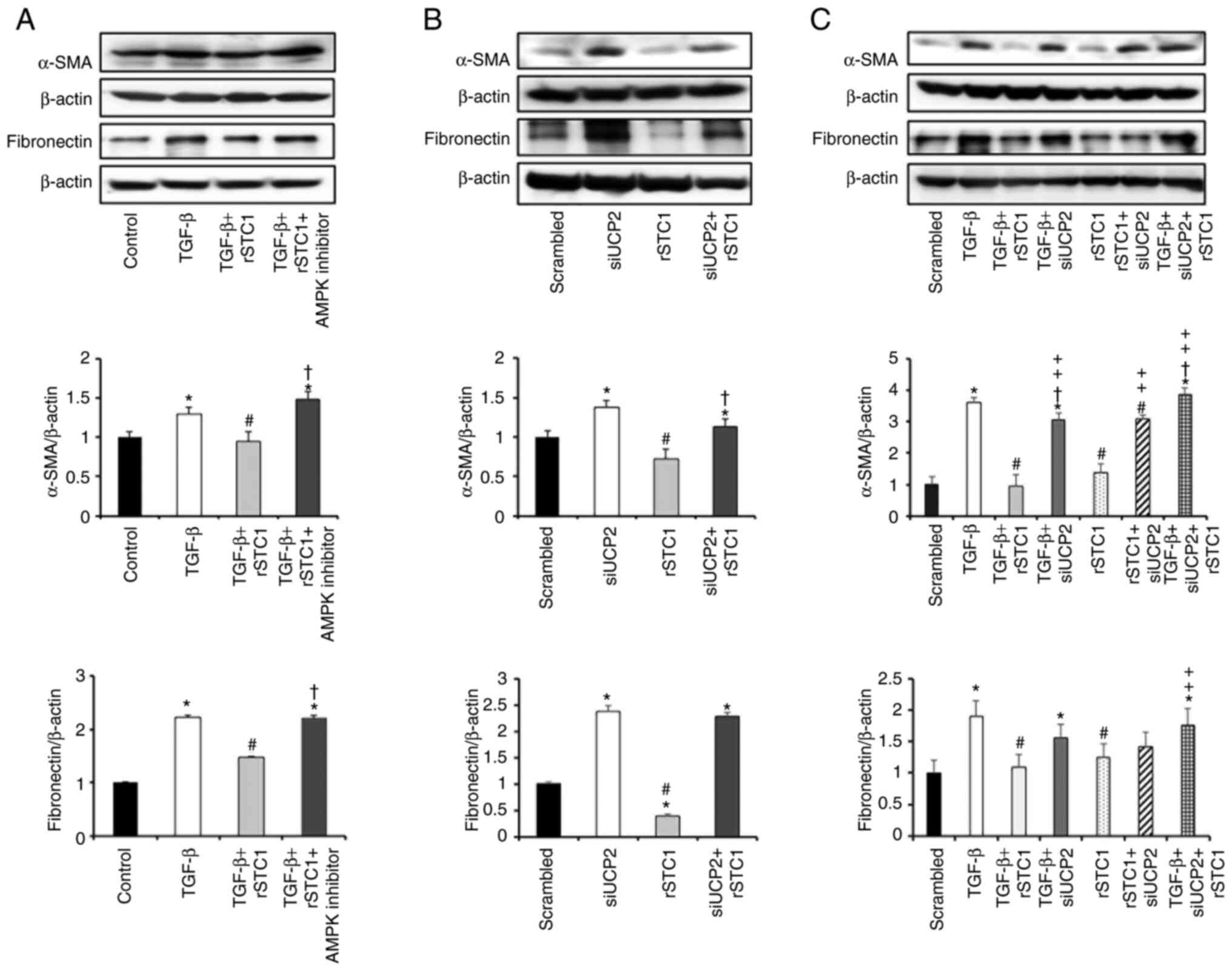
UCP2 inhibition decreases the effects
of STC1 on TGF-β-induced fibrosis in HK2 cells
STC1 regulates renal AMPK-UCP2 activity (19), and AMPK activation may inhibit
renal fibrosis (22). To
determine whether rSTC1 inhibits fibrotic progression via an
AMPK-UCP2-dependent pathway, cells were treated with an AMPK
inhibitor. The STC1-mediated attenuation of TGF-β-induced
upregulation of α-SMA and fibronectin was reversed by AMPK
inhibition (Fig. 3A). To reveal
the function of UCP2 in suppressing fibrosis, UCP2 was knocked down
using a siRNA (siUCP2). HK2 cells were transfected with either
scrambled siRNA or siUCP2. siUCP2 transfection resulted in the
upregulation of α-SMA and fibronectin compared with
scrambled-siRNA-transfected cells. Treatment of siUCP2-transfected
cells with rSTC1 increased the expression levels of α-SMA and
fibronectin compared with those in cells treated with rSTC1 alone
(Fig. 3B). The treatment of
siUCP2-transfected cells with rSTC1 and TGF-β together increased
the expression levels of α-SMA and fibronectin compared with the
levels in scrambled siRNA-transfected cells or cells treated with
rSTC1 and TGF-β (Fig. 3C). These
data suggest that the STC1-mediated reduction in fibrosis occurs
via UCP2.
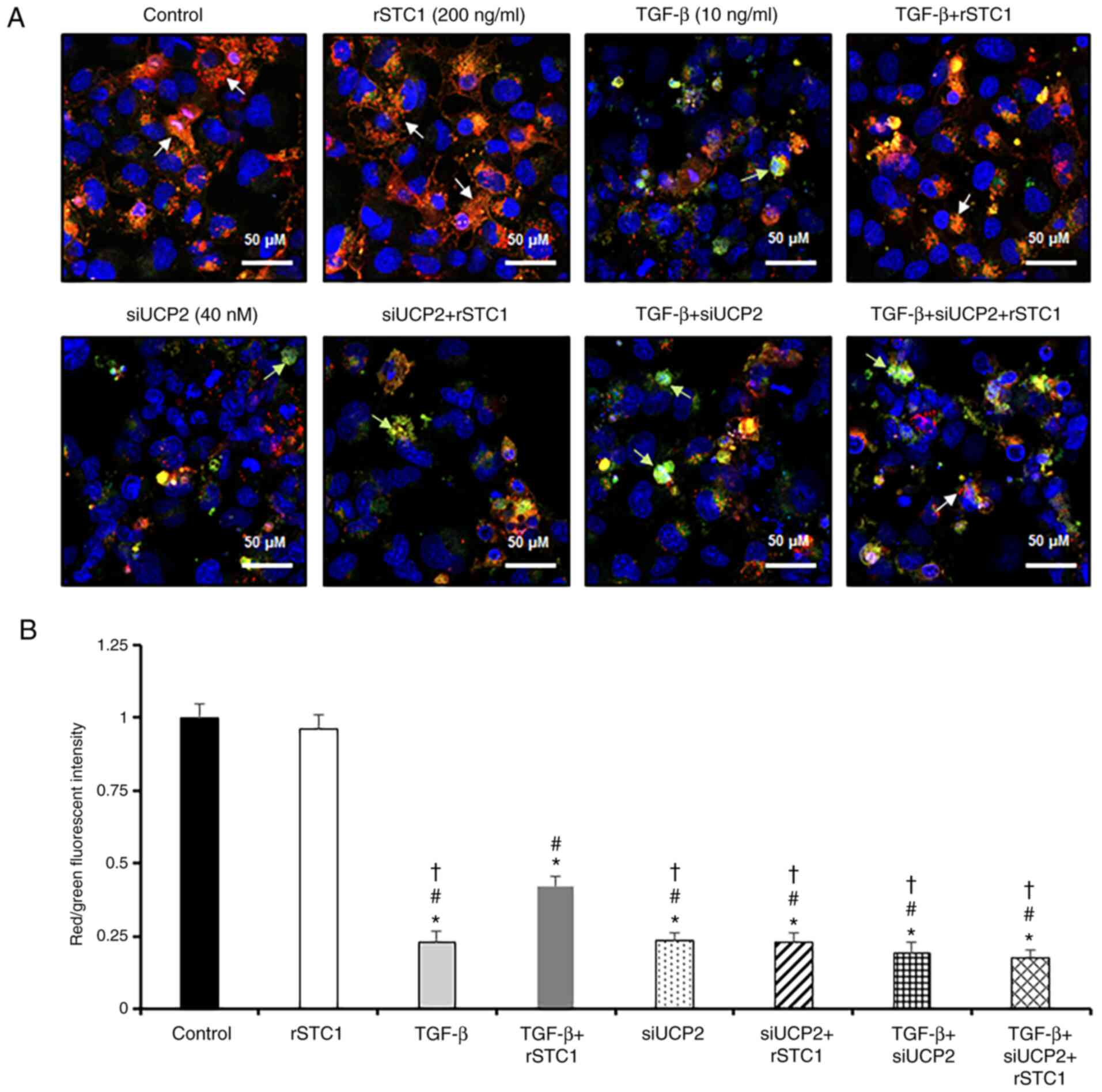
rSTC1 treatment attenuates the
TGF-β-induced decrease in MMP and increase in mitochondrial ROS
generation in HK2 cells
Mitochondria account for the majority of cellular
ROS production, and UCP2 serves an important role in restoring MMP
and dissipating metabolic energy to prevent oxidative stress
(23). Since UCP2 expression was
induced by rSTC1 and attenuated by TGF-β treatment, we hypothesized
that STC1 might be involved in UCP2-dependent regulation of MMP and
ROS generation in mitochondria. Treatment of HK2 cells with TGF-β
reduced MMP levels compared with the control, as indicated by a
decrease in the ratio of red/green fluorescence intensity (Fig. 4). In comparison with levels in
cells treated with TGF-β alone, the present study revealed that
after treatment with rSTC1 and TGF-β, the MMP level was restored,
as indicated by an increase in the ratio of red/green fluorescence
intensity (42% vs. 23%). To further elucidate the role of UCP2, HK2
cells pre-treated with STC1 alone or in combination with TGF-β were
transfected with siUCP2. Treatment of siUCP2-transfected cells with
rSTC1 and TGF-β significantly decreased the intensity of red
fluorescence compared with that in cells with rSTC1 + TGF-β
treatment. These data suggest that rSTC1 attenuates TGF-β-induced
reductions in MMP, and this effect is dependent on UCP2.
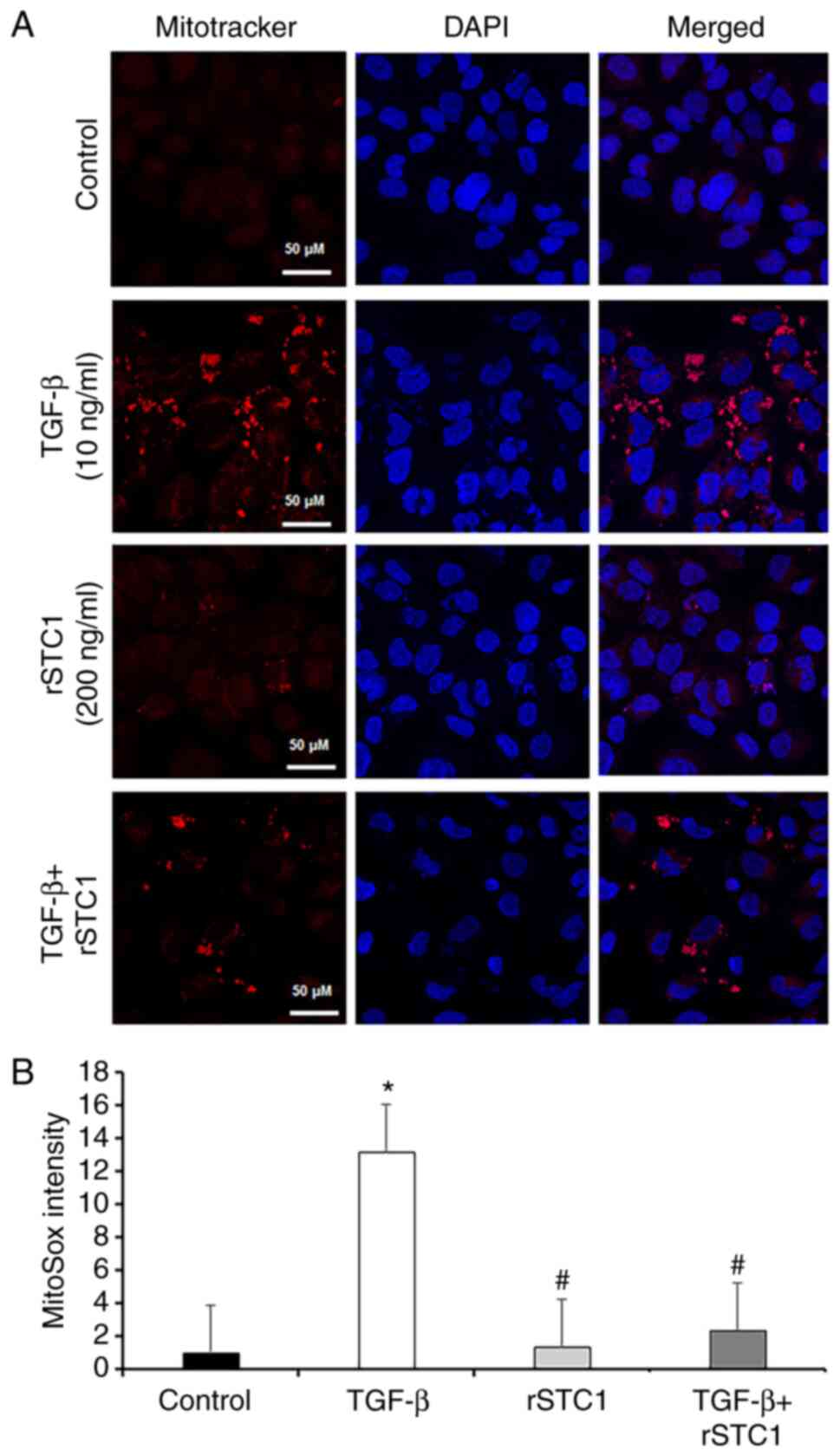
Mitochondrial UCP2 regulates oxidative stress (24) and STC1 is an effective ROS
scavenger (16,17). To identify the effect of STC1 on
mitochondrial ROS generation, the mitochondria-specific probe
MitoSOX was used. As shown in Fig.
5B, mitochondrial ROS production was 12.9-fold higher in
TGF-β-only-treated HK2 cells than in control cells. However,
treatment with TGF-β and rSTC1 decreased mitochondrial ROS
production to 17.5% of the levels in cells treated with TGF-β alone
(Fig. 5).
Discussion
The present results suggest that STC1 protects
against cellular fibrosis in kidney tubular epithelial cells based
on three lines of evidence. First, TGF-β-induced fibrosis and
elevated ROS production were ameliorated by rSTC1 treatment in HK2
cells. Second, the effects of rSTC1 treatment were mediated via the
AMPK-UCP2 signaling pathway in HK2 cells. Third, STC1-mediated
activation of the AMPK-UCP2 signaling pathway antagonized
mitochondrial ROS production and the MMP decrease in HK2 cells.
Therefore, the present results indicate that exogenous STC1 is a
potential agent for the management of cellular fibrosis in kidney
epithelial cells.
Fibrosis represents the final pathway shared by
nearly all progressive CKDs (4).
Although there are no targeted therapies to slow fibrosis, previous
advances in CKD research have clarified the cellular and molecular
mechanisms underlying the disease (5,25,26). However, few studies have
evaluated the anti-fibrotic function of STC1 (27,28). Ono et al (27) revealed that STC1 ameliorates
pulmonary fibrosis by reducing oxidative stress in a dose-dependent
manner and reduces endoplasmic reticulum (ER) stress and TGF-β1
synthesis in alveolar macrophages in bleomycin-induced pulmonary
fibrosis. STC1 may modulate UCP2 expression in lung cells and
uncoupling respiration reduces MMP. In addition, STC1 reduce ER
stress through the reduction of ROS, ameliorating pulmonary
fibrosis (28). The present
study revealed that STC1 upregulated AMPK and UCP2 and that an
inhibitor of AMPK reduced the expression levels of UCP2 in tubular
epithelial cells, suggesting that STC1 may regulate renal AMPK-UCP2
activity, as reported by Pan et al (19). In the present study, treatment
with STC1 reduced the expression levels of fibrosis markers, such
as α-SMA and fibronectin, in TGF-β-stimulated HK-2 cells.
However, the mechanism underlying the STC1-mediated
inhibition of fibrosis in the kidney is unclear. We hypothesized
that AMPK-UCP2 activation could rescue fibrosis by regulating MMP.
AMPK is highly expressed in the kidneys, where it is considered to
be involved in a variety of physiological and pathological
processes (29), and a reduction
in AMPK activity has been proposed as a mechanism underlying renal
fibrosis in different experimental models of CKD (30–33). Mechanisms underlying the
inhibition of fibrosis by AMPK activation have been reported
(31,32). TGF-β1, which serves a prominent
role in fibrosis, diminishes AMPK phosphorylation and increases
fibrosis (22,31). By contrast, the pharmacologic
activation of AMPK reverses the fibrogenic response induced by
TGF-β (32). Similar to the
results of a previous study by Pan et al (19), the present study revealed that
AMPK inhibition restored the STC1-mediated attenuation of
TGF-β-induced upregulation of α-SMA and fibronectin in HK2 cells.
This result suggested that the anti-fibrotic effect of STC1 is
mediated by AMPK activity.
AMPK increases the expression of several antioxidant
genes, such as superoxide dismutase (16), UCP2 and nuclear factor
erythroid-2-related factor (34). Among them, UCP2 regulates the
production of mitochondrial ROS (27,28). The optimal production of
mitochondrial ROS is essential for mitochondrial biogenesis;
however, increased ROS production exceeding local antioxidant
capacities is associated with mitochondrial dysfunction,
mitochondrial DNA damage and aberrant metabolism (35,36). Mitochondrial damage triggers cell
signaling pathways that can cause ROS overproduction, resulting in
oxidative stress, which serves crucial roles in the pathogenesis of
renal fibrosis (37,38). The role of UCP2 in renal disease
has been explored in the context of acute kidney injury and
diabetic nephropathy (24,39). UCP2 ameliorates mitochondrial
dysfunction and oxidative stress in lipopolysaccharide-induced
acute kidney injury (39) and
UCP2 deletion aggravates tubular injury in ischemia-reperfusion
injury by inducing ROS overproduction (40), which highlights the importance of
these transporters in ROS dissipation. Additionally, under disease
conditions, the deletion of UPC2 may contribute to the ameliorate
of kidney fibrosis by inhibiting macrophage infiltration (41,42). The present results revealed that
the upregulation of UCP2 by STC1 reduced fibrosis markers induced
by TGF-β in HK2 cells and UCP2 silencing in cells treated with
rSTC1 and TGF-β increased the levels of fibrosis markers, such as
α-SMA and fibronectin. The effect of STC1 in cellular fibrosis was
diminished by UCP2 suppression. Therefore, we hypothesize that the
inhibitory effect of STC1 on cellular fibrosis is mediated via the
AMPK-UCP2 signaling pathway.
Mitochondrial STC1 suppresses ROS generation through
the induction of UCP expression (16,18) and serves an important role in the
regulation of mitochondrial function (43). Increased UCP2 expression in the
tubular epithelium of STC1 transgenic animal kidneys is associated
with lower superoxide generation (18). Excessive production of
mitochondrial ROS is linked to mitochondrial dysfunction, which
causes cellular damage and the progression of renal disease
(5,6). TGF-β, a key driver of renal
fibrosis, induces oxidative stress and mitochondrial dysfunction
(6), as evidenced by the marked
decrease in MMP and increase in mitochondrial ROS production in HK2
cells (44). Mitochondrial
dysfunction-induced renal fibrosis has attracted immense research
attention since the early 2000s (5). The kidneys have high energy
requirements, and thus, kidney cells contain several mitochondria.
Mitochondrial dysfunction serves a crucial role in the pathogenesis
of CKD, and studies have demonstrated that mitochondrial
dysfunction is involved in the pathological development of renal
fibrosis (5,11,45). Mitochondrial dysfunction promotes
inflammation and fibrotic responses, which induces
tubulointerstitial fibrosis and various forms of CKD, including
diabetic and IgA nephropathy (6,35). In the present study,
TGF-β-mediated fibrosis involved the enhanced generation of
mitochondrial ROS and loss of MMP via the AMPK-UCP2 signaling
pathway. The observed changes in MMP may lead to mitochondrial
dysfunction and occur as a consequence of ROS generation by TGF-β
(46). The total force driving
protons into the mitochondria is a combination of the MMP and
mitochondrial pH gradient (47).
The proton electrochemical gradient potential provides the charge
gradient required for mitochondrial Ca2+ sequestration
and regulates ROS production; thus, it is also a central regulator
of cellular health (47).
TGF-β-induced MMP loss was reduced by STC1, and this effect was
diminished by knocking down UCP2 expression using siUCP2.
The present study demonstrated a protective role of
exogenous STC1 in TGF-β-induced cellular fibrosis in HK2 cells.
STC1 treatment attenuated TGF-β-induced fibrosis by reducing
mitochondrial ROS production via the AMPK-UCP2 pathway. To
establish the role of STC1 in renal fibrosis, further research
using animals may be required to confirm the results of the present
study.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Individual Basic Science and
Engineering Research Program of the Ministry of Education of the
Republic of Korea and National Research Foundation of Korea (grant
no. NRF-2016R1D1A1B03933207), the National Research Foundation of
Korea (NRF) funded by the Korean government (MSIT) (grant nos.
NRF-2019R1A2C2086276 and NRF-2020R1A2C1003310), and the grant of
Chonnam National University Hospital Biomedical Research Institute
(grant no. BCRI-22079).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SWK conceived and designed the experiments. EMY, JSP
and SYJ performed the experiments. EHB and JSP analyzed the data
and confirm the authenticity of all the raw data. SKM contributed
to interpretation of data. EMY wrote the paper. SWK and SKM
contributed to critical revisions of the paper. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jha V, Garcia-Garcia G, Iseki K, Li Z,
Naicker S, Plattner B, Saran R, Wang AYM and Yang CW: Chronic
kidney disease: Global dimension and perspectives. Lancet.
382:260–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vallianou NG, Mitesh S, Gkogkou A and
Geladari E: Chronic kidney disease and cardiovascular disease: Is
there any relationship? Curr Cardiol Rev. 15:55–63. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bello AK, Alrukhaimi M, Ashuntantang GE,
Basnet S, Rotter RC, Douthat WG, Kazancioglu R, Köttgen A, Nangaku
M, Powe NR, et al: Complications of chronic kidney disease: Current
state, knowledge gaps, and strategy for action. Kidney Int Suppl
(2011). 7:122–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He J, Xu Y, Koya D and Kanasaki K: Role of
the endothelial-to-mesenchymal transition in renal fibrosis of
chronic kidney disease. Clin Exp Nephrol. 17:488–497. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quadri MM, Fatima SS, Che RC and Zhang AH:
Mitochondria and renal fibrosis. Adv Exp Med Biol. 1165:501–524.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhargava P and Schnellmann RG:
Mitochondrial energetics in the kidney. Nat Rev Nephrol.
13:629–646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ishimoto Y and Inagi R: Mitochondria: A
therapeutic target in acute kidney injury. Nephrol Dial Transplant.
31:1062–1069. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaushal GP, Chandrashekar K and Juncos LA:
Molecular interactions between reactive oxygen species and
autophagy in kidney disease. Int J Mol Sci. 20:37912019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coughlan MT and Sharma K: Challenging the
dogma of mitochondrial reactive oxygen species overproduction in
diabetic kidney disease. Kidney Int. 90:272–279. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsuji N, Tsuji T, Ohashi N, Kato A,
Fujigaki Y and Yasuda H: Role of mitochondrial DNA in septic AKI
via toll-like receptor 9. J Am Soc Nephrol. 27:2009–2020. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Che R, Yuan Y, Huang S and Zhang A:
Mitochondrial dysfunction in the pathophysiology of renal diseases.
Am J Physiol Renal Physiol. 306:367–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yeung BH, Law AY and Wong CK: Evolution
and roles of stanniocalcin. Mol Cell Endocrinol. 349:272–280. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ishibashi K and Imai M: Prospect of a
stanniocalcin endocrine/paracrine system in mammals. Am J Physiol
Renal Physiol. 282:F367–F375. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McCudden CR, James KA, Hasilo C and Wagner
GF: Characterization of mammalian stanniocalcin receptors.
Mitochondrial targeting of ligand and receptor for regulation of
cellular metabolism. J Biol Chem. 277:45249–4558. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ellard JP, McCudden CR, Tanega C, James
KA, Ratkovic S, Staples JF and Wagner GF: The respiratory effects
of stanniocalcin-1 (STC-1) on intact mitochondria and cells: STC-1
uncouples oxidative phosphorylation and its actions are modulated
by nucleotide triphosphates. Mol Cell Endocrinol. 264:90–101. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Huang L, Abdelrahim M, Cai Q,
Truong A, Bick R, Poindexter B and Sheikh-Hamad D: Stanniocalcin-1
suppresses superoxide generation in macrophages through induction
of mitochondrial UCP2. J Leukoc Biol. 86:981–988. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheikh-Hamad D: Mammalian stanniocalcin-1
activates mitochondrial antioxidant pathways: New paradigms for
regulation of macrophages and endothelium. Am J Physiol Renal
Physiol. 298:F248–F254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang L, Belousova T, Chen M, DiMattia G,
Liu D and Sheikh-Hamad D: Overexpression of stanniocalcin-1
inhibits reactive oxygen species and renal ischemia/reperfusion
injury in mice. Kidney Int. 82:867–877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan JS, Huang L, Belousova T, Lu L, Yang
Y, Reddel R, Chang A, Ju H, DiMattia G, Tong Q, et al:
Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an
AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol.
26:364–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sivandzade F, Bhalerao A and Cucullo L:
Analysis of the mitochondrial membrane potential using the cationic
JC-1 dye as a sensitive fluorescent probe. Bio Protoc.
9:e31282019.PubMed/NCBI
|
|
22
|
Kim H, Moon SY, Kim JS, Baek CH, Kim M,
Min JY and Lee SK: Activation of AMP-activated protein kinase
inhibits ER stress and renal fibrosis. Am J Physiol Renal Physiol.
308:F226–F236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Donadelli M, Dando I, Fiorini C and
Palmieri M: UCP2, a mitochondrial protein regulated at multiple
levels. Cell Mol Life Sci. 71:1171–1190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Humphreys BD: Mechanisms of renal
fibrosis. Annu Rev Physiol. 80:309–326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Djudjaj S and Boor P: Cellular and
molecular mechanisms of kidney fibrosis. Mol Aspects Med. 65:16–36.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ono M, Ohkouchi S, Kanehira M, Tode N,
Kobayashi M, Ebina M, Nukiwa T, Irokawa T, Ogawa H, Akaike T, et
al: Mesenchymal stem cells correct inappropriate
epithelial-mesenchyme relation in pulmonary fibrosis using
stanniocalcin-1. Mol Ther. 23:549–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohkouchi S, Ono M, Kobayashi M, Hirano T,
Tojo Y, Hisata S, Ichinose M, Irokawa T, Ogawa H and Kurosawa H:
Myriad functions of stanniocalcin-1 (STC1) cover multiple
therapeutic targets in the complicated pathogenesis of idiopathic
pulmonary fibrosis (IPF). Clin Med Insights Circ Respir Pul Med.
9:91–96. 2015.PubMed/NCBI
|
|
29
|
Juszczak F, Caron N, Mathew AV and
Declèves AE: Critical role for AMPK in metabolic disease-induced
chronic kidney disease. Int J Mol Sci. 21:79942020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma K: Obesity, oxidative stress, and
fibrosis in chronic kidney disease. Kidney Int Suppl (2011).
4:113–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thakur S, Viswanadhapalli S, Kopp JB, Shi
Q, Barnes JL, Block K, Gorin Y and Abboud HE: Activation of
AMP-activated protein kinase prevents TGF-β1-induced
epithelial-mesenchymal transition and myofibroblast activation. Am
J Pathol. 185:2168–2180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao J, Ye J, Ying Y, Lin H and Luo Z:
Negative regulation of TGF-β by AMPK and implications in the
treatment of associated disorders. Acta Biochim Biophys Sin
(Shanghai). 50:523–531. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Borges CM, Fujihara CK, Malheiros D, de
Ávila VF, Formigari GP and Lopes de Faria JB: Metformin arrests the
progression of established kidney disease in the subtotal
nephrectomy model of chronic kidney disease. Am J Physiol Renal
Physiol. 318:F1229–F1236. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trewin AJ, Berry BJ and Wojtovich AP:
Exercise and mitochondrial dynamics: Keeping in shape with ROS and
AMPK. Antioxidants (Basel). 7:72018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bhatia D, Capili A and Choi ME:
Mitochondrial dysfunction in kidney injury, inflammation, and
disease: Potential therapeutic approaches. Kidney Res Clin Pract.
39:244–258. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Czajka A, Ajaz S, Gnudi L, Parsade CK,
Jones P, Reid F and Malik AN: Altered mitochondrial function,
mitochondrial DNA and reduced metabolic flexibility in patients
with diabetic nephropathy. EBioMedicine. 2:499–512. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su H, Wan C, Song A, Qiu Y, Xiong W and
Zhang C: Oxidative stress and renal fibrosis: Mechanisms and
therapies. Adv Exp Med Biol. 1165:585–604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lv W, Booz GW, Fan F, Wang Y and Roman RJ:
Oxidative stress and renal fibrosis: Recent isights for the
development of novel therapeutic strategies. Front Physiol.
9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding Y, Zheng Y, Huang J, Peng W, Chen X,
Kang X and Zeng Q: UCP2 ameliorates mitochondrial dysfunction,
inflammation, and oxidative stress in lipopolysaccharide-induced
acute kidney injury. Int Immunopharmacol. 71:336–349. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou Y, Cai T, Xu J, Jiang L, Wu J, Sun Q,
Zen K and Yang J: UCP2 attenuates apoptosis of tubular epithelial
cells in renal ischemia-reperfusion injury. Am J Physiol Renal
Physiol. 313:F926–F937. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wynn TA and Vannella KM: Macrophages in
tissue repair, regeneration, and fibrosis. Immunity. 44:450–462.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Anders HJ and Ryu M: Renal
microenvironments and macrophage phenotypes determine progression
or resolution of renal inflammation and fibrosis. Kidney Int.
80:915–925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Y, Shan P, Srivastava A, Li Z and
Lee PJ: Endothelial stanniocalcin 1 maintains mitochondrial
bioenergetics and prevents oxidant-induced lung injury via
toll-like receptor 4. Antioxid Redox Signal. 30:1775–1796. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Abe Y, Sakairi T, Beeson C and Kopp JB:
TGF-β1 stimulates mitochondrial oxidative phosphorylation and
generation of reactive oxygen species in cultured mouse podocytes,
mediated in part by the mTOR pathway. Am J Physiol Renal Physiol.
305:F1477–F1490. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Galvan DL, Green NH and Danesh FR: The
hallmarks of mitochondrial dysfunction in chronic kidney disease.
Kidney Int. 92:1051–1057. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ribeiro A, Bronk SF, Roberts PJ, Urrutia R
and Gores GJ: The transforming growth factor beta(1)-inducible
transcription factor TIEG1, mediates apoptosis through oxidative
stress. Hepatology. 30:1490–1497. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Perry SW, Norman JP, Barbieri J, Brown EB
and Gelbard HA: Mitochondrial membrane potential probes and the
proton gradient: A practical usage guide. Biotechnique. 50:98–115.
2011. View Article : Google Scholar : PubMed/NCBI
|