Introduction
p53 is the most frequently mutated tumour suppressor
gene in cancers. About 50% of tumours harbour mutations in the p53
gene, and the remainder have disruptions in the p53 pathway
(1). p53 is normally a short-lived
protein that is maintained at low levels in resting cells by its
negative regulator HDM2. Upon DNA damage induced, for instance, by
ionizing radiation (IR), p53 transiently stabilizes and accumulates
in the nucleus where it acts as a transcription factor regulating
multiple genes important for the regulation of cell cycle arrest,
senescence and apoptosis. The precise mechanisms of p53 activation
are not fully understood, but the activation is clearly dependent
on post-translational modifications like ubiquitination,
phosphorylation and acetylation (2–4). p53
was the first non-histone protein described to be acetylated
(5). Upon various types of stress,
acetylation of p53 is dramatically induced, indicating the
importance of this specific post-translational modification. There
is also a direct competition between acetylation and ubiquitination
on specific lysine residues in the C-terminal domain of p53.
Acetylation of p53 physically blocks the ubiquitin sites preventing
ubiquitination by HDM2 and subsequent degradation (6). Acetylation is also important for
recruiting CBP/p300 and PCAF to promoter regions for activation of
p53-targeted genes such as p21, HDM2 and PUMA. Histone acetyl
transferases (HATs) CBP/p300, PCAF and TIP60 can all acetylate p53
(5,7,8),
while histone deacetylases (HDACs) can remove acetyl moieties from
ε-N-acetylated lysine residues of histones and non-histone proteins
such as p53. Six C-terminal residues (K305, K372, K373, K381, K382
and K386) and one DNA binding domain (DBD) residue (K164) are
acetylated by CBP/p300 (5,7–10),
whereas K320 is acetylated by PCAF (7,8).
HDAC inhibitors are also reported to induce acetylation in
non-histone proteins (5). The
HDAC1 inhibitor TSA leads to acetylation of p53 on K373 primarily
under conditions in which cells are subjected to IR, whereas the
SIRT1 deacetylase is known to induce acetylation of p53 on K382
(8).
cAMP is a second messenger important in multiple
physiological and pathological settings (11). This signal transducer is generated
by adenylyl cyclases subsequent to stimulation of certain G
protein-coupled receptors (GPCRs). Our lab has previously reported
that elevation of cAMP in lymphoid cells leads to arrest in the G1
phase of the cell cycle (12–14),
arrest in the S phase and inhibition of apoptosis by anticancer
agents (15). Using B cell
precursor acute lymphoblastic leukaemia (BCP-ALL) cells as a model
system, we also reported that the inhibitory effect of cAMP on
apoptosis is p53-dependent (16),
and that cAMP antagonizes the disruption of p53-HDM2 interaction by
DNA damage (17). It was therefore
of interest to further investigate the mechanism by which cAMP
abolishes IR-induced p53 stability and apoptosis in these cells,
and in particular, reveal how cAMP signaling promotes the
interaction between p53 and HDM2. Because cAMP exerted only a
slight inhibitory effect on IR-induced phosphorylation of S15, T18
and S20 on p53 (17), we directed
our attention towards the effect of cAMP on p53 acetylation. Our
results indicate that cAMP, through inhibition of p53 acetylation,
attenuates the IR-induced dissociation of p53 from HDM2 and,
thereby, prevents stabilization of p53 and apoptosis.
Materials and methods
Reagents and antibodies
Forskolin (F), PGE2, propidium iodide (PI),
cycloheximide (CHX) and nicotinamide (NIA) were obtained from
Sigma-Aldrich, 8-CPT-cAMP was purchased from Biolog and
Trichostatin A (TSA) was obtained from Calbiochem. Antibodies were:
total p53 (DO-1, FL-393, Bp53-12), HDM2 (SMP14) and actin (C2) from
Santa Cruz Biotechnology; HDM2 (IF2) from Calbiochem; HDM2 (4B2)
was a kind gift from Dr A. Levine (Princeton University, NJ);
acetyl-p53 (K373) and acetyl-p53 (K382) were from Epitomics and
Cell Signaling, respectively.
Cell cultures, radiation treatment and
cell death analysis
The BCP-ALL cell line Reh (18) was cultured as previously described
(15). For treatment of cells with
γ-radiation, cells were exposed to a 137 Cs source at a dose rate
of 4.3 Gy/min using a Gammacell irradiator from MSD Nordion. To
analyze cell death, cells were incubated with PI (20 μg/ml)
at room temperature for 10 min before examination for PI uptake by
flow cytometry.
Two-dimensional SDS-PAGE
For two-dimensional (2D) gel electrophoresis, cells
were washed twice in saline and lysed in 7% trichloro-acetic acid
(TCA) for 30 min on ice. After homogenization and centrifugation,
the precipitated proteins were washed once in 5% TCA and three
times in water-saturated ether to remove salts, each time followed
by centrifugation at 13,000 rpm for 20 min at 4°C. The protein
pellet was resuspended in sample buffer for 2D gel electrophoresis
(7 M urea, 2 M thiourea, 100 mM dithiotreitol, 1.5% ampholyte 3–10,
0.5% ampholyte 5–6, 4% CHAPS). The protein concentration was
measured by use of the Bradford method (19). Protein sample (100 μg) was
diluted in rehydration buffer (7 M urea, 2 M thiourea, 100 mM
dithiotreitol, 1.5% ampholyte 3–10, 0.5% ampholyte 5–6, 4% CHAPS,
Bromophenol blue) to a final volume of 170 μl. Isoelectric
focusing was performed using 7 cm pH 3.0–10.0 (Zoom Strip,
Invitrogen Corp., Carlsbad, CA, USA) isoelectric focusing gel
strips. Following rehydration of the strips in rehydration buffer,
the strips were subjected to the electrophoresis program: 200 V for
40 min, 450 V for 30 min, 750 V for 30 min and 1,000 V for 1 h.
Reduction and acetylation of sulfide bindings were performed
according to the manufacturer’s procedure. Second dimension gel
electrophoresis was run for 1 h at 200 V. Following
electrophoresis, proteins were transferred to polyvinylidene
fluoride membrane (Amersham Biosciences AB, Uppsala, Sweden) by
standard electroblotting. p53 protein was detected using primary
Bp53-12 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
and secondary HRP-conjugated mouse antibody (Jackson
ImmunoResearch, West Grove, PA, USA). Visualization of the proteins
was performed using the Supersignal West Pico or Femto
Chemiluminescent Substrate system (Pierce Biotechnology Inc.,
Rockford, IL, USA) and Kodak Image Station 2000R (Eastman Kodak
Company, Rochester, NY, USA).
Immunoblot analysis and
immunoprecipitation
For immunoblot analysis, cells were lysed in
radioimmunoprecipitation buffer [RIPA; 50 mM Tris-HCl (pH 7.4), 150
mM NaCl, 1% NP-40, 50 mM NaF, 10 mM β-glycerophosphate, 0.1% SDS,
0.5% EDTA, 1 mM Na3VO4, 0.2 mM PMSF, 10
μg/ml leupeptin, 0.5% aprotinin). Equal amounts of protein
were separated on a 10% SDS-PAGE. After transfer to a
nitrocellulose membrane (GE Healthcare, Amersham, UK), proteins
were detected by use of standard immunoblotting procedures.
For immunoprecipitation of HDM2 in complex with p53,
cells were lysed in NP-40 lysis buffer [50 mM Tris (pH 7.5), 150 mM
NaCl, 0.5% NP-40, 10 mM NaF, 1 mM Na3VO4, 1
mM phenylmethanesulfonyl fluoride, 10 mg/ml leupeptin and 0.5%
aprotinin]. Lysates containing 600 μg of protein were
immunoprecipitated with the p53 antibody FL-393 followed by 50
μl of a 1:1 slurry of protein G-agarose (Upstate, Temecula,
CA, USA). Beads were washed four times in lysis buffer, eluted in
boiling 1X SDS buffer, and subjected to immunoblot analysis. For
densitometric analysis, blots were analyzed using Genetools
analysis (SynGene).
Statistical analysis
In all figures, the histograms represent the mean
values, with error bars corresponding to SEM values.
Results
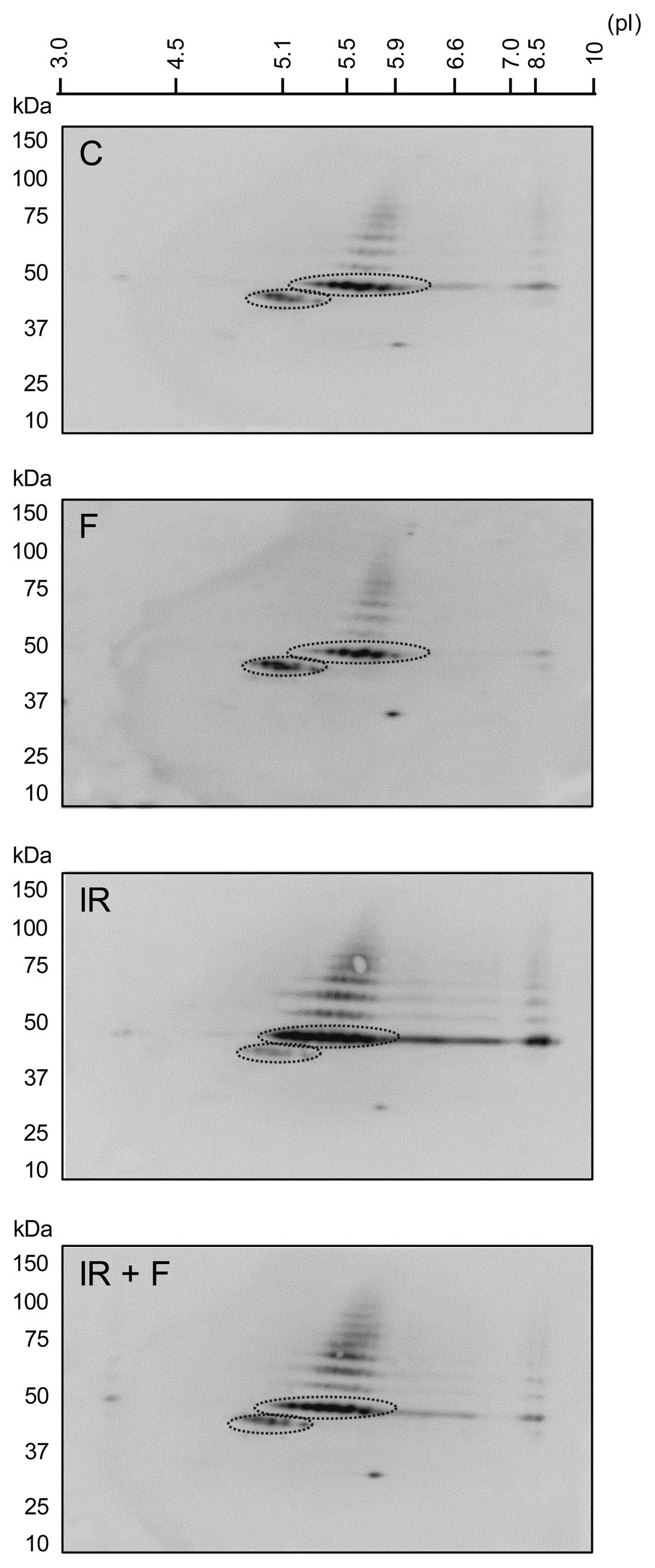
cAMP inhibits p53 accumulation and
isoelectric point shift in IR-treated BCP-ALL cells on 2D-PAGE
We have previously shown that stimulation of cAMP
signalling inhibits DNA damage-induced accumulation of p53 by
facilitating its interaction with HDM2 (17). Because the interaction between p53
and HDM2 is known to be regulated by post-translational
modifications of p53, we decided to examine whether cAMP affected
the post-translational modifications of p53 in IR-treated cells. To
this end, we performed 2D-immunoblotting with anti-p53 antibodies
on lysates of Reh cells that were treated with IR in the absence or
presence of forskolin. Reh is a BCP-ALL cell line that expresses wt
p53 and forskolin is a plant-derived diterpene known to induce
intracellular cAMP levels by activating adenylyl cyclase. As shown
in Fig. 1, IR not only led to an
increase in the abundance of p53, but it also led to accumulation
of more acidic forms of p53, indicating that IR leads to
modification of p53 by phosphorylation or acetylation.
Interestingly, forskolin was found to inhibit the IR-induced
accumulation and acidification of p53, suggesting that cAMP
protects p53 from IR-induced post-translational modifications.
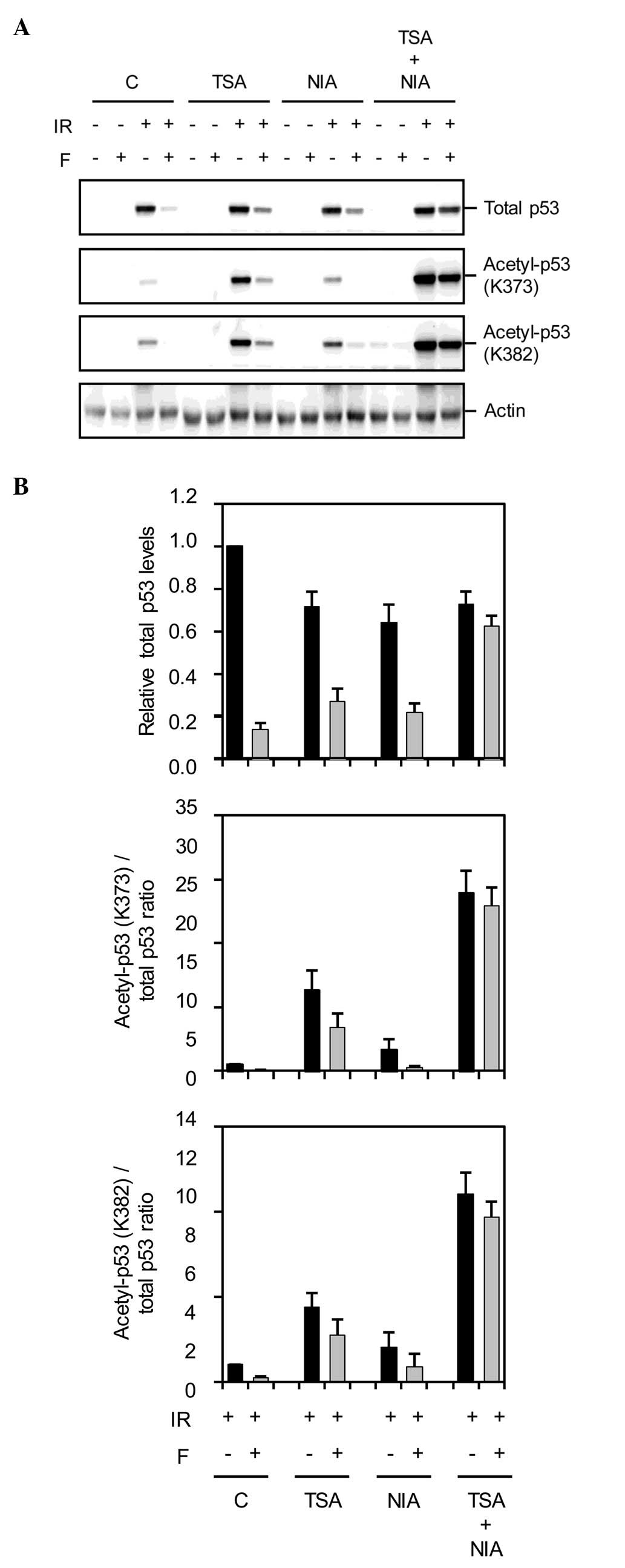
cAMP affects the DNA damage-induced
acetylation of K373 and K382 on p53
Our recent result showing that cAMP has only a
slight inhibitory effect on the IR-mediated phosphorylation of p53
(17), suggested that inhibition
of acetylation might account for the ability of cAMP to reduce the
IR-induced acidification of p53. To assess this assumption, Reh
cells that were exposed to IR in the absence or presence of
forskolin were harvested at 4 h post-IR and subjected to immunoblot
analysis with antibodies specific for p53 acetylated at K373 and
K382. Acetylation of p53 at these two residues is
characteristically induced by IR (4). As shown in Fig. 2, IR led to an increase in
acetylation of p53 at K373 and K382, and importantly, activation of
the cAMP signalling pathway by forskolin inhibited this
acetylation.
Following DNA damage, p53 is acetylated through the
activity of HATs such as p300/CBP and pCAF, an event that is
tightly regulated by the deacetylase activity of enzymes such as
HDACs and SIRT1 (4). The cAMP
signalling pathway has been linked to both SIRT activation and
subcellular localization of HDACs (20–23),
and we therefore hypothesized that cAMP antagonizes the IR-mediated
acetylation of p53 through HDACs and/or SIRT1. To test this
possibility, we examined the effects of the deacetylase inhibitors
trichostatin A (TSA) and nicotineamide (NIA) on the ability of cAMP
to abrogate the IR-induced acetylation of p53. TSA is an inhibitor
of HDACs whereas NIA inhibits the NAD+-dependent
deacetylases such as SIRT1. To this end, Reh cells were pretreated
with TSA and/or NIA before exposure to IR in the absence or
presence of forskolin. Four hours post-IR, cells were harvested and
subjected to immunoblot analysis sequentially with antibodies
directed against p53 acetylated at K373, K382 as well as total p53.
Treatment of cells with TSA or NIA alone or the combination of the
two had minor effects on p53 acetylation under unstressed
conditions (Fig. 2). In accordance
with previous findings (16,24),
exposure of cells to IR in the presence of TSA or NIA increased the
IR-mediated acetylation of p53. However, pretreatment of cells with
both TSA and NIA substantially further enhanced the p53 acetylation
induced by IR. Interestingly, Fig. 2A
and B also show that pretreatment of IR-exposed cells with both
TSA and NIA almost completely abrogated the effect of forskolin on
acetylated p53 levels, suggesting that the inhibitory effect of
cAMP on IR-induced acetylation of p53 depends on HDACs and SIRT1
activities.
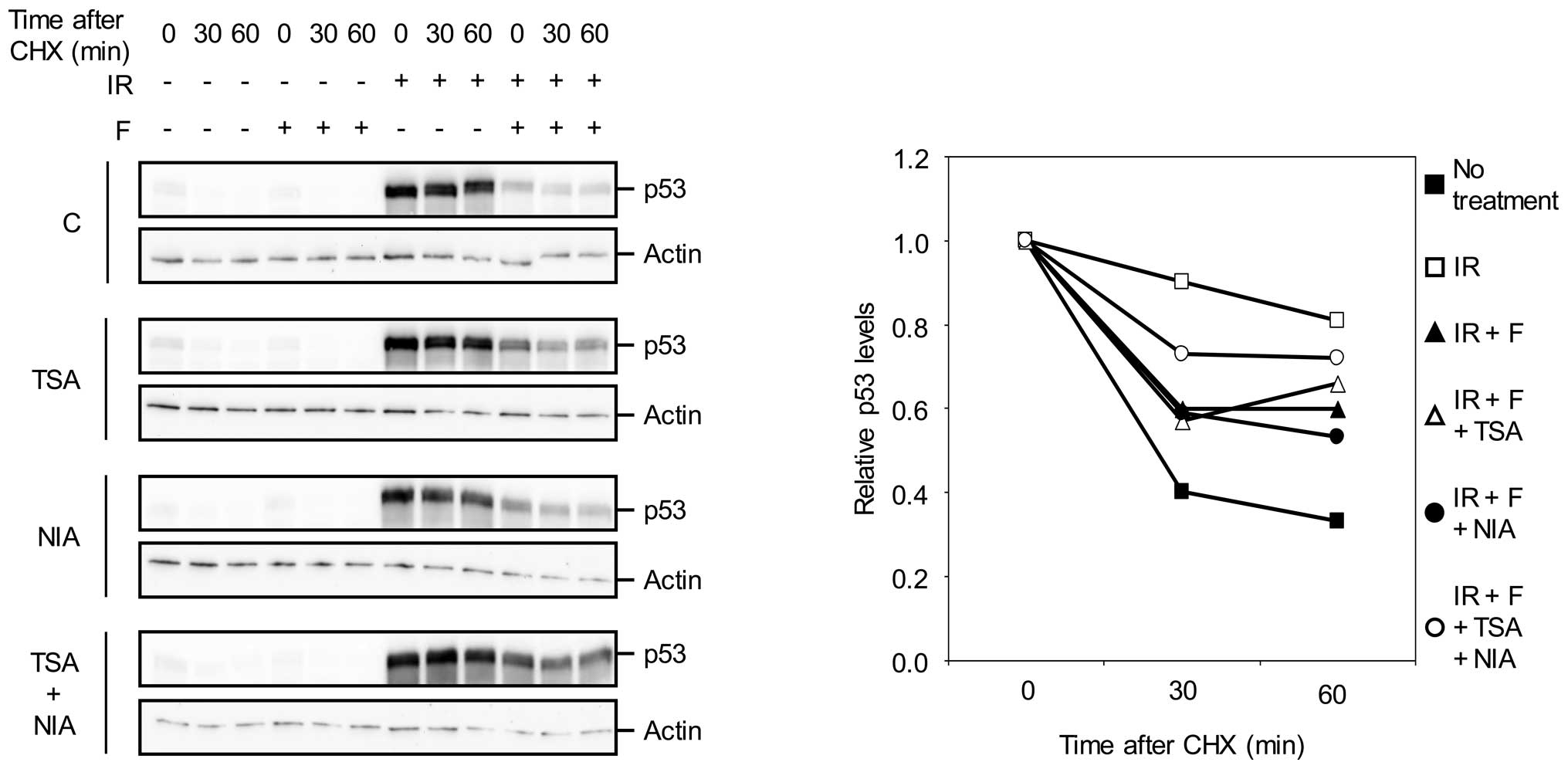
cAMP destabilizes IR-induced p53 through
acetylation inhibition
The immunoblot shown in Fig. 2A revealed that in addition to
reversal of forskolin-induced deacetylation of p53, the combination
of TSA and NIA also abrogated the inhibitory effect of forskolin on
IR-induced accumulation of p53. Given our previous finding that
cAMP facilitated the degradation of p53, we wished to examine
whether TSA and NIA abolished the inhibitory effect of cAMP on
IR-mediated accumulation of p53 by increasing the stability of p53.
To this end, Reh cells treated with IR in the absence or presence
of forskolin were exposed to the protein synthesis inhibitor
cycloheximide (CHX) and then analyzed for the level of p53 by
western blot analysis.
In agreement with our previous finding, forskolin
substantially decreased the half-life of p53 in IR-treated cells
(Fig. 3). Interestingly,
combination of TSA and NIA alleviated the inhibitory effect of
forskolin on p53 stability in IR-treated cells. This result
indicates that simultaneous inhibition of HDACs and SIRT1
antagonizes the destabilizing effect of cAMP on p53, and implicates
the modulation of p53 acetylation as the means by which cAMP
regulates the stability of p53.
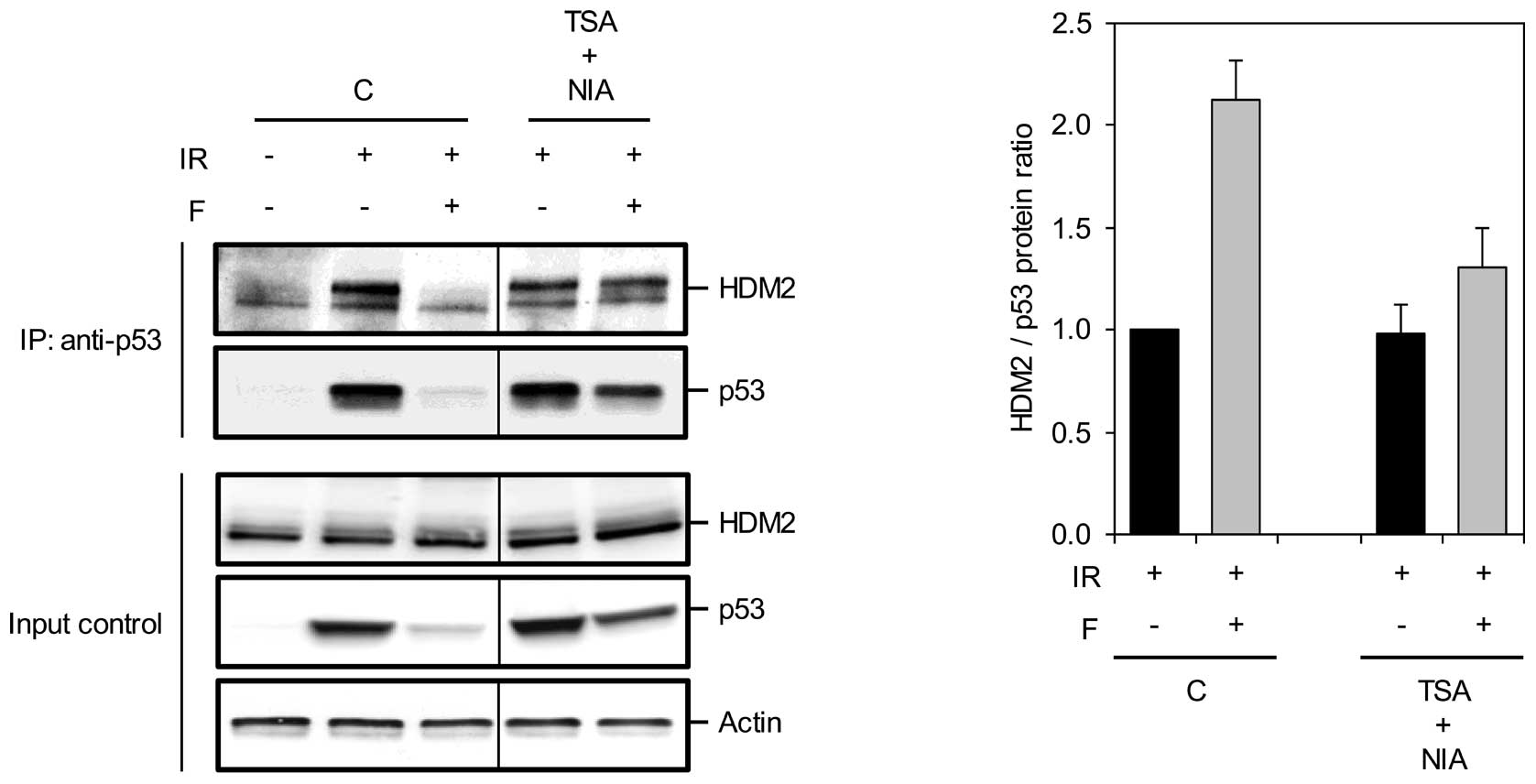
cAMP facilitates p53-HDM2 binding through
p53 acetylation
We have shown that cAMP abrogates the DNA
damage-induced stabilization of p53 by promoting its interaction
with HDM2 (17). This, together
with the finding that p53 acetylation has an inhibitory effect on
its association with HDM2 (6,25)
suggested deacetylation of p53 as the mechanism by which cAMP
enhances the p53-HDM2 interaction in IR-treated cells. To assess
this hypothesis, we examined the formation of p53-HDM2 complexes
under conditions in which the inhibitory effect of cAMP on
IR-induced acetylation of p53 is blocked. For this purpose, Reh
cells were pretreated with TSA or NIA alone or in combination
before exposure to IR in the absence or presence of forskolin.
Cells were harvested at 4 h after IR, and the lysates were
subjected to immunoprecipitation with anti-p53 antibodies followed
by immunoblot analysis with antibodies against p53 and HDM2. In
accordance with our previous results, pretreatment of cells with
forskolin prevented the IR-induced dissociation of p53 from HDM2
(Fig. 4). Importantly, TSA and NIA
together alleviated the facilitating effect of forskolin on the
p53-HDM2 interaction and reduced the level of HDM2 in complex with
p53 to a level comparable to that found in IR-only treated cells.
This result suggests that cAMP counteracts the IR-induced p53-HDM2
dissociation through inhibition of p53 acetylation.
cAMP attenuates the IR-induced apoptosis
by inhibiting p53 acetylation
Initiation of a p53-dependent apoptotic program is
dependent on acetylation of p53 (10). Given that inhibition of HDACs and
SIRT1 abrogates the ability of cAMP to suppress the IR-induced
acetylation of p53, we wished to examine whether blocking the HDACs
and SIRT1 activities could alleviate the inhibitory effect of cAMP
on IR-mediated apoptosis. To this end, Reh cells were pretreated
with TSA and NIA before exposure to IR in the absence or presence
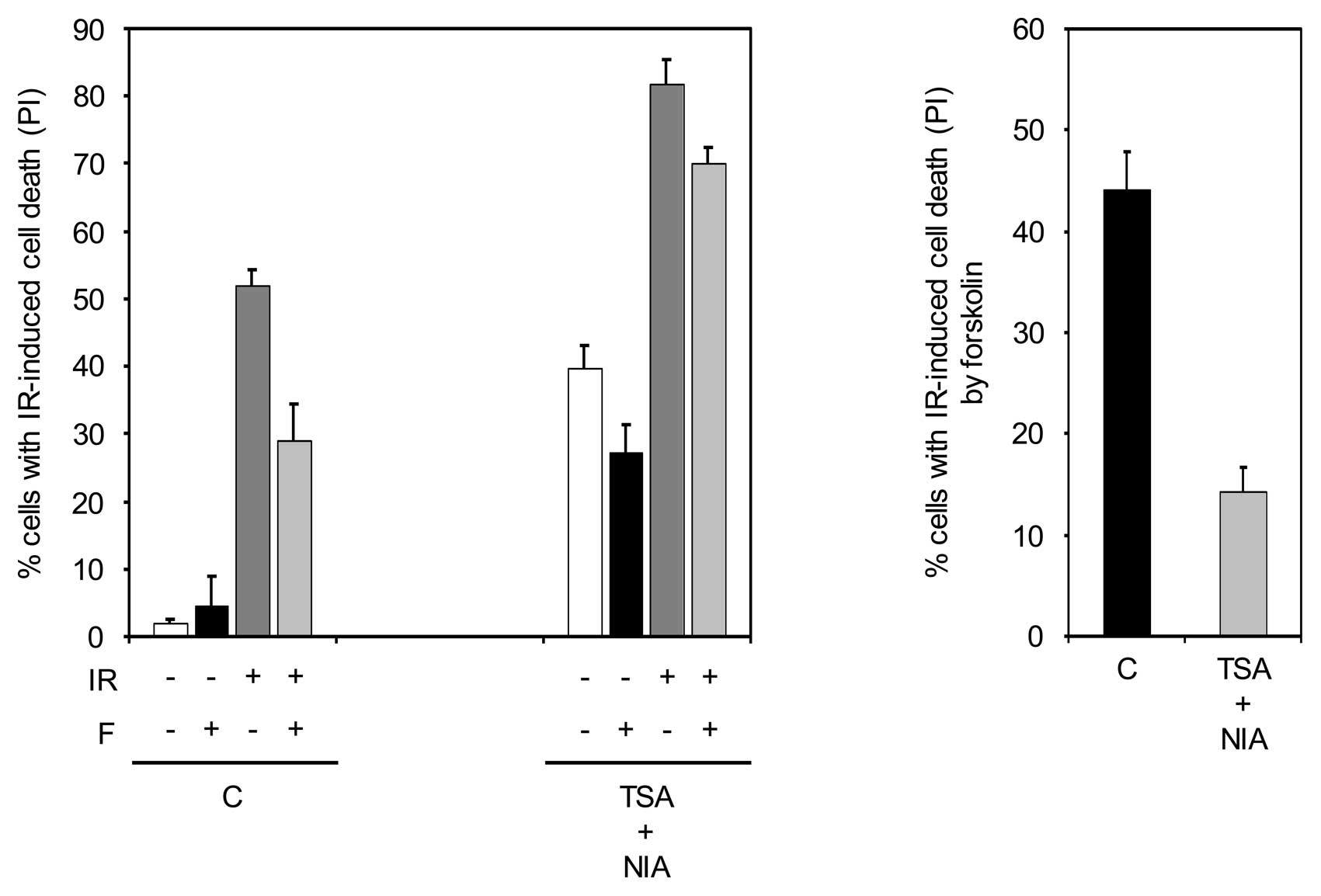
of forskolin and then examined for cell death. As shown in Fig. 5, the combined inhibition of HDACs
and SIRT1 opposed the inhibiting effect of forskolin on IR-induced
cell death, suggesting that inhibition of p53 acetylation plays an
important role in the ability of cAMP to inhibit IR-mediated cell
death.
Discussion
Elevation of intracellular cAMP levels has an
inhibitory effect on DNA damage-induced apoptosis in normal
lymphoid cells as well as BCP-ALL cells (16). We have previously shown that this
inhibitory effect of cAMP is mediated through its ability to
abrogate the DNA damage-induced p53-HDM2 dissociation, leading to
restoration of p53 degradation in DNA-damaged cells (17). Here, we provide evidence for the
mechanism by which cAMP promotes the interaction of p53 with HDM2
and thereby prevents DNA damage-induced cell death. We show that
activation of cAMP signalling attenuates the acetylation of p53
after DNA damage, thus facilitating the p53-HDM2 association.
The p53 levels are tightly regulated by its
HDM2-mediated ubiquitination and the ensuing proteasomal
degradation. Because ubiquitnation and acetylation of p53 occur at
the same lysine residues, the acetylation of p53 at lysine residues
in its C-terminus inhibits the HDM2-mediated ubiquitination and
degradation of p53 (6,26). Furthermore, p53 acetylation has
also been shown to block its interaction with HDM2 (10). Thus, acetylation of p53 plays a
central role in regulation of p53 stability and activity following
DNA damage by modulating its interaction with HDM2. Acetylation of
p53 is carried out by a number of acetyl transferases, among which
p300/CBP is responsible for acetylating the C-terminal lysine
residues of p53 (5,7–9). The
steady-state level of acetylated p53 is achieved by HDAC1 and SIRT1
deacetylases. Indeed, deacetylation of p53 by these two enzymes is
required for restraining hyper-activation of p53 after DNA damage
(4). Our finding that cAMP
inhibits DNA damage-induced acetylation of p53 provides a
mechanistic explanation for the ability of cAMP to abrogate a
p53-dependent response following DNA damage. By inducing the
deacetylation of p53, cAMP favours p53-HDM2 interaction and thus
abrogates the DNA damage-mediated stabilization of p53. This
ability of cAMP requires deacetylation of p53 at both HDAC- and
SIRT1-targeted sites, because only simultaneous inhibition of both
HDACs and SIRT1 reverses the destabilizing effect of cAMP on p53.
In addition, through induction of p53 deacetylation, cAMP
attenuates DNA damage-induced events downstream of p53. This
conclusion is supported by the observation that inhibition of HDACs
and SIRT1 reverses the inhibitory effect of cAMP on the DNA
damage-mediated apoptosis. Thus, cAMP, through modulation of p53
acetylation, inhibits stabilization of p53 and prevents subsequent
cell death induced by DNA damage.
In theory, cAMP could inhibit DNA damage-induced
acetylation of p53 by two distinct mechanisms: (i) by inhibiting
the enzymes that acetylate p53 or (ii) by stimulating the
deacetylases that target p53. Supported by studies showing that
cAMP signalling stimulates the deacetylase activity of SIRT1 and
promotes the nuclear retention of HDACs (20–23),
we favour the second possibility and suggest that cAMP, by
utilizing HDACs and SIRT1, maintains p53 in a hypoacetylated state,
thus leading to its HDM2-dependent degradation even in the face of
DNA damage. Given the cytotoxic effect of histone deacetylase
inhibition (27,28), we are tempted to suggest that
combination of inhibitors of cAMP signalling with histone
deacetylase inhibitors might prove beneficial for increasing the
antitumour activity of histone deacetylase inhibitors.
Acknowledgements
We thank the University of Oslo, the
Norwegian Cancer Society, Anders Jahre’s Research Foundation and
the Blix Family Foundation for generously supporting this
study.
References
|
1.
|
Dai C and Gu W: p53 post-translational
modification: deregulated in tumorigenesis. Trends Mol Med.
16:528–536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Appella E and Anderson CW:
Post-translational modifications and activation of p53 by genotoxic
stresses. Eur J Biochem. 268:2764–2772. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Brooks CL and Gu W: Ubiquitination,
phosphorylation and acetylation: the molecular basis for p53
regulation. Curr Opin Cell Biol. 15:164–171. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Brooks CL and Gu W: The impact of
acetylation and deacetylation on the p53 pathway. Protein Cell.
2:456–462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gu W and Roeder RG: Activation of p53
sequence-specific DNA binding by acetylation of the p53 C-terminal
domain. Cell. 90:595–606. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Li M, Luo J, Brooks CL and Gu W:
Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol
Chem. 277:50607–50611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Liu L, Scolnick DM, Trievel RC, Zhang HB,
Marmorstein R, Halazonetis TD and Berger SL: p53 sites acetylated
in vitro by PCAF and p300 are acetylated in vivo in response to DNA
damage. Mol Cell Biol. 19:1202–1209. 1999.PubMed/NCBI
|
|
8.
|
Sakaguchi K, Herrera JE, Saito S, Miki T,
Bustin M, Vassilev A, Anderson CW and Appella E: DNA damage
activates p53 through a phosphorylation-acetylation cascade. Genes
Dev. 12:2831–2841. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wang YH, Tsay YG, Tan BC, Lo WY and Lee
SC: Identification and characterization of a novel p300-mediated
p53 acetylation site, lysine 305. J Biol Chem. 278:25568–25576.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Tang Y, Zhao W, Chen Y, Zhao Y and Gu W:
Acetylation is indispensable for p53 activation. Cell. 133:612–626.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Torgersen KM, Vang T, Abrahamsen H, Yaqub
S and Tasken K: Molecular mechanisms for protein kinase A-mediated
modulation of immune function. Cell Signal. 14:1–9. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Blomhoff HK, Blomhoff R, Stokke T, deLange
DC, Brevik K, Smeland EB, Funderud S and Godal T: cAMP-mediated
growth inhibition of a B-lymphoid precursor cell line Reh is
associated with an early transient delay in G2/M, followed by an
accumulation of cells in G1. J Cell Physiol. 137:583–587. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Blomhoff HK, Smeland EB, Beiske K,
Blomhoff R, Ruud E, Bjoro T, Pfeifer-Ohlsson S, Watt R, Funderud S
and Godal T: Cyclic AMP-mediated suppression of normal and
neoplastic B cell proliferation is associated with regulation of
myc and Ha-ras protooncogenes. J Cell Physiol. 131:426–433. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Naderi S, Gutzkow KB, Christoffersen J,
Smeland EB and Blomhoff HK: cAMP-mediated growth inhibition of
lymphoid cells in G1: rapid down-regulation of cyclin D3 at the
level of translation. Eur J Immunol. 30:1757–1768. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Naderi S, Wang JY, Chen TT, Gutzkow KB and
Blomhoff HK: cAMP-mediated inhibition of DNA replication and S
phase progression: involvement of Rb, p21Cip1, and PCNA. Mol Biol
Cell. 16:1527–1542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Naderi EH, Findley HW, Ruud E, Blomhoff HK
and Naderi S: Activation of cAMP signaling inhibits DNA
damage-induced apoptosis in BCP-ALL cells through abrogation of p53
accumulation. Blood. 114:608–618. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Naderi EH, Jochemsen AG, Blomhoff HK and
Naderi S: Activation of cAMP signaling interferes with
stress-induced p53 accumulation in ALL-derived cells by promoting
the interaction between p53 and HDM2. Neoplasia. 13:653–663.
2011.PubMed/NCBI
|
|
18.
|
Rosenfeld C, Goutner A, Choquet C, Venuat
AM, Kayibanda B, Pico JL and Greaves MF: Phenotypic
characterisation of a unique non-T, non-B acute lymphoblastic
leukaemia cell line. Nature. 267:841–843. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Cai R, Kwon P, Yan-Neale Y, Sambuccetti L,
Fischer D and Cohen D: Mammalian histone deacetylase 1 protein is
post-translationally modified by phosphorylation. Biochem Biophys
Res Commun. 283:445–453. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Du M, Perry RL, Nowacki NB, Gordon JW,
Salma J, Zhao J, Aziz A, Chan J, Siu KW and McDermott JC: Protein
kinase A represses skeletal myogenesis by targeting myocyte
enhancer factor 2D. Mol Cell Biol. 28:2952–2970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gerhart-Hines Z, Dominy JE Jr, Blattler
SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP and
Puigserver P: The cAMP/PKA pathway rapidly activates SIRT1 to
promote fatty acid oxidation independently of changes in NAD(+).
Mol Cell. 44:851–863. 2011.PubMed/NCBI
|
|
23.
|
Ha CH, Kim JY, Zhao J, Wang W, Jhun BS,
Wong C and Jin ZG: PKA phosphorylates histone deacetylase 5 and
prevents its nuclear export, leading to the inhibition of gene
transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci
USA. 107:15467–15472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Luo J, Nikolaev AY, Imai S, Chen D, Su F,
Shiloh A, Guarente L and Gu W: Negative control of p53 by Sir2alpha
promotes cell survival under stress. Cell. 107:137–148. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wang X, Taplick J, Geva N and Oren M:
Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett.
561:195–201. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Li M, Chen D, Shiloh A, Luo J, Nikolaev
AY, Qin J and Gu W: Deubiquitination of p53 by HAUSP is an
important pathway for p53 stabilization. Nature. 416:648–653. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Batty N, Malouf GG and Issa JP: Histone
deacetylase inhibitors as anti-neoplastic agents. Cancer Lett.
280:192–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|