Introduction
Gliomas are the most common malignant primary brain
tumors. Despite aggressive surgery, radiation and chemotherapy, the
median survival is only 12–15 months for glioblastoma multiforme
(GBM) (1). Therefore, it is
crucial to investigate the mechanism involved in the development
and progression of glioma and to find new therapeutic targets.
MicroRNAs are small, endogenous, nonprotein-coding
RNA molecules which have a functional role as negative gene
regulators through complementarity to the 3′-untranslated region
(3′-UTR) of mRNAs (2), it plays
critical regulatory roles in the diverse biological processes of
development, differentiation and apoptosis. MiRNAs show pleiotropic
and pivotal effects on a variety of pathological and physiological
cellular processes (3,4). Furthermore, each miRNA can
potentially regulate hundreds of mRNAs and more than one-third of
human genes might be miRNA targets (5). A considerable amount of evidence
indicates that miRNAs are involved in human cancer (6,7).
Deregulated miRNAs exhibit oncogenic or tumor suppressor
properties. However, only a small fraction of miRNAs has already
been investigated. In glioma, apart from upregulated miRNAs such as
miR-21, the functions of downregulated miRNAs are relatively
unknown.
As a stemness gene, Nanog has been reported to be
significant in ESCs (embryonic stem cells), in which the level and
activity are the pluripotent status indicators of ESCs (8). Nanog, a transcription factor, is not
only known to play an essential role in ESC maintenance and
differentiation by reprogramming gene sets that include OCT4, SOX2,
c-myc and Klf4, but is also involved in the reprogramming of
differentiated cells towards the induction of pluripotent stem
(iPS) cells (9,10). Several tumor cell types have been
reported to express Nanog (11,12).
In 2009, You et al (13)
reported that Nanog was abnormally overexpressed in human embryonic
carcinoma NCCIT cells. Downregulation of Nanog by histone
deacetylase inhibitor apicidin could lead to cell cycle arrest,
differentiation and apoptosis in NCCIT cells. Subsequently, Zbinden
et al demonstrated the function of Nanog in human
glioblastomas and its relationship with HH-GLI activity (14). Our preliminary research has
demonstrated overexpression of Nanog in glioma tissues and brain
tumor stem cells (BTSCs) compared with normal brain tissues,
indicating that Nanog may contribute to the existence of BTSCs and
may be related to tumorigenesis of the cerebrum by maintaining the
undifferentiated state of glioma cells (15).
MiR-134 is a brain-enriched microRNA which can
promote vertebrate central nervous system development (including
neuron, cylindraxile and dendrite) (16–19).
Reporter assay with 3′-UTR of Nanog cloned downstream of the
luciferase gene showed reduced luciferase activity in the presence
of miR-134, strongly supporting miR-134 was a direct regulator of
Nanog (16). It is worth noting
that miR-134 is essential in the promotion of embryonic stem cell
differentiation by its direct translational attenuation of Nanog.
Data revealed that miR-134 alone can enhance the differentiation of
mESCs to ectodermal lineages and modulate mESC (mouse embryonic
stem cells) differentiation through its capacity to target and
regulate multiple mRNAs, especially Nanog (16).
In this study, we focused on the downregulated
miR-134, in human glioma tissues and in glioblastoma cell line U87
then compared it to normal brain tissues, finding that ectopic
expression of miR-134 reduced the level of Nanog expression,
causing inhibition of proliferation, invasiveness and migration, it
also increased apoptosis in glioblastoma cells. These findings
suggested that miR-134 could act as a biomarker in glioma and its
restoration might be a possible therapeutic approach aimed at
Nanog, that deserve further investigation in glioma and BTSCs.
Materials and methods
Cell lines and human tissue samples
The U87 glioblastoma cells and 293T cells (Chinese
Academy of Sciences Type Culture Collection) were cultured as a
monolayer of cells in Dulbecco’s modified Eagle’s medium: nutrient
mixture F-12 (Ham’s) (1:1) (DMEM/F-12) (Gibco, USA), supplemented
with 10% fetal bovine serum (FBS) (Hyclone, USA), 100 U/ml
penicillin/streptomycin (Gibco).
Clinical sample collection
Glioma samples were obtained from 42 patients with
primary gliomas who underwent surgical treatment at the Department
of Neurosurgery, Anhui Provincial Hospital Affiliated to Anhui
Medial University, between October 2010 and September 2011 in
accordance with the national regulation of clinical sampling in
China. Eleven adult normal brain tissues as normal controls were
obtained from the patients with severe traumatic brain injury (TBI)
who needed post-trauma surgery after informed consent. This study
was approved by the hospital institutional review board and written
informed consent was obtained from all patients. Tumor specimens
were immediately sectioned from the resected glioma tissues, frozen
in liquid nitrogen and stored at −80°C until RNA/protein
extraction. All of the glioma samples were verified by pathological
analysis and classified according to the WHO 2007 classification.
There were 13 low-grade (WHO grades I and II) and 31 high-grade
tumors (WHO grades III and IV). None of the patients had received
chemotherapy, immunotherapy and radiotherapy prior to specimen
collection. Informed consent was obtained from all patients before
surgery as advocated by the regional ethics committee.
RNA extraction, real-time PCR
Total RNA was extracted from the cultured cells or
the human glioma samples with TRIzol reagent (Invitrogen, USA).
Relative levels of miR-134 were examined using SYBR green real-time
quantitative reverse transcription-PCR (real-time PCR) and
normalized with U6 snRNA. cDNA was synthesized by using miScript II
RT kit (Qiagen, USA). The primers of miR-134 and U6 snRNA and
miScript SYBR Green PCR kit were also purchased from Qiagen for the
reaction system of real-time PCR. The real-time PCR reactions were
performed on a 7500 Fast System real-time PCR cycler (Applied
Biosystems, USA) for 40 cycles. The procedure for PCR was 95°C for
15 min; 94°C for 15 sec, 55°C for 30 sec, 70°C for 30 sec. All
procedures were performed according to the instructions provided by
the manufacturer. The fold-change of each miRNA was calculated
using the 2ΔΔCt method (20).
RT-PCR
Reverse transcription-polymerase chain reaction
(RT-PCR) was performed as described before (15). The primer sequences were as
follows: Nanog (403 bp), 5′-ATGCCTGTGATTTGTGGGCC-3′ (forward) and
5′-GCCAGTTGTTTTTCTGCCAC-3′ (reverse); β-actin (252 bp),
5′-ATGGATGATGATATCGCCGCGCTC-3′ (forward) and
5′-TTTCTCCATGTCGTCCCAGTTGG-3′ (reverse). β-actin was used as the
internal control.
Constructs and cell lines
Mature miR-134 sequence (UGUGACUGGUUGACCAGAGGGG) was
transformed into premiRNA sequence that could be more suitable for
expression. A genomic sequence spanning pre-miR-134 was amplified
using primers (miR-134-F:
5′-TGCTGTGTGACTGGTTGACCAGAGGGGGTTTTGGCCACTGACTGACCCCCTCTGCAACCAGTCACA-3′;
miR-134-R:
5′-CCTGTGTGACTGGTTGCAGAGGGGGTCAGTCAGTGGCCAAAACCCCCTCTGGTCAACCAGTCACAC-3′)
and then cloned into the pLenti6.3/V5-DEST Gateway Vector
(Invitrogen); lentiviruses were packaged in HEK293T cells according
to the manufacturer’s instructions and were used to infect U87
glioblastoma cells with polybrene (Sigma, USA). Cells were then
subcultured to 10% confluence in medium containing 10 μg/ml
of blasticidin (Sigma). The process was similar to a previous assay
(21). The cells expressing
pLenti-miR-134-GFP/pLenti-GFP were termed
U87-miR-134/vector-control, respectively, while U87 cells without
any treatment were the blank group. The expression of miR-134 was
detected by real-time PCR.
Western blotting
Western blotting and immunohistochemistry assays
were performed as previously described (15). Membranes were probed with mouse
polyclonal antibodies against human Nanog (1:100 dilution) (R&D
Systems, USA) at 4°C overnight or mouse monoclonal anti-β-actin
antibody (1:1,000 dilution) (Beyotime, China) for 1 h at room
temperature followed by the horseradish peroxidase (HRP)-conjugated
goat anti-mouse IgG antibody (ZSGB-BIO, China). Immunoblots were
visualized by chemiluminescence using the ECL detection system
(BeyoECL Plus, Beyotime). The intensity of the bands was determined
using the Image-pro plus 6.0 software (Japan).
Proliferation assay
MTT assay was used to quantitate cell viability of
glioblastoma cells as previously described (22). The absorbance values of each well
were measured with a micro-plate spectrophotometer (Molecular
Devices, USA) at 570 nm at 24, 48, 72 and 96 h. All proliferation
assays were repeated as independent experiments at least three
times.
Tumor growth in vivo
Glioma tumor xenografts were established in female
BALB/c athymic mice (Cancer Institute of The Chinese Academy of
Medical Science) by subcutaneous injection totals of
5×106 stable U87-miR-134, vector-control cells, while
U87 glioblastoma cells as blank group (8 mice per group). All
experimental procedures were performed according to Anhui Medical
University policies. Tumor growth was measured serially and the
tumor volume was measured twice a week with the formula: volume =
length × width2/2. Paraffin sections of xenograft tumors
were subjected to immunohistochemical staining.
Immunohistochemistry
Immunohistochemical studies were performed as
previously described (15).
Endogenous peroxidase was neutralized with 3%
H2O2 in methanol (10 min) after antigen
retrieval in 0.1 M citrate buffer (pH 5.8) at 95°C for 5 min and
cooled at 25°C for 1 h. Sections were blocked with normal goat
serum (10 min), then treated with the following primary antibodies
overnight at 4°C; NANOG (1:100 dilution). Negative control sections
were incubated with PBS instead of the primary antibody. After
treatment with biotinylated secondary antibody, color reactions
were performed with diaminobenzidine (DAB) (Sigma) and
counterstained with Mayer’s hematoxylin. The immunohistochemical
staining results were scored by two pathologists.
Transwell assay and Transwell matrix
penetration assay
Cells (4×104) in 200 μl serum-free
DMEM were plated on the upper compartment of a Transwell device
(without Matrigel for Transwell assay) or plated on the top side of
polycarbonate Transwell filter coated with Matrigel (for Transwell
matrix penetration assay) in the upper chamber of the BioCoat™
Invasion Chambers (BD, USA) and incubated at 37°C for 24 h,
followed by removal of cells inside the upper chamber with cotton
swabs. Migrated and invaded cells on the lower membrane surface
were fixed in 1% paraformaldehyde, stained with 0.1% crystal violet
and counted (8 random 200x fields per well). Cell counts were
expressed as the mean number of cells per field of view.
Wound scraping assay
U87-miR-134 and vector-control cells were plated in
6-well plates and were grown to 80–90% confluence. Then, the
monolayer of cells was scraped with a standard 200-μl
sterile micropipette tip to create a denuded gap of constant width.
The cells were washed with PBS subsequently and then maintain in
serum-free medium. After 48 h, the cells migrated into the gap were
observed under a phase microscope qualitatively.
Apoptosis assay
With DAPI staining method, transfected cells were
fixed for 20 min in 4% (v/v) paraformaldehyde at 4°C and then
incubated with DAPI dye (Sigma) (10 μg/ml) for 15 min. After
washing with PBS, cells were observed using an inverted
fluorescence microscope (IX70; Olympus, Japan). Transfected cells
were detached from culture flask, washed, suspended in PBS and
concentrated by centrifugation. Cell samples were fixed in 2.5%
(w/v) glutaraldehyde, postfixed in 2% (w/v) buffered osmium
tetroxide for 2 h and dehydrated in ethanol. Specimens for
transmission electron microscopy were embedded in Epon. Sections
were cut using an ultra-microtome and double-stained with uranyl
acetate and lead citrate. Electron micrography was performed
(JEM-2000EX; Jeol, Japan) using an operating voltage of 80 kV
(23). In the flow cytometry
assay, transfected cells were dual stained with the viability dye
7-amino-actinomycin D (7AAD) and Annexin V-PE using an Annexin
V-PE/7-AAD apoptosis detection kit (KeyGen Biotech, China)
according to the manufacturer’s protocol. Stained cells were
immediately analysed with a flow cytometer (Cell Lab Quanta SC;
Beckman Coulter, USA).
Statistical analysis
Experimental data are presented as mean ± standard
deviation (SD). All statistical analyses were performed using a
two-tailed Student’s t-test or one-way ANOVA (SPSS 17.0). P<0.05
was considered statistically significant. All experiments were
repeated three times.
Results
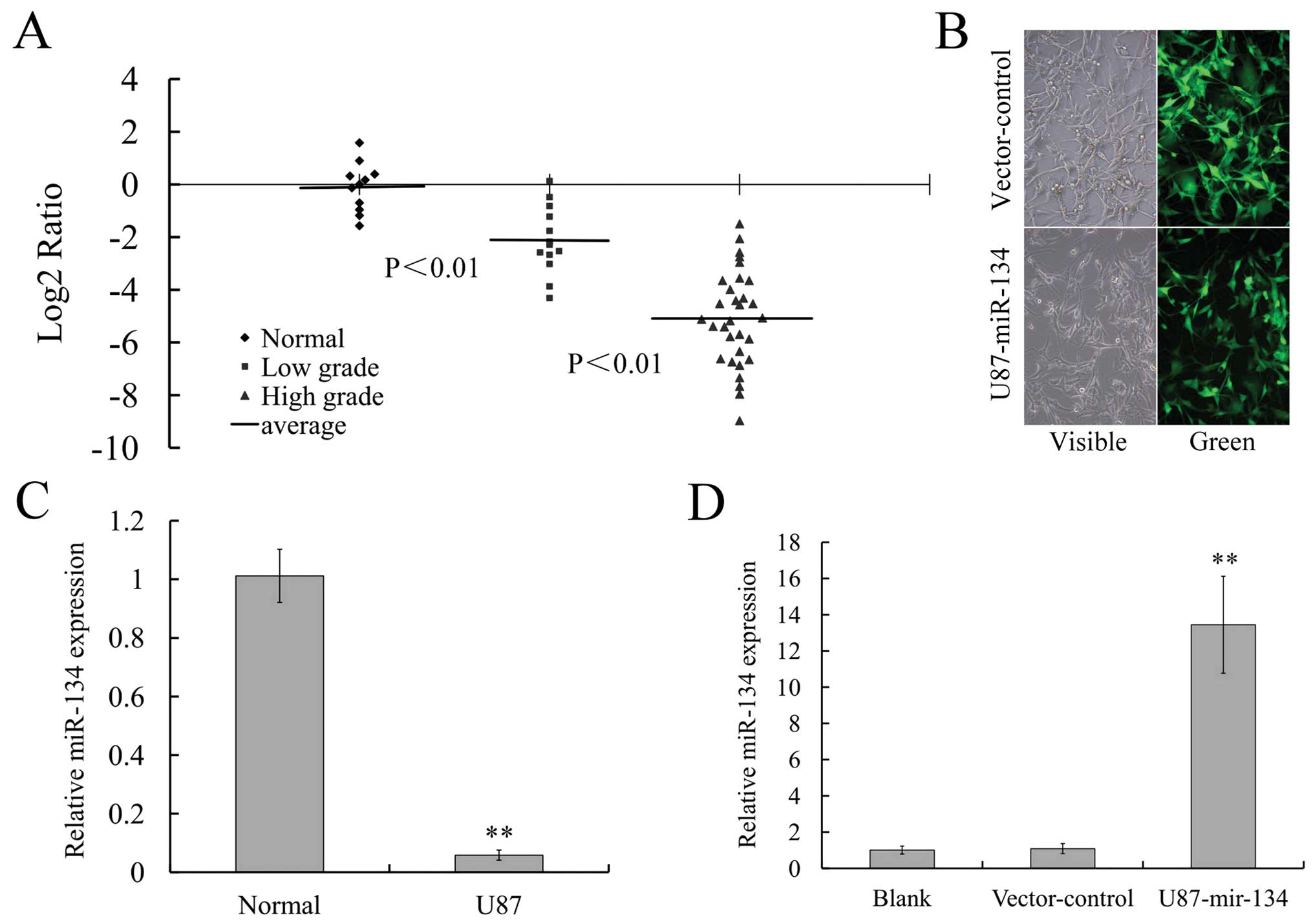
Downregulation of miR-134 in human glioma
tissues and glioblastoma cell line U87
Real-time PCR analysis revealed that the expression
of miR-134 was significantly lower in glioma samples compared with
normal brain tissues (P<0.01) and was lower in grade III and IV
gliomas compared to grade I and II tumors (P<0.01; Fig. 1A). Remarkable downregulation of
miR-134 could also be observed in glioblastoma cell line U87
(P<0.01; Fig. 1C). These
results suggested that miR-134 could be closely related to human
glioma and the low level of miR-134 might be related to glioma
oncogenesis and its invasive propensity.
Ectopic expression of miR-134 reduced the
expression of its miRNA target NANOG in U87 glioblastoma cells
MiR-134 expressing the U87 stable cell line was
generated for the effect of microRNA-134 on NANOG protein level
with lentivirus transfection method. As shown in Fig. 1B, green fluorescent signal was
detected in >98% GFP-labeled miR-134-transfected U87 cells,
indicating that RNA oligonucleotides could readily gain access to
the cells. The expression of miR-134 in U87 cells was detected by
real-time PCR. We determined that the expression of miR-134 was
significantly increased in U87-miR-134 cells, while in
vector-control cells it did not change obviously (Fig. 1D).
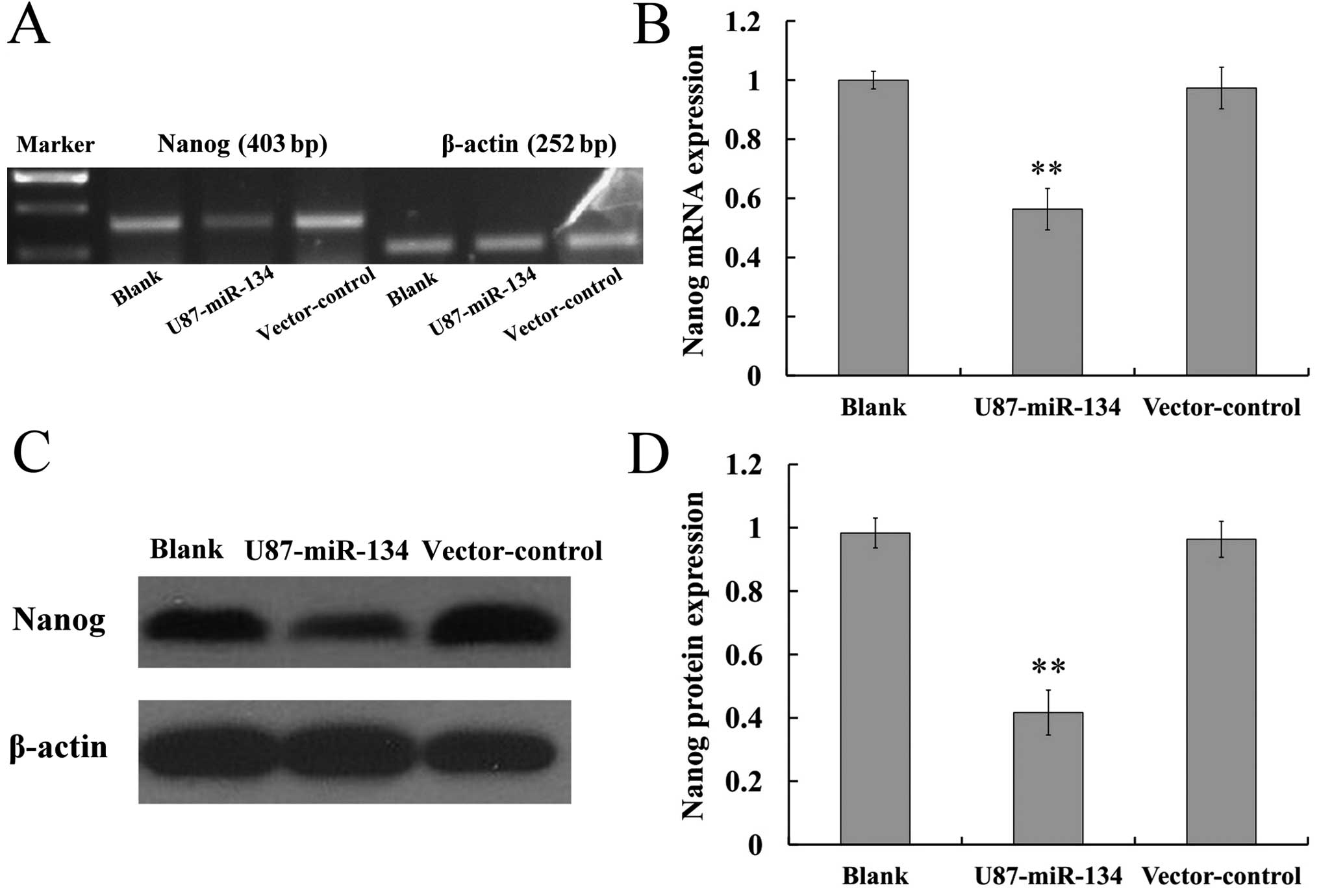
Nanog is a comfirmed miR-134 target with 3′-UTR
luciferase assays reported by Tay et al (16). We therefore determined whether the
Nanog mRNA and protein levels could be affected by ectopic
expression of miR-134 in glioblastoma cells by RT-PCR and western
blotting, respectively. Results revealed that miR-134 can restrain
the mRNA and protein expression of Nanog in glioblastoma cells
(P<0.01) (Fig. 2). The
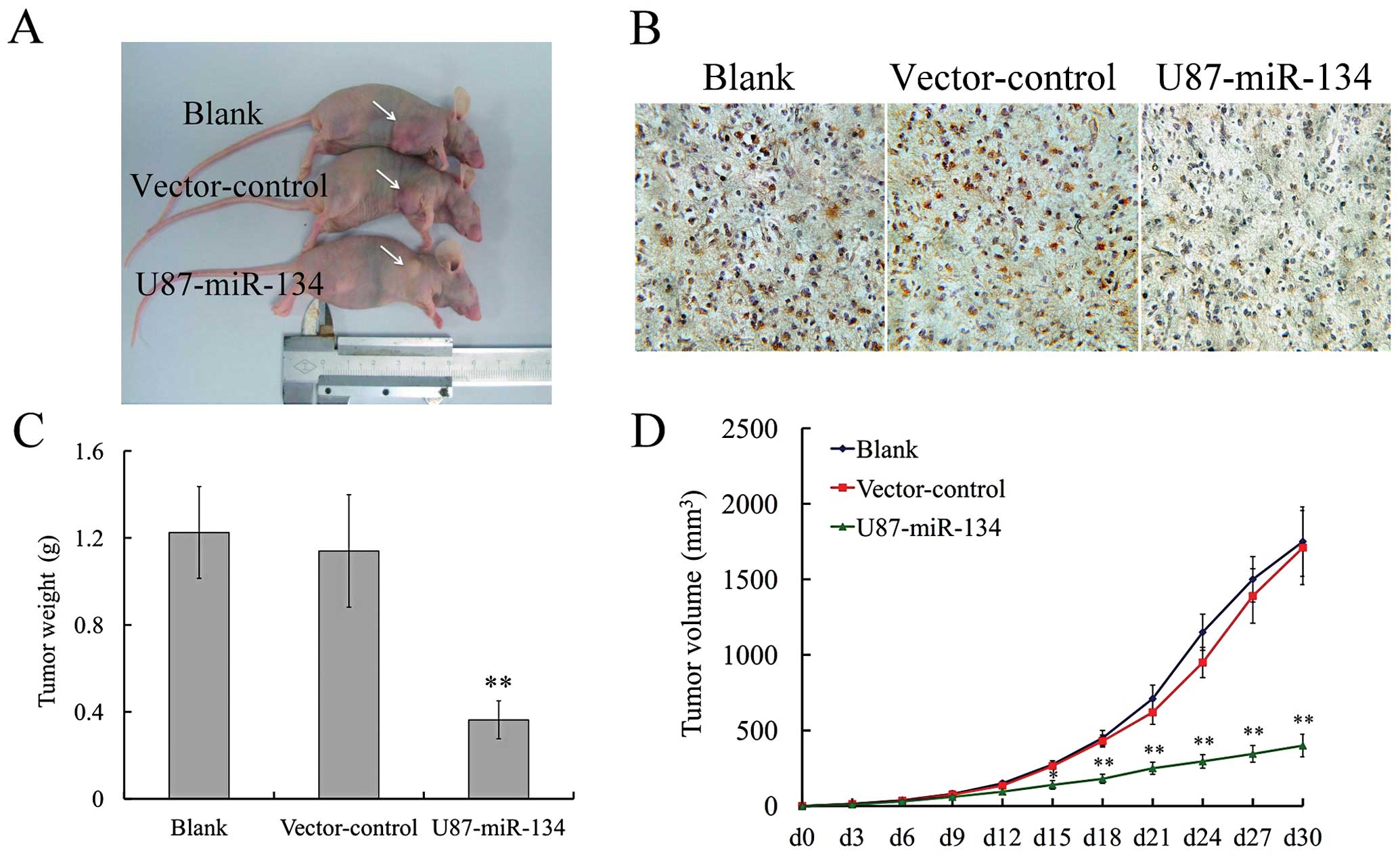
immunohistochemical staining results (Fig. 5B) also showed that the glioma
xenografts of U87-miR-134 group expressed less Nanog than the
tumors in the blank and vector-control group (P<0.01).
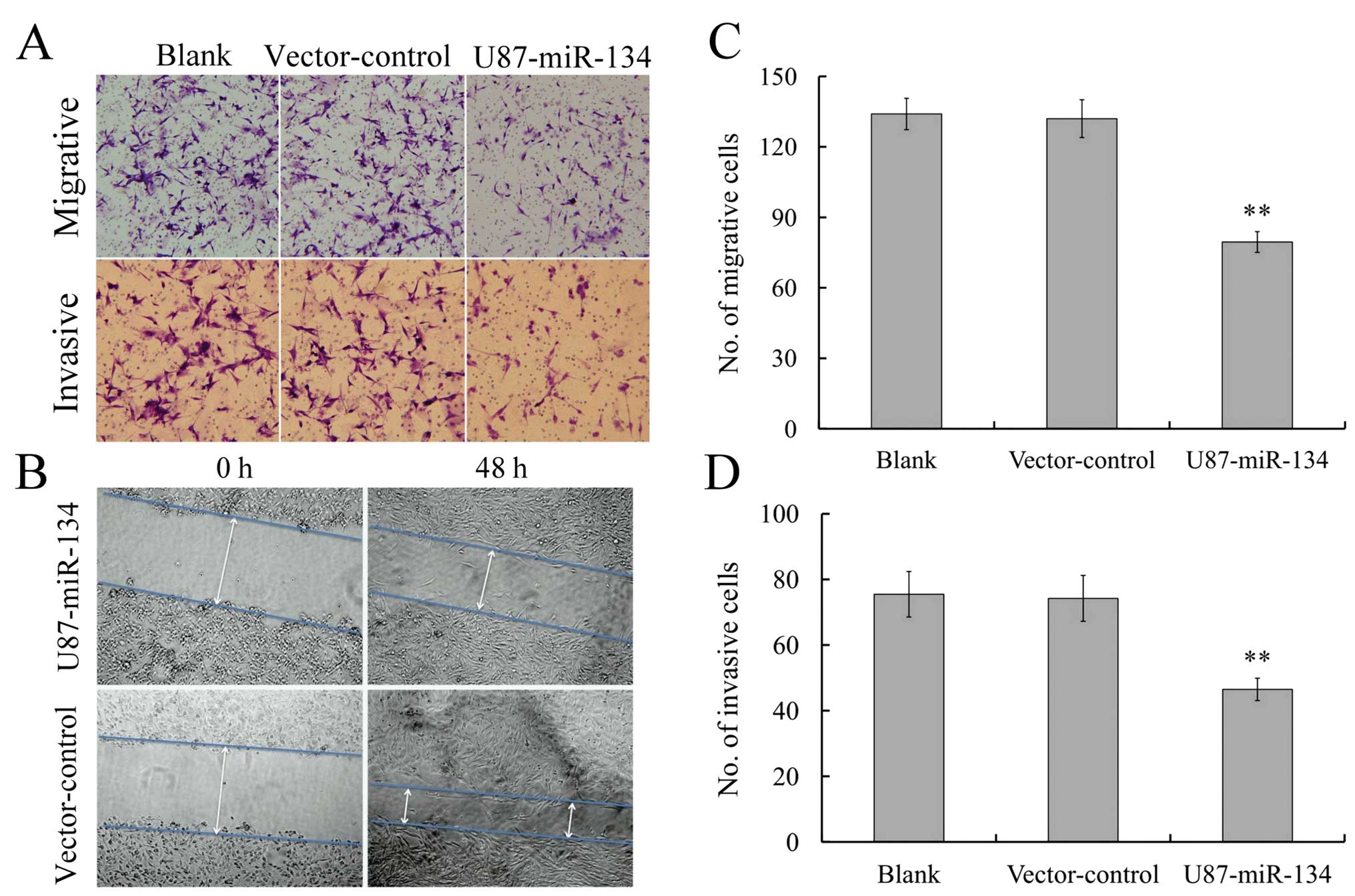
MiR-134 affects the invasiveness and
migration capability in U87 glioblastoma cells
Since invasiveness is one of the pathophysiological
features of glioblastoma, we asked whether miR-134 overexpression
was associated with the invasiveness of gliomas. Transwell assay
(without Matrigel) showed that the migratory speed of U87
glioblastoma cells highly expressing miR-134 was markedly slower
than that of control cells (Fig. 3A
and C). Furthermore, Transwell matrix penetration (coated with
Matrigel) assay showed that the upregulation of miR-134
dramatically reduced the invasiveness of U87 glioblastoma cells
(Fig. 3A and D). Wound healing
assay showed that miR-134 expression inhibited wound closure speed
of U87 cells (Fig. 3B). These
findings suggested that miR-134 could substantially inhibit
migration and invasion of U87 glioblastoma cells in
vitro.
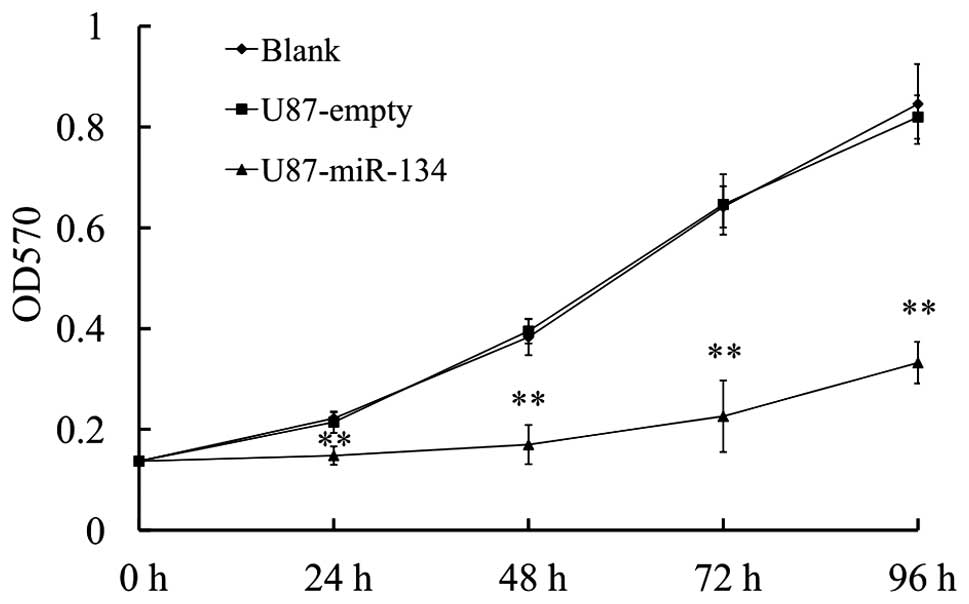
MiR-134 affects the proliferation of U87
glioblastoma cells in vitro and vivo
We further detected the effect of miR-134
overexpression on the proliferative ability of glioblastoma cells
by MTT assay. The result revealed that U87-miR-134 cells showed a
significant reduction in cell viability compared to vector-control
or the blank group (P<0.01) (Fig.
4).
To confirm the effect of miR-134 in vivo,
tumor xenograft animal model was performed. Thirty days after
subcutaneous inoculation of U87-miR-134, vector-control or blank
group cells into nude mice, each cell inoculation developed into
solid tumor xenografts (Fig. 5).
The growth curve of tumor xenografts showed that high miR-134 level
significantly slowed down tumor growth in vivo, while there
was no statistical difference between vector-control and blank
groups (Fig. 5D). The experimental
mice were euthanized on day 36. The average tumor weight in the
vector-control group was significantly higher than in the
U87-miR-134 group (Fig. 5C). These
findings indicated that exogenous miR-134 was able to inhibit
glioma growth in vitro and in vivo.
MiR-134 promotes apoptosis in U87
glioblastoma cells
Nanog knockdown induced cell cycle arrest, and
apoptosis, whereas suppressed proliferation and telomerase activity
in mESCs (24). To confirm the
effect of miR-134 overexpression in U87 glioblastoma cell, flow
cytometry, DAPI staining and transmission electron microscopy
methods were used. Phosphatidylserine externalization was assayed
by flow cytometry using Annexin V-PE/7-amino-actinomycin D
double-stained U87 cells. As shown in Fig. 6A, the percentage of the apoptotic
cells were significantly higher in U87-miR-134 (17.75±3.71%)
compared with the vector-control (4.18±1.42%) and blank group
(1.37±0.35%) (P<0.01, ANOVA). Typical apoptotic morphological
changes were found in U87-miR-134 cells including shrinkage,
deformation and detachment from culture dishes. Nuclear
condensation and chromatin margination were observed using an
inverted fluorescence microscope (Fig.
6B) and chromatin margination, nuclear condensation were noted
under transmission electron microscopy (Fig. 6C). These statistical results and
morphological changes elucidated the apoptotic inducing ability of
miR-134 to repress tumor proliferation.
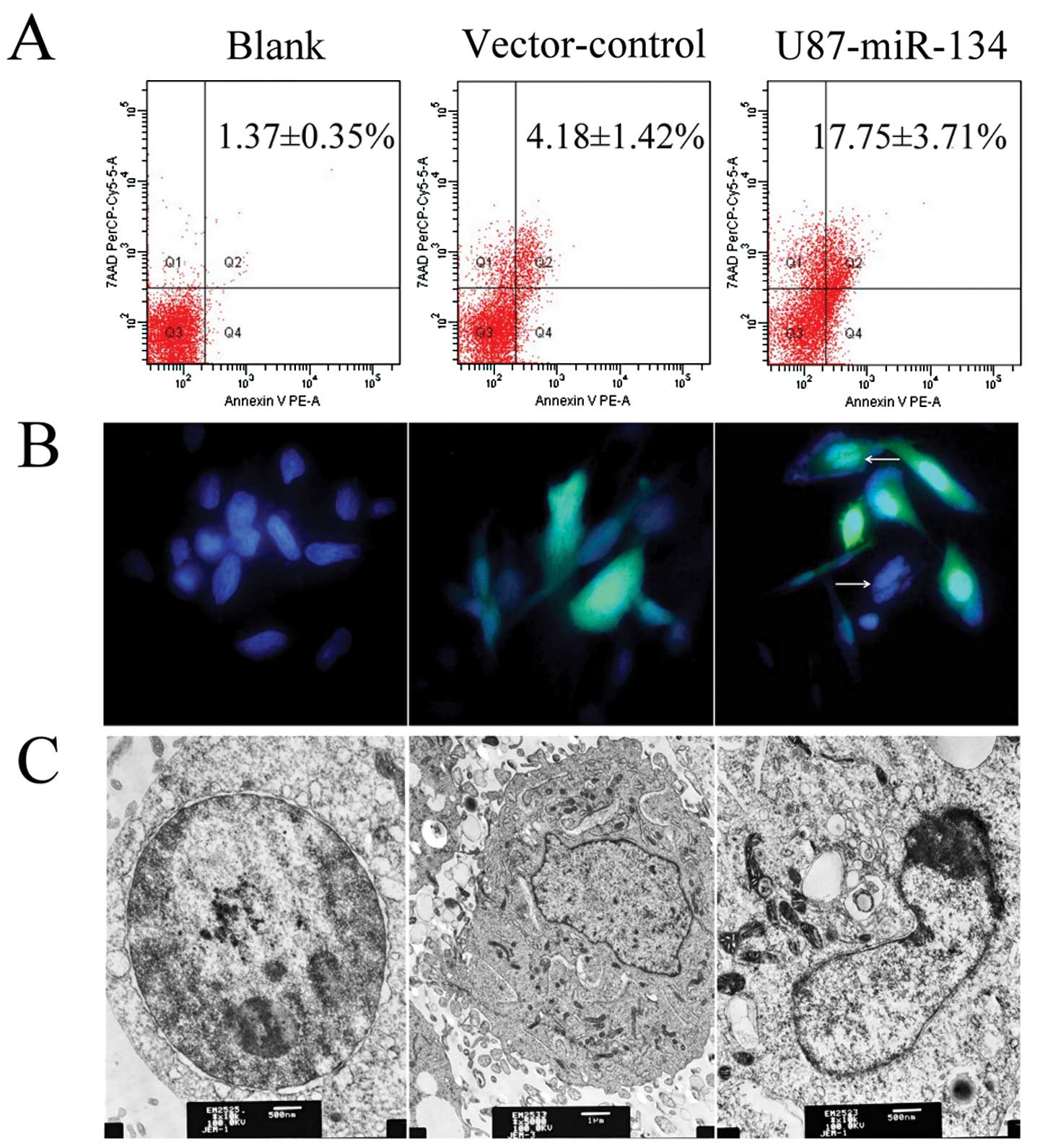 | Figure 6MiR-134 overexpression induced
apoptosis in U87 glioblastoma cells. (A) The percentage of
apoptotic cells were determined by flow cytometry. (B) DAPI
staining. White arrows indicate apoptotic cells in U87-miR-134
cells. Original magnification, ×400. (C) Transmission electron
microscopy. Left, blank cells; original magnification, ×10,000;
bar, 500 nm; middle, vector-control cells; original magnification,
×5,000; bar, 1 μm; right, U87-miR-134 cells; original
magnification, ×10,000; bar, 500 nm. |
Discussion
Accumulated evidence shows that miRNAs have an
important impact on tumor gene expression and it might play a key
role in tumorigenesis due to their widespread dysregulation.
MiR-134 is located in a very large cluster of brain-specific miRNAs
at chromosome 14q32.31 (25). It
is worth noting that miR-134 is detected in low expression in
oligodendrogliomas (ODG) and glioblastomas (GBM) (26). Whereas Nanog, which could be
post-transcriptionally downregulated by miR-134, is upregulated in
glioma tissues and BTSCs (14,15).
However, the association between pathological grade of glioma and
miR-134 expression, downregulation of Nanog expression by miR134 in
glioblastoma cells and knockdown of Nanog impacting the
proliferation and progression of glioblastoma cells are still
unclear.
In the present study, we investigated the expression
of miR-134 in glioma tissues and U87 glioblastoma cells and found
miR-134 was downregulated in glioma and U87 cell line compared to
normal brain tissues. It is noteworthy that miR-134 expression was
significantly lower in grade III and IV glioma compared to grade I
and II tumors. It was suggested that miR-134 might be a novel
specific biomarker for gliomas. In addition the loss of miR-134
could be involved in glioma development. The miR-134 ectopic
expression slowed down tumor cells growth in vitro and in
vivo, compared to vector-control and blank groups. Furthermore,
miR-134 overexpression in vitro inhibited migration and
invasion and significantly increased apoptosis in U87 glioblastoma
cells. We suggest that miR-134 may play a critical role in brain
tumorigenesis and progression.
Nanog overexpression has already been detected in a
number of human tumors, including glioma cells and it participated
in some oncogenic pathways (11–14).
Nanog, Stat-3 and miR-21 could form a functional signaling axis
that provides therapeutic targets to cause tumor cell apoptosis and
overcome cisplatin chemoresistance (12,27).
Moreover, Nanog and GLI1 are able to form a positive functional
loop modulated by p53; Nanog expression depends on endogenous
HH-GLI activity and its function is required for glioblastoma
growth in vivo (14). We
further confirmed that Nanog mRNA and protein expressions were
substantially downregulated by ectopic miR-134 in U87 glioblastoma
cells. Our tumor growth assays also indicated that Nanog was
associated with GBM tumorigenicity in vivo. However, whether
miR-134 can function independently of Nanog and if miR-134 has
other targets that affect glioma cell growth are still unknown.
Therefore, further research aimed at Nanog is needed for glioma
carcinogenesis.
In conclusion, comprehensive analysis indicated that
loss of miR-134 expression is a common event in glioma and miR-134
overexpression reduced the proliferation, invasiveness and
migration capabilities. MiR-134 overexpression also promoted
apoptosis in glioblastoma cells. Restoration of miR-134 expression
may represent a novel therapeutic approach in multi-modal therapy
for poorly differentiated glioblastoma. However, more
investigations are required to determine the possible promising
roles of miR-134 and Nanog in Nanog-Stat-3-miR-21 signaling axis
and functional Gli-Nanog-P53 network (14,28).
Further testing of miR-134 in preclinical models of glioblastoma in
conjunction with various delivery strategies will help define its
ultimate therapeutic potential for treatment of glioblastoma.
Acknowledgements
Grant support for this study was
provided by The National Natural Science Foundation of China (no.
81172407). We thank all surgeons at the Neurosurgery Department of
Anhui Provincial Hospital affiliated with Anhui Medical University
for helping us collect tumor samples.
References
|
1.
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Stefani G and Slack FJ: Small non-coding
RNAs in animal development. Nat Rev Mol Cell Biol. 9:219–230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ruvkun G: The perfect storm of tiny RNAs.
Nat Med. 14:1041–1045. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar
|
|
6.
|
Babashah S and Soleimani M: The oncogenic
and tumour suppressive roles of microRNAs in cancer and apoptosis.
Eur J Cancer. 47:1127–1137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Seca H, Almeida GM, Guimaraes JE, et al:
MiR signatures and the role of miRs in acute myeloid leukaemia. Eur
J Cancer. 46:1520–1527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Loh YH, Wu Q, Chew JL, et al: The Oct4 and
Nanog transcription network regulates pluripotency in mouse
embryonic stem cells. Nat Genet. 38:431–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yu J, Vodyanik MA, Smuga-Otto K, et al:
Induced pluripotent stem cell lines derived from human somatic
cells. Science. 318:1917–1920. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ezeh UI, Turek PJ, Reijo RA, et al: Human
embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are
expressed in both seminoma and breast carcinoma. Cancer.
104:2255–2265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Bourguignon LY, Peyrollier K, Xia W, et
al: Hyaluronan-CD44 interaction activates stem cell marker Nanog,
Stat-3-mediated MDR1 gene expression, and ankyrin-regulated
multidrug efflux in breast and ovarian tumor cells. J Biol Chem.
283:17635–17651. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
You JS, Kang JK, Seo DW, et al: Depletion
of embryonic stem cell signature by histone deacetylase inhibitor
in NCCIT cells: involvement of Nanog suppression. Cancer Res.
69:5716–5725. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zbinden M, Duquet A, Lorente-Trigos A, et
al: NANOG regulates glioma stem cells and is essential in vivo
acting in a cross-functional network with GLI1 and p53. EMBO J.
29:2659–2674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Niu CS, Li DX, Liu YH, et al: Expression
of NANOG in human gliomas and its relationship with
undifferentiated glioma cells. Oncol Rep. 26:593–601.
2011.PubMed/NCBI
|
|
16.
|
Tay YM, Tam WL, Ang YS, et al:
MicroRNA-134 modulates the differentiation of mouse embryonic stem
cells, where it causes post-transcriptional attenuation of Nanog
and LRH1. Stem Cells. 26:17–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Gaughwin P, Ciesla M, Yang H, et al:
Stage-specific modulation of cortical neuronal development by
Mmu-miR-134. Cereb Cortex. 21:1857–1869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Schratt GM, Tuebing F, Nigh EA, et al: A
brain-specific microRNA regulates dendritic spine development.
Nature. 439:283–289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wayman GA, Davare M, Ando H, et al: An
activity-regulated microRNA controls dendritic plasticity by
down-regulating p250GAP. Proc Natl Acad Sci USA. 105:9093–9098.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Hao J, Song X, Song B, et al: Effects of
lentivirus-mediated HIF-1alpha knockdown on hypoxia-related
cisplatin resistance and their dependence on p53 status in
fibrosarcoma cells. Cancer Gene Ther. 15:449–455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Zhou X, Ren Y, Moore L, et al:
Downregulation of miR-21 inhibits EGFR pathway and suppresses the
growth of human glioblastoma cells independent of PTEN status. Lab
Invest. 90:144–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Wang P, Zhen X, Jiang X, et al: Boron
neutron capture therapy induces apoptosis of glioma cells through
Bcl-2/Bax. BMC Cancer. 10:6612010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Chen T, Du J and Lu G: Cell growth arrest
and apoptosis induced by Oct4 or Nanog knockdown in mouse embryonic
stem cells: a possible role of Trp53. Mol Biol Rep. 39:1855–1861.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Fiore R, Khudayberdiev S, Christensen M,
et al: Mef2-mediated transcription of the miR379-410 cluster
regulates activity-dependent dendritogenesis by fine-tuning
Pumilio2 protein levels. EMBO J. 28:697–710. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lages E, Guttin A, El Atifi M, et al:
MicroRNA and target protein patterns reveal physiopathological
features of glioma subtypes. PLoS One. 6:e206002011. View Article : Google Scholar
|
|
27.
|
Bourguignon LY, Earle C, Wong G, et al:
Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21
expression and chemoresistance in hyaluronan/CD44-activated head
and neck squamous cell carcinoma cells. Oncogene. 31:149–160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Po A, Ferretti E, Miele E, et al: Hedgehog
controls neural stem cells through p53-independent regulation of
Nanog. EMBO J. 29:2646–2658. 2010. View Article : Google Scholar : PubMed/NCBI
|