Introduction
Neuroblastoma (NB) is the most common extracranial
pediatric tumor. NB patients diagnosed over 18 months of age are
often in the later stages of the disease with widespread
dissemination and often possess MYCN tumor gene amplification, a
significant predictor of poor outcome (1–4).
MYCN is a transcription factor that regulates the expression of a
number of genes involved in cell differentiation, proliferation,
migration, and invasion (5–9).
Ornithine decarboxylase (ODC) is a rate-limiting enzyme in the
biosynthesis of polyamines and its expression is regulated by MYCN.
Inhibiting ODC in NB cells produces many deleterious effects
including G1 cell cycle arrest, inhibition of cell
proliferation, and decreased tumor growth, making ODC a promising
target for drug interference.
Our previous studies have shown that
α-difluoromethylornithine (DFMO; also referred to as eflornithine)
treatment induced significant accumulation of the cyclin-dependent
kinase inhibitor p27Kip1 protein and caused
p27Kip1/Rb-coupled G1 cell cycle arrest in
MYCN-amplified NB tumor cells (10). In addition, we found that the
anti-proliferative effect of DFMO includes p27Kip1
phosphorylation at residues Ser10 (nuclear export) and Thr198
(protein stabilization). Furthermore, DFMO activates an opposing
pathway that promotes NB cell survival via Akt/PKB (11). The p27Kip1 protein,
encoded by the CDKN1B gene, is a member of the Cip/Kip family of
CDK-binding cell cycle inhibitors, and is well known for its tumor
suppressor function. The CDK inhibitor function of
p27Kip1 has been well characterized and the CDK/cyclin
binding domains in regulating this nuclear process are well
documented (12–16). Interestingly, recent studies have
revealed an additional, cytoplasmic function of p27Kip1
in the regulation of cell migration and invasion, and metastasis
(17–24). However, the role of
p27Kip1 in these processes is controversial because it
has also been shown to inhibit migration and invasion in some cell
types, and promote these processes in other cell types. The
phosphorylation of p27Kip1 at Ser10 and Thr198 residues
has been implicated in cell migration and invasion by regulating
the interaction between p27Kip1 and the RhoA small
GTPases, thereby modulating RhoA activity. In addition, the
C-terminal end of p27Kip1 was identified as a ‘scatter’
domain involved in cell migration that binds and regulates activity
of the Rac1 small GTPase (20,25–27).
Finally, p27Kip1 has been shown to regulate cell
migration by binding the microtubule stabilizing protein, stathmin
(17,28). Therefore, the phosphorylation and
localization of p27Kip1 could control whether this
protein regulates cell proliferation or cell migration (19,20,25,27,29–33).
Together, these facts demonstrate how p27Kip1 regulates
multiple roles in the cell depending on its expression,
localization, and phosphorylation status.
The aim of the present study was to investigate the
clinical relevance of p27Kip1 expression in NB and to
examine its role in NB migration and invasion. We demonstrate that
high p27Kip1 mRNA expression in tumors correlates with
increased overall survival of NB patients and is predictive of
decreased bone and bone marrow metastasis. Furthermore, we examine
the mechanism by which polyamines regulate p27Kip1, and
subsequently modulate NB migration and invasion. Interestingly, the
effects of DFMO were significantly greater in NB cells with MYCN
overexpression. The results from the present study suggest that
both p27Kip1 mRNA and p27Kip1 protein
expression correlate with the survival probability of NB patients
by activating mechanisms that not only inhibit tumor growth, but
also decrease NB metastasis. Finally, this study presents new
evidence of the role of polyamines in cancer, and specifically the
malignant progression of NB.
Materials and methods
Cell lines and treatment of cultured
cells
The human NB cell line MYCN2 (provided by Jason
Shohet, TX, USA) were maintained in DMEM (Biosource, Rockeville,
MD, USA) containing 10% (v/v) tetracycline-free, heat-inactivated
fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), and
hygromycin (100 μg/ml) (34). Cells in early log-phase were seeded
and doxycyline (100 ng/ml) was added 3 h before treatment with 5.0
mM α-difluoromethylornithine (DFMO) and incubated for an additional
72 h. NB cells were cultured at 37°C, in a humidified atmosphere
containing 5% CO2. DFMO was kindly provided by Dr
Patrick Woster (Medical University of South Carolina, Charleston,
SC, USA). Putrescine (put), spermidine (spd), and spermine (spm)
were from Sigma-Aldrich (St. Louis, MO, USA).
Flow cytometry
Cells were seeded and treated with 5.0 mM DFMO ± 10
μM spermidine or left untreated. Cells were trypsinized,
washed twice in phosphate-buffered saline (PBS), counted, and
1–2×105 cells suspended in 0.1 ml of PBS. Cells were
stained with 5 μl propidium iodide for 30 min in the dark at
room temperature. Assay buffer (0.4 ml) was added, and 5,000 cells
analyzed using a FACScan flow cytometry instrument
(Becton-Dickinson, San Jose, CA, USA). The CellQuest program
(Becton Dickinson) was used for data analysis.
Western blot analysis
Cell lysates were prepared in RIPA buffer [20 mM
Tris-HCl, pH 7.5, 0.1% (w/v) sodium lauryl sulfate, 0.5% (w/v)
sodium deoxycholate, 135 mM NaCl, 1% (v/v) Triton X-100, 10% (v/v)
glycerol, 2 mM EDTA], supplemented with Complete protease inhibitor
cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA), and
phosphatase inhibitors sodium fluoride (NaF) (20 mM) and sodium
vanadate (Na3VO4) (0.27 mM). Western blot
analysis was performed as previously described (10). The total protein concentration was
determined using the protein assay dye reagent from Bio-Rad
Laboratories (Hercules, CA, USA). Cell lysates in SDS-sample buffer
were boiled for 5 min and equal amounts of total protein analyzed
by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and
western blotting. The antibodies used in this study are: rabbit
polyclonal phospho-GSK3-β (Ser9) (1:1,000), rabbit polyclonal
phospho-Akt/PKB (Ser473) (1:1,000), rabbit polyclonal anti-tubulin
(1:1000) from Cell Signaling Technology, Inc. (Beverly, MA, USA);
rabbit polyclonal anti-MYCN (1:500) and rabbit polyclonal
anti-p27Kip1 (1:500) from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Secondary anti-mouse (1:5,000) and
anti-rabbit (1:5,000) antibodies coupled to horseradish peroxidase
(HRP) were from Amersham Biosciences (Piscataway, NJ, USA).
Proteins were detected using the ECL Plus reagents (Amersham
Biosciences) and Kodak BioMax XAR film (Fisher Scientific,
Pittsburgh, PA, USA). Membranes were stripped at 50°C for 30 min
with ECL stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, 100 mM
2-mercaptoethanol) and sequentially probed. Quantification was
performed as described previously using a Bio-Rad Fluor-S Multi
Imager and Quantity One Quantitation Software, Version 4 (Bio-Rad
Laboratories).
Polyamine pool analysis
Intracellular polyamine (putrescine, spermidine,
spermine) pools were measured in human MYCN2 cells treated with
DFMO using a previously described method (35). The samples were normalized in 0.2 N
sodium hydroxide and the amount of total protein per sample was
measured using the Bio-Rad assay. Polyamine measurements are
presented as nmol per mg of total cellular protein.
Cell proliferation assay
The CellTiter 96® AQueous One
Solution Cell Proliferation Assay is a colorimetric method for
determining the number of viable cells in proliferation or
cytotoxicity assays (Promega, San Luis Obispo, CA, USA). The
CellTiter 96® AQueous One Solution Reagent contains the
tetrazolium compound MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt] and was used to determine the proliferation rate of
cells treated with DFMO ± putrescine, spermidine or spermine and
compared to untreated control cells. Briefly, cells were seeded at
a density of 1,000 cells/well on a transparent, flat-bottom 96-well
plate, in a total volume of 100 μl. After cell treatments,
20 μl of CellTiter 96 AQueous One Solution Reagent was added
to wells, and incubated for 1–4 h at 37°C. The absorbance was
measured at 490 nm using a 96-well microplate reader.
Knockdown experiments with siRNA
Cells were transfected with siRNA from Santa Cruz
Biotechnology, Inc. The p27-specific siRNA (sc-29429) used in this
study was as follows: sense: AAGUACGAGUGGCAAGAGGUG and antisense:
CACCUCUUGCCACUCGUACUU. The scrambled control siRNA (sc-36869) was
from Santa Cruz. Briefly, siRNA (40–80 pmol) and Lipofectamine 2000
reagent (10 μl) were diluted, in separate vials, in
serum-free DMEM. After 5-min incubation at room temperature, the
siRNA and Lipofectamine 2000 were mixed together and incubated at
room temperature for an additional 30 min, then added to the cells.
The medium was exchanged with DMEM supplemented with 10% FBS after
overnight incubation. The cells were analyzed at 48 h
post-transfection.
Wound healing assay
NB cells were grown to confluence and serum-starved
overnight. A uniform scratch was placed through the confluent cell
monolayer. Cells were washed 3 times and then allowed to settle for
2 h. The width of the wound was measured and recorded as t=0. The
cells were then allowed to migrate back into the wounded area, and
the closing of the wound was measured over time (t=8–12 h).
Boyden chamber assay
Migration and invasion assays were performed as
outlined in the manufacturer’s protocol (Trevigen, Gaithersburg,
MD, USA). Briefly, transwell plates were either coated with 0.7X
basement membrane extract (BME) solution and allowed to incubate
for 4 h at 37°C in a CO2 incubator, or left uncoated.
Serum-starved cells (5×104 cells) were seeded into the
top chamber in medium without FBS, while medium with FBS was
present in the bottom chamber. The cells were incubated for 24 h.
The media and remaining cells were aspirated from the top chamber
and washed 2 times with 1X wash buffer. The bottom chamber was
aspirated and washed 2 times with 1X wash buffer. Calcein-AM in
cell dissociation solution was added to the bottom chamber, the
cell migration/invasion device re-assembled, and incubated for 1 h.
The top chamber was removed and the fluorescence intensity
(calcein-AM labeled cells) was measured at 485 nm excitation (520
nm emission) in a plate reader.
Gene annotation
NCBI Gene_IDs and NCBI Gene Names for the genes
analyzed were respectively: ODC: 4953, ODC1 (ornithine
decarboxylase) and CDKN1B: 1027, CDKN1B (cyclin-dependent kinase
inhibitor 1B; p27, Kip1).
Affymetrix DNA micro-array hybridization
and analysis
The Affymetrix NB tumor dataset NB88 contains the
expression profiles of 88 NB tumors with documented genetic and
clinical features as previously reported (34). This set is called ‘NB88’. Total RNA
was extracted from frozen NBs containing >95% tumor cells, and
Affymetrix HG-U133 Plus 2.0 micro-array analysis (Affymetrix, Santa
Clara, CA, USA) was performed as described (36). The NB88 set has been deposited for
public access in a MIAME-compliant format through the Gene
Expression Omnibus (GEO) database at the NCBI website under no.
GSE16476. Normalized expression data from the public GEO data-sets
for the Jagannathan-100 (GSE19274) and Oberthuer-251 set
(E-TABM-38; EMBL-EBI ArrayExpress) series were downloaded and
analyzed as described (36).
Annotations and clinical data for these series are available from
http://www.ncbi.nlm.nih.gov/geo/query/ or http://www.ebi.ac.uk/arrayexpress/ through their
GEO or EMBL-EBI ID’s, respectively. Affymetrix probe-sets were
selected using the R2 bio-informatic platform (see below). All gene
transcript levels were determined from data image files using
GeneChip operating software (MAS5.0 and GCOS1.0, from Affymetrix).
Samples were scaled by setting the average intensity of the middle
96% of all probe-set signals to a fixed value of 100 for every
sample in the data-set, allowing comparisons between micro-arrays.
The probe-sets selected for a gene showed the highest expression in
samples containing a present call for that gene. The TranscriptView
genomic analysis and visualization tool was used to check if the
probe-set selected had an anti-sense position in an exon of the
gene (http://r2.amc.nl). The Affymetrix probe-sets
selected were 200790_at (ODC) and 209112_at (CDKN1B). All analyses
were performed using R2, an Affymetrix analysis and visualization
platform developed in the Department of Oncogenomics at the
Academic Medical Center at the University of Amsterdam. R2 can be
accessed at: http://r2.amc.nl.
Microscopy
For immunofluorescence micrographs, cells were
washed twice in PBS, fixed in 4% (w/v) paraformaldehyde for 10 min,
and exposed to 0.1% (v/v) Triton X-100 for 3 min. Fixed cells were
washed again, blocked in 1% (w/v) bovine serum albumin in PBS for
30 min, and incubated with p27Kip1 antibody (1:100) for
1 h. After incubation, cells were washed twice with PBS and
incubated for 1 h with anti-rabbit Alexa Fluor 488 antibody
(1:5,000). Washed coverslips were mounted using ProLong Gold
Antiface mounting medium (Invitrogen) containing
4’,6-diamidino-2-phenylindole and samples were analyzed with a
Leica laser-scanning confocal microscope equipped with a digital
camera.
Results
Expression of p27Kip1 in NB
tumors
To examine the expression of p27Kip1
(CDKN1B) and its clinical relevance in NB, a set of Affymetrix mRNA
expression profiles for 88 NB tumors was prepared (named NB88)
using the genome-wide HG-U133 Plus 2.0 platform. The genetic and
clinical features were available from patient files for all 88 NB
tumors (Academic Medical Center at the University of Amsterdam).
Using Kaplan-Meier scan analysis, a correlation between
p27Kip1 mRNA expression and NB patient survival was
found (Fig. 1A). While the most
significant P-value was calculated when the NB88 set was divided
into a group of 79 with high, and of 9 with low expression of
p27Kip1 mRNA, the expression of p27Kip1 mRNA
was also significantly indicative for survival in other groupings
(Fig. 1A). Survival of patients
with high p27Kip1 expression (n=79) was ∼70% for up to
216 months, while that for patients with low p27Kip1
expression (n=9) dropped to 0% within 30 months
(P=4.4×10−7).
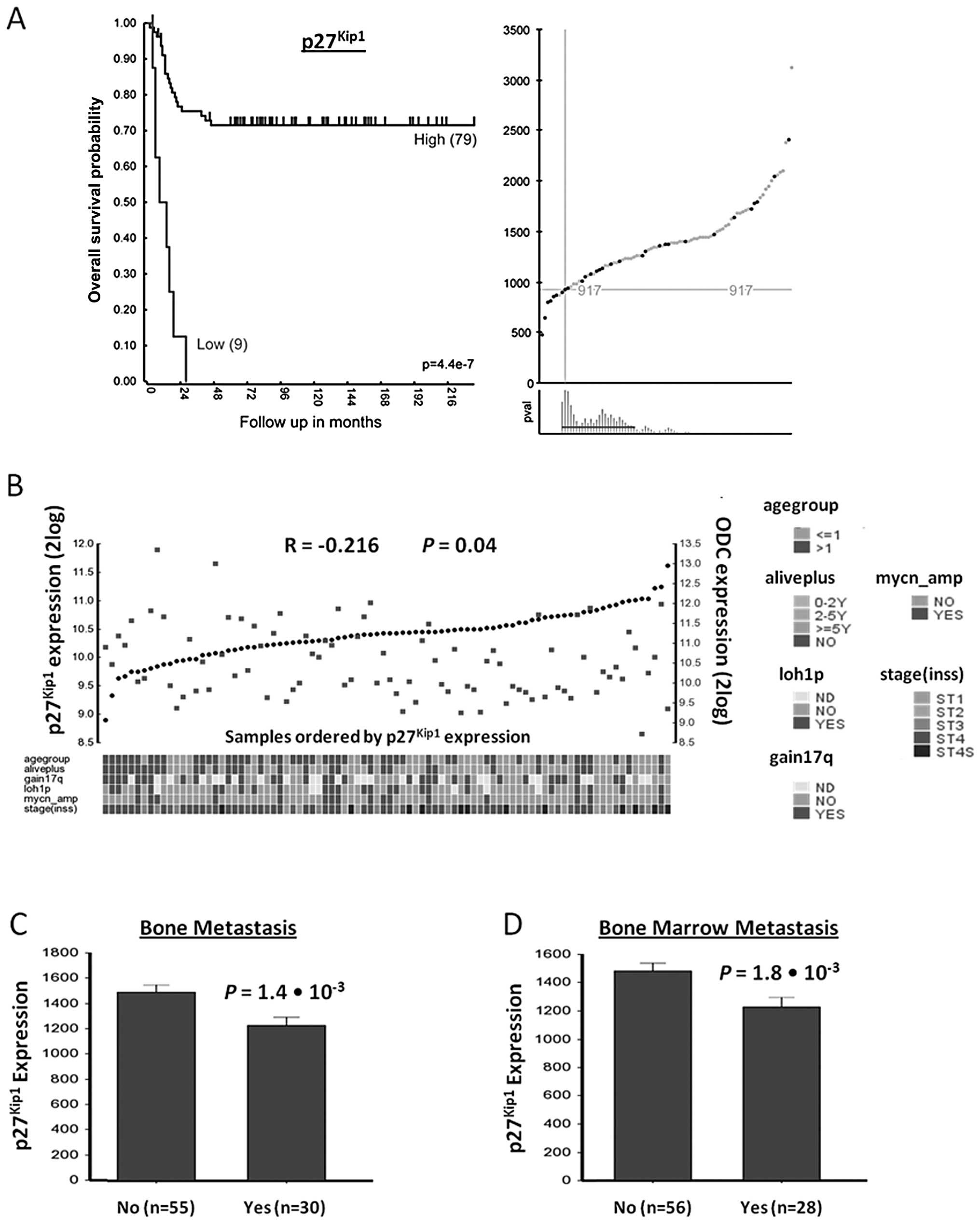 | Figure 1p27Kip1 gene expression
correlation with patient prognosis, ODC expression, and metastasis
in NB. (A) Correlation of p27Kip1 gene expression with
NB patient survival prognosis. The Kaplan-Meier graph represents
the survival prognosis of 88 NB patients based on high or low
expression levels of p27Kip1 (A, left graph). The
survival probability of NB patients (follow-up over 216 months)
with low p27Kip1 expression is significantly lower than
of patients with high p27Kip1 expression. For the
Kaplan-Meier analysis, the P-values were calculated for all 72
groups tested. For p27Kip1, the 9 ‘low’ versus the 79
‘high’ group represents the most significant P-value
(P=4.4×10−7), but the P-value was <0.05 for all
groups from 8 low/80 high to 30 low/58 high. Also when the 88
tumors were divided using the median or average p27Kip1
expression, the P-value was <0.05, showing that
p27Kip1 expression has a robust correlation with
survival as shown in the expression curves and P-value ranges for
the Kaplan-Meier graph (A, right graphs). Upper graph, X-axis shows
tumors ranked from left to right according to their
p27Kip1 gene expression level, green dots and red dots
represent tumors from patients that were still alive or were dead
from disease at the time of this analysis, respectively. Y-axis
shows Affymetrix p27Kip1 expression levels. Crosshairs
mark the expression cut-off used for the grouping shown in the
Kaplan-Meier curve. Lower graph, X-axis shows tumor groupings
tested. Y-axis shows P-value of each grouping (with a P-value of 1
at the bottom). Red horizontal line represents the P<0.05
cut-off. Statistical analysis was performed with the log-rank test.
(B) p27Kip1 and ODC expression correlation in NB tumors.
Visual representation of p27Kip1 and ODC expression in
all 88 NB tumors, ranked horizontally from left to right according
to their p27Kip1 expression. p27Kip1 and ODC
expression values for each tumor are visualized with black circles
and red rectangles, respectively. The correlation between
p27Kip1 and ODC expression is r = −0.216, with P=0.04
(2log Pearson). Below the graph is the clinical annotation of all
88 tumor samples, the annotation legend is to the right of the
graph. (C and D) Correlation of p27Kip1 expression
levels with NB metastasis. Bone and bone marrow metastasis are
significant predictors of poor outcome in NB. (C) Correlation with
bone metastasis. Children with NB metastasized to the bone (n=30)
show significantly lower p27Kip1 tumor expression than
infants without metastasis (55 samples; P=1.4×10−3, data
available for 85 of 88 tumors). (D) Correlation with bone
metastasis. Children with NB metastasized to the bone marrow (n=28)
show significantly lower p27Kip1 tumor expression than
infants without metastasis (56 samples; P=1.8×10−3, data
available for 84 of 88 tumors). Statistical analysis of (C) and (D)
was performed using the non-parametric Kruskal-Wallis tests, but
for reasons of representation, the bar plots show actual expression
values. For expression value calculations in all panels, see
Materials and methods. |
Next, we examined whether there was a correlation
between p27Kip1 and ODC mRNA expression in the NB88
tumor set. Fig. 1B shows a visual
representation of p27Kip1 and ODC mRNA expression
measured with Affymetrix profiling for every tumor in the NB88 set.
An inverse relation between p27Kip1 and ODC expression
(r = −0.216, P=0.04) was observed. This suggests that ODC
expression and intracellular polyamine levels may inhibit
p27Kip1 expression or decrease p27 Kip1 mRNA
stability.
NB metastasis to bone and bone marrow is predictive
of poor patient outcome. We therefore investigated whether
p27Kip1 expression correlates with bone and bone marrow
metastasis in the NB88 set. Indeed, there was a significantly
higher expression of p27Kip1 in tumors without bone
metastasis (P=1.4–1.8×10−3 Kruskal-Wallis t test) or
bone marrow metastasis (P=2.2×10−3 Kruskal-Wallis t
test) (Fig. 1C and D). These
results suggest that p27Kip1 expression correlates with
decreased metastasis and increased survival of NB patients.
DFMO-induced polyamine depletion inhibits
NB proliferation and induces G1 cell cycle arrest
Our previous studies have shown that DFMO depletes
intracellular polyamine pools in MYCN gene-amplified LAN-1 and
NMB-7 NB cells (10). To examine
the effect of DFMO in NB cells with and without MYCN
overexpression, we used MYCN2 NB cells which contain a
doxycycline-inducible MYCN transgene. The cells were grown either
in the presence or absence of doxycycline [with or without MYCN
overexpression, called MYCN2 (+) or MYCN2 (−), respectively], and
then treated with 5 mM DFMO ± 10 μM spermidine, or left
untreated for 72 h. The intracellular polyamine levels were
measured to determine the efficacy of 5 mM DFMO on MYCN2 cells.
DFMO depleted intracellular putrescine (put), spermidine (spd) and
spermine (spm) levels significantly in both MYCN2 (+) and MYCN2 (−)
cells (Fig. 2A). Supplementing the
medium with exogenous spd during DFMO treatment restored
intracellular polyamines in both cell lines to levels comparable to
that of control cells treated with vehicle (water) without DFMO
(Fig. 2A), confirming the effect
of DFMO on polyamine biosynthesis. The results clearly show that 5
mM DFMO effectively decreased intracellular polyamine levels in
MYCN2 cells, independent of MYCN expression.
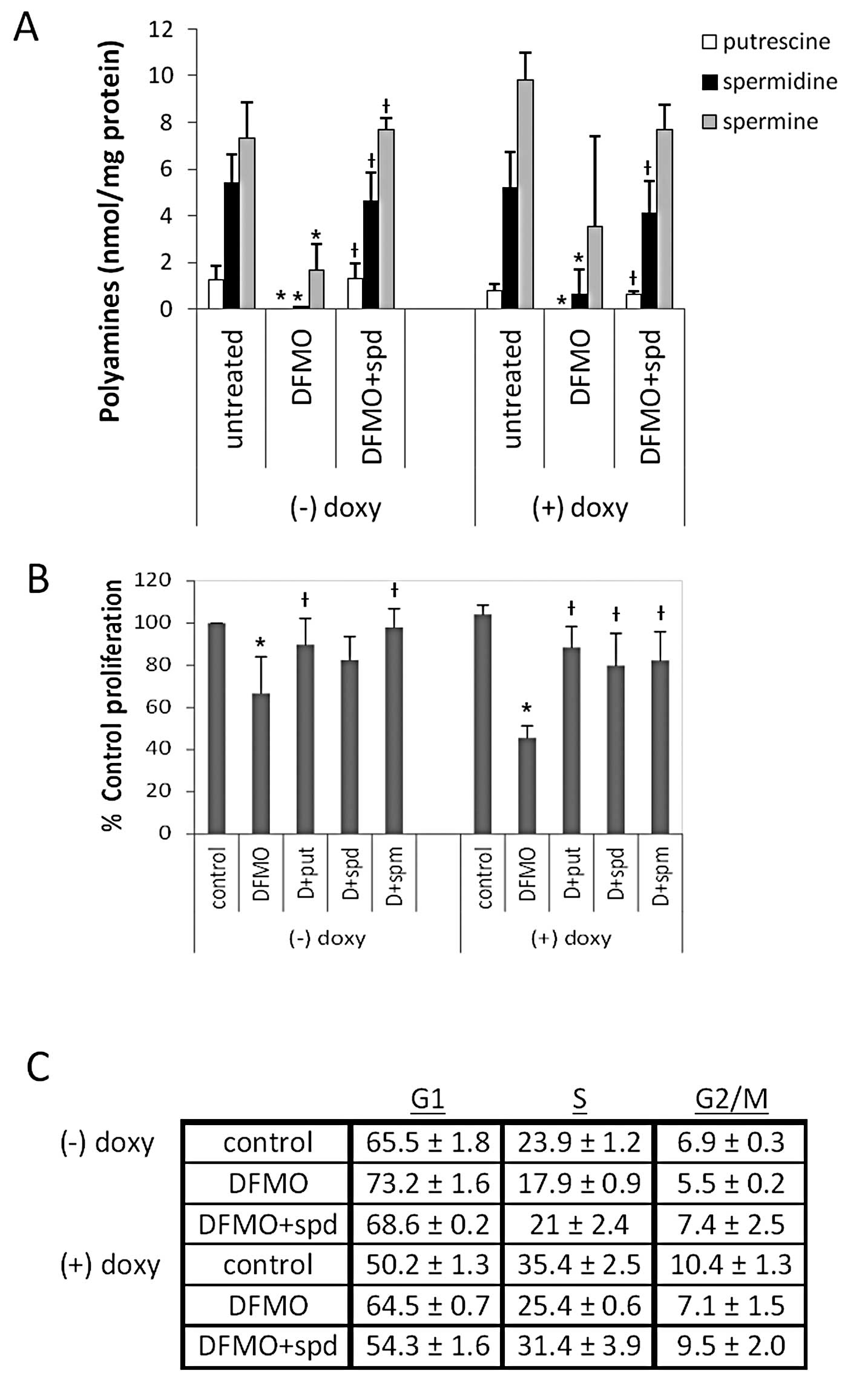 | Figure 2DFMO inhibits polyamine biosynthesis,
cell proliferation, and induces G1 cell cycle arrest in
tetracycline-inducible MYCN overexpressing NB cells (MYCN2). (A)
Cells were treated with doxycycline ± 5 mM DFMO ± 10 μM
spermidine or left untreated for 72 h, and intracellular polyamine
levels were measured. DFMO treatment depleted intracellular
polyamine levels, and supplemental spermidine in culture media
reversed the effects of DFMO. (B) Cells were treated with
doxycycline ± 5 mM DFMO ± 10 μM putrescine, 10 μM
spermidine or 10 μM spermine, or left untreated for 72 h,
and cell proliferation was measured using the MTS assay. DFMO
significantly inhibited proliferation in NB cells with and without
MYCN overexpression. Supplementing external media with polyamines
reversed the effects of DFMO. Results of (A) and (B) are
represented as mean ± SD, n=3. *Statistically
significant difference between values obtained from DFMO-treated
vs. untreated cells. †Statistically significant
difference between values obtained from DFMO-treated cells and
cells treated with both DFMO and spermidine (P<0.05). (C) Cells
were treated with doxycycline ± 5 mM DFMO ± 10 μM spermidine
or left untreated for 72 h, and flow cytometry was performed with
propidium iodide to quantify the percentage of cells in each phase
of the cell cycle (G1, S, and G2/M). DFMO
increased the percentage of cells in the G1 phase of the
cell cycle, and supplementing external media with spermidine
reversed the effects of DFMO. Results are represented as mean ± SD,
n=3. Doxy, doxycyline; put, putrescine; spd, spermidine; spm,
spermine. |
To determine whether MYCN overexpression influences
the efficacy of DFMO in NB, cell proliferation was measured in
MYCN2 cells utilizing the tetrazolium based MTS assay. While DFMO
treatment significantly inhibited the proliferation in both MYCN2
(−) and MYCN2 (+) compared to untreated cells (Fig. 2B), the inhibitory effect of DFMO
was significantly enhanced when MYCN was overexpressed in NB cells.
Polyamine (put, spd or spm) supplementation attenuated this DFMO
effect in both cell lines (Fig.
2B).
To further examine the influence of DFMO on NB cell
proliferation in the absence or presence of MYCN, we determined the
cell cycle distribution of MYCN2 (−) and MYCN2 (+) cells. The
results revealed that DFMO caused a perturbation in the cell cycle
distribution of these cells by increasing the fraction of MYCN2 (−)
and MYCN2 (+) cells in the G1 phase of the cell cycle
and by decreasing the fraction of MYCN2 (−) and MYCN2 (+) cells in
the S and G2/M phases of the cell cycle, compared to
control untreated cells (Fig. 2C).
The inhibitory effects of DFMO were greater in MYCN2 (+) cells
compared to MYCN2 (−). The addition of spd reversed the effects of
DFMO, indicating that these effects were due to polyamine
depletion. These results show that DFMO induces G1 cell
cycle arrest, an effect that is more pronounced in NB cells with
MYCN over-expression.
DFMO inhibits cell migration in MYCN2
cells
To examine the regulation of NB cell migration by
polyamines and MYCN, MYCN2 (−) and MYCN2 (+) cells were treated
with DFMO ± put, spd or spm, or left untreated for 72 h, and NB
migration was measured using the wound healing assay (Fig. 3A). As early as 6–8 h, a significant
difference between cell migration of untreated control cells
compared to DFMO-treated cells was observed. DFMO inhibited cell
migration by 73 and 72% in MYCN2 (−) and MYCN2 (+) cells,
respectively (Fig. 3A).
Supplementing the medium with polyamines (put, spd or spm)
alleviated the effects of DFMO in MYCN2 (−) cells and MYCN2 (+)
cells (Fig. 3A). These results
suggest that DFMO-induced polyamine depletion exerted an inhibitory
effect on cell migration in MYCN2 cells, and supplementing DFMO
treatment with any of the three polyamines was able to partially
reverse this inhibitory effect.
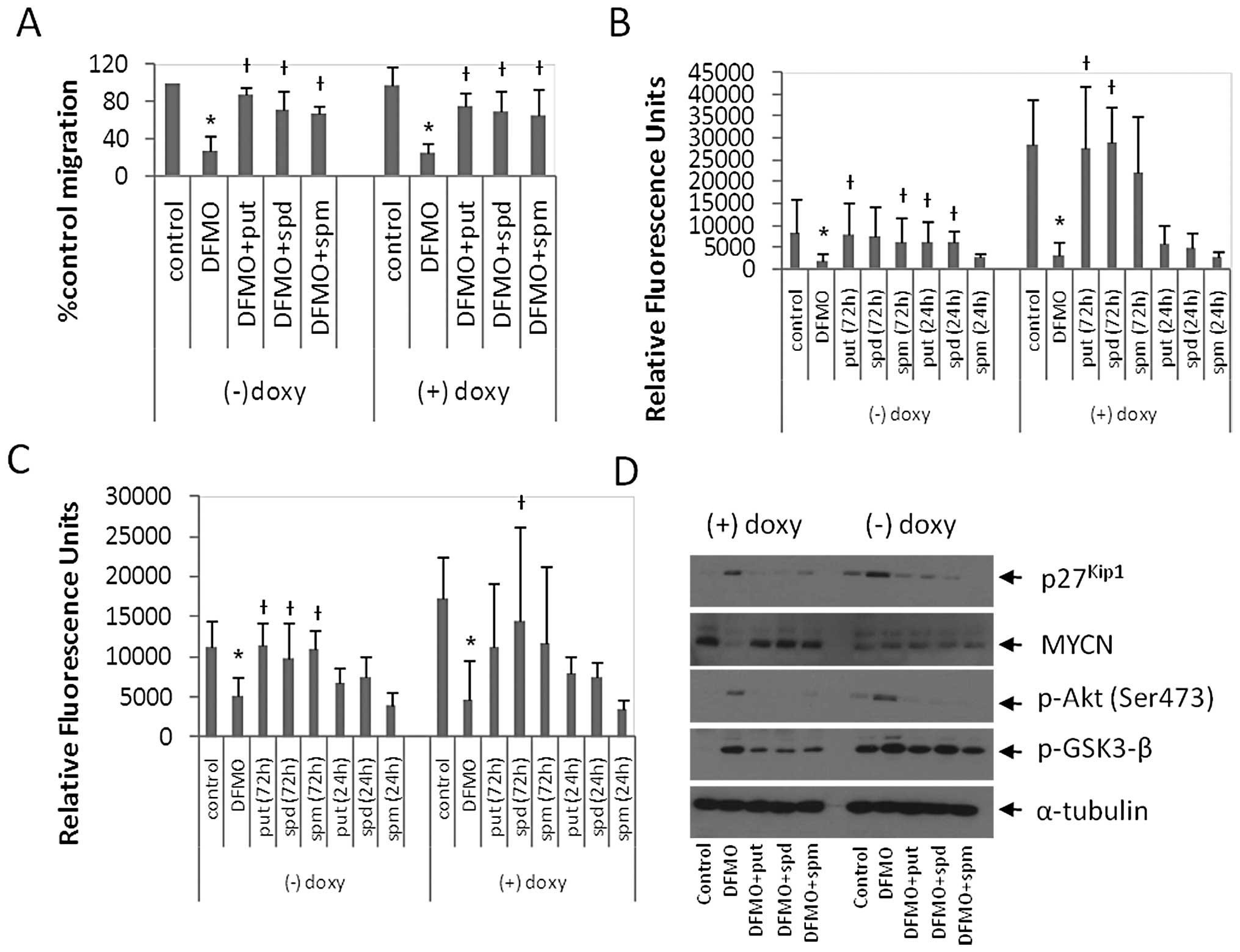 | Figure 3DFMO inhibits cell migration and
invasion, increases p27Kip1 expression, and increases
the phosphorylation of Akt (Ser473) and GSK3-β (Ser9). (A) MYCN2
cells were treated with or without doxycycline and 5 mM DFMO ± 10
μM putrescine, 10 μM spermidine or 10 μM
spermine for 72 h and the wound healing assay was performed. DFMO
inhibited cell migration. Supplementing the external media with
putrescine, spermidine or spermine reversed the effects of DFMO.
Results are represented as mean ± SD, n=3. (B and C) MYCN2 cells
were treated with doxycycline ± 5 mM DFMO. The external medium was
supplemented with ± 10 μM putrescine, 10 μM
spermidine or 10 μM spermine or left untreated for 72 h of
DFMO treatment or the last 24 h of DFMO treatment. Transwell
migration (B) and invasion (C) assays were performed. DFMO
inhibited NB migration and invasion. Supplementing external media
with polyamines for 72 h of DFMO treatment reversed the effects of
DFMO. Supplementing external media with polyamines for the last 24
h of DFMO treatment only partially reversed the effects of DFMO.
(A–C) Results are represented as mean ± SD, n=3 in duplicates.
*Statistically significant difference between values
obtained from DFMO-treated vs. untreated cells.
†Statistically significant difference between values
obtained from DFMO-treated cells and cells treated with both DFMO
and putrescine, spermidine or spermine (P<0.05). (D) MYCN2 cells
were treated with or without doxycycline and 5 mM DFMO ± 10
μM putrescine, 10 μM spermidine or 10 μM
spermine for 72 h. DFMO treatment induced p27Kip1
accumulation, downregulation of MYCN expression, and increased the
phosphorylation of Akt (Ser473) and GSK3-β (Ser9). Tubulin was used
as a loading control. Analysis was performed in three independent
experiments (n=3). Doxy, doxycyline; put, putrescine; spd,
spermidine; spm, spermine. |
To extend these results, we examined the effects of
DFMO on NB invasion using the Boyden chamber transwell assay. MYCN2
cells were treated with DFMO or left untreated for 72 h. The
culture medium of the DFMO-treated cells was supplemented with
polyamines (put, spd or spm), either for 72 h of DFMO treatment or
for only the last 24 h of the DFMO treatment. In MYCN2 (−) cells,
DFMO inhibited migration by 56% compared to untreated control cells
(Fig. 3B). The presence of
exogenously added polyamines for 72 h attenuated this effect to
levels comparable to untreated control cells. The addition of
polyamines for the last 24 h of the 72-h DFMO incubation period
only partially reversed the DFMO effects. Interestingly, NB
migration was ∼1.5-fold higher in MYCN2 (+) cells than in MYCN2 (−)
cells. In MYCN2 (+) cells DFMO inhibited migration by 73% compared
to untreated cells (Fig. 3B). The
addition of polyamines to the medium of DFMO-treated cells during
the 72 h of treatment attenuated the DFMO effect to levels
comparable to untreated cells (Fig.
3B). Supplementing DFMO treatment with polyamines for the last
24 h of DFMO treatment only partially reversed the DFMO effects on
NB migration (Fig. 3B).
To determine whether polyamines also regulate NB
invasive migration, the transwell assay was repeated. However, in
this experiment, the porous filter which separates the top and
bottom chambers of the assay plate was coated with extracellular
matrix molecules. In order to examine the role of MYCN in this
process, MYCN2 (−) and MYCN2 (+) cells were treated as described
above. In MYCN2 (−) cells, DFMO inhibited invasion by 77% compared
to untreated cells (Fig. 3C).
Polyamine (put, spd, spm) supplementation for 72 h of the DFMO
treatment reversed the drug-induced effect (Fig. 3C). Supplementing DFMO-treatment
with polyamines for the last 24 h of treatment partially attenuated
the DFMO effects (Fig. 3C). MYCN
overexpression increased NB invasion by ∼3.5-fold compared to NB
cells without MYCN overexpression. In MYCN2 (+) cells, DFMO
inhibited NB invasion by 89% compared to untreated cells (Fig. 3C) and polyamine supplementation for
72 h of DFMO-treatment reversed the drug-induced effect (Fig. 3C). Supplementing DFMO-treated cells
with polyamines for the last 24 h of DFMO treatment did not reverse
the drug-induced effects (Fig.
3C). These results suggest that polyamines regulate NB invasion
and that DFMO suppresses invasiveness most effectively in MYCN
overexpressing NB cells.
To identify the cellular signaling pathways involved
in DFMO action, MYCN2 (+) and MYCN2 (−) cells were treated with
DFMO ± polyamines (put, spd or spm), or left untreated for 72 h.
Whole cell lysates were prepared and analyzed by Western blotting.
Doxycycline-induced MYCN overexpression was confirmed by the
increased intensity of the 67-kDa band representing the MYCN
protein (Fig. 3D). Remarkably,
DFMO treatment significantly decreased MYCN protein levels in MYCN2
(+) cells but not in MYCN2 (−) cells that express only low levels
of MYCN. DFMO also induced the accumulation of p27Kip1
protein (Fig. 3D). The
DFMO-induced downregulation of MYCN and accumulation of
p27Kip1 were reversed by the addition of polyamines
(Fig. 3D).
As p27Kip1 is a downstream target of
Akt/PKB (37,38), the effect of DFMO on Akt/PKB was
also examined. DFMO treatment indeed induced an increase in Akt/PKB
phosphorylation at Ser473, indicative of Akt/PKB activation
(Fig. 3D). DFMO also induced an
increase in glycogen synthase kinase-3β (GSK3-β) phosphorylation at
Ser9, a downstream target of Akt/PKB (Fig. 3) (39). Polyamine supplementation reversed
the effects of DFMO on the phosphorylation of these signaling
proteins (Fig. 3D). Together,
these results suggest that p27Kip1 is involved in the
regulation of polyamine-dependent NB migration and/or invasion and
that the Akt/PKB-GSK3-β pathway is also involved in these
regulatory processes.
DFMO-induced inhibition of NB migration
involves p27Kip1
DFMO-induced polyamine depletion was previously
shown to induce p27Kip1 protein accumulation, and
p27Kip1 phosphorylation at Ser10 and Thr198 (10,11).
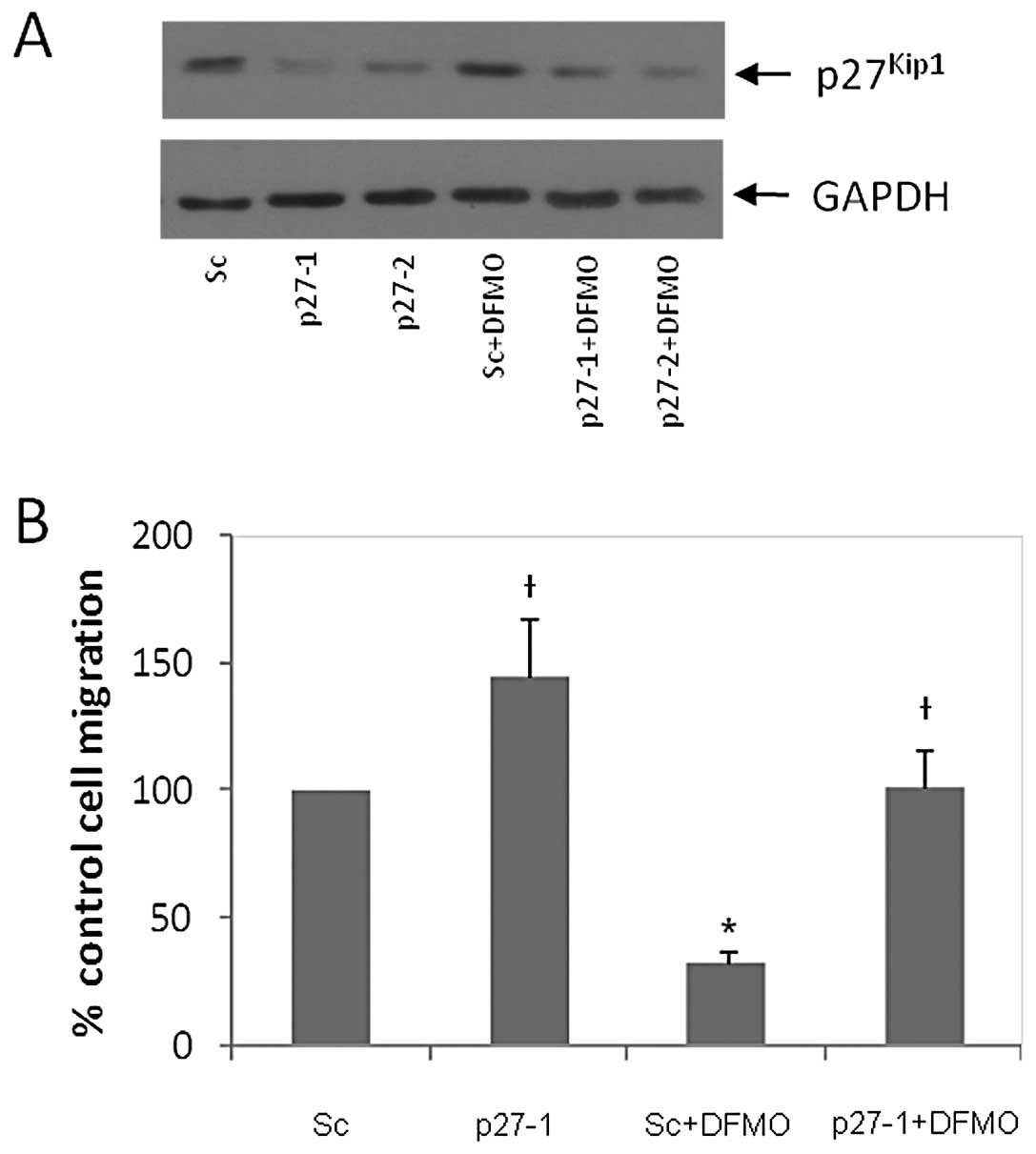
To determine the role of p27Kip1 in DFMO-induced
inhibition of NB migration, p27Kip1 specific siRNA was
used to downregulate p27Kip1 protein expression in NB
cells. Scrambled siRNA was used as a control. The
p27Kip1 siRNA (p27-1 and p27-2 represent 40 and 80 pmol
siRNA, respectively) and scrambled siRNA (designated ‘sc’) were
transfected into MYCN2 (−) cells. These transfected cells were
either exposed to DFMO or left untreated for 72 h. Western blot
analysis of whole cell lysates revealed that p27Kip1
siRNA downregulated p27Kip1 protein levels compared to
sc siRNA (Fig. 4A). As expected,
DFMO increased p27Kip1 levels in cells transfected with
sc, but not in those transfected with p27-1 or p27-2 siRNA
(Fig. 4A). Cells transfected with
p27Kip1 or sc control siRNA were also used to measure
cell migration using the wound healing assay. Fig. 4B shows that downregulation of
p27Kip1 increased cell migration by 45% compared to the
scrambled control. DFMO treatment inhibited cell migration by 68%
in scrambled control cells. However, p27Kip1
downregulation completely reversed DFMO-induced inhibition of cell
migration to that of untreated (no DFMO) scrambled cells (Fig. 4B). These results show that
p27Kip1 plays a key role in regulating NB migration.
Expression and subcellular localization
of p27Kip1 in NB cells treated with DFMO
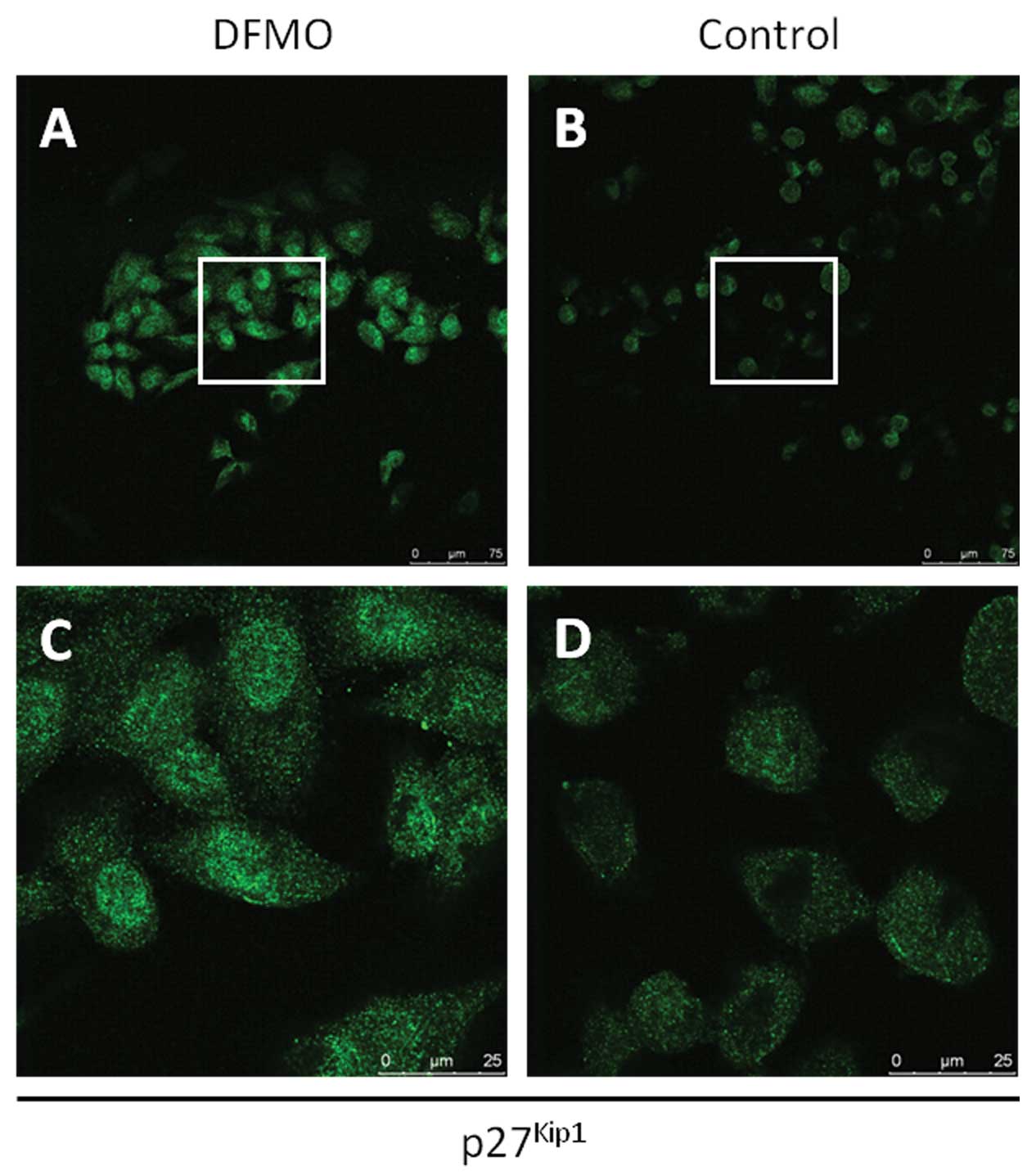
To further examine the role of p27Kip1 in
NB cell migration and invasion, the subcellular localization of
p27Kip1 was examined in NB cells. MYCN2 (+) cells were
treated with DFMO or left untreated for 72 h. Confocal laser
microscope analysis of DFMO-treated and untreated NB cells revealed
that DFMO treatment resulted in an increase of p27Kip1
protein levels in NB cells compared to untreated cells.
Importantly, DFMO treatment led to the accumulation of
p27Kip1 protein in both the nucleus and cytoplasm
(Fig. 5), suggesting that
cytoplasmic p27Kip1 may function to inhibit NB migration
and invasion, a function that may be separate and distinct from its
role in cell cycle regulation.
Discussion
This study analyzed the role of polyamines and
p27Kip1 in NB metastasis. First, we determined the
clinical relevance of p27Kip1 in NB. High
p27Kip1 expression correlated with increased patient
survival in the NB88 set presented in this study. This result was
confirmed in the only other publicly available NB tumor cohort with
Kaplan-Meier survival data, the Oberthuer-251 set (results not
shown), and these findings are consistent with those from an
earlier study (40). We also
investigated the correlation between p27Kip1 expression
and metastasis of the bone and bone marrow. The expression of
p27Kip1 correlated with fewer occurrences of bone and
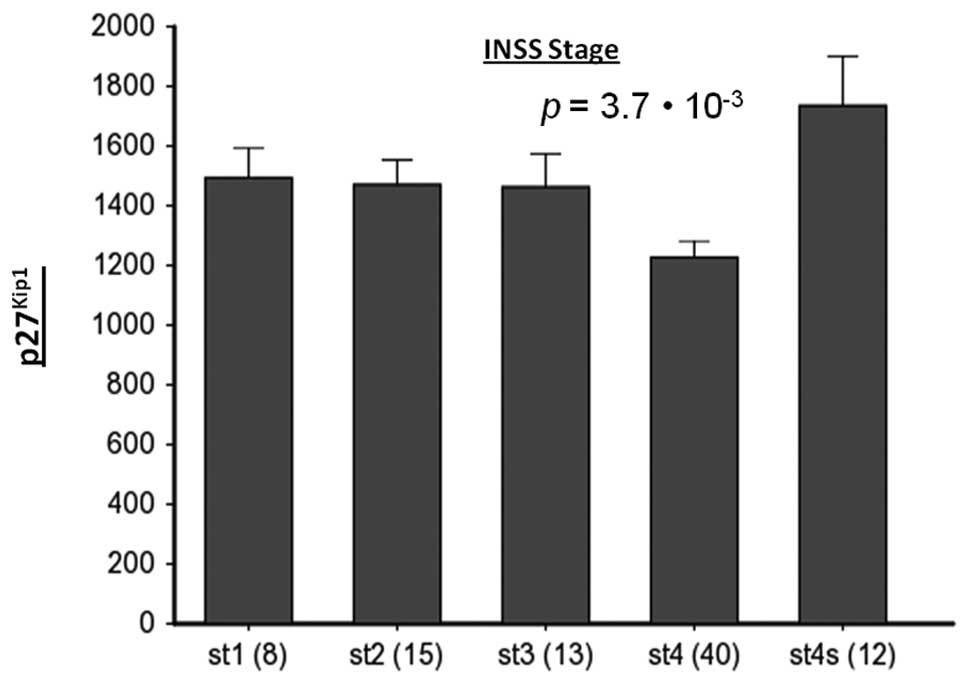
bone marrow metastasis. Interestingly, NB patients in stages 1, 2
and 3 have higher p27Kip1 expression than those in stage
4, which is the metastasizing stage (Fig. 6). Finally, we observed an inverse
correlation between p27Kip1 and ODC mRNA expression
suggesting that ODC and possibly intra-cellular polyamines
regulates metastasis through a process that involves modulation of
p27Kip1 expression.
Our previous studies have shown that the ODC
inhibitor DFMO depletes intracellular polyamine levels, induces
p27Kip1 protein accumulation and G1 cell
cycle arrest, downregulates MYCN protein, but also promotes cell
survival by modulating the phosphorylation of Akt/PKB via the
PI3K/Akt signaling pathway in MYCN-amplified NB cells (10,11).
In the present study, we found that DFMO inhibits the migration of
NB cells. The results from the wound healing assay suggested that
MYCN overexpression had no effect on NB migration and DFMO
inhibited migration to the same degree, regardless of the MYCN
status. The results from the transwell migration assay suggested
that MYCN overexpression enhanced NB migration, and the inhibitory
effect of DFMO was augmented in MYCN2 (+) cells. In addition, MYCN
overexpression increased NB invasion by 3.5-fold and DFMO was much
more potent at inhibiting invasion in MYCN2 (+) cells than MYCN2
(−) cells. The different effects observed in the wound healing
assay compared to the transwell assay may be due to the differences
in the assay design. The wound healing assay in two-dimensional
cell culture allows for chemokinetic migration, or random
migration, but not chemotaxis. In contrast, the design of the
transwell assay with chemoattractant in the bottom chamber allows
for both chemo-kinetic and chemotactic migration. The results from
this set of experiments showed that polyamines promote NB migration
and invasion, and DFMO is a potent inhibitor of these processes. In
addition, MYCN overexpression significantly increased NB invasion
and DFMO inhibited migration and invasion more effectively in MYCN
over-expressing cells relative to controls.
Interestingly, each individual polyamine appeared to
be equally effective at regulating NB proliferation, migration, and
invasion. In addition, the reversal of the DFMO-induced effects by
polyamines appeared to be a time-dependent process. However, other
polyamine-metabolizing enzymes are still active and the addition of
any polyamines can be converted or retro-converted into the other
polyamines. Therefore, it is still unclear whether one particular
polyamine plays a larger role than the other polyamines in
regulating NB proliferation, migration or invasion.
Next, we examined the role of p27Kip1 in
NB migration and invasion and discovered that downregulation of
p27Kip1 expression attenuated the inhibitory effect of
DFMO on NB migration and invasion. Furthermore, DFMO induced
translocation of p27Kip1 from the nucleus to the
cytoplasm of NB cells. The results from these experiments suggest
that polyamines regulate NB migration and invasion by modulating
the expression and localization of p27Kip1, and that
DFMO inhibits NB migration and invasion by inducing
p27Kip1 accumulation. Our previous results showed that
DFMO induces Ser10 phosphorylation of p27Kip1, which has
been shown to be a nuclear export signal (11). Phosphorylation at this site was
also shown to induce nuclear accumulation in some cell types
(41). The results from this study
showed that DFMO induces p27Kip1 accumulation in both
the nucleus and cytoplasm suggesting a dual role for
p27Kip1. While nuclear p27Kip1 is a
well-established cell cycle regulator, nuclear and/or cytoplasmic
p27Kip1 may play a role in regulating cell migration and
invasion.
Previous studies have shown that polyamines regulate
cell migration and metastasis in other cell types (42–50).
The present study examined the role of polyamines in NB migration
and invasion and provides evidence that p27Kip1 plays a
key role in regulating these processes, in addition to its
well-characterized role as a cell cycle regulator. In other
studies, DFMO has been shown to regulate these processes via
p21Cip1. However, in NB cells we have shown that DFMO
effects are mediated via p27Kip1. Therefore,
p27Kip1 expression is not only a prognostic indicator of
NB, but also an indicator for NB metastasis. This is particularly
interesting since although p27Kip1 is a well-known tumor
suppressor gene, its mRNA expression in a number of different
cancer types is not lower in cancerous than in corresponding normal
tissues (data not shown). In NB, however, p27Kip1
expression is in fact lower in patients with stage 4, meta-static
NB than those patients with lower stage, non-metastatic disease.
This was not just found for the NB set described in this study, but
also for two other NB tumor sets in the public domain (Fig. 6). In addition, this study revealed
that there is an inverse relationship between p27Kip1
and ODC expression suggesting that the function of
p27Kip1 in tumor progression, metastasis and overall
survival may be regulated by intracellular polyamines. We also
showed that DFMO exhibits anti-migratory properties, and it will be
interesting to see whether this is also true of NB metastasis,
in vivo. Importantly, the potency of DFMO on NB migration
and invasion was greater in NB cells with MYCN overexpression.
NB cells stabilize MYCN protein, for example, by
inhibition of MYCN degradation. Akt/PKB-mediated inactivation of
GSK3-β (through phosphorylation on its Ser9 residue) prevents
GSK3-β from phosphorylating MYCN on Thr58, which normally leads to
proteasomal degradation of MYCN (51). Our results show that DFMO can
reduce MYCN protein levels in MYCN2 (+) cells, in spite of Akt/PKB
activation, as shown by p27Kip1 accumulation, providing
a molecular rationale for the use of DFMO in NB tumors with high
MYCN expression. Together, these results suggest that DFMO may be
particularly effective as part of a combination treatment to
prevent metastasis of NB, and for the treatment of advanced stage,
NB patients with MYCN-amplification. Recent in vivo studies
with NB tumor-bearing mice using the transgenic TH-MYCN model
revealed significant antitumor effects of DFMO (52,53).
Importantly, a phase I human clinical trial with DFMO alone and
combined with etoposide in relapsed/refractory NB is near
completion (ClinicalTrials. gov Identifier NCT01059071) and a phase
II preventative trial with DFMO in patients with high-risk NB in
remission is now open for participant enrollment at several
Neuroblastoma and Medulloblastoma Research Consortium (NMTRC)
Children’s Hospitals throughout the US (ClinicalTrials.gov Identifier NCT01586260).
Acknowledgements
This study was supported by NIH grants
from the National Cancer Institute R01 CA-111419 (André S.
Bachmann) and R01 Supplement CA-111419-S1 (André S. Bachmann), R01
CA-018138 (Anthony E. Pegg, David J. Feith), the Alex’s Lemonade
Stand Foundation (ALSF) grant 439744 (Dana-Lynn T. Koomoa), the
Dutch Cancer Society (KWF Kankerbestrijding) UVA2003-2849 and
UVA2005-3665 (Dirk Geerts). We thank Dr Patrick Woster (Medical
University of South Carolina) for providing the ODC inhibitor DFMO,
Dr Jason Shohet (Texas Children’s Hospital) for providing the NB
cell line MYCN2, Suzanne Sass-Kuhn for polyamine HPLC analysis, and
Risha Mishima and Noah Yuen (University of Hawaii Cancer Center)
for their technical assistance.
References
|
1
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM: Recent advances in
neuroblastoma. N Engl J Med. 362:2202–2211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwab M, Westermann F, Hero B and
Berthold F: Neuroblastoma: biology and molecular and chromosomal
pathology. Lancet Oncol. 4:472–480. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Canete A, Gerrard M, Rubie H, et al: Poor
survival for infants with MYCN-amplified metastatic neuroblastoma
despite intensified treatment: the International Society of
Paediatric Oncology European Neuroblastoma Experience. J Clin
Oncol. 27:1014–1019. 2009. View Article : Google Scholar
|
|
6
|
De Bernardi B, Gerrard M, Boni L, et al:
Excellent outcome with reduced treatment for infants with
disseminated neuroblastoma without MYCN gene amplification. J Clin
Oncol. 27:1034–1040. 2009.PubMed/NCBI
|
|
7
|
Johnsen JI, Segerstrom L, Orrego A, et al:
Inhibitors of mammalian target of rapamycin downregulate MYCN
protein expression and inhibit neuroblastoma growth in vitro and in
vivo. Oncogene. 27:2910–2922. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Otto T, Horn S, Brockmann M, et al:
Stabilization of N-Myc is a critical function of Aurora A in human
neuroblastoma. Cancer Cell. 15:67–78. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ushmorov A, Hogarty MD, Liu X, Knauss H,
Debatin KM and Beltinger C: N-myc augments death and attenuates
protective effects of Bcl-2 in trophically stressed neuroblastoma
cells. Oncogene. 27:3424–3434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wallick CJ, Gamper I, Thorne M, et al: Key
role for p27Kip1, retinoblastoma protein Rb, and MYCN in
polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified
human neuroblastoma cells. Oncogene. 24:5606–5618. 2005.PubMed/NCBI
|
|
11
|
Koomoa DL, Yco LP, Borsics T, Wallick CJ
and Bachmann AS: Ornithine decarboxylase inhibition by
alpha-difluoromethylornithine activates opposing signaling pathways
via phosphorylation of both Akt/protein kinase B and
p27Kip1 in neuroblastoma. Cancer Res. 68:9825–9831.
2008. View Article : Google Scholar
|
|
12
|
Chiarle R, Pagano M and Inghirami G: The
cyclin dependent kinase inhibitor p27 and its prognostic role in
breast cancer. Breast Cancer Res. 3:91–94. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chu IM, Hengst L and Slingerland JM: The
Cdk inhibitor p27 in human cancer: prognostic potential and
relevance to anticancer therapy. Nat Rev Cancer. 8:253–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Esposito V, Baldi A, De Luca A, et al:
Prognostic role of the cyclin-dependent kinase inhibitor p27 in
non-small cell lung cancer. Cancer Res. 57:3381–3385.
1997.PubMed/NCBI
|
|
15
|
Lee J and Kim SS: The function of
p27Kip1 during tumor development. Exp Mol Med.
41:765–771. 2009.
|
|
16
|
Ravanko K, Jarvinen K, Paasinen-Sohns A
and Holtta E: Loss of p27Kip1 from cyclin
E/cyclin-dependent kinase (CDK) 2 but not from cyclin D1/CDK4
complexes in cells transformed by polyamine biosynthetic enzymes.
Cancer Res. 60:5244–5253. 2000.PubMed/NCBI
|
|
17
|
Baldassarre G, Belletti B, Nicoloso MS, et
al: p27(Kip1)-stathmin interaction influences sarcoma cell
migration and invasion. Cancer Cell. 7:51–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Itoh Y, Masuyama N, Nakayama K, Nakayama
KI and Gotoh Y: The cyclin-dependent kinase inhibitors p57 and p27
regulate neuronal migration in the developing mouse neocortex. J
Biol Chem. 282:390–396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Z, Jiao X, Wang C, et al: Cyclin D1
induction of cellular migration requires p27(Kip1).
Cancer Res. 66:9986–9994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McAllister SS, Becker-Hapak M, Pintucci G,
Pagano M and Dowdy SF: Novel p27(Kip1) C-terminal
scatter domain mediates Rac-dependent cell migration independent of
cell cycle arrest functions. Mol Cell Biol. 23:216–228. 2003.
|
|
21
|
See WL, Heinberg AR, Holland EC and Resh
MD: p27 deficiency is associated with migration defects in
PDGF-expressing gliomas in vivo. Cell Cycle. 9:1562–1567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stehr W, Mercer TI, Bernal NP, Erwin CR
and Warner BW: Opposing roles for p21(waf1/cip1) and
p27(Kip1) in enterocyte differentiation, proliferation,
and migration. Surgery. 138:187–194. 2005.PubMed/NCBI
|
|
23
|
Sun J, Marx SO, Chen HJ, Poon M, Marks AR
and Rabbani LE: Role for p27(Kip1) in vascular smooth muscle cell
migration. Circulation. 103:2967–2972. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Woods TC: Regulation of cell migration by
mTOR is mediated through changes in p27(Kip1) phosphorylation. Cell
Cycle. 9:2057–2058. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Assoian RK: Stopping and going with
p27Kip1. Dev Cell. 6:458–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Besson A, Gurian-West M, Schmidt A, Hall A
and Roberts JM: p27Kip1 modulates cell migration through
the regulation of RhoA activation. Genes Dev. 18:862–876. 2004.
|
|
27
|
Larrea MD, Wander SA and Slingerland JM:
p27 as Jekyll and Hyde: regulation of cell cycle and cell motility.
Cell Cycle. 8:3455–3461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iancu-Rubin C and Atweh GF:
p27(Kip1) and stathmin share the stage for the first
time. Trends Cell Biol. 15:346–348. 2005.
|
|
29
|
Denicourt C, Saenz CC, Datnow B, Cui XS
and Dowdy SF: Relocalized p27Kip1 tumor suppressor
functions as a cytoplasmic metastatic oncogene in melanoma. Cancer
Res. 67:9238–9243. 2007.PubMed/NCBI
|
|
30
|
Kossatz U, Vervoorts J, Nickeleit I, et
al: C-terminal phosphorylation controls the stability and function
of p27Kip1. EMBO J. 25:5159–5170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sicinski P, Zacharek S and Kim C: Duality
of p27Kip1 function in tumorigenesis. Genes Dev.
21:1703–1706. 2007. View Article : Google Scholar
|
|
32
|
Supriatno, Harada K, Kawaguchi S, Yoshida
H and Sato M: Effect of p27Kip1 on the ability of
invasion and metastasis of an oral cancer cell line. Oncol Rep.
10:527–532. 2003.
|
|
33
|
Wang XQ, Lui EL, Cai Q, et al:
p27Kip1 promotes migration of metastatic hepatocellular
carcinoma cells. Tumour Biol. 29:217–223. 2008.
|
|
34
|
Geerts D, Koster J, Albert D, et al: The
polyamine metabolism genes ornithine decarboxylase and antizyme 2
predict aggressive behavior in neuroblastomas with and without MYCN
amplification. Int J Cancer. 126:2012–2024. 2010.
|
|
35
|
Koomoa DL, Borsics T, Feith DJ, et al:
Inhibition of S-adenosylmethionine decarboxylase by inhibitor
SAM486A connects polyamine metabolism with p53-Mdm2-Akt/protein
kinase B regulation and apoptosis in neuroblastoma. Mol Cancer
Ther. 8:2067–2075. 2009. View Article : Google Scholar
|
|
36
|
Revet I, Huizenga G, Chan A, et al: The
MSX1 homeobox transcription factor is a downstream target of PHOX2B
and activates the Delta-Notch pathway in neuroblastoma. Exp Cell
Res. 314:707–719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Motti ML, Califano D, Troncone G, et al:
Complex regulation of the cyclin-dependent kinase inhibitor
p27Kip1 in thyroid cancer cells by the PI3K/AKT pathway:
regulation of p27Kip1 expression and localization. Am J
Pathol. 166:737–749. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Motti ML, De Marco C, Califano D, Fusco A
and Viglietto G: Akt-dependent T198 phosphorylation of
cyclin-dependent kinase inhibitor p27Kip1 in breast
cancer. Cell Cycle. 3:1074–1080. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
van Weeren PC, de Bruyn KM, de Vries-Smits
AM, van Lint J and Burgering BM: Essential role for protein kinase
B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation.
Characterization of dominant-negative mutant of PKB. J Biol Chem.
273:13150–13156. 1998.PubMed/NCBI
|
|
40
|
Bergmann E, Wanzel M, Weber A, Shin I,
Christiansen H and Eilers M: Expression of P27(Kip1) is
prognostic and independent of MYCN amplification in human
neuroblastoma. Int J Cancer. 95:176–183. 2001.
|
|
41
|
Borriello A, Bencivenga D, Criscuolo M, et
al: Targeting p27(Kip1) protein: its relevance in the
therapy of human cancer. Expert Opin Ther Targets. 15:677–693.
2011.
|
|
42
|
Richert MM, Phadke PA, Matters G, et al:
Metastasis of hormone-independent breast cancer to lung and bone is
decreased by alpha-difluoromethylornithine treatment. Breast Cancer
Res. 7:R819–R827. 2005. View Article : Google Scholar
|
|
43
|
Manni A, Washington S, Craig L, et al:
Effects of alpha-difluoromethylornithine on local recurrence and
pulmonary metastasis from MDA-MB-435 breast cancer xenografts in
nude mice. Clin Exp Metastasis. 20:321–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jun JY, Griffith JW, Bruggeman R, et al:
Effects of polyamine depletion by alpha-difluoromethylornithine on
in vitro and in vivo biological properties of 4T1 murine mammary
cancer cells. Breast Cancer Res Treat. 105:29–36. 2007. View Article : Google Scholar
|
|
45
|
Ohmori T, Okada K, Tabei R and Shibata T:
Effects on tumor induction, growth, metastasis and histology of
concurrent administration of putrescine and its metabolizing
inhibitor alpha-difluoromethylornithine in nickel tumorigenesis in
soft tissue. Carcinogenesis. 15:647–652. 1994. View Article : Google Scholar
|
|
46
|
Zirvi KA, Dasmahapatra KS, Atabek U and
Lyons MA: alpha-Difluoromethylornithine inhibits liver metastasis
produced by intrasplenic injection of human tumor cells into nude
mice. Clin Exp Metastasis. 7:591–598. 1989. View Article : Google Scholar
|
|
47
|
Kubota S, Ohsawa N and Takaku F: Effects
of DL-alpha-difluoromethylornithine on the growth and metastasis of
B16 melanoma in vivo. Int J Cancer. 39:244–247. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sunkara PS and Rosenberger AL:
Antimetastatic activity of DL-alpha-difluoromethylornithine, an
inhibitor of polyamine biosynthesis, in mice. Cancer Res.
47:933–935. 1987.PubMed/NCBI
|
|
49
|
Meyskens FL, Kingsley EM, Glattke T,
Loescher L and Booth A: A phase II study of
alpha-difluoromethylornithine (DFMO) for the treatment of
metastatic melanoma. Invest New Drugs. 4:257–262. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Klein S, Miret JJ, Algranati ID and de
Lustig ES: Effect of alpha-difluoromethylornithine in lung
metastases before and after surgery of primary adenocarcinoma
tumors in mice. Biol Cell. 53:33–36. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yaari S, Jacob-Hirsch J, Amariglio N,
Haklai R, Rechavi G and Kloog Y: Disruption of cooperation between
Ras and MycN in human neuroblastoma cells promotes growth arrest.
Clin Cancer Res. 11:4321–4330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hogarty MD, Norris MD, Davis K, et al:
ODC1 is a critical determinant of MYCN oncogenesis and a
therapeutic target in neuroblastoma. Cancer Res. 68:9735–9745.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rounbehler RJ, Li W, Hall MA, Yang C,
Fallahi M and Cleveland JL: Targeting ornithine decarboxylase
impairs development of MYCN-amplified neuroblastoma. Cancer Res.
69:547–553. 2009. View Article : Google Scholar : PubMed/NCBI
|