Introduction
The principal issue derived from prostate cancer
(PCa) is its inclination to metastasize to bone, which occurred in
as many as 90% of patients with advanced PCa (1). However, the exact mechanisms of bone
metastasis of PCa need further to be elucidated.
As a transcription factor, the tumor suppressor p53
mediates changes in gene expression that promote apoptosis,
senescence or a reversible and protective cell cycle arrest
(2,3). In about half of all human cancers,
p53 is either lost or mutated. Loss of wild-type p53 (WT-p53)
function is well known to influence cell cycle checkpoint controls
and apoptosis (4) and gain of
function of mutant p53 is involved in development and progression
of many cancers (4–6). Importantly, emerging evidence has
shown that WT-p53 also plays a role in regulating key stages of
metastatic progression (4,6), but how it functions as
metastasis/invasion suppressor is just beginning to be understood
(7). In PCa, it also remains
elusive whether and how WT-p53 regulates bone metastasis although
mutant p53 may promote bone metastasis (5).
Recent studies have found that two important
mechanisms by which p53 regulates metastasis, repression of
migration and invasion of cancer cells through modulating
epithelial-mesenchymal transition (EMT) and suppression of cancer
stem cell (CSC) properties (4,8–11).
EMT is a key step of the progression of tumor cell metastasis
(12). It also has been identified
as an important step in bone metastasis of PCa (13). E-cadherin plays a critical role in
EMT which is regulated by transcription factors including Snail,
Slug, Twist and Zeb1/2 (14). By
targeting these transcription factors, p53 regulates EMT (6). Furthermore, the CSCs are the cells
within tumors that possess the ability of self-renewal,
immortalized proliferation and differentiate into the heterogeneous
lineages of cancer cells which consist of the whole tumor (15,16).
These capabilities of cancer stem cells have formed the basic
definition of ‘stemness’ (17).
Accumulating evidence suggests that cancer cell stemness is
associated with the metastasis of tumors (16,17).
Recent studies indicated that p53 has crucial influence on cancer
cell stemness by regulating key stemness genes (8,11).
Emerging evidence has demonstrated that miRNAs are
components of the cellular signaling circuitry that regulates the
EMT program (18), such as miR-200
family (19,20), miR-34 family (21,22)
and miR-205 (19). These miRNAs
directly target transcription factors Snail, Slug and Zeb1/2, and
regulate the EMT of cancer cells. Furthermore, miRNAs also played a
pivotal role in regulating the properties of CSCs by negatively
regulating the expression of certain key genes, such as CD44, Oct4,
Sox2, c-Myc and Klf4 (23).
Moreover, some miRNAs are transcriptionally regulated by p53
(24). Importantly, several
miRNAs, such as miR-200c and miR-34 family mediate p53 regulation
of EMT (8–11) and stem cell properties in cancers
(21).
Our previous studies have demonstrated that miR-145
is associated with bone metastasis of PCa by suppressing EMT and
stemness of cancer cells (25,26).
Also microRNA-145 is directly regulated by WT-p53 (27–30),
and the loss of WT-p53 function occurs in many PCa, therefore, we
reasoned that WT-p53 may play a role in regulating EMT and cancer
cell stemness of PCa cells and miR-145 may mediate the function of
WT-p53.
To test the hypothesis, we upregulated expression of
WT-p53 in p53-null PC-3 cells derived from PCa bone metastasis and
found that ectopic expression of WT-p53 inhibited migration,
invasion, EMT and stemness of PC-3 cells, and the inhibitory
effects of WT-p53 on EMT and cancer cell stemness of PC-3 cells
were reversed by anti-miR-145. Our findings demonstrate that WT-p53
represses EMT and stemness of PC-3 cells at least partially by
mediating the miR-145.
Materials and methods
Cell culture
The bone metastatic PCa cell line PC-3 was purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA) and
grown in Ham’s F-12 culture medium (HyClone, Logan, UT, USA)
supplemented with 10% fetal bovine serum (HyClone). Cells were
grown at a humidified atmosphere of 5% CO2 at 37°C.
Plasmids and transient transfection
Plasmids expressing WT-P53 and miR-145-antagomiR
were purchased from RiboBio Co. Ltd (Ribo, China). The cloning
sequence of WT-P53 was from 203 to 1,384 in the CDS region and
miR-145 in pMSCV was constructed as described previously (25). Before transfection,
2×105 cells were seeded into each well of 6-well plates.
After 24 h incubation in growth medium, the cells were transiently
transfected with using Lipofectamine 2000 (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer’s protocol. Transfection
medium is the Opti-Mem medium which is used for transient
transfection. The transfection medium was removed after 4–6 h, and
the cells were incubated for an additional 48 h in 10% fetal bovine
serum medium (2 ml per well).
Quantitative reverse
transcription-PCR
The procedure was carried out according to the
manuscript of All-in-One™ miRNA qRT-PCR detection kit (GeneCopoeia,
Rockville, MD, USA). Total RNAs were extracted from cells by using
RNeasy kit (Qiagen). Total RNA was reverse transcribed by adding
poly-A sequence, and real-time PCR analysis was performed with
specific primer to WT-p53 and hsa-miR-145 (GeneCopoeia). Each
sample was analyzed in triplicate. No template, and no reverse
transcription were included as negative controls. U6 snRNA was used
as normalization control. Relative expression values from three
independent experiments were calculated following the
2−ΔΔCt method of Schmittgen and Livak (31).
Western blot analysis
For the analysis of expression of related proteins,
western blot assay was performed. The cells were seeded in 6-well
plates. After 24–48 h, cells were washed with pre-chilled PBS and
at confluence of 60–70% harvested in sample buffer [62.5 mmol/l
Tris-HCl (pH 6.8), 2% SDS, 10% glycerol and 5% β-mercaptoethanol].
Equal amounts of protein from the supernatant were loaded per lane
and resolved by SDS-polyacrylamide electrophoresis. Then, protein
was transferred onto PVDF membrane (Millipore), blocked by 5%
non-fat milk for 1 h at room temperature, and probed with primary
antibodies (1:1,000) overnight at 4°C, including rabbit anti-P53,
Oct4, c-Myc, Klf4, CD44 and mouse anti-E-cadherin, vimentin (Cell
Signaling Technology); rabbit anti-N-cadherin (Millipore); mouse
anti-fibronectin (BD Biosciences) and mouse anti-ZEB2 (Sigma, St.
Louis, MO, USA). Membranes were washed three times (10 min each) in
TBS-T buffer and incubated for 40 min at room temperature with
horseradish peroxidase-conjugated anti-mouse or anti-rabbit
secondary antibodies. Membranes were washed thrice (10 min each) in
TBS-T and developed using the ECL system. Protein loading was
normalized by reprobing the blots with rabbit anti-β-actin (Cell
Signaling Technology).
Wound healing assay
PC-3 cells were trypsinized and seeded equivalently
into 6-well tissue culture plates 24 h before scratching, and grew
to reach almost total confluence in 24 h. Non-serum starvation
lasted for 24 h. After cell monolayers formed, a wound was
scratched onto the mono-layer with a sterile 100 μl tip
(Axygen, Union City, CA, USA). After scratching, the cells were
washed with PBS twice and cultured in 10% fetal bovine serum media.
Images of PC-3 cells migrating into the wound were captured at time
points of 0, 6 and 12 h by an inverted microscope (×40).
Invasion assay
The invasion assay was performed by using Transwell
chamber consisting of 8-mm membrane filter inserts (Corning) coated
with Matrigel (BD Biosciences). Briefly, the cells were trypsinized
and suspended in serum-free medium. Then 1.5×105 cells
were added to the upper chamber, and lower chamber was filled with
the medium with 10% FBS. After incubated for 24–48 h, cells passed
through the coated membrane to the lower surface, in which cells
were fixed with 4% paraformaldehyde and stained with hematoxylin.
The cell count was done under a microscope (×100).
Adhesion assay
Briefly, 96-well plates were coated with 50
μl fibronectin (50 μg/ml) at cell incubator for 1 h.
After washed with warm PBS, the plates were blocked with 1% BSA at
37°C for 1 h and washed twice. After trypsinization, suspended
cells were seeded to each well with serum-free media at a density
of 1.5×104 cells per well. The cells were incubated for
30 min, non-adherent cells were removed and plates were gently
washed twice with PBS. Adherent cells were fixed in 4%
paraformaldehyde for 20 min at room temperature, then stained with
hematoxylin and counted under an inverted microscope (×100).
Colony formation assay
The cells were trypsinized as single cells and
suspended in serum-free medium. Cells (300 cells/ml) were seeded
into each well of 6-well plates for 1–2 weeks, and colonies were
dyed with crystal violet. Plating efficiency = number of colonies
(≥50 cells per colony) per input cells × 100%. Colony morphology
was determined, by scoring under a light microscope.
Self-renewing spheroid formation
assay
Cells (500 cells/well and 50–100 cells/well) were
plated respectively onto 6-well and 24-well Ultra Low Cluster plate
(Corning) and were cultured in suspension in serum-free DMEM-F12
(BioWhittaker), supplemented with B27 (1:50, Invitrogen), 20 ng/ml
EGF (BD Biosciences), 0.4% bovine serum albumin (Sigma), and 4
mg/ml insulin (Sigma) for 10–14 days. After 10–14 days, the number
of PC-3 cell spheres (tight, spherical, non-adherent masses >50
μm in diameter) were counted, and image of the spheres were
captured under an inverse microscope. Sphere formation efficiency =
colonies/input cells × 100%.
Statistical analyses
Statistical significance of the studies was analyzed
by Student’s t-test or one-way ANOVA. Statistical significance was
accepted at p<0.05.
Results
WT-p53 repressed invasiveness of PC-3
cells and anti-miR-145 rescued the effect
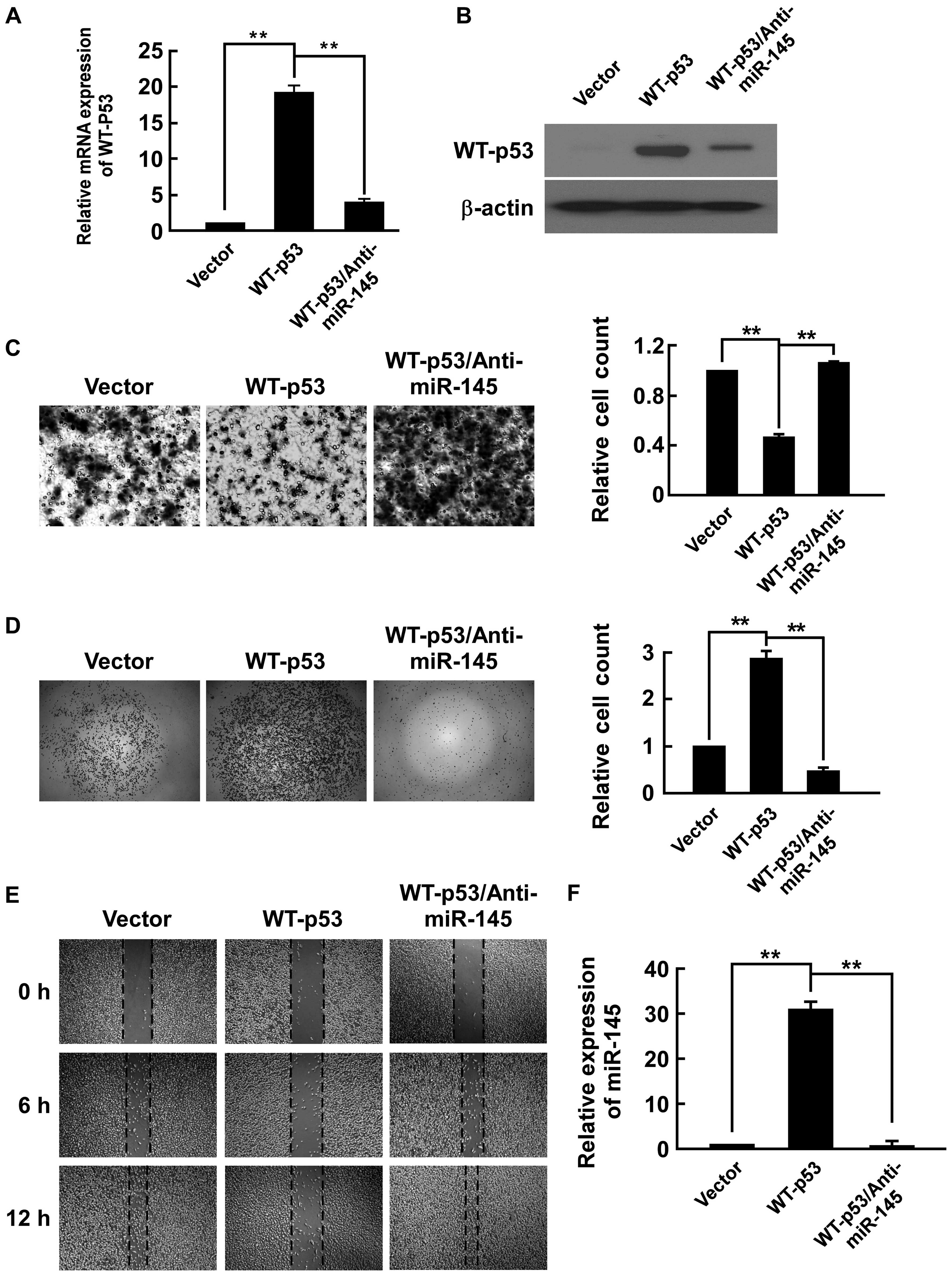
To investigate the role of WT-p53 in the development
and progression of PCa metastasis, we upregulated WT-p53 by
transfecting the plasmid of WT-p53 expression in PC-3 cells, in
which p53 is null. The expression of WT-p53 was confirmed by
real-time PCR in mRNA level (Fig.
1A) and western blot analysis in protein level (Fig. 1B). We investigated whether WT-p53
was able to regulate invasion, migration and adhesion in PC-3
cells. By using Transwell Matrigel invasion assay to assess the
invasive ability of cells, we found that WT-p53 repressed invasive
ability to 45.7% of PC-3/vector (Fig.
1C). In adhesion assay, WT-p53 increased adhesive ability
2.87-fold that of PC-3/vector (Fig.
1D). As shown in Fig. 1E, in
cell migration observed by wound healing assay, WT-p53 decreased
the healing speed of the cell wound compared to the
PC-3/vector.
Because miR-145 is transcriptionally regulated by
WT-p53 (27–30), determined whether inhibiting
miR-145 could rescue the effects of WT-p53. After upregulating
WT-p53, we applied miR-145-antagomiR in PC-3 cells. The expression
of miR-145 was confirmed by real-time PCR. We found that WT-p53
upregulated the expression of miR-145 31.0-fold that of the
PC-3/vector (Fig. 1F). The
anti-miR-145 repressed WT-p53 expression in mRNA and protein
levels. It also completely counteracted expression of WT-p53 in
PC-3 cells as anti-miR-145 increased the cell migration speed and
invasive ability that was reduced by WT-p53 expression, and also
reduced miR-145 expression and the adhesive ability that was
increased by WT-p53 (Fig. 1).
These results indicated that WT-p53 inhibited invasiveness of PC-3
cells through modulation of miR-145.
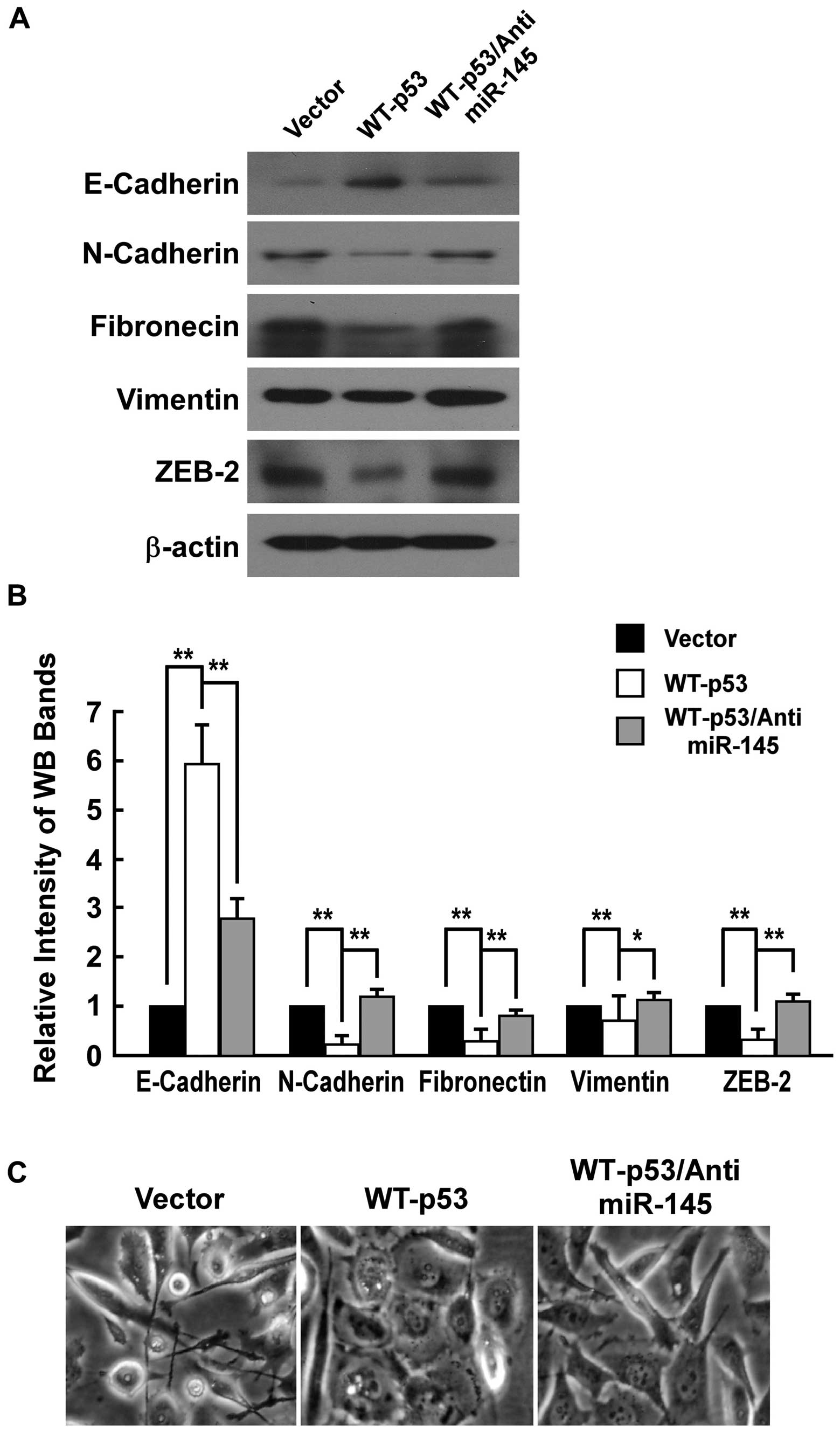
WT-p53 represses EMT and anti-miR-145
rescues the effect in PC-3 cells
To investigate whether miR-145 regulated
invasiveness by repressing EMT, we examined the influence of
ectopic expression of WT-p53 on expressions of E-cadherin,
N-cadherin, fibronectin, vimentin and ZEB2 of PC-3 cells by western
blot analysis. E-cadherin, which is one of key epithelial markers
and supposed to be downregulated during EMT, was increased in PC-3
cells with WT-p53 expression. N-cadherin, fibronectin and vimentin,
which are kind of mesenchymal markers and should be upregulated
during EMT, were repressed in PC-3 cells with WT-p53 expression.
ZEB2, which is a transcription factor upregulating EMT, was
repressed in PC-3 cells with WT-p53 expression (Fig. 2A and B). Further, we detected the
change of morphology of PC-3 cell with characteristics of EMT. The
PC-3 cells with ectopic expression of WT-p53 converted the
predominant mechenchymal phenotype to an evident epithelial
phenotype i.e. from a stick-like or long spindle-shaped mesenchymal
profile to a cobblestone-like or a short spindle-shaped epithelial
morphology (Fig. 2C).
We determined whether inhibiting miR-145 would
rescue the effects of expressing WT-p53 in PC-3 cell. As Fig. 2 shows, the anti-miR-145 completely
counteracted the effects of WT-p53 expression in PC-3 cells as
anti-miR-145 reduced the level of the epithelial cell marker
E-cadherin that was increased by p53 expression, and restored
expression levels of the mesenchymal cell marker N-cadherin,
fibronectin, vimentin and ZEB2 that was reduced by WT-p53
expression. The anti-miR-145 also was able to convert the
predominant epithelial phenotype that was changed by WT-p53
expression to an evident mechenchymal phenotype. These observations
indicated that WT-p53 repressed EMT through modulation of miR-145
in PC-3 cells.
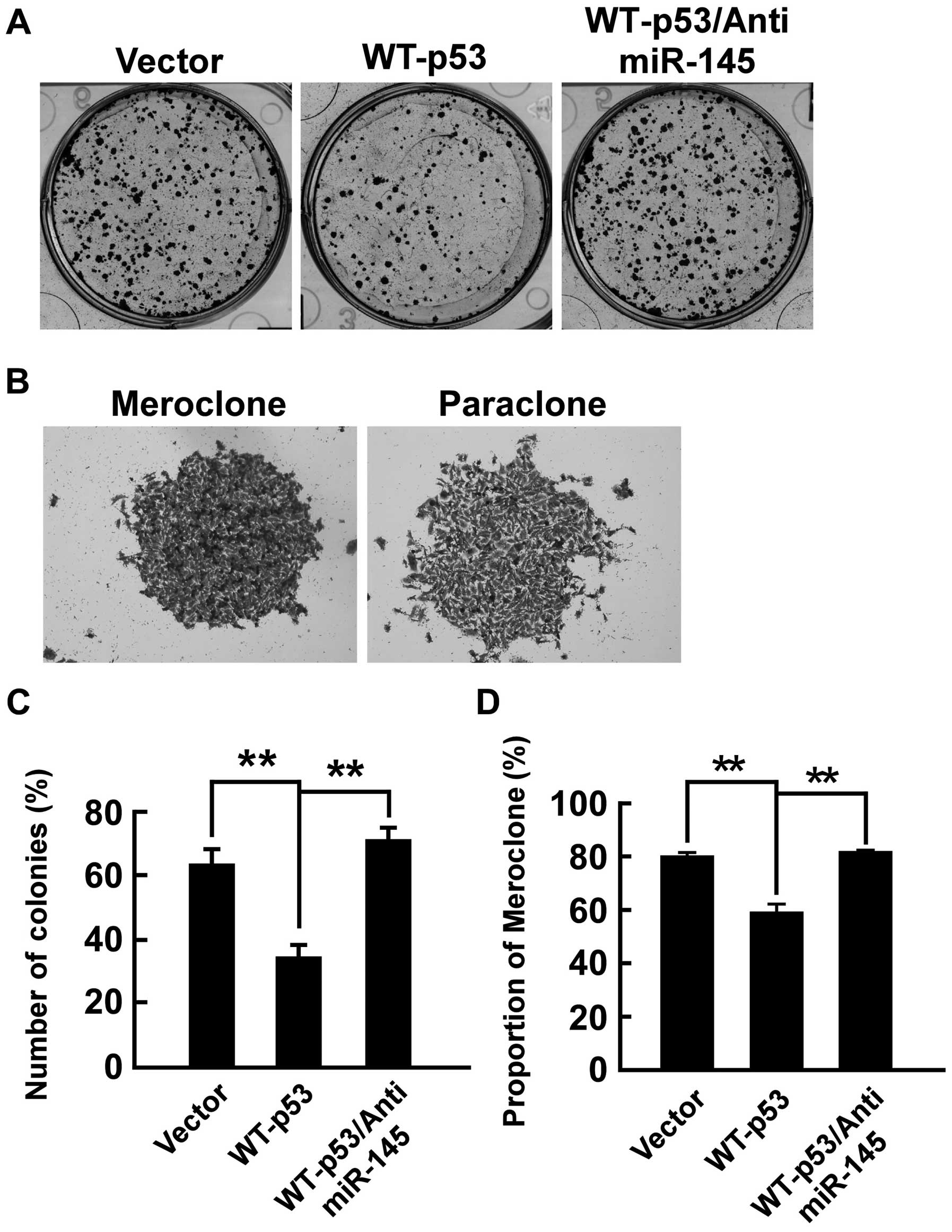
WT-p53 inhibits colony formation of PC-3
cells and anti-miR-145 rescues the effect
We determined efficiency of WT-p53 inhibiting
colony-forming of PC-3 cells in vitro. The colony-forming
assay was performed. The number of colonies (% plating efficiency)
were 34.3% in PC-3/WT-p53 and 63.7% in PC-3/vector and
significantly decreased in PC-3/WT-p53 compared with PC-3/vector
(p<0.01, respectively) (Fig.
3). Colonies with different morphologies in vitro are
classified as holoclones, meroclones and paraclones (32). Holoclones are generally more round
and tightly packed; paraclones are irregular in composition and
often contain more elongated or flattened cells; and meroclones are
an intermediate phenotype. We did not find typical holoclones in
PC-3 cells. The proportion of meroclones was 80.1% in PC-3/vector
and 58.9% in PC-3/WT-p53, and WT-p53 significantly decreased the
proportion of meroclones of PC-3 cells (p<0.01, respectively)
(Fig. 3). Furthermore, we
determined whether WT-p53 regulates colony-forming by modulating
miR-145 in PC-3 cells. As shown in Fig. 3, the anti-miR-145 completely
counteracted the effect of expressing WT-p53 in PC-3 cells as
anti-miR-145 restored colony-forming capability that was diminished
by WT-p53 expression. The number of colonies was 71.4% and the
proportion of meroclones was 81.4% in PC-3/WT-p53/anti-miR-145.
These data indicated that WT-p53 repressed cancer cell
colony-forming by modulation of miR-145 in PC-3 cells.
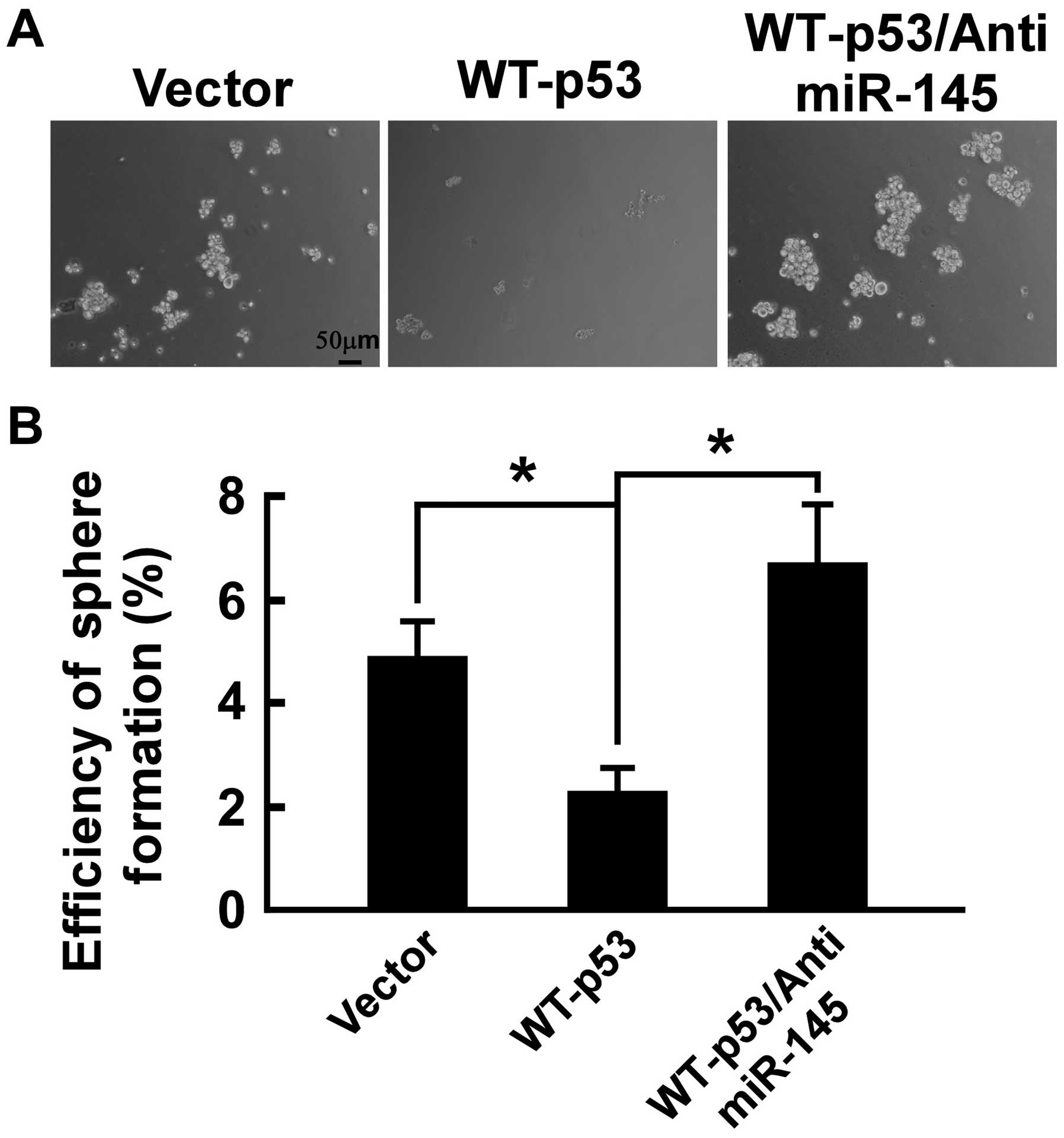
WT-p53 inhibits tumor spheroid formation
of PC-3 cells and anti-miR-145 rescues the effect
The ability to grow as non-adherent spheroids in the
sphere medium has been widely used to assess the self-renewal
capability of CSCs and is one of the properties of prostate CSCs
(33). To confirm that WT-p53 can
inhibit the self-renewal capability of PC-3 cells, prostasphere
formation of PC-3 cells was studied. As shown in Fig. 4, after culturing for 12 days under
non-adherent conditions, there were prostaspheres in all the cell
types. The spheroid formation efficiency was 4.8% in PC-3/vector
and 2.3% in PC-3/WT-p53, confirming the presence of the
self-renewal cell in PC-3/vector and PC-3/WT-p53, and WT-p53
suppressed significantly prostasphere formation (p<0.05).
Furthermore, we determined whether WT-p53 regulates tumor spheroid
formation by modulating miR-145 in PC-3 cells. As shown in Fig. 4, the anti-miR-145 completely
counteracted the effect of WT-p53 expression in PC-3 cells as
anti-miR-145 restored the capability of tumor spheroid formation
that was diminished by WT-p53 expression. The spheroid formation
efficiency was 6.7% in PC-3/WT-p53/anti-miR-145. These data
indicated that WT-p53 repressed the self-renewal capability of
cancer cells by modulating miR-145 in PC-3 cells.
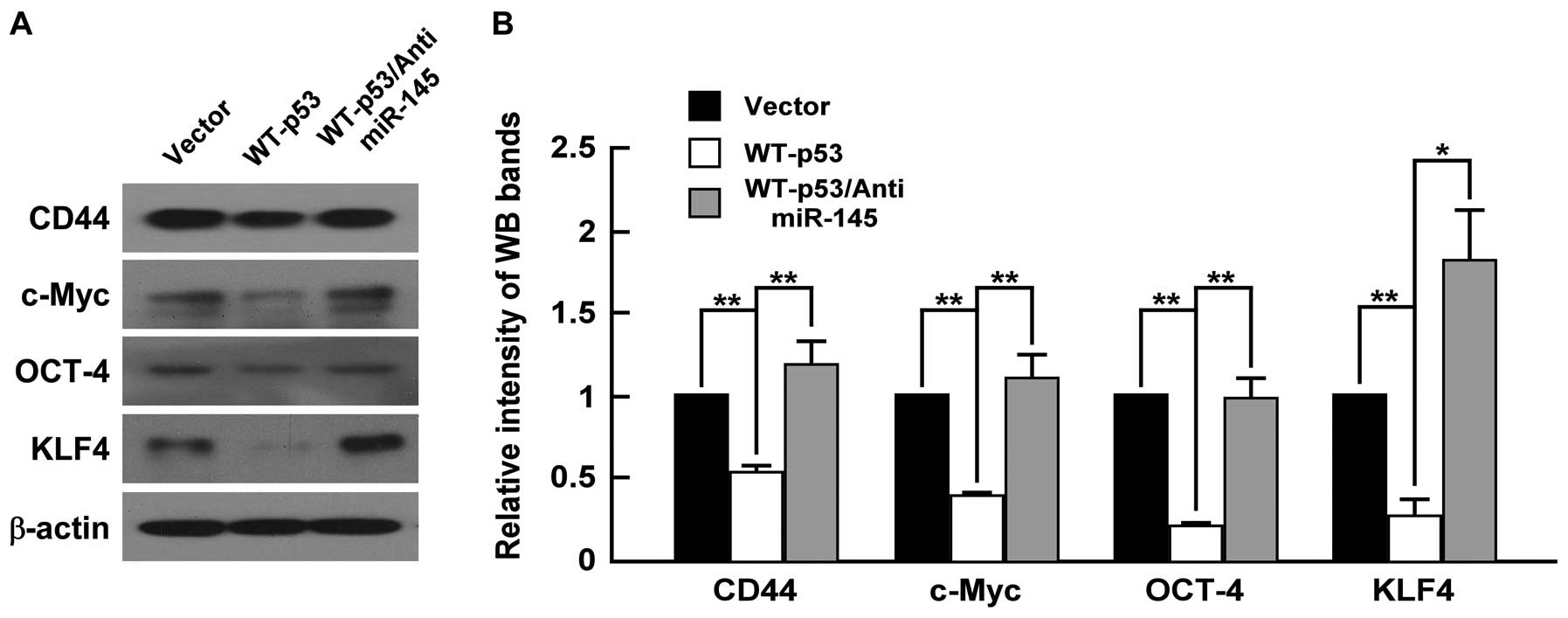
WT-p53 inhibits CD44, c-Myc, Oct4 and
Klf4 expression and anti-miR-145 restores their expression in PC-3
cells
To elucidate whether WT-p53 also have an influence
on CSC marker and ‘stemness’ factor expression of PCa cells, we
upregulated WT-p53 in PC-3 cells by transfecting the plasmid of
WT-p53 expression and detected the expression of stem cell
properties-associated factors and markers, including c-Myc, Oct4,
Klf4 and CD44. As shown in Fig. 5,
WT-p53 reduced the expression of CD44, which has been described as
prostate CSC marker based on clinical investigations and in
vitro studies of prostate cancer cell lines (21), and downregulated the expression of
Oct4, c-Myc and Klf4, which are the key ‘stemness’ factors, and are
required for maintaining self-renewal and pluripotency of stem
cells (34). Furthermore, we
determine whether WT-p53 regulates the stemness factors by
modulating miR-145 in PC-3 cells, and found that anti-miR-145
counteracted the effects of WT-p53 expression as anti-miR-145
restored expression levels of CD44, Oct4, c-Myc and Klf4 that was
reduced by WT-p53 expression (Fig.
5). These data indicated that WT-p53 repressed ‘stemness’
factors through modulation of miR-145 in PC-3 cells.
Discussion
We have previously identified that miR-145 may
repress bone metastasis of PCa and is involved in regulating EMT
and stemness of PCa cells (25,26).
In the present study, we found that the ectopic expression of
WT-p53 inhibited migration, invasion, EMT, colony formation, tumor
sphere formation and expression of CD44, Oct4, c-Myc and Klf4 in
PC-3 PCa cells, and enhanced miR-145 expression in PC-3 cells.
Importantly, the anti-miR-145 was able to reverse the
above-mentioned inhibitory effects of WT-p53. Our findings
demonstrate that WT-p53 repressed EMT and cancer cell stemness of
PC-3 cells at least partially through regulation of miR-145.
Many studies have demonstrated that p53 plays a key
role in the function of cell cycle, apoptosis, senescence,
DNA-repair mechanisms and autophagy (3,4).
However, the emerging evidence demonstrates that p53 also plays a
crucial role in regulating key stages of the metastatic progression
(4,6). In PCa, although some studies have
showed that mutant p53 gain of function promoted cancer development
and metastasis (5,35–38),
there are hardly any studies on the role of WT-p53 in PCa
metastasis.We found that WT-p53 repressed invasion and migration,
and suppressed EMT, which is a key step of the progression of tumor
cell metastasis, and cancer stem cells properties in PC-3 cells,
which might be the critical drivers of tumor progression and
metastasis (16,17). Zhou et al(39,40)
have found that the combined deficiency for p53 and Rb in prostate
epithelium results in invasive and highly metastatic prostate
carcinogenesis, and preferential malignant transformation of
prostate stem cell compartment by combined deficiency by p53 and Rb
indicates a critical role of these genes in the regulation of
prostate stem cells during ontogenesis. Moreover, the loss of
Pten/TP53 in prostate epithelium led to transformation of
multipotential progenitors and EMT (41). These findings provide evidences
that loss of WT-p53 may promote the metastasis of PCa by elevating
migration, invasion, EMT and stemness of cancer cells.
Recently studies have found that miRNAs contribute
to downregulation of mRNA and protein expression observed after p53
activation (24) and several
miRNAs, which were the direct transcriptional targets of p53,
played a critical role in mediating p53 regulation of EMT in cancer
progression (8–11). The miR-34a, a tumor suppressor
which directly targets Snail, was decreased due to the absence of
WT-p53 function, thus Snail-dependent EMT was activated in colon,
breast and lung carcinoma cells (22). The p53 also played a role in
regulating EMT through transcriptional activation of miR-200c
(8). Inhibition or overexpression
of the miR-200 family affected p53-regulated EMT by altering ZEB1
and ZEB2 expression (10). Mutant
p53 gain-of-function induces EMT through modulation of the
miR-130b-ZEB1 axis in endometrial cancer (42). Previous studies have demonstrated
that miR-145 is transcriptionally and post-transcriptionally
regulated by WT-p53 (27–30), and miR-145 augments the effects of
p53 by suppressing the inhibitors of p53 in cervical cancer cells
(29). In this study, we found
that WT-p53 enhanced miR-145 expression in PC-3 cells and
anti-miR-145 reversed EMT features of PC-3 cells which were
inhibited by ectopic expression of WT-p53. Importantly, miR-145 is
upregulated by WT-p53, but not with mutant p53 in PC-3 cells
(30), the anti-miR-145 also
repressed the expression of WT-p53, and miR-145 itself also
represses EMT in PC-3 cells (25).
Collectively, these findings indicate that miR-145 is a mediator of
WT-p53-regulated EMT.
Although miR-34a and miR-200c mediated p53-regulated
EMT of cancer cells mainly through targeting EMT regulators Snail1,
ZEB1 and ZEB2 (8,10,42),
our results suggested that miR-145 might mediate p53-regulated EMT
of PC-3 cells by targeting several pathways. We found that WT-p53
repressed expression of the mesenchymal markers N-cadherin, which
was one of miR-145 targets (43),
and ZEB2, which was speculated as one of miR-145 targets and a
transcription factor promoting EMT, and the inhibitory effects of
WT-p53 on expression of N-cadherin and ZEB2 in PC-3 cells were
reversed by anti-miR-145. We also found that metastasis-promoting
protein HEF1 was a direct target of miR-145 and it promoted EMT of
PC-3 cells (the results not shown). Therefore, miR-145 may mediate
WT-p53-regulated EMT of PCa cells by targeting N-cadherin, ZEB2 and
HEF1.
Besides regulating EMT, miRNAs which are the direct
transcriptional targets of p53 played a role in mediating p53
regulation of cancer cell stemness in cancer progression. Liu et
al(21) found that miR-34a
inhibited prostate cancer stem cells and metastasis by directly
repressing CD44. Furthermore, p53 regulated stem cell properties
through modulating miR-200c by regulating KLF4 and BMI1 (8). Our previous results showed that
miR-145 repressed stemness of PC-3 cells by suppressing CD44, Oct4,
c-Myc and Klf4 (26). In present
study, we showed that anti-miR-145 reversed colony formation and
tumor sphere formation of PC-3 cells which were inhibited by
ectopic expression of WT-p53. Moreover, WT-p53 also suppressed
Oct4, c-Myc and Klf4 in PC-3 cells, which were directly targeted by
miR-145 (27,31), CD44, which was speculated as one of
miR-145 putative targets (miRWalk), and the inhibitory effect of
WT-p53 on the above-mentioned ‘stemness’ factors is reversed by
anti-miR-145. All these findings indicate that miR-145 also is a
mediator of WT-p53 regulation of cancer cell stemness and suggest
that miR-145 may target CD44, Oct4, c-Myc and Klf4 in PC-3
cells.
Recent evidence has demonstrated that the EMT can
generate cancer cells with properties of stem cells (44,45).
This important finding implies a direct link between EMT and cancer
stem cells. A previous study found that miR-200 played a critical
role in linking EMT phenotype with stem cell signatures by
regulating the expression of Lin28B and Notch1 in PCa cells
(45). We found that p53/miR-145
pathway regulated both EMT and stemness of PC-3 cells. These
finding suggested that the p53/miR-145 pathway might be the new
link between EMT and cancer cell stemness in PC-3 cells. Thus, the
discovery of molecular targets which are regulated by p53/miR-145
axis and linked to EMT and CSC properties are required.
In conclusion, our findings demonstrate that WT-p53
suppresses migration, invasion, EMT and stemness of PC-3 PCa cells
at least partially through modulating miR-145. These results
suggest that loss of WT-p53 might promote bone metastasis of PCa at
least partially through repressing miR-145 to elevate EMT and
stemness of cancer cells. Therefore, the activation of the
p53/miR-145 regulatory axis may function as a therapeutic
alternative for bone metastasis of PCa.
Abbreviations:
|
PCa
|
prostate cancer;
|
|
miRNAs
|
microRNAs;
|
|
EMT
|
epithelial-mesenchymal transition;
|
|
CSCs
|
cancer stem cells;
|
|
WT-p53
|
wild-type p53
|
Acknowledgements
We thank NSFC funding, China (no.
81272938); Science and Technology planning project of Guangdong
Province, China (no. 2010B010800019); and Science and Technology
Planning Project of Guangzhou, China (no. 11C22060772) for
supporting this study.
References
|
1
|
Carlin BI and Andriole GL: The natural
history, skeletal complications, and management of bone metastases
in patients with prostate carcinoma. Cancer. 88:2989–2994. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vousden KH and Prives C: Blinded by the
light: the growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goh AM, Coffill CR and Lane DP: The role
of mutant p53 in human cancer. J Pathol. 223:116–126. 2011.
View Article : Google Scholar
|
|
4
|
Muller PA, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Navone NM, Labate ME, Troncoso P, Pisters
LL, Conti CJ, von Eschenbach AC and Logothetis CJ: p53 mutations in
prostate cancer bone metastases suggest that selected p53 mutants
in the primary site define foci with metastatic potential. J Urol.
161:304–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang SP, Wang WL, Chang YL, et al: p53
controls cancer cell invasion by inducing the MDM2-mediated
degradation of Slug. Nat Cell Biol. 11:694–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boominathan L: The guardians of the genome
(p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs
network. Cancer Metastasis Rev. 29:613–639. 2010. View Article : Google Scholar
|
|
8
|
Chang CJ, Chao CH, Xia W, et al: p53
regulates epithelial-mesenchymal transition and stem cell
properties through modulating miRNAs. Nat Cell Biol. 13:317–323.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim NH, Kim HS, Li XY, et al: A
p53/miRNA-34 axis regulates Snail1-dependent cancer cell
epithelial-mesenchymal transition. J Cell Biol. 195:417–433. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim T, Veronese A, Pichiorri F, et al: p53
regulates epithelial-mesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schubert J and Brabletz T: p53 spreads out
further: suppression of EMT and stemness by activating miR-200c
expression. Cell Res. 21:705–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sethi S, Macoska J, Chen W and Sarkar FH:
Molecular signature of epithelial-mesenchymal transition (EMT) in
human prostate cancer bone metastasis. Am J Transl Res. 3:90–99.
2010.PubMed/NCBI
|
|
14
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Monteiro J and Fodde R: Cancer stemness
and metastasis: therapeutic consequences and perspectives. Eur J
Cancer. 46:1198–1203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bracken CP, Gregory PA, Khew-Goodall Y and
Goodall GJ: The role of microRNAs in metastasis and
epithelial-mesenchymal transition. Cell Mol Life Sci. 66:1682–1699.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar
|
|
21
|
Liu C, Kelnar K, Liu B, et al: The
microRNA miR-34a inhibits prostate cancer stem cells and metastasis
by directly repressing CD44. Nat Med. 17:211–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Siemens H, Jackstadt R, Hünten S, Kaller
M, Menssen A, Götz U and Hermeking H: miR-34 and SNAIL form a
double-negative feedback loop to regulate epithelial-mesenchymal
transitions. Cell Cycle. 10:4256–4271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hatfield S and Ruohola-Baker H: microRNA
and stem cell function. Cell Tissue Res. 331:57–66. 2008.
View Article : Google Scholar
|
|
24
|
Hermeking H: MicroRNAs in the p53 network:
micromanagement of tumour suppression. Nat Rev Cancer. 12:613–626.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peng X, Guo W, Liu T, et al:
Identification of miRs-143 and -145 that is associated with bone
metastasis of prostate cancer and involved in the regulation of
EMT. PLoS One. 6:e203412011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang S, Guo W, Tang Y, Ren D, Zou X and
Peng X: miR-143 and miR-145 inhibit stem cell characteristics of
PC-3 prostate cancer cells. Oncol Rep. 28:1831–1837.
2012.PubMed/NCBI
|
|
27
|
Sachdeva M, Zhu S, Wu F, et al: p53
represses c-Myc through induction of the tumor suppressor miR-145.
Proc Natl Acad Sci USA. 106:3207–3212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto
T, Kato S and Miyazono K: Modulation of microRNA processing by p53.
Nature. 460:529–533. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi M, Du L, Liu D, et al: Glucocorticoid
regulation of a novel HPV-E6-p53-miR-145 pathway modulates invasion
and therapy resistance of cervical cancer cells. J Pathol.
228:148–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suh SO, Chen Y, Zaman MS, et al:
MicroRNA-145 is regulated by DNA methylation and p53 gene mutation
in prostate cancer. Carcinogenesis. 32:772–778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schmittgen TD and Livak KJ: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C (T)) method. Methods. 25:402–408. 2011.
|
|
32
|
Pfeiffer MJ and Schalken JA: Stem cell
characteristics in prostate cancer cell lines. Eur Urol.
57:246–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bisson I and Prowse DM: WNT signaling
regulates self-renewal and differentiation of prostate cancer cells
with stem cell characteristics. Cell Res. 19:683–697. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu N, Papagiannakopoulos T, Pan G, Thomson
JA and Kosik KS: MicroRNA-145 regulates OCT-4, SOX2, and KLF4 and
represses pluripotency in human embryonic stem cells. Cell.
137:647–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Downing S, Bumak C, Nixdorf S, Ow K,
Russell P and Jackson P: Elevated levels of prostate-specific
antigen (PSA) in prostate cancer cells expressing mutant p53 is
associated with tumor metastasis. Mol Carcinog. 38:130–140. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stapleton AM, Timme TL, Gousse AE, et al:
Primary human prostate cancer cells harboring p53 mutations are
clonally expanded in metastases. Clin Cancer Res. 3:1389–1397.
1997.PubMed/NCBI
|
|
37
|
Kogan-Sakin I, Tabach Y, Buganim Y, et al:
Mutant p53 (R175H) upregulates Twist1 expression and promotes
epithelial-mesenchymal transition in immortalized prostate cells.
Cell Death Differ. 18:271–281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vinall RL, Chen JQ, Hubbard NE, Sulaimon
SS, Shen MM, Devere White RW and Borowsky AD: Initiation of
prostate cancer in mice by Tp53R270H: evidence for an alternative
molecular progression. Dis Model Mech. 5:914–920. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou Z, Flesken-Nikitin A, Corney DC, Wang
W, Goodrich DW, Roy-Burman P and Nikitin AY: Synergy of p53 and Rb
deficiency in a conditional mouse model for metastatic prostate
cancer. Cancer Res. 66:7889–7898. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou Z, Flesken-Nikitin A and Nikitin AY:
Prostate cancer associated with p53 and Rb deficiency arises from
the stem/progenitor cell-enriched proximal region of prostatic
ducts. Cancer Res. 67:5683–5690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Martin P, Liu YN, Pierce R, et al:
Prostate epithelial Pten/TP53 loss leads to transformation of
multipotential progenitors and epithelial to mesenchymal
transition. Am J Pathol. 179:422–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong P, Karaayvaz M, Jia N, et al: Mutant
p53 gain-of-function induces epithelial-mesenchymal transition
through modulation of the miR-130b-ZEB1 axis. Oncogene. Jul
30–2012.(Epub ahead of print). View Article : Google Scholar
|
|
43
|
Gao P, Xing AY, Zhou GY, et al: The
molecular mechanism of microRNA-145 to suppress invasion-metastasis
cascade in gastric cancer. Oncogene. 32:491–501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kong D, Banerjee S, Ahmad A, Li Y, Wang Z,
Sethi S and Sarkar FH: Epithelial to mesenchymal transition is
mechanistically linked with stem cell signatures in prostate cancer
cells. PLoS One. 5:e124452010. View Article : Google Scholar : PubMed/NCBI
|