Contents
Introduction
Hyperthermia
Cisplatin
Gemcitabine
Temozolomide
Halogenated pyrimidines
PARP1 inhibitors
Discussion
Conclusion
Introduction
The treatment of cancer by ionizing radiation is
frequently combined with chemotherapy as well as with other agents,
in an effort to increase effectiveness. The selection of these
combinations has been considerably based on experimental studies
with cells in culture and experimental tumours in animals. These
studies were designed to obtain insights into the mechanisms of
interaction and to derive quantitative information on potential
methods of enhancing effectiveness, either through a decrease in
cell survival in vitro or an increase in tumour response
in vivo.
The results of experiments on combined treatments of
cells or tumours are generally expressed in a single sensitisation
or enhancement factor (ER), as calculated from dose-effect
relationships for the endpoints assessed, following treatment with
or without the combined agents. However, such a single
sensitisation factor provides only part of the information that can
be derived from the complete experimentally assessed dose-effect
relationships. Different plating conditions were investigated.
Cells were plated prior to irradiation or combined treatment (ppi),
plated immediately after irradiation or combined treatment (ip) or
plated with a 24-h delay (dp) to establish potentially lethal
damage repair (PLDR).
Quantitative information derived from numerous
studies on cultured cells, tumours and normal tissues in animals,
can be conveniently analysed in terms of mathematical dose-effect
relationships based on the linear-quadratic (LQ) model of cell
reproductive death as a function of the radiation dose (1–4). The
LQ formula for cell reproductive death, the surviving fraction
(Sd) of cells exposed to radiation dose (d), compared to
the survival of unirradiated cells (S0) is described by
an inverse exponential approximation: Sd/S0 =
e−(αd+βd2) and contains two parameters, α
(Gy−1) and β (Gy−2). The initial slope of
cell survival curves and the effectiveness at low doses is
determined by α, while β represents the increasing contribution
from cumulative damage, presumably due to the interaction of two or
more lesions induced by separate ionizing radiations (1). DNA double-strand breaks (DSBs) are
generally assumed to be the most relevant lesions.
The α/β ratio represents the dose at which the two
terms contribute equally to the total effect. Data from numerous
studies on cells, tumours and normal tissues have demonstrated that
the values of α/β usually range between 3 and 10 Gy. Since the dose
fractions applied in cancer radiotherapy are mostly in the 1.5–2.5
Gy range, it seems clear that the clinical effect of radiotherapy
on tumours is largely determined by the linear parameter α.
However, in experimental studies on the enhancement
of radiation treatments by chemical or physical agents, the
enhancement ratios are typically calculated as the ratio of doses
required to obtain equal effects at dose levels for which the
effects can be most easily assessed experimentally, namely at doses
between 5 and 10 Gy. At these high-fraction doses, the
effectiveness is largely determined by the quadratic term
βd2 and not by the linear term αd. The common use of a
single ER is based on the implicit assumption that any
radiosensitiser changes both LQ parameters equally, although this
assumption may not hold for all radiosensitising agents. It is
therefore of interest to analyse enhancement factors for the linear
and quadratic parameters separately, to evaluate their impact at
the doses commonly applied in radiotherapy. The parameters α and β
were determined from survival curves using SPSS statistical
software performing a fit to the data according to the LQ formula
by multiple regression analysis.
In this review, radiosensitisation data from our own
laboratory are presented and subsequently discussed and compared to
the data from the literature. In a number of former studies
conducted by our laboratory, radiation sensitisation by a variety
of agents in different types of mammalian cells has been
investigated and cell survival curves have been analysed using the
LQ model (5–11). Radiation enhancement could thus be
assessed in separate α- and β-values and in the α/β ratio. The
results for various radiosensitising agents are presented in the
following sections.
Hyperthermia
Radiosensitisation by hyperthermia
Hyperthermia (HT) refers to heat treatment of cancer
cells or tumours by increasing the temperature to a level between
39 and 45°C. It is used in combination with chemo- and/or
radiotherapy and it is has been shown to enhance their anticancer
effects experimentally and clinically (12–16).
A number of in vitro studies on the combination of HT and
radiation have demonstrated a synergistic interaction between the
two modalities, particularly at higher temperatures (>42°C)
(17–19). This interaction possibly results
from the inhibition of the repair of radiation-induced DNA damage
by HT (20,21). The sequence of combined radiation
and HT treatment is important. Optimal sensitisation is achieved
when radiation and HT are applied simultaneously or within a short
time interval (22). Radiotherapy
with concomitant HT is not always feasible in clinical practice.
Therefore, in our experiments, HT was also applied sequentially,
immediately following radiation treatment.
Despite the clinical goal to realise cytotoxic
temperatures as high as 43°C, in practice, tumour temperature
distributions are heterogeneous. In large areas of the tumour,
temperatures are often <43°C. Nonetheless, satisfactory results
have been obtained in locally advanced cervical cancers treated
with radiotherapy plus mild HT <43°C (13). Mild temperatures may have more
subtle effects than high temperatures, such as tumour reoxygenation
(23–26). We recently discovered that mild HT
(42°C for 1 h) transiently breaks down the BRCA2 protein (27). In the following sections, the
effects of HT for 1 h at 41 or 43°C on the LQ parameters are
summarized. Several different cell types have been studied.
Effect of HT on radiosensitivity of SiHa
and RKO cells
The SiHa cell line is derived from a human cervical
carcinoma. The cells were plated prior to treatment. Mild HT alone
(41°C for 1 h) had almost no effect and resulted in a surviving
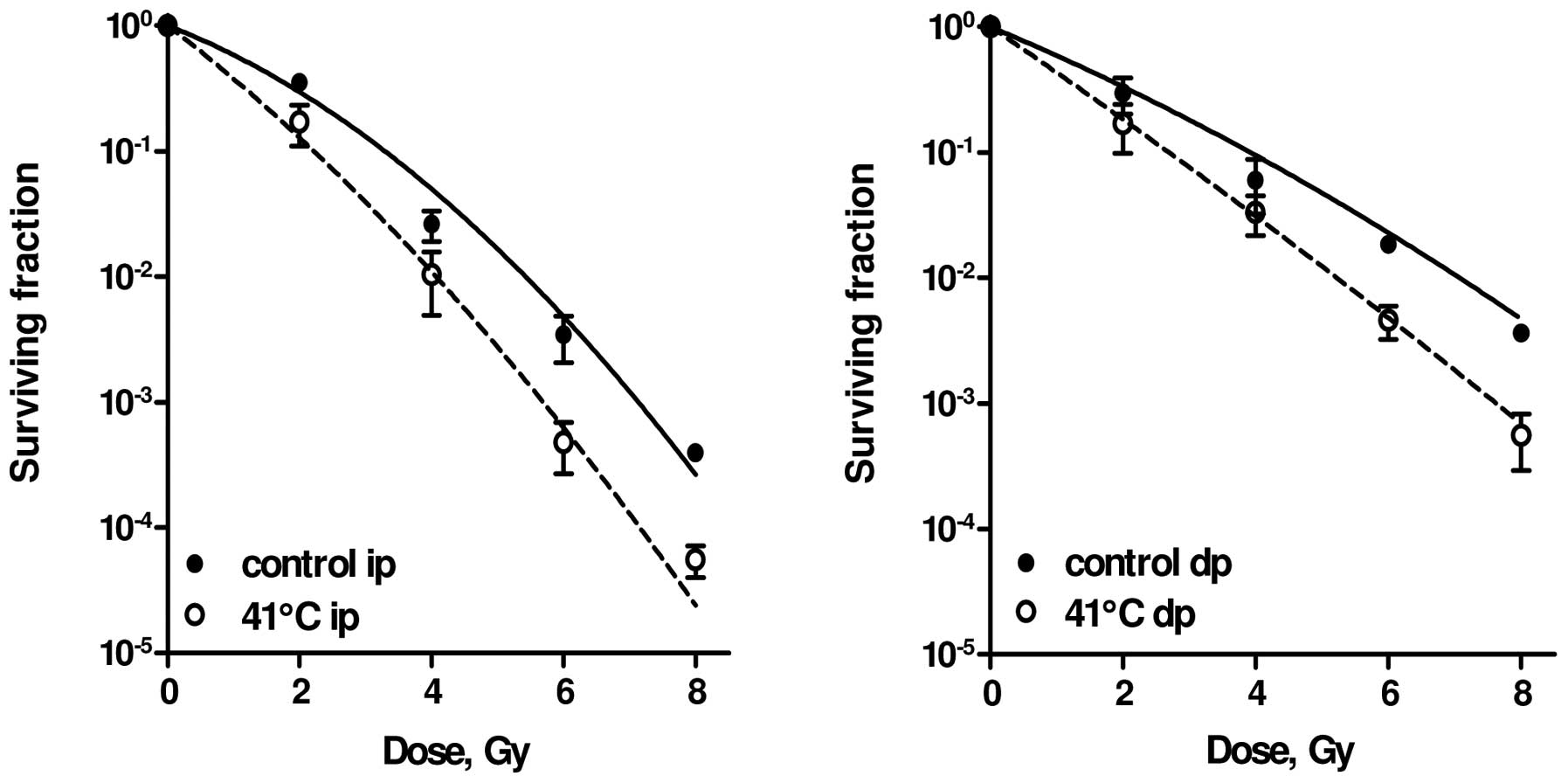
fraction of 0.95±0.2. As can be observed in Fig. 1 and Table I, 1 h at 41.0°C exclusively
enhanced the quadratic parameter, β, by a factor of 3.9. The value
of the linear parameter, α, was hardly influenced. HT treatment at
43°C for 1 h significantly increased the values of both
parameters.
 | Table IValues of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and following combined
radiation and hyperthermia (HT) treatment. |
Table I
Values of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and following combined
radiation and hyperthermia (HT) treatment.
| Cells | Treatment °C
(h) | α (Gy−1)
control | β (Gy−2)
control | α/β | α-EF | β-EF |
|---|
| SiHa ppi | Sham | 0.33±0.06 | 0.02±0.01 | 13.8±6.2 | | |
| HT 41 (1) | 0.31±0.05 | 0.09±0.02 | 3.3±0.7 | 0.9±0.2 | 3.9±1.6 |
| HT 43 (1) | 0.76±0.04 | 0.09±0.01 | 8.7±0.8 | 1.4±0.1 | 2.7±0.5 |
| RKO ip | Sham | 0.55±0.09 | 0.02±0.01 | 27.5±14.1 | | |
| HT 41 (1) | 0.93±0.09a | 0.05±0.02 | 18.6±7.7 | 1.7±0.3 | 2.5±1.6 |
| RKO dp | Sham | 0.47±0.09 | 0.01±0.01 | 47.0±47.6b | | |
| HT 41 (1) | 0.83±0.08a | 0.07±0.02 | 11.9±3.6 | 1.8±0.4 | 7.0±7.3 |
| SW-1573 ip | Sham | 0.21±0.02 | 0.06±0.02 | 3.5±1.2 | | |
| HT 41 (1) | 0.06±0.02 | 0.11±0.03 | 0.6±0.2 | 0.3±0.1 | 1.8±0.8 |
| HT 43 (1) | 0.49±0.04a | 0.12±0.03 | 4.1±1.1 | 2.3±0.3 | 2.0±0.8 |
| SW-1573 dp | Sham | 0.09±0.02 | 0.06±0.02 | 1.5±1.6 | | |
| HT 41 (1) | 0.05±0.02 | 0.08±0.02 | 0.6±0.6 | 0.6±0.3 | 1.3±0.6 |
| HT 43 (1) | 0.40±0.04a | 0.11±0.03 | 3.6±1.1 | 4.4±1.1 | 1.8±0.8 |
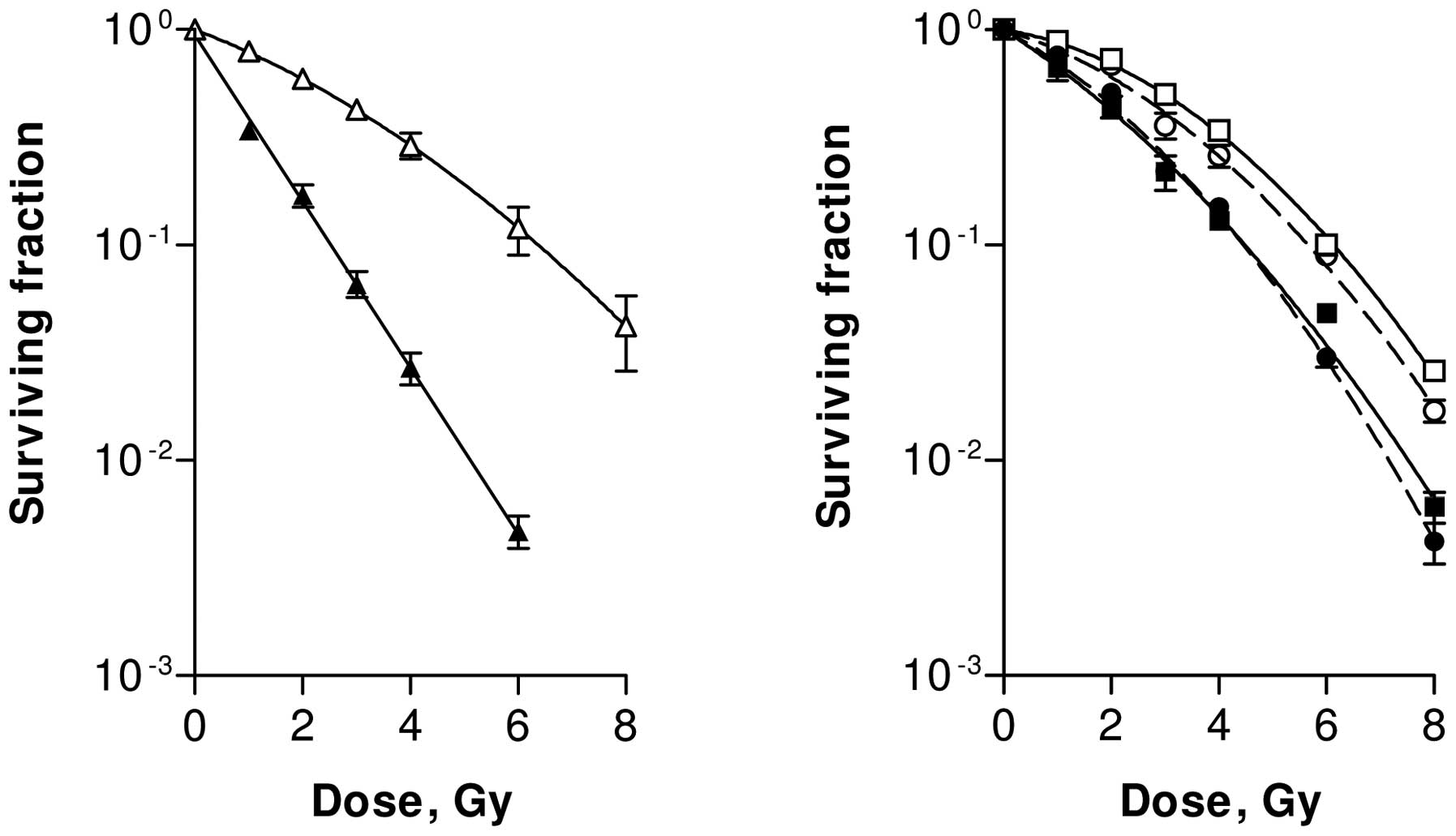
The RKO cell line, derived from a human colon
cancer, is relatively sensitive to HT treatment. HT alone for 1 h
at 43°C decreased the relative survival to <0.01 and combination
with radiation doses in excess of 5 Gy always resulted in the
complete absence of colony formation. Mild HT alone (41°C for 1 h)
had little effect and resulted in a surviving fraction of 0.8±0.4
in immediately plated (ip) and of 0.9±0.1 in delayed plated (dp)
cells. When the cells were heated to 41°C for 1 h immediately prior
to irradiation, a significant (P<0.001) enhancement of cellular
radiosensitivity was observed in both ip (Fig. 1, left panel) and dp (Fig. 1, right panel) cells (25,28).
The effects of HT on the LQ parameters are
summarized in Table I. The value
of α increased by a factor of 1.7 to 1.8, while the value of β
increased by a factor as high as 2.5 to 7.0. One must bear in mind
that the quadratic component βd2 in this cell line is
quite small and small absolute changes can result in large relative
changes of the numerical values of β.
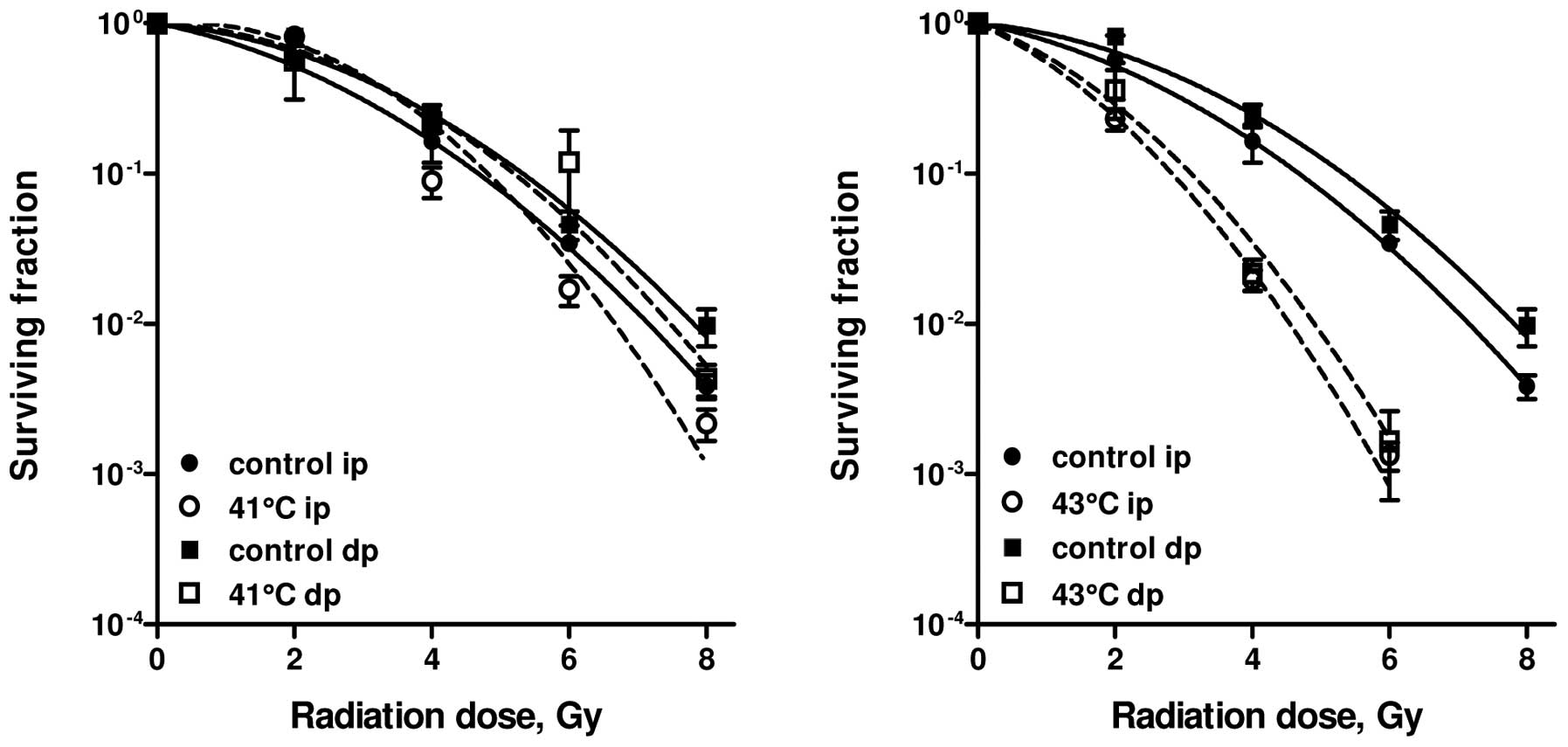
Effect of HT on radiosensitivity of
SW-1573 cells
SW-1573 cells are derived from a human lung tumour
and are much less sensitive to HT than RKO cells. Studies have been
carried out to evaluate whether pre-treatment with HT at 41 or 43°C
can enhance the radiosensitivity of SW-1573 cells (25). HT at 41°C for 1 h without
irradiation did not result in a further decrease of the surviving
fraction for ip and dp cells, compared to irradiation alone.
One-hour HT at 43°C decreased survival to 0.5±0.1 for ip and to
0.4±0.2 for dp cells. Pre-treatment of cells at 41°C for 1 h did
not affect cellular radiosensitivity of either ip or dp cells
(Fig. 2, left panel). However, 1-h
treatment at 43°C resulted in a significant radiation enhancement
in both ip and dp cells (p<0.001; Fig. 2, right panel). In Table I, the values of the LQ parameters
for radiation alone and for combined treatments are summarised. HT
for 1 h at 41°C resulted in an increase of the β-value by a factor
of 1.3 to 1.8, while the α-value was decreased. HT treatment for 1
h at 43°C resulted in an increase of the α-value by a factor of 2.3
to 4.4, while the β-value was increased by a factor of 1.8 to
2.0.
Cisplatin
Cisplatin is a widely used anticancer drug that is
often combined with radiotherapy (29). Cisplatin-based chemoradiotherapy
has become standard treatment for, among others, locally advanced
cervical carcinoma (30) and
locally advanced non-small-cell lung cancer (NSCLC) (31). There have been numerous studies on
the radiation-sensitising effect of cisplatin; however, the results
vary from a clear cisplatin-induced radio-sensitisation (24,25,32–34)
to a merely additive effect on cell survival (35). Cisplatin and radiation share a
common cellular target, DNA (36).
Cisplatin causes DNA damage by inducing the
formation of inter- and intrastrand adducts (37). The cisplatin-DNA adducts may cause
cell cycle arrest, inhibition of DNA replication and transcription
and eventually, apoptosis (38).
Repair inhibition of DNA has also been implicated (39). The most important repair pathways
reported to be involved in cisplatin-induced DNA damage repair are
nucleotide excision repair (NER) and/or homologous recombination
(HR) (40,41). An additional route for the repair
of cisplatin-DNA interstrand adducts is the
post-replication/translation repair pathway which assists the cell
in tolerating or bypassing the lesion (42).
Irradiation causes repairable (potentially lethal)
and non-repairable (lethal) DNA lesions, which are independently
induced. The ultimate effect of the repairable lesions depends on
the competing processes of repair and misrepair. The PLDR is
reflected by the difference in survival between ip and dp cells.
The inhibition of PLDR has been implicated in cisplatin-induced
radiation sensitisation (25).
More specifically, cisplatin-induced radiation sensitisation is
caused by the inhibition of the non-homologous end joining (NHEJ)
pathway and recombination repair (38,40,43).
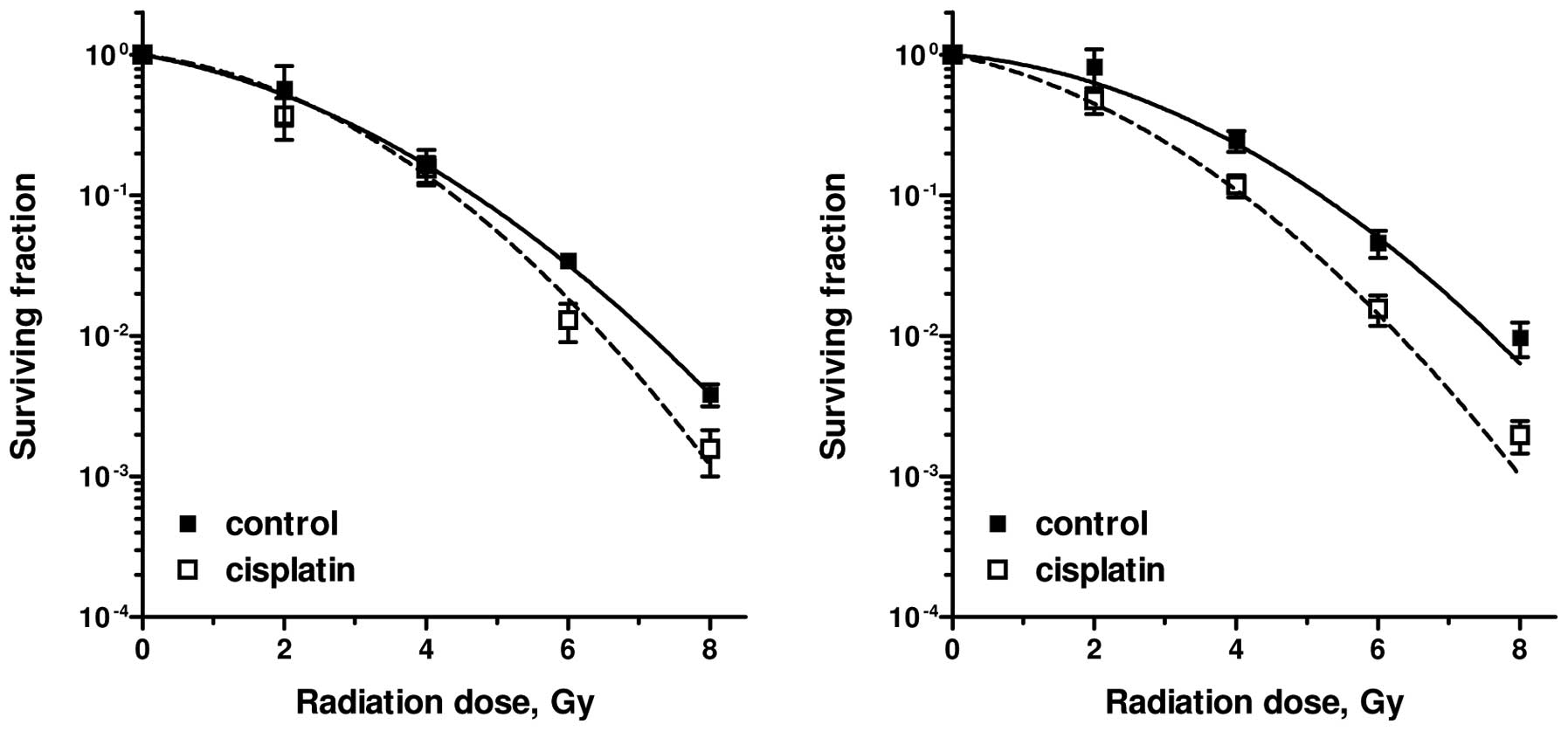
In this section, the radiation sensitisation of
cisplatin on the SW-1573 lung tumour cell line and the SiHa
cervical tumour cell line is quantified by changes in the LQ
parameters (25). Survival curves
for SW-1573 lung tumour cells following radiation alone and
radiation combined with cisplatin (1 μM for 1 h) are
presented in Fig. 3. Cisplatin was
added to the cultures immediately prior to irradiation. The
survival curves were obtained directly (ip) and 24 h after (dp)
treatment to determine PLDR. A slight, but statistically
significant effect of cisplatin on radiosensitivity was only
observed in the dp cells (P=0.02). This was also described by an
increase in the α- and β-values (Table
II). An increase in the α-value by a factor of 2.5 was achieved
in the dp cells by cisplatin treatment, whereas an increase in the
β-value by a factor of 1.2 was observed under both plating
conditions. The effects on the LQ parameters of different plating
conditions, 1-h incubation with 1 or 5 μM cisplatin and
continuous incubation with cisplatin during the complete duration
of the clonogenic assay, are also presented in Table II. The radiosensitizing effects are
more evident in the SiHa cervical tumour cell line with 1 μM
continous cisplatin incubation compared to the SW-1573 lung tumour
cell line.
| Table IIValues of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from SW-1573
and SiHa cells treated with ionizing radiation only and following
combined radiation and cisplatin (1 μM for 1 h; 1 μM
continuously; 5 μM continuously) treatment. |
Table II
Values of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from SW-1573
and SiHa cells treated with ionizing radiation only and following
combined radiation and cisplatin (1 μM for 1 h; 1 μM
continuously; 5 μM continuously) treatment.
| Cells | Treatment | α (Gy−1)
control | β (Gy−2)
control | α/β | α-EF | β-EF |
|---|
| SW-1573 ip | Sham | 0.21±0.09 | 0.061±0.016 | 3.4±1.7 | | |
| 1 μM
cisplatin (1 h) | 0.21±0.08 | 0.072±0.018 | 2.9±1.3 | 1.0±0.6 | 1.2±0.4 |
| SW-1573 dp | Sham | 0.10±0.09 | 0.063±0.016 | 1.6±1.5 | | |
| 1 μM
cisplatin (1 h) | 0.25±0.09a | 0.077±0.017 | 3.3±1.4 | 2.5±2.4 | 1.2±0.4 |
| SW-1573 ppi | Sham | 0.37±0.12 | 0.014±0.034 | 26.4±64.8b | | |
| 1 μM
cisplatin (cont) | 0.41±0.08 | 0.019±0.025 | 21.6±28.7b | 1.1±0.4 | 1.4±3.8 |
| 5 μM
cisplatin (cont) | 0.58±0.20a | 0.030±0.008a | 19.3±8.4 | 1.6±0.7 | 2.1±5.2 |
| SiHa ppi | Sham | 0.41±0.04 | 0.01±0.01 | 41.0±41.2b | | |
| 1 μM
cisplatin (cont) | 0.81±0.12a | 0.02±0.02 | 40.5±41.0b | 2.0±0.4 | 2.0±2.8 |
Gemcitabine
Gemcitabine (dFdC, difluorodeoxycytidine) is a
deoxycytidine analogue with clinical activity in NSCLC and
pancreatic cancer (44–47). It requires phosphorylation to its
active metabolites, gemcitabine-diphosphate (dF-dCDP) and
gemcitabine-triphosphate (dF-dCTP), with the initial
phosphorylation by deoxycytidine kinase (dCK) being the
rate-limiting step (48,49). dF-dCTP inhibits ribonucleotide
reductase, the enzyme regulating the production of
deoxy-nucleotides, which are necessary for DNA synthesis and repair
(50). Deoxynucleotide depletion
leads to the increased incorporation of dF-dCTP into DNA, thereby
blocking DNA synthesis (masked chain termination). Following the
incorporation of dF-dCTP into DNA, an increase in the number of DNA
single-strand breaks (SSBs), chromosome breaks and micronuclei has
been observed (51).
In vitro and in vivo studies have
demonstrated that gemcitabine is a potent radiosensitiser (39,49,52–59).
However, in a previous study on NSCLC patients, radiotherapy with
concurrent gemcitabine resulted in unacceptable pulmonary toxicity,
due to the large amount of radiation delivered to the lungs
(60). Phase I trials have
demonstrated that radiotherapy combined with gemcitabine at lower
doses is feasible without severe pulmonary toxicity (45,61).
Its unique mechanism of action, lack of overlapping toxicity and
favourable toxicity profile make gemcitabine an ideal candidate for
combination therapy (45). There
are numerous ongoing randomized studies in which radiotherapy is
combined with gemcitabine.
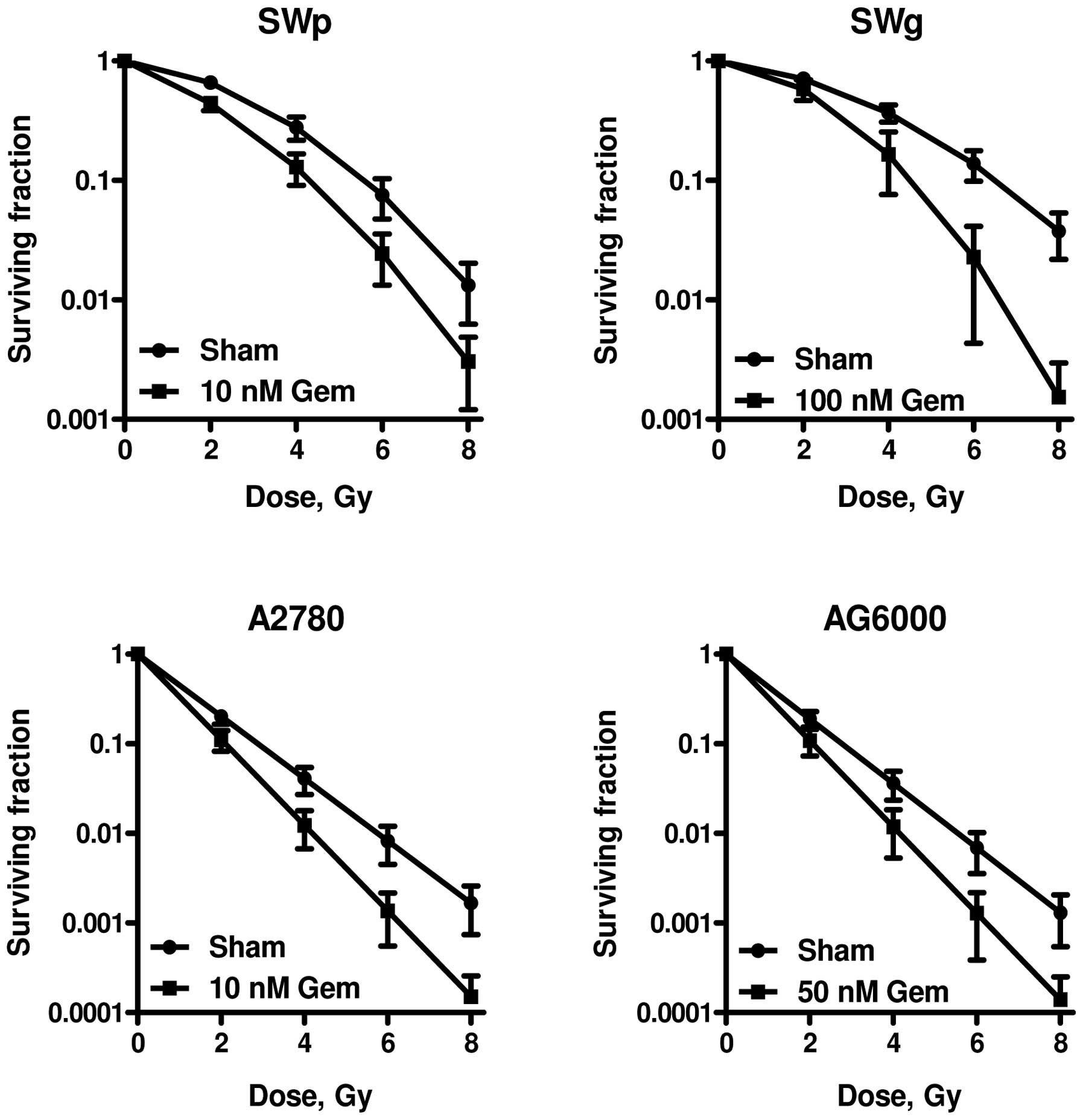
Gemcitabine radiosensitisation has been investigated
in gemcitabine-sensitive and -resistant human lung tumour cells,
SWp and SWg, respectively, as well as in gemcitabine-sensitive and
-resistant human ovarian tumour cells, A2780 and AG6000,
respectively (62–64). Gemcitabine was administered 24 h
prior to radiation treatment (64). The SWp cell line is similar to the
SW-1573 cell line described above. It is termed SWp to distinguish
it from SWg, the gemcitabine-resistant counterpart which was
developed by van Bree et al(64). Lung tumour cells exhibit different
sensitivities to radiation alone as compared to ovarian cancer
cells (62–64).
Table III
summarizes the LQ parameters of the different cell lines obtained
following analyses of the radiation dose-survival curves for
irradiation alone and following combined irradiation and
gemcitabine. SWp and SWg cells were almost equally sensitive to
ionizing radiation alone with respect to the low-dose region
described by the α-value. A slight increase in survival was
observed in the SWg cells under the high-radiation dose region
(>4 Gy), which was reflected by a slightly lower β-value
(0.040±0.006 vs. 0.055±0.008). The A2780 human ovarian carcinoma
cell line and its gemcitabine-resistant variant, AG6000, were
equally sensitive to ionizing radiation. The surviving fractions of
the different cell lines following incubation with gemcitabine
alone were as follows: SWp cells: 10 nM, 0.52±0.06; SWg cells: 10
μM, 0.95±0.03; 100 μM, 0.24±0.11; A2780 cells: 2 nM,
0.82±0.08; 10 nM, 0.21±0.08; AG6000 cells: 20 μM, 0.62±0.07;
50 μM, 0.22±0.04.
| Table IIIValues of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and gemcitabine-sensitised
radiation dose-survival curves of gemcitabine-sensitive (SWp and
A2780) and gemcitabine-resistant (SWg and AG6000) cells. |
Table III
Values of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and gemcitabine-sensitised
radiation dose-survival curves of gemcitabine-sensitive (SWp and
A2780) and gemcitabine-resistant (SWg and AG6000) cells.
| Cells | Treatment | α (Gy−1)
control | β (Gy−2)
control | α/β | α-EF | β-EF |
|---|
| SWp | Sham | 0.10±0.03 | 0.055±0.008 | 1.8±0.6 | | |
| 10 nM
gemcitabine | 0.30±0.06a | 0.053±0.007 | 5.7±1.4 | 3.0±2.8 | 0.96±0.2 |
| SWg | Sham | 0.09±0.02 | 0.040±0.006 | 2.3±0.6 | | |
| 100 μM
gemcitabine | 0.09±0.03 | 0.090±0.041b | 1.0±0.6 | 1.0±0.5 | 2.3±1.1 |
| A2780 | Sham | 0.80±0.10 | Na | | | |
| 10 nM
gemcitabine | 1.10±0.15a | Na | | 1.4±0.3 | |
| AG6000 | Sham | 0.83±0.13 | Na | | | |
| 50 μM
gemcitabine | 1.11±0.20b | Na | | 1.3±0.3 | |
As depicted in Fig.
4 and Table III,
radiosensitisation is observed in both gemcitabine-sensitive and
gemcitabine-resistant cells. However, much higher gemcitabine doses
were required for the radiation sensitisation of
gemcitabine-resistant cells to result in similar cytotoxicity. Both
gemcitabine-sensitive cell lines (SWp and A2780) were sensitised by
incubation with 10 nM of gemcitabine for 24 h prior to irradiation,
while the SWg and AG6000 cell lines were not radiosensitised by
this dose of gemcitabine. Radiosensitisation of the two
gemcitabine-sensitive cell lines was reflected by an increase in
the α-values by a factor of 3 and 1.4, respectively, whereas the
β-values were not significantly altered. Higher concentrations of
gemcitabine (50 and 100 nM) were required to sensitise the
gemcitabine-resistant AG6000 and SWg cells to irradiation. For the
SWg cells, the radiosensitisation was reflected by an increase in
the β-value by a factor of 2.25 in, whereas in the AG6000 cells,
only the α-value was increased by a factor of 1.3.
Temozolomide
The combination of fractionated radiotherapy with
temozolomide (TMZ) has significantly improved the survival of
patients with newly diagnosed glioblastoma multiforme (GBM)
(65,66). The combination of radiotherapy and
TMZ has become standard therapy for GBM patients. The benefits of
TMZ are most prominent for tumours with a methylated
O6-methylguanine-DNA methyltransferase (MGMT) promoter:
methylation of the MGMT promoter has been associated with a longer
overall survival of GBM patients treated with radiotherapy and TMZ,
compared to radiotherapy alone (67,68).
Van Nifterik et al(10)
demonstrated a relatively lower cell survival in methylated GBM
cell lines following treatment with radiotherapy and TMZ, which
suggests an interaction between TMZ and irradiation.
TMZ is a chemotherapeutic prodrug that transforms
under physiological conditions into its active unstable methylating
metabolite, 5-(3-methyl-1-triazeno)imidazole-4-carboxamide (MTIC).
Methylation of DNA by MTIC results in the formation of
O6-methylguanine adducts. These adducts are considered
to be responsible for the cytotoxic effects of TMZ (69,70).
O6-methylguanine adducts can result in failure of the
mismatch repair system, leading to DNA double-strand breakage and
eventually, cell death (71,72).
O6-methylguanine-DNA methyltransferase is
a cytoprotective DNA repair protein that can remove the methyl
group from the O6 position of guanine. Therefore,
presence of this repair protein may undo, in part, the cytotoxic
effect of alkylating agents, hence resulting in tumour resistance
to TMZ (73,74). Hypermethylation of the CpG islands
in the promoter region of the MGMT gene has been found to be
associated with transcriptional silencing (74,75)
and, subsequently, with a good clinical response to alkylating
agents in glioma patients (76,77).
Few studies have been published on the
radiosensitising potential of TMZ for glioma cell lines using
different treatment protocols. In certain cell lines, an
enhancement of the radiation effect has been demonstrated, whereas
other cell lines have shown no interaction, but merely an additive
effect (78–83).
In this review, we present the results of combined
TMZ-radiation treatment on three long-term primary TMZ-sensitive
glioma cell lines (Table IV).
These three cell lines contain a MGMT promoter region that is for
the most part, methylated and do not express the MGMT protein
(10,84,85).
| Table IVValues of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and temozolomide-sensitised
radiation dose-survival curves of three glioma cell lines AMC-3046,
VU-109 and VU-122 with different sensitivities to temozolomide
(10). |
Table IV
Values of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors from cells
treated with ionizing radiation only and temozolomide-sensitised
radiation dose-survival curves of three glioma cell lines AMC-3046,
VU-109 and VU-122 with different sensitivities to temozolomide
(10).
| Cell line | Treatment | α
(Gy−1) | β
(Gy−2) | α/β (Gy) | α-EF | β-EF |
|---|
| AMC-3046 | Sham | 0.014±0.033 | 0.065±0.007 | 0.22±0.06 | | |
| Temozolomide | 0.43±0.025a | 0.009±0.005 | 47.6±26.7 | 30.7±7.5 | 0.14±0.8 |
| VU-109 | Sham | 0.14±0.031 | 0.037±0.006 | 3.8±1.0 | | |
| Temozolomide | 0.19±0.038 | 0.032±0.008 | 6.0±1.9 | 1.4±0.4 | 0.9±0.3 |
| VU-122 | Sham | 0.11±0.025 | 0.063±0.005 | 1.8±0.4 | | |
| Temozolomide | 0.21±0.047b | 0.067±0.010 | 3.1±0.8 | 1.9±0.6 | 1.1±0.2 |
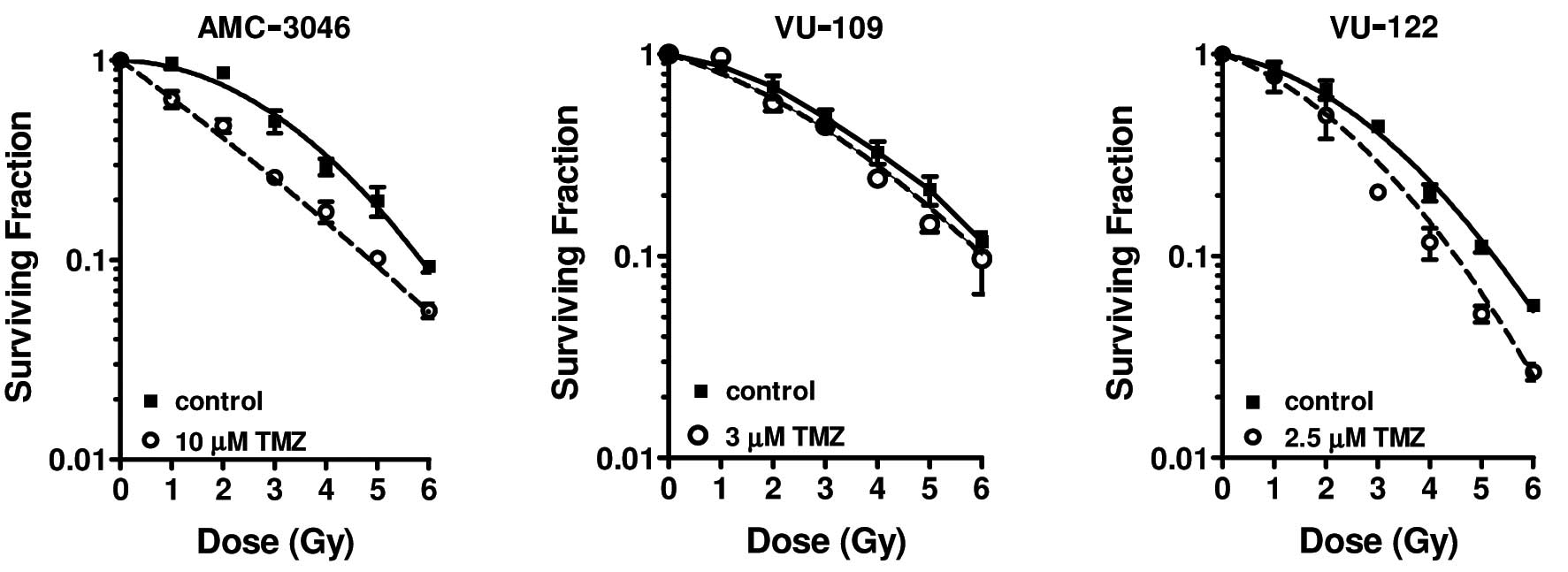
The cells were exposed to isotoxic doses of TMZ for
96 h prior to γ-irradiation. A significant radiosensitising effect
(P<0.05) of TMZ was demonstrated in the AMC-3046 glioma cell
line (Fig. 5, left panel). The
shoulder of the survival curve for irradiated cells disappeared as
a result of pre-treatment with TMZ. This was also reflected by
TMZ-induced changes in both the α and β parameters of the LQ model
(Table IV). No radiosensitisation
was observed in the VU-109 glioma cells (P=0.054; Fig. 5, middle panel), as demonstrated by
the unaffected α and β parameters (Table IV). The VU-122 glioma cells
displayed a small but significant radiosensitising effect of TMZ
(P<0.05; Fig. 5, right panel),
which was most obvious in the lower-radiation dose range. This
difference was reflected by an increase in the α parameter without
any change in the β parameter (Table
IV).
Halogenated pyrimidines
Incorporation of halogenated pyrimidines (HPs),
chloro-, bromo- and iodo-deoxyuridine (CldUrd, BrdUrd, IdUrd) into
DNA is known to sensitise cells to ionizing radiation (6,8,11,86–93).
The induced radiosensitisation increases with the degree of
thymidine replacement. The mechanism of radiosensitisation by the
HPs has been suggested to be due to an increase in the amount of
DNA damage induced by radiation, an influence on repair of
sublethal damage (SLD), or an enhanced expression of potentially
lethal damage (PLD) (6,94). Since different processes are
involved in these phenomena, several mechanisms may contribute to
the radiosensitisation.
HPs have been suggested to provide an advantage in
radiotherapy as radiosensitisers of cells in rapidly growing
tumours, particularly under clinical conditions in which critical
normal tissues show limited proliferation, and as a consequence,
take up less HP. Labelling depends on the growth fraction, cell
loss, cell cycle time and potential doubling time. Of particular
importance for sensitisation is the rate at which non-cycling cells
are recruited into the proliferative compartment during exposure to
HPs and a course of radiotherapy. However, even in rapidly growing
tumours, cells may, following proliferative cycles, retreat into a
non-proliferative state. This may compromise the degree of
radiation sensitisation, since resting cells are less affected by
HPs, or are better able to cope with additional damage by PLDR.
In this review, we present the results of
radiosensitisation following incubation with 4 μM of IdUrd
for 72 h. IdUrd-induced radiosensitisation was observed in all the
studied cell lines, SW-1573, RUCII (rat ureteral carcinoma), R1
(rat rhabdomyosarcoma) and V79 (Chinese hamster lung cells), in
exponentially growing and in plateau-phase cells. Values of α and β
derived by LQ analysis of the survival curves of exponentially
growing and plateau-phase cells are summarized in Table V. Fig.
6 depicts the survival curves of SW and V79 cell lines. The
plating conditions of the V79 cells, i.e., exponentially growing
cells plated prior to or after irradiation (ppi or pai,
respectively), and plateau-phase cells, plated immediately or 6–24
h after irradiation (ip or dp, respectively), had no influence on
the enhancement factor of the α-value. It is demonstrated that the
α-value can be enhanced by a factor of 1.9 to 7.5 and that, in
general, low α-values are more enhanced than higher α-values. The
value of β is less enhanced and its enhancement factor ranges from
0.7 to 2.4.
| Table VValues of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors of several
cell lines treated with ionizing radiation only and after
sensitisation with iododeoxyuridine (IdUrd) (incubation with 4
μM of IdUrd for 72 h). |
Table V
Values of the linear-quadratic
parameters α and β, α/β ratio and enhancement factors of several
cell lines treated with ionizing radiation only and after
sensitisation with iododeoxyuridine (IdUrd) (incubation with 4
μM of IdUrd for 72 h).
| Cell line | α (Gy−1)
control | β (Gy−2)
control | α (Gy−1)
IdUrd-sens | β (Gy−2)
IdUrd-sens | α/β control | α/β IdUrd-sens | α-EF | β-EF |
|---|
SW-1573
cells
Exp growing ip | 0.22±0.01 | 0.022±0.001 | 0.83±0.06 | Na | 10.0±0.6 | Na | 3.8±0.3 | Na |
SW-1573
cells
Plateau-phase ip | 0.17±0.03 | 0.042±0.004 | 0.31±0.03 | 0.047±0.005 | 4.1±0.8 | 6.6±1.0 | 1.8±0.4 | 1.1±0.2 |
SW-1573
cells
Plateau-phase dp | 0.09±0.02 | 0.046±0.002 | 0.37±0.04 | 0.033±0.006 | 2.0±0.4 | 11.2±2.4 | 4.1±1.0 | 0.7±0.1 |
RUCII
cells
Exp growing ppi | 0.008±0.007 | 0.025±0.001 | 0.06±0.02 | 0.026±0.001 | 0.3±0.3 | 2.3±0.8 | 7.5±7.0 | 1.0±0.1 |
R1 cells
Exp growing ppi | 0.23±0.01 | 0.068±0.003 | 0.44±0.05 | 0.075±0.016 | 3.4±0.2 | 5.9±1.4 | 1.9±0.3 | 1.1±0.2 |
V79 cells
Exp growing ip | 0.18±0.02 | 0.017±0.003 | 0.38±0.04 | 0.023±0.007 | 10.6±2.2 | 16.5±5.3 | 2.1±0.3 | 1.4±0.5 |
V79 cells
Exp growing ppi | 0.15±0.02 | 0.013±0.003 | 0.29±0.03 | 0.016±0.004 | 11.5±3.1 | 18.1±4.9 | 1.9±0.3 | 1.2±0.4 |
V79 cells
Plateau-phase ip | 0.09±0.03 | 0.026±0.004 | 0.17±0.02 | 0.062±0.005 | 3.5±1.3 | 2.7±0.4 | 1.9±0.7 | 2.4±0.4 |
V79 cells
Plateau-phase dp | 0.07±0.02 | 0.020±0.002 | 0.30±0.03 | 0.024±0.004 | 3.5±1.1 | 12.5±2.4 | 4.3±1.3 | 1.2±0.2 |
The direct comparison between immediate and delayed
plating of plateau-phase cells and between plateau-phase and
exponentially growing cells shows significant quantitative
differences. The data on the LQ parameters presented herein provide
various new insights into the interpretation of radio-sensitisation
of dp plateau-phase cells. It is demonstrated that in dp
HP-sensitised plateau-phase cells PLD is not abolished.
PARP1 inhibitors
The effect of inhibition of poly(ADP-ribose)
polymerase-1 (PARP1) by olaparib on the LQ parameters was examined
in mouse embryonic fibroblasts (MEFs). PARP1 is an enzyme which is
involved in the repair of DNA SSBs. The DNA SSBs induced by
ionizing radiation are mostly repaired by the base excision repair
(BER) system, whereas the DNA DSBs are repaired by NHEJ or by HR.
Inhibiting PARP1 activity reduces the repair of SSBs (95). Apart from its role in BER, PARP1 is
further involved in a number of nuclear processes, such as DNA
replication, transcription, DSB repair, apoptosis and genome
stability (95–97). It was recently hypothesised that
cells deficient in BRCA2 or BRCA1 are particularly sensitive to
PARP1 inhibition (27,96). SSBs are induced during DNA
replication. In the absence of PARP1, these SSBs transform into
DSBs. These DSBs are repaired with HR. Therefore, cells deficient
in HR (e.g., BRCA1 or BRCA2 tumours) may be sensitive to PARP1
inhibitors. Since PARP1 is involved in numerous DNA repair
processes, PARP1 inhibitors may function effectively as
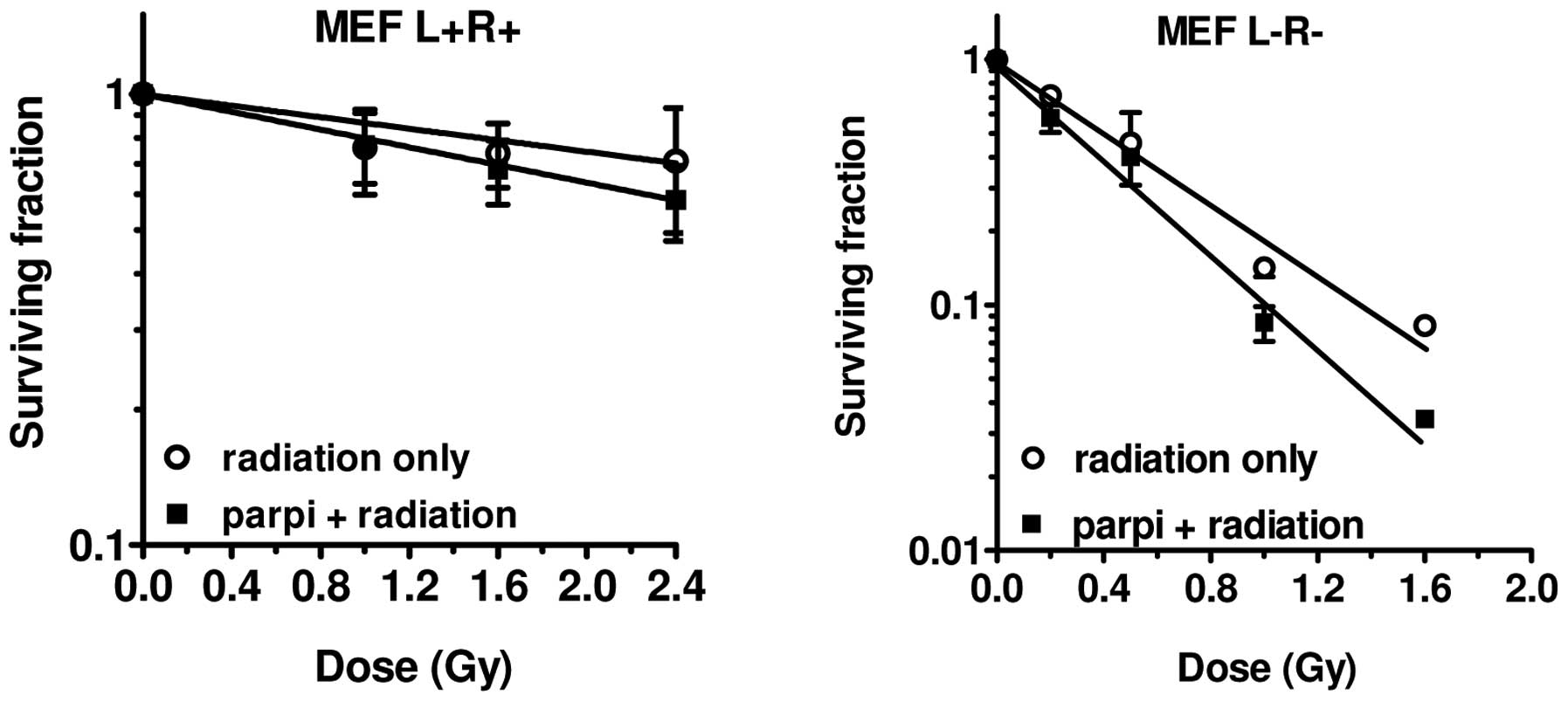
radiosensitisers (97). As can be
observed in Fig. 7, we achieved a
modest sensitisation effect by the PARP1 inhibitor NU-1025 in the
MEF cell lines. The increase of the α-value in the repair-deficient
cell line was greater than in the repair-proficient cell line, 1.4
vs. 1.2, respectively (Table VI).
The radiation dose-survival curves of these MEF cells did not
exhibit a shoulder and therefore the quadratic parameter β could
not be determined.
| Table VIValues of the linear parameter α and
the enhancement factors from repair-proficient and repair-deficient
MEF cells. |
Table VI
Values of the linear parameter α and
the enhancement factors from repair-proficient and repair-deficient
MEF cells.
| MEF cells | Treatment with
PARPi | α
(Gy−1) | β
(Gy−2) | α-EF |
|---|
|
LigIV+/+,
Rad54+/+ | No | 0.28±0.01 | Na | |
|
LigIV+/+,
Rad54+/+ | Yes | 0.33±0.03 | Na | 1.2 |
|
LigIV−/−,
Rad54−/− | No | 1.59±0.18 | Na | |
|
LigIV−/−,
Rad54−/− | Yes | 2.28±0.42 | Na | 1.4 |
Discussion
Radiosensitisation by a variety of chemotherapeutic
agents is in most cases reflected by an increase of the linear or α
component of the LQ model, which corresponds to an enhanced direct
PLD at low radiation doses (1,4–7,98,99).
The β component, which presumably depends on the interaction of
repairable SLD, is affected by HT treatment. Furthermore, it
appears that radiosensitisation is more pronounced in
radioresistant than in radiosensitive cell lines. In addition, it
can be concluded that the extent of radiosensitisation also depends
on cell cycle stage (plateau or exponentially growing phase) and
post-treatment plating conditions.
Hyperthermia
Hyperthermia (HT) is a very potent radiosensitiser,
already effective at mild temperatures. HT for 1 h at 41°C without
radiation exerted only a slight cytotoxic effect in both
heat-sensitive and heat-resistant cell lines. This is in agreement
with the general idea of cell kill induction at temperatures ≥42°C
for 1 h or more (23). HT at 43°C
for 1 h did not have a significant cytotoxic effect in
heat-resistant SW-1573 cells. Radiosensitisation by HT at 41°C was
observed in SiHa and RKO, but not in SW-1573 cells. The ability of
mild HT (40–42°C) to increase radiosensitivity of human tumour
cells has been shown to be cell line-dependent (8,26,100–105). In a study by Xu et al,
pre-treatment of cells at 41.1°C for 1 h did not induce
radiosensitisation, whereas treatment for 2 h or more resulted in
radiosensitisation in the HT-resistant but not in the HT-sensitive
cell line (106). However,
simultaneous treatment of the sensitive cell line with 1-h 41.1°C
HT combined with irradiation increased cellular radiosensitivity
(107). In vivo
radiosensitisation by mild HT is usually attributed to
reoxygenation of tumours by an increase in blood flow (108–110). We recently discovered that the
BRCA2 protein is transiently inhibited by mild HT (27). Translocation of the Mre11 DSB
repair protein from the nucleus to the cytoplasm has also been
implicated (106,111). However, disappearance of Mre11
protein foci at the sites of irradiation-induced DNA DSBs was not
observed by pre-incubation of cells at 41°C (24,27).
A role for mitotic catastrophe, occurring as a result of G2/M
checkpoint abrogation, has also been suggested (112). It has been demonstrated that
radiosensitisation by HT at 41–43°C correlates with an increased
number of chromosomal fragments, but not of colour junctions, 24 h
after treatment, compared to radiation alone (101). HT at clinically reachable
temperatures mainly enhances the quadratic parameter, β, which
represents the frequency of induction-repairable SLD. The fact that
HT breaks down the repair protein BRCA2 and in this way influences
DNA DSB repair correlates well with the effect on the repairable
factor β.
Cisplatin
Cisplatin causes radiosensitisation as measured by
clonogenic survival, but only after allowing a PLDR time of 24 h.
These results are in agreement with those of Wilkins et
al(113), who investigated
the effect of cisplatin and radiation on PLDR in confluent cultures
of two different brain tumour cell lines (113). Wilkins et al did not
observe radiosensitisation by cisplatin in ip cells, whereas a
cisplatin-induced radiosensitisation was observed in dp cells 8 h
following irradiation (114).
Their results indicate that the radiosensitising effect of
cisplatin is caused by the inhibition of post-irradiation recovery.
The strongest inhibition of PLDR was achieved when cisplatin was
administered shortly before or after irradiation (113,114). In our experiments, cells were
irradiated with cisplatin present in the medium.
Results from studies using exponentially growing
cell cultures vary from a cisplatin-induced radiosensitisation
(31–33,115), to a merely additive effect
(29,31,116–118). The effect of cisplatin treatment
on radiosensitivity may depend on the cell type used. Loprevite
et al(31) observed
synergism in a squamous lung carcinoma cell line when exposed to
cisplatin, whereas an adenocarcinoma of the lung was not sensitised
by cisplatin (31). Even cell
lines derived from a single biopsy can differ in their response to
combined treatment with cisplatin and irradiation (116).
Although dependence on cell cycle phase (119,120), cisplatin incubation time and
sequence of treatment modalities have been implicated (29,119,120), there is currently no consensus to
account for the varying response of cells to cisplatin and
irradiation.
The mechanism of cisplatin-induced
radiosensitisation may be due to the inhibition of the DNA repair
NHEJ and HR pathways (38,43). The Ku protein complex, which plays
an important role in NHEJ, was demonstrated to show a reduced
ability to translocate on DNA containing cisplatin-DNA adducts
compared with undamaged DNA. This resulted in a decreased
interaction between Ku and DNA-dependent protein kinase catalytic
subunit (DNA-PKcs) (121).
However, the biochemical processes that cisplatin undergoes in the
cell are complex and its intracellular fate may be linked to copper
transport (122). Therefore,
other processes, such as the formation of peroxy complexes inside
the cell, may be involved in cisplatin-induced radiosensitisation
(123). Bergs et
al(34) demonstrated an
increase in the induction of apoptosis 24 h after combined
treatment as compared to radiation or cisplatin alone. This was
confirmed by several other studies (124,125). These apoptotic effects observed
by Bergs et al correlated with clonogenic survival (34). Fujita et al(126) also observed an inhibitory effect
of the combination of cisplatin and radiation on the survival of
lung tumour cells and ascribed this effect to the induction of
tumour cell apoptosis (126).
In conclusion, the radiosensitising effect of
cisplatin on cell survival was observed in confluent cultures when
cells were replated after a 24-hour incubation period during which
PLDR was allowed to occur. By contrast, cisplatin did not induce a
significant radiosensitisation after immediate plating.
Gemcitabine
A number of previous studies have demonstrated that
gemcitabine is a potent sensitiser to ionizing radiation (49,55,127). Among other proposed mechanisms of
action, the effect of gemcitabine on cell cycle distribution may be
the most important (55,57). In our study, the
gemcitabine-sensitive cell lines SWp and A2780 were sensitised to
irradiation following administration of cytotoxic gemcitabine
treatments. The radiosensitisation was accompanied by a clear
arrest of cells in early S phase, which has been argued to be vital
for gemcitabine-induced radiosensitisation (54). Both cell lines showed an increase
in the α-value, indicating the efficacy of gemcitabine-induced
radiosensitisation in the clinically relevant dose range. The
gemcitabine-resistant cells were also sensitised, although with
only much higher gemcitabine doses. In the resistant AG6000 ovarian
carcinoma cell line, this was demonstrated by an increase in the
α-value. By contrast, in the gemcitabine-resistant lung tumour cell
line an increase in the β-value was observed, while the α-value was
not affected. In both gemcitabine-resistant cell lines the
sensitivity to ionizing radiation alone was not altered. It is
reported that gemcitabine-resistant tumours are cross-resistant to
related drugs like Ara-C (128,129). In the AG6000 and SWg
gemcitabine-resistant cell lines, this was indeed the case
(64). Moreover, the AG6000 cells
were more resistant to cisplatin and taxoids as well (62). However, no altered sensitivity was
observed in SWg cells for cisplatin, paclitaxel, methotrexate (MTX)
and 5-fluorouracil (5-FU), while AG6000 cells were 2.5-fold more
sensitive to MTX (62). These
findings indicate that patients previously treated with gemcitabine
may still benefit from radiotherapy combined with cisplatin or
paclitaxel.
Temozolomide
The potential of TMZ to enhance the radiation
response in long-term primary GBM cell lines has been clearly
demonstrated (10) and therewith,
the rationale for the clinical use of this drug concomitantly with
radiotherapy. A distinct increase of the α parameter is shown
following treatment with TMZ. The three MGMT promoter methylated
cell lines discussed above responded differently to the combination
treatment, even though they were treated at similar TMZ sensitivity
levels. The combined effect of TMZ and radiation was found to be
synergistic and is at least additive.
Halogenated pyrimidines
Radiosensitisation by halogenated pyrimidines (HPs)
is mainly due to an increase in the linear parameter α. The
quadratic parameter β is rarely influenced. Different
radiosensitisation mechanisms induced by HPs have been described.
Wang et al(130) suggested
that increased DNA damage was the major component of
radiosensitisation in exponentially growing cells, while in
plateau-phase cells, radiosensitisation occurred through inhibited
repair and/or enhanced fixation of PLD (6,90).
The increase of the α-values for exponentially growing cells, as
presented in our study, indicates an increase in the number of
directly lethal events due to the HPs. This is in agreement with
the observations of Webb et al(131) and Jones et al(94), which suggested that an important
mechanism of radiosensitisation involves an increase of effective
DNA DSBs (87,89,130). Miller et al(92,93)
suggested that radiation-induced damage in cells which have HPs
incorporated into the DNA after low-LET radiation resembles the
damage produced by high-LET radiation. In plateau-phase cells
plated immediately after irradiation, the increase of α might be
due to the same mechanism as involved in exponentially growing
cells. In these cells an increase of β was also observed,
indicating that accumulation of SLD was a major contributor
(2). Due to the immediate plating
after irradiation this SLD may be fixated.
The most significant increases in the α value were
observed in dp plateau-phase cells. This radiosensitisation may be
interpreted as an enhanced fixation of PLD due to immediate DNA
damage and/or to damaged DNA repair function in these cells,
expressed during the interval before delayed plating. The value of
β in these cells returned to values found in cells not containing
HPs. This demonstrates that SLD was repaired in HP-containing
plateau-phase cells.
PARP1 inhibitors
Since PARP1 has been implicated in several DNA
repair processes, PARP1 inhibitors may be good radiosensitisers.
Several studies have already demonstrated the radiosensitising
effect of PARP1 inhibitors (27,95,132). Löser et al(97) concluded that the effects of PARP1
inhibitors are more pronounced on rapidly dividing and/or DNA
repair-deficient cells (95). In
our study, at the time of treatment most of the cells in culture
were accumulated in G1 phase. Therefore, radiosensitisation effects
were modest. However, the increase in the α-value in
repair-deficient cells was more pronounced following PARP1
inhibition than in repair-proficient cells.
Conclusion
The increase in the α parameter by the various
radiosensitising agents yields promising perspective for clinical
practice. The radiation tolerance dose is generally expressed
quantitatively in terms of the biologically effective dose (BED),
as defined by the LQ model. BED takes total dose, dose per
fraction, dose rate and overall treatment time into account. By
definition, BED is the total dose required to obtain an equal
biological effect E (isoeffect) for a certain endpoint, e.g., few
log cell kill (e.g., 10−2 cell survival), a normal
tissue effect (e.g., 1% complication rate) or tumour response
(e.g., tumour cure rate of 50%) when applying an infinite number
(∞) of tiny dose fractions (∼0). At this point, the α parameter is
inversely proportional to BED [BED = E/α; (133)]. With increasing α, BED is
decreasing, resulting in a lower radiation tolerance dose for a
certain isoeffect.
The radiosensitising effect of HT on the LQ
parameters seems to be temperature-dependent. HT for 1 h at 41°C
increases the β-value while HT for 1 h at 43°C increases both the
α- and β-values. Increase of the β-value consequently lowers the
α/β ratio, which makes the tumour more sensitive to higher fraction
doses. The effects of HT on BED remain to be elucidated.
Radiosensitising agents that selectively sensitise
tumour cells and not normal tissue cells will be of therapeutic
benefit. This is due to the increase of the α parameter of tumour
cells only and, as a consequence, the decrease in tumour response
dose proportionally to the relative increase in α. This effect
could be further exploited using smaller sized fractions in
external beam radiotherapy or by lowering the dose rate in
brachytherapy. With lower fraction size or lower dose rate, normal
tissue cells with low α/β ratios will tolerate a higher total dose.
Tumour tissues with high α/β ratios will not exhibit an increased
sparing effect, since an increase in α by a radio-sensitising agent
will further increase the α/β ratio, resulting in even less
sensitivity to a modification in fraction size. However, since an
increase in the number of fractions or a lower dose rate might
increase the overall treatment time, tumour cell repopulation rate
should be taken into account.
Acknowledgements
We thank Jan Sijbrands for his
technical support. The authors are thankful for financial support
from several foundations. The Maurits and Anna de Kock and the
Nijbakker Morra foundations are acknowledged for sponsoring
laboratory equipment. The Dutch Cancer Foundation (grant nos. UVA
2006-3484, UVA 2008-4019 and UVA 2012-5540) and the Stichting
Vanderes are acknowledged for financing personnel support.
References
|
1.
|
Barendsen GW: Dose fractionation, dose
rate and iso-effect relationships for normal tissue responses. Int
J Radiat Oncol Biol Phys. 8:1981–1997. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Barendsen GW: Mechanisms of cell
reproductive death and shapes of radiation dose-survival curves of
mammalian cells. Int J Radiat Biol. 57:885–896. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Barendsen GW: The relationships between
RBE and LET for different types of lethal damage in mammalian
cells: biophysical and molecular mechanisms. Radiat Res.
139:257–270. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Barendsen GW: Parameters of
linear-quadratic radiation dose-effect relationships: dependence on
LET and mechanisms of reproductive cell death. Int J Radiat Biol.
71:649–655. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Barendsen GW, van Bree C and Franken NAP:
Importance of cell proliferative state and potentially lethal
damage repair on radiation effectiveness: Implications for combined
tumor treatments (Review). Int J Oncol. 19:257–256. 2001.
|
|
6.
|
Franken NAP, van Bree C, Kipp JB and
Barendsen GW: Modification of potentially lethal damage in
irradiated Chinese hamster V79 cells after incorporation of
halogenated pyrimidines. Int J Radiat Biol. 72:101–109. 1997.
View Article : Google Scholar
|
|
7.
|
Franken NAP, Ten Cate R, van Bree C and
Haveman J: Induction of the early response protein EGR-1 in human
tumour cells after ionizing radiation is correlated with a
reduction of repair of lethal lesions and an increase of repair of
sublethal lesions. Int J Oncol. 24:1027–1031. 2004.
|
|
8.
|
Franken NAP, van Bree C, Veltmaat MA,
Ludwików G, Kipp JB and Barendsen GW: Increased chromosome exchange
frequencies in iodo-deoxyuridine-sensitized human SW-1573 cells
after γ-irradiation. Oncol Rep. 6:59–63. 1999.PubMed/NCBI
|
|
9.
|
Franken NAP, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nature
Protoc. 1:2315–2319. 2006. View Article : Google Scholar
|
|
10.
|
van Nifterik KA, van den Berg J, Stalpers
LJA, Lafleur MV, Leenstra S, Slotman BJ, Hulsebos TJ and Sminia P:
Differential radiosensitizing potential of temozolomide in MGMT
promoter methylated glioblastoma multiforme cell lines. Int J
Radiat Oncol Biol Phys. 69:1246–1253. 2007.PubMed/NCBI
|
|
11.
|
van Bree C, Franken NAP, Bakker PJ,
Klomp-Tukker LJ, Barendsen GW and Kipp JB: Hyperthermia and
incorporation of halogenated pyrimidines: radiosensitization in
cultured rodent and human tumor cells. Int J Radiat Oncol Biol
Phys. 39:489–496. 1997.PubMed/NCBI
|
|
12.
|
González González D, Van Dijk JD and Blank
LE: Radiotherapy and hyperthermia. Eur J Cancer. 31A:1351–1355.
1995.
|
|
13.
|
van der Zee J, González González D, van
Rhoon GC, van Dijk JD, van Putten WL and Hart AA: Comparison of
radiotherapy alone with radiotherapy plus hyperthermia in locally
advanced pelvic tumours: a prospective, randomised, multicentre
trial. Dutch Deep Hyperthermia Group. Lancet. 355:1119–1125.
2000.
|
|
14.
|
van der Zee J, Treurniet-Donker AD, The
SK, Helle PA, Seldenrath JJ, Meerwaldt JH, Wijnmalen AJ, van de
Berg AP, van Rhoon GC, Broekmeyer-Reurink MP, et al: Low dose
reirradiation in combination with hyperthermia: a palliative
treatment for patients with breast cancer recurring in previously
irradiated areas. Int J Radiat Oncol Biol Phys. 15:1407–1413.
1988.PubMed/NCBI
|
|
15.
|
van der Zee J and González GD: The Dutch
Deep Hyperthermia Trial: results in cervical cancer. Int J
Hyperthermia. 18:1–12. 2002.Erratum in: Int J Hyperthermia 19:213,
2003.
|
|
16.
|
Crezee J, Barendsen GW, Westermann AM,
Hulshof MC, Haveman J, Stalpers LJ, Geijsen ED and Franken NAP:
Quantification of the contribution of hyperthermia to results of
cervical cancer trials: in regard to Plataniotis and Dale (Int J
Radiat Oncol Biol Phys 73: 1538–1544, 2009). Int J Radiat Oncol
Biol Phys. 75:6342009.PubMed/NCBI
|
|
17.
|
Dewey WC, Sapareto SA and Betten DA:
Hyperthermic radio-sensitization of synchronous Chinese hamster
cells: relationship between lethality and chromosomal aberrations.
Radiat Res. 76:48–59. 1978. View Article : Google Scholar
|
|
18.
|
Roti Roti JL: Introduction:
radiosensitization by hyperthermia. Int J Hyperthermia. 20:109–114.
2004.PubMed/NCBI
|
|
19.
|
Raaphorst GP, Feeley MM, Danjoux CE,
DaSilva V and Gerig LH: Hyperthermia enhancement of radiation
response and inhibition of recovery from radiation damage in human
glioma cells. Int J Hyperthermia. 7:629–641. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kampinga HH and Dikomey E: Hyperthermic
radiosensitization: mode of action and clinical relevance. Int J
Radiat Biol. 77:399–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Hildebrandt B, Wust P, Ahlers O, Dieing A,
Sreenivasa G, Kerner T, Felix R and Riess H: The cellular and
molecular basis of hyperthermia. Crit Rev Oncol Hematol. 43:33–56.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hall EJ and Giaccia AJ: Hyperthermia.
Radiobiology for the Radiologist. Chapter 28.6th edition.
Lippincott Williams & Wilkins; Philadelphia, PA: pp. 469–490.
2006
|
|
23.
|
Dewhirst MW, Vujaskovic Z, Jones E and
Thrall D: Re-setting the biologic rationale for thermal therapy.
Int J Hyperthermia. 21:779–790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bergs JWJ: Hyperthermia, cisplatin and
radiation trimodality treatment: In vitro studies on interaction
mechanisms. PhD Thesis, University of Amsterdam. 2007
|
|
25.
|
Bergs JWJ, Haveman J, Ten Cate R, Medema
JP, Franken NAP and van Bree C: Effect of 41°C and 43°C on
cisplatin radio-sensitization in two human carcinoma cell lines
with different sensitivities for cisplatin. Oncol Rep. 18:219–226.
2007.
|
|
26.
|
Bergs JWJ, Franken NAP, Haveman J, Geijsen
ED, Crezee J and van Bree C: Hyperthermia, cisplatin and radiation
trimodality treatment: a promising cancer treatment? A review from
preclinical studies to clinical application. Int J Hyperthermia.
23:329–341. 2007. View Article : Google Scholar
|
|
27.
|
Krawczyk PM, Eppink B, Essers J, Stap J,
Rodermond H, Odijk H, Zelensky A, van Bree C, Stalpers LJ, Buist
MR, Soullié T, Rens J, Verhagen HJ, O’Connor MJ, Franken NAP, Ten
Hagen TL, Kanaar R and Aten JA: Mild hyperthermia inhibits
homologous recombination, induces BRCA2 degradation, and sensitizes
cancer cells to poly(ADP-ribose) polymerase-1 inhibition. Proc Natl
Acad Sci USA. 108:9851–9856. 2011. View Article : Google Scholar
|
|
28.
|
Franken NAP, van Bree C, Ten Cate R, van
Oven CH and Haveman J: Importance of TP53 and RB in the repair of
potentially lethal damage and induction of color junctions after
exposure to ionizing radiation. Radiat Res. 158:707–714. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Gorodetsky R, Levy-Agababa F, Mou X and
Vexler AM: Combination of cisplatin and radiation in cell culture:
effect of duration of exposure to drug and timing of irradiation.
Int J Cancer. 75:635–642. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Dueñas-Gonzalez A, Cetina L, Mariscal I
and de la Garza J: Modern management of locally advanced cervical
carcinoma. Cancer Treat Rev. 29:389–399. 2003.
|
|
31.
|
Loprevite M, Favoni RE, de Cupis A, Pirani
P, Pietra G, Bruno S, Grossi F, Scolaro T and Ardizzoni A:
Interaction between novel anticancer agents and radiation in
non-small cell lung cancer cell lines. Lung Cancer. 33:27–39. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Begg AC, van der Kolk PJ, Dewit L and
Bartelink H: Radiosensitization by cisplatin of RIF1 tumour cells
in vitro. Int J Radiat Biol Relat Stud Phys Chem Med. 50:871–884.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Nakamoto S, Mitsuhashi N, Takahashi T,
Sakurai H and Niibe H: An interaction of cisplatin and radiation in
two rat yolk sac tumour cell lines with different
radiosensitivities in vitro. Int J Radiat Biol. 70:747–753. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Bergs JWJ, Franken NAP, Ten Cate R, van
Bree C and Haveman J: Effects of cisplatin and gamma-irradiation on
cell survival, the induction of chromosomal aberrations and
apoptosis in SW-1573 cells. Mutat Res. 594:148–154. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Fehlauer F, Barten-Van Rijbroek AD,
Stalpers LJ, Leenstra S, Lindeman J, Tjahja I, Troost D, Wolbers
JG, van der Valk P and Sminia P: Additive cytotoxic effect of
cisplatin and X-irradiation on human glioma cell cultures derived
from biopsy-tissue. J Cancer Res Clin Oncol. 126:711–716. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Crul M, van Waardenburg RC, Beijnen JH and
Schellens JH: DNA-based drug interactions of cisplatin. Cancer
Treat Rev. 28:291–303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Myint WK, Ng C and Raaphorst GP: Examining
the nonhomologous repair process following cisplatin and radiation
treatments. Int J Radiat Biol. 78:417–424. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Lawrence TS, Blackstock AW and McGinn C:
The mechanism of action of radiosensitization of conventional
chemotherapeutic agents. Semin Radiat Oncol. 13:13–21. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Haveman J, Castro Kreder N, Rodermond HM,
van Bree C, Franken NAP, Stalpers LJ, Zdzienicka MZ and Peters GJ:
Cellular response of X-ray sensitive hamster mutant cell lines to
gemcitabine, cisplatin and 5-fluorouracil. Oncol Rep. 12:187–192.
2004.PubMed/NCBI
|
|
41.
|
De Silva IU, McHugh PJ, Clingen PH and
Hartley JA: Defects in interstrand cross-link uncoupling do not
account for the extreme sensitivity of ERCC1 and XPF cells to
cisplatin. Nucleic Acids Res. 30:3848–3856. 2002.PubMed/NCBI
|
|
42.
|
Dronkert ML and Kanaar R: Repair of DNA
interstrand cross-links. Mutat Res. 486:217–247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Dolling JA, Boreham DR, Brown DL,
Raaphorst GP and Mitchel RE: Cisplatin-modification of DNA repair
and ionizing radiation lethality in yeast, Saccharomyces
cerevisiae. Mutat Res. 433:127–136. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Fossella FV, Lipmann SM, Shin DM,
Tarassoff P, Calayag-Jung M, Perez-Soler R, Lee JS, Murphy WK,
Glisson B, Rivera E and Hong WK: Maximum-tolerated dose defined for
single-agent gemcitabine: a phase I dose-escalation study in
chemotherapy-naive patients with advanced non-small-cell lung
cancer. J Clin Oncol. 15:310–316. 1997.PubMed/NCBI
|
|
45.
|
Manegold C, Zatloukal P, Krejcy K and
Blatter J: Gemcitabine in non-small lung cancer (NSCLC). Invest New
Drugs. 18:29–42. 2000. View Article : Google Scholar
|
|
46.
|
Shewach DS and Lawrence TS: Gemcitabine
and radiosensitization in human tumor cells. Invest New Drugs.
14:257–263. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Castro Kreder N, van Bree C, Franken NAP
and Havenman J: Effects of gemcitabine on cell survival and
chromosome aberrations after pulsed low dose-rate irradiation. J
Radiat Res. 45:111–118. 2004.PubMed/NCBI
|
|
48.
|
Heinemann V, Xu YZ, Chubb S, Sen A, Hertel
LW, Grindey GB and Plunkett W: Cellular elimination of
2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of
self-potentiation. Cancer Res. 52:533–539. 1992.
|
|
49.
|
Shewach DS, Hahn TM, Chang E, Hertel LW
and Lawrence TS: Metabolism of 2′,2′-difluoro-2′-deoxycytidine and
radiation sensitization of human colon carcinoma cells. Cancer Res.
54:3218–3223. 1994.
|
|
50.
|
Plunkett W, Huang P and Gandhi V:
Preclinical characteristics of gemcitabine. Anticancer Drugs.
6(Suppl 6): 7–13. 1995. View Article : Google Scholar
|
|
51.
|
Auer H, Oehler R, Lindner R, Kowalski H,
Sliutz G, Orel L, Kucera E, Simon MM and Glössl J: Characterisation
of genotoxic properties of 2′,2′-difluorodeoxycytidine. Mutat Res.
393:165–173. 1997.
|
|
52.
|
Rockwell S and Grindey GB: Effect of
2′,2′-difluorodeoxycytidine on the viability and radiosensitivity
of EMT6 cells in vitro. Oncol Res. 4:151–155. 1992.
|
|
53.
|
Shewach DS and Lawrence TS:
Radiosensitization of human solid tumor cell lines with
gemcitabine. Semin Oncol. 23(Suppl 10): 65–71. 1996.PubMed/NCBI
|
|
54.
|
Latz D, Fleckenstein K, Eble M, Blatter J,
Wannenmacher M and Weber KJ: Radiosensitizing potential of
gemcitabine (2′,2′-difluoro-2′-deoxycytidine) within the cell cycle
in vitro. Int J Radiat Oncol Biol Phys. 41:875–882. 1998.
|
|
55.
|
Gregoire V, Hittelman WN, Rosier JF and
Milas L: Chemoradiotherapy: Radiosensitizing nucleoside analogues
(Review). Oncol Rep. 6:949–957. 1999.PubMed/NCBI
|
|
56.
|
Milas L, Fujii T, Hunter N, Elshaikh M,
Mason K, Plunkett W, Ang KK and Hittelman W: Enhancement of tumor
radioresponse in vivo by gemcitabine. Cancer Res. 59:107–114.
1999.PubMed/NCBI
|
|
57.
|
van Putten JWG, Groen HJM, Smid K, Peters
GJ and Kampinga HH: End-joining deficiency and radiosensitization
induced by gemcitabine. Cancer Res. 61:1585–1591. 2001.PubMed/NCBI
|
|
58.
|
Wachters FM, van Putten JWG, Maring JG,
Zdzienicka MZ, Groen HJ and Kampinga HH: Selective targeting of
homologous DNA recombination repair by gemcitabine. Int J Radiat
Oncol Biol Phys. 57:553–562. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Castro Kreder N, van Bree C, Franken NAP
and Haveman J: Colour junctions as predictors of radiosensitivity:
X-irradiation combined with gemcitabine in a lung carcinoma cell
line. J Cancer Res Clin Oncol. 129:597–603. 2003.PubMed/NCBI
|
|
60.
|
Scalliet P, Goor C, Galdermans J, et al:
Gemzar (gemcitabine) with thoracic radiotherapy - a phase II pilot
study in chemo-naive patients with advanced non-small-cell lung
cancer (NSCLC) (Abstract). Proc ASCO. 17:499a1998.
|
|
61.
|
Blackstock AW, Lesser GJ, Fletcher-Steede
J, Case LD, Tucker RW, Russo SM, White DR and Miller A: Phase I
study of twice-weekly gemcitabine and concurrent thoracic radiation
for patients with locally advanced non-small cell lung cancer. Int
J Radiat Oncol Biol Phys. 51:1281–1289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Bergman AM, Giaccone G, van Moorsel CJ,
Mauritz R, Noordhuis P, Pinedo HM and Peters GJ: Cross-resistance
in the 2′,2′-difluorodeoxycytidine (gemcitabine)-resistant human
ovarian cancer cell line AG6000 to standard and investigational
drugs. Eur J Cancer. 36:1974–1983. 2000.
|
|
63.
|
Bergman AM, Pinedo HM, Jongsma AP, Brouwer
M, Ruiz van Haperen VW, Veerman G, Leyva A, Eriksson S and Peters
GJ: Decreased resistance to gemcitabine
(2′,2′-difluorodeoxycitidine) of cytosine arabinoside-resistant
myeloblastic murine and rat leukemia cell lines: role of altered
activity and substrate specificity of deoxycytidine kinase. Biochem
Pharmacol. 57:397–406. 1999.
|
|
64.
|
van Bree C, Castro Kreder N, Loves WJ,
Franken NAP, Peters GJ and Haveman J: Sensitivity to ionizing
radiation and chemotherapeutic agents in gemcitabine-resistant
human tumor cell lines. Int J Radiat Oncol Biol Phys. 54:237–244.
2002.PubMed/NCBI
|
|
65.
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A,
Lacombe D, Cairncross JG, Eisenhauer E and Mirimanoff RO; European
Organisation for Research and Treatment of Cancer Brain Tumor and
Radiotherapy Groups; National Cancer Institute of Canada Clinical
Trials Group: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, et al: Effects of radiotherapy with
concomitant and adjuvant temozolomide versus radiotherapy alone on
survival in glioblastoma in a randomised phase III study: 5-year
analysis of the EORTC-NCIC trial. Lancet Oncol. 10:459–466.
2009.
|
|
67.
|
Hegi ME, Diserens AC, Godard S, Dietrich
PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N and
Stupp R: Clinical trial substantiates the predictive value of
O-6-methylguanine-DNA methyltransferase promoter methylation in
glioblastoma patients treated with temozolomide. Clin Cancer Res.
10:1871–1874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC and
Stupp R: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Brennand J and Margison GP: Reduction of
the toxicity and mutagenicity of alkylating agents in mammalian
cells harboring the Escherichia coli alkyltransferase gene.
Proc Natl Acad Sci USA. 83:6292–6296. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Wedge SR, Porteus JK, May BL and Newlands
ES: Potentiation of temozolomide and BCNU cytotoxicity by
O(6)-benzylguanine: a comparative study in vitro. Br J Cancer.
73:482–490. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Karran P, Macpherson P, Ceccotti S,
Ceccotti S, Dogliotti E, Griffin S and Bignami M:
O6-methylguanine residues elicit DNA repair synthesis by
human cell extracts. J Biol Chem. 268:15878–15886. 1993.
|
|
72.
|
Ochs K and Kaina B: Apoptosis induced by
DNA damage O6-methylguanine is Bcl-2 and caspase-9/3
regulated and Fas/caspase-8 independent. Cancer Res. 60:5815–5824.
2000.PubMed/NCBI
|
|
73.
|
Gerson SL: MGMT: its role in cancer
aetiology and cancer therapeutics. Nat Rev Cancer. 4:296–307. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Hotta T, Saito Y, Fujita H, Mikami T,
Kurisu K, Kiya K, Uozumi T, Isowa G, Ishizaki K and Ikenaga M:
O6-alkylguanine-DNA alkyltransferase activity of human
malignant glioma and its clinical implications. J Neurooncol.
21:135–140. 1994.
|
|
75.
|
Qian XC and Brent TP: Methylation hot
spots in the 5′ flanking region denote silencing of the
O6-methylguanine-DNA methyltransferase gene. Cancer Res.
57:3672–3677. 1997.
|
|
76.
|
Watts GS, Pieper RO, Costello JF, Peng YM,
Dalton WS and Futscher BW: Methylation of discrete regions of the
O6-methylguanine DNA methyltransferase (MGMT) CpG island
is associated with heterochromatinization of the MGMT transcription
start site and silencing of the gene. Mol Cell Biol. 17:5612–5619.
1997.PubMed/NCBI
|
|
77.
|
Paz MF, Yaya-Tur R, Rojas-Marcos I, Reynes
G, Pollan M, Aguirre-Cruz L, García-Lopez JL, Piquer J, Safont MJ,
Balaña C, Sanchez-Cespedes M, García-Villanueva M, Arribas L and
Esteller M: CpG island hypermethylation of the DNA repair enzyme
methyltransferase predicts response to temozolomide in primary
gliomas. Clin Cancer Res. 10:4933–4938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Donson AM, Addo-Yobo SO, Handler MH, Gore
L and Foreman NK: MGMT promoter methylation correlates with
survival benefit and sensitivity to temozolomide in pediatric
glioblastoma. Pediatr Blood Cancer. 48:403–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Chalmers AJ, Ruff EM, Martindale C,
Lovegrove N and Short SC: Cytotoxic effects of temozolomide and
radiation are additive- and schedule-dependent. Int J Radiat Oncol
Biol Phys. 75:1511–1519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Esteller M, Garcia-Foncillas J, Andion E,
Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB and Herman JG:
Inactivation of the DNA-repair gene MGMT and the clinical response
of gliomas to alkylating agents. N Engl J Med. 343:1350–1354. 2000.
View Article : Google Scholar
|
|
81.
|
Chakravarti A, Erkkinen MG, Nestler U,
Stupp R, Mehta M, Aldape K, Gilbert MR, Black PM and Loeffler JS:
Temozolomide-mediated radiation enhancement in glioblastoma: a
report on underlying mechanisms. Clin Cancer Res. 12:4738–4746.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
van Rijn J, Heimans JJ, van den Berg J,
van der Valk P and Slotman BJ: Survival of human glioma cells
treated with various combinations of temozolomide and X-rays. Int J
Radiat Oncol Biol Phys. 47:779–784. 2000.PubMed/NCBI
|
|
83.
|
Wedge SR, Porteous JK, Glaser MG, Marcus K
and Newlands ES: In vitro evaluation of temozolomide combined with
X-irradiation. Anticancer Drugs. 8:92–97. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
van Nifterik KA, van den Berg J, van der
Meide WF, Ameziane N, Wedekind LE, Steenbergen RD, Leenstra S,
Lafleur MV, Slotman BJ, Stalpers LJ and Sminia P: Absence of the
MGMT protein as well as methylation of the MGMT promoter predict
the sensitivity for temozolomide. Br J Cancer. 103:29–35.
2010.PubMed/NCBI
|
|
85.
|
van Nifterik KA, van den Berg J, Slotman
BJ, Lafleur MV, Sminia P and Stalpers LJ: Valproic acid sensitizes
human glioma cells for temozolomide and γ-radiation. J Neurooncol.
107:61–67. 2012.
|
|
86.
|
Franken NAP, van Bree C, Streefkerk J,
Kuper I, Rodermond H, Kipp JB and Barendsen GW: Radiosensitization
by iodo-deoxyuridine in cultured SW-1573 human lung tumor cells:
Effects on α and β of the linear-quadratic model. Oncol Rep.
4:1073–1076. 1997.PubMed/NCBI
|
|
87.
|
Franken NAP, Ruurs P, Ludwików G, van Bree
C, Kipp JB, Darroudi F and Barendsen GW: Correlation between cell
reproductive death and chromosome aberrations assessed by FISH for
low and high doses of radiation and sensitization by
iododeoxyuridine in human SW-1573 cells. Int J Radiat Biol.
75:293–299. 1999. View Article : Google Scholar
|
|
88.
|
Iliakis G, Kurtzman S, Pantelias G and
Okayasu R: Mechanism of radiosensitisation by halogenated
pyrimidines: effect of BrdU on radiation induction of DNA and
chromosome damage and its correlation with cell killing. Radiat
Res. 119:286–304. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
89.
|
Iliakis G, Wang Y, Pantelias GE and
Metzger L: Mechanism of radiosensitisation of halogenated
pyrimidines: effect of BrdU on repair of DNA breaks, interphase
chromatin breaks and potentially lethal damage in plateau-phase CHO
cells. Radiat Res. 129:202–211. 1992. View Article : Google Scholar
|
|
90.
|
Iliakis G, Wright E and Ngo FQ: Possible
importance of PLD repair in the modulation of BrdUrd and
IdUrd-mediated radiosensitisation in plateau-phase C3H10T1/2 mouse
embryo cells. Int J Radiat Biol Relat Stud Phys Chem Med.
51:541–548. 1987. View Article : Google Scholar
|
|
91.
|
Iliakis G, Pantelias G and Kurtzman S:
Mechanism of radiosensitisation by halogenated pyrimidines: effect
of BrdU on cell killing and interphase chromosome breakage in
radiation-sensitive cells. Radiat Res. 25:56–64. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Miller EM, Fowler JF and Kinsella TJ:
Linear-quadratic analysis of radiosensitisation by halogenated
pyrimidines. I Radiosensitisation of human colon cancer cells by
iododeoxyuridine. Radiat Res. 131:81–89. 1992. View Article : Google Scholar
|
|
93.
|
Miller EM, Fowler JF and Kinsella TJ:
Linear-quadratic analysis of radiosensitisation by halogenated
pyrimidines. II Radiosensitisation of human colon cancer cells by
bromodeoxyuridine. Radiat Res. 131:90–97. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
94.
|
Jones GD, Ward JF, Limoli CL, Moyer DJ and
Aguilera JA: Mechanisms of radiosensitization in
iododeoxyuridine-substituted cells. Int J Radiat Biol. 67:647–653.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
95.
|
Bouchard VJ, Rouleau M and Poirier GG:
PARP-1, a determinant of cell survival in response to DNA damage.
Exp Hematol. 31:446–454. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
96.
|
Rouleau M, Patel A, Hendzel MJ, Kaufmann
SH and Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev
Cancer. 10:293–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97.
|
Löser DA, Shibata A, Shibata AK, Woodbine
LJ, Jeggo PA and Chalmers AJ: Sensitization to radiation and
alkylating agents by inhibitors of poly(ADP-ribose) polymerase is
enhanced in cells deficient in DNA double-strand break repair. Mol
Cancer Ther. 9:1775–1787. 2010.PubMed/NCBI
|
|
98.
|
Cate RT, Krawczyk P, Stap J, Aten JA and
Franken NAP: Radiosensitizing effect of the histone
acetyltransferase inhibitor anacardic acid on various mammalian
cell lines. Oncol Lett. 1:765–769. 2010.PubMed/NCBI
|
|
99.
|
Rodermond HM, Ten Cate R, Haveman J, van
Kuilenburg A, Medema JP, van Bree C and Franken NAP:
Cyclopentenylcytosine does not enhance cisplatin-induced
radiosensitization in human lung tumour cells. Oncol Lett.
1:537–540. 2010.PubMed/NCBI
|
|
100.
|
Ryu S, Brown SL, Kim SH, Khil MS and Kim
JH: Preferential radiosensitization of human prostatic carcinoma
cells by mild hyperthermia. Int J Radiat Oncol Biol Phys.
34:133–138. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
101.
|
Bergs JWJ, Ten Cate R, Haveman J, Medema
JP, Franken NAP and van Bree C: Chromosome fragments have the
potential to predict hyperthermia-induced radio-sensitization in
two different human tumor cell lines. J Radiat Res. 49:465–472.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
102.
|
van Bree C, Savonije JH, Franken NAP,
Haveman J and Bakker PJ: The effect of p53-function on the
sensitivity to paclitaxel with or without hyperthermia in human
colorectal carcinoma cells. Int J Oncol. 16:739–744.
2000.PubMed/NCBI
|
|
103.
|
van Bree C, van der Maat B, Ceha HM,
Franken NAP, Haveman J and Bakker PJ: Inactivation of p53 and of
pRb protects human colorectal carcinoma cells against
hyperthermia-induced cytotoxicity and apoptosis. J Cancer Res Clin
Oncol. 125:549–555. 1992.PubMed/NCBI
|
|
104.
|
Larsson C and Ng CE: p21+/+
(CDKN1A+/+) and p21−/− (CDKN1A−/−)
human colorectal carcinoma cells display equivalent amounts of
thermal radiosensitization. Radiat Res. 160:205–209. 2003.
|
|
105.
|
Murthy AK, Harris JR and Belli JA:
Hyperthermia and radiation response of plateau phase cells.
Potentiation and radiation damage repair. Radiat Res. 70:241–247.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
106.
|
Xu M, Myerson RJ, Xia Y, Whitehead T,
Moros EG, Straube WL and Roti Roti JL: The effects of 41 degrees C
hyperthermia on the DNA repair protein, MRE11, correlate with
radiosensitization in four human tumor cell lines. Int J
Hyperthermia. 23:343–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107.
|
Xu M, Wright WD, Higashikubo R, Wang LL
and Roti Roti JL: Thermal radiosensitization of human tumour cell
lines with different sensitivities to 41.1 degrees C. Int J
Hyperthermia. 15:279–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
108.
|
Vujaskovic Z and Song CW: Physiological
mechanisms underlying heat-induced radiosensitization. Int J
Hyperthermia. 20:163–174. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
109.
|
Oleson JR: Eugene Robertson Special
Lecture. Hyperthermia from the clinic to the laboratory: a
hypothesis. Int J Hyperthermia. 11:315–322. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
110.
|
Song CW, Shakil A, Osborn JL and Iwata K:
Tumour oxygenation is increased by hyperthermia at mild
temperatures. Int J Hyperthermia. 12:367–373. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
111.
|
Xu M, Myerson RJ, Straube WL, Moros EG,
Lagroye I, Wang LL, Lee JT and Roti Roti JL: Radiosensitization of
heat resistant human tumour cells by 1 hour at 41.1 degrees C and
its effect on DNA repair. Int J Hyperthermia. 18:385–403. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
112.
|
Mackey MA and Ianzini F: Enhancement of
radiation-induced mitotic catastrophe by moderate hyperthermia. Int
J Radiat Biol. 76:273–280. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
113.
|
Wilkins DE, Ng CE and Raaphorst GP:
Cisplatin and low dose rate irradiation in cisplatin resistant and
sensitive human glioma cells. Int J Radiat Oncol Biol Phys.
36:105–111. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
114.
|
Wilkins DE, Heller DP and Raaphorst GP:
Inhibition of potentially lethal damage recovery by cisplatin in a
brain tumor cell line. Anticancer Res. 13:2137–2142.
1993.PubMed/NCBI
|
|
115.
|
Huang H, Huang SY, Chen TT, Chen JC, Chiou
CL and Huang TM: Cisplatin restores p53 function and enhances the
radiosensitivity in HPV16 E6 containing SiHa cells. J Cell Biochem.
91:756–765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
116.
|
Britten RA, Peacock J and Warenius HM:
Collateral resistance to photon and neutron irradiation is
associated with acquired cis-platinum resistance in human ovarian
tumour cells. Radiother Oncol. 23:170–175. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
117.
|
Britten RA, Evans AJ, Allalunis-Turner MJ
and Pearcey RG: Effect of cisplatin on the clinically relevant
radiosensitivity of human cervical carcinoma cell lines. Int J
Radiat Oncol Biol Phys. 34:367–374. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
118.
|
Monk BJ, Burger RA, Parker R, Radany EH,
Redpath L and Fruehauf JP: Development of an in vitro
chemo-radiation response assay for cervical carcinoma. Gynecol
Oncol. 87:193–199. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
119.
|
Meyn RE, Meistrich ML and White RA:
Cycle-dependent anticancer drug cytotoxicity in mammalian cells
synchronized by centrifugal elutriation. J Natl Cancer Inst.
64:1215–1219. 1980.PubMed/NCBI
|
|
120.
|
Krishnaswamy G and Dewey WC: Cisplatin
induced cell killing and chromosomal aberrations in CHO cells:
treated during G1 or S phase. Mutat Res. 293:161–172. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
121.
|
Turchi JJ, Henkels KM and Zhou Y:
Cisplatin-DNA adducts inhibit translocation of the Ku subunits of
DNA-PK. Nucleic Acids Res. 28:4634–4641. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
122.
|
Muggia FM and Fojo T: Platinums: extending
their therapeutic spectrum. J Chemother. 16(Suppl 4): 77–82. 2004.
View Article : Google Scholar
|
|
123.
|
Dewit L: Combined treatment of radiation
and cisdiamminedichloroplatinum (II): a review of experimental and
clinical data. Int J Radiat Oncol Biol Phys. 13:403–426. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
124.
|
Kumala S, Niemiec P, Widel M, Hancock R
and Rzeszowska-Wolny J: Apoptosis and clonogenic survival in three
tumour cell lines exposed to gamma rays or chemical genotoxic
agents. Cell Mol Biol Lett. 8:655–665. 2003.PubMed/NCBI
|
|
125.
|
Guchelaar HJ, Vermes I, Koopmans RP,
Reutelingsperger CP and Haanen C: Apoptosis- and necrosis-inducing
potential of cladribine, cytarabine, cisplatin, and 5-fluorouracil
in vitro: a quantitative pharmacodynamic model. Cancer Chemother
Pharmacol. 42:77–83. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
126.
|
Fujita M, Fujita T, Kodama T, Tsuchida T
and Higashino K: The inhibitory effect of cisplatin in combination
with irradiation on lung tumor cell growth is due to induction of
tumor cell apoptosis. Int J Oncol. 17:393–397. 2000.PubMed/NCBI
|
|
127.
|
Ostruszka LJ and Shewach DS: The role of
cell cycle progression in radiosensitization by
2′,2′-difluoro-2′-deoxycytidine. Cancer Res. 60:6080–6088.
2000.
|
|
128.
|
Ruiz van Haperen VW, Veerman G, Eriksson
S, Boven E, Stegmann AP, Hermsen M, Vermorken JB, Pinedo HM and
Peters GJ: Development and molecular characterization of a
2′,2′-difluorodeoxycytidine-resistant variant of the human ovarian
carcinoma cell line A2780. Cancer Res. 54:4138–4143. 1994.
|
|
129.
|
Peters GJ, Ruiz van Haperen VW, Bergman
AM, Veerman G, Smitskamp-Wilms E, van Moorsel CJ, Kuiper CM and
Braakhuis BJ: Preclinical combination therapy with gemcitabine and
mechanisms of resistance. Sem Oncology. 23(Suppl 10): 16–24.
1996.PubMed/NCBI
|
|
130.
|
Wang Y, Pantelias GE and Iliakis G:
Mechanism of radiosensitization by halogenated pyrimidines: the
contribution of excess DNA and chromosome damage in BrdU
radiosensitization may be minimal in plateau-phase cells. Int J
Radiat Biol. 66:133–142. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
131.
|
Webb CF, Jones GD, Ward JF, Moyer DJ,
Aguilera JA and Ling LL: Mechanisms of radiosensitisation in
bromodeoxyuridine-substituted cells. Int J Radiat Biol. 64:695–705.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
132.
|
Albert JM, Cao C, Kim KW, Willey CD, Geng
L, Xiao D, Wang H, Sandler A, Johnson DH, Colevas AD, Low J,
Rothenberg ML and Lu B: Inhibition of poly(ADP-ribose) polymerase
enhances cell death and improves tumor growth delay in irradiated
lung cancer models. Clin Cancer Res. 13:3033–3042. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
133.
|
Douglas BG and Fowler JF: The effect of
multiple small doses of x-rays on skin reactions in the mouse and a
basic interpretation. Radiat Res. 66:401–426. 1976. View Article : Google Scholar
|