Introduction
Proteasome inhibition has emerged as an important
therapeutic strategy for multiple myeloma (MM). For many years, the
combination of oral melpharan and prednisolone (MP) has been a
conventional treatment for MM patients. Bortezomib (Velcade, BZ)
became the first-in-class proteasome inhibitor to be introduced
into clinic treatment 10 years ago and is now predominantly used in
combination regimens such as VMP (consisting of BZ and MP) and VTD
(BZ plus thalidomide and dexamethasone) in MM patients (1–4).
Although regimen involving BZ has contributed to substantial
improvement in survival in MM compared to conventional MP therapy,
MM is still an incurable neoplasm with median survival ranging from
3 to 6 years (3–5). Therefore, further therapeutic
improvement remains a crucial issue.
Constitutive nuclear factor (NF)-κB activity in MM
cells mediates survival, as well as resistance to chemotherapy and
radiotherapy, by inducing the expression of anti-apoptotic
proteins, adhesion molecules and autocrine growth factors such as
interleukin-6 (6,7). Since IκBα is a substrate of the
proteasome, the initial rationale for using BZ in MM was to inhibit
NF-κB (8,9). However, recent reports demonstrated
instead that BZ activates the canonical pathway of NF-κB in MM and
lymphoma cells; therefore, the inhibition of NF-κB activity is not
involved in the therapeutic effect of BZ (10–14).
It was reported that BZ-induced calpain-dependent IκBα degradation
facilitated p65 nuclear translocation and increased NF-κB activity
(12). It was also demonstrated
that BZ treatment promoted IκBα phosphorylation, ubiquitination and
degradation via the autophagy-lysosome degradation system,
resulting in increased NF-κB nuclear translocation and
transcription activity in diffuse large B-cell lymphoma cells.
Therefore, blocking autophagy with chloroquine prevented BZ-induced
NF-κB activation by reducing IκBα degradation and enhanced
BZ-induced killing of lymphoma cells (13). Immunohistochemistry using anti-p65
antibody in MM cells derived from 60 samples of MM patients
confirmed that NF-κB was almost exclusively expressed in the
cytoplasm, which indicated its inactive form. In addition, BZ
exhibited consistent antitumor activity against MM cells,
regardless of NF-κB localization (14). All these data suggest the existence
of another molecular mechanism underlying BZ-mediated
cytotoxicity.
Increasing lines of evidence indicate that
inhibition of the 26S proteasome by BZ leads to the accumulation of
misfolded proteins in the endoplasmic reticulum (ER) (10,15–19).
This results in ER stress followed by a coordinated cellular
response known as unfolded protein response (UPR). Since MM is
characterized by the uncontrolled cell growth of monoclonal
antibody-producing plasma cells, large quantities of unfolded or
misfolded immunoglobulin production itself triggers ER stress. ER
stress is caused by an imbalance between the amount of unfolded or
misfolded protein in the ER lumen and the capacity of the ER
machinery to refold these proteins (20). The main functions of UPR are to
reduce the amount of protein that enters the ER by suppuration of
translational rate and to increase the folding capacity of the ER
via translational activation of chaperon proteins. Additionally, if
proteins cannot be folded correctly in the ER, they are
retrotranslocated to the cytoplasm for degradation via the
ubiquitin-proteasome pathway, a process termed ER-associated
degradation (ERAD) for adaptation. However, if these strategies for
adaptation fail, apoptosis is triggered with the induction of a
proapoptotic transcription factor CHOP and with the IRE1 involved
in signaling via caspase-12 (20–22).
Thus, therapeutic manipulation of this pathway using BZ and other
reagents might interfere with the ability to deal with high protein
loads, cellular stress and might result in induction of MM cell
death (10,18).
Macroautophagy (hereafter, autophagy) is a highly
conserved cellular process in eukaryotes. Intracellular proteins
and organelles including the ER are engulfed in a double-membrane
vesicle called an autophagosome and are delivered to lysosomes for
degradation by lysosomal hydrolases (23,24).
Autophagy has been regarded as a bulk non-selective degradation
system for long-lived proteins and organelles, in contrast to the
specific degradation of polyubiquitinated short-lived proteins by
proteasome. However, recent reports revealed the selective
degradation pathway of ubiquitinated protein through autophagy via
docking proteins such as p62 and the related protein NBR1, having
both a microtubule-associated protein 1 light chain 3
(LC3)-interacting region and a ubiquitin-associated domain
(25,26). LC3 is essential for autophagy and
is associated with autophagosome membranes after processing
(27). By binding ubiquitin via
their C-terminal ubiquitin-associated domains, p62-mediated
degradation of ubiquitinated cargo occurs by selective autophagy.
Thus the two major intracellular degradation systems are directly
linked (25,26). We have reported on the inhibition
of autophagy using the autophagy inhibitor bafilomycin
A1 enhanced BZ-induced apoptosis by burdening ER stress
in MM cell lines (10). It was
also reported that macrolide antibiotics such as clarithromycin
(CAM) and azithromycin (AZM) attenuated or blocked autophagy flux,
probably mediated through inhibiting the lysosomal function
(28,29).
We therefore investigated whether simultaneous
inhibition of protein degradation systems such as the
ubiquitin-proteasome system by BZ and the autophagy-lysosome system
by a macrolide antibiotic enhances the loading of ER-stress and
ER-stress-mediated CHOP induction, followed by transcriptional
activation for proapoptotic genes. In the present study, we clearly
demonstrate that the combination of BZ and a macrolide such as AZM,
CAM, or erythromycin (EM) enhances ER stress-mediated cytotoxicity
via transcriptional activation of CHOP. Our data suggest that BZ
and a macrolide antibiotic is a promising combination for MM
therapy.
Materials and methods
Reagents
BZ was purchased from Toronto Research Chemical Inc.
(North York, Ontario, Canada). BZ was dissolved in dimethyl
sulfoxide (DMSO) at a concentration of 1 mM as a stock solution.
CAM, bafilomycin A1 and concanamycin A were purchased
from Wako Pure Chemical Industries (Osaka, Japan), EM was purchased
from Sigma-Aldrich (St. Louis, MO) and AZM was purchased from Tokyo
Chemical Industry (Tokyo, Japan). Bafilomycin A1,
concanamycin A and AZM were dissolved in DMSO to make stock
solutions of 10 μM, 10 μM, and 10 mg/ml,
respectively. CAM and EM were dissolved in ethanol to make stock
solutions of 5 and 10 mg/ml. E-64d and pepstatin A, which are
inhibitors of lysosomal proteases, were purchased from Peptide
Institute (Osaka, Japan).
Cell lines and culture conditions
For this study, MM cell lines IM-9, U266 and
RPMI8226 cells were obtained from the American Type Culture
Collection (ATCC) (Manassas, VA). A CHOP−/− MEF cell
line (CHOP-KO-DR) established from a 13.5-day-old
CHOP−/− mouse embryo by SV-40 immortalization and a
CHOP+/+ MEF cell line (DR-wild-type) established by
SV-40 immortalization as a control cell line for CHOP-KO-DR were
also obtained from ATCC. IM-9, U266 and RPMI8226 cells were
maintained in continuous culture in RPMI-1640 medium (Gibco, Grand
Island, NY) supplemented with 10% FBS (PAA Laboratories, Austria),
2 mM L-glutamine, penicillin (100 U/ml) and streptomycin (100
μg/ml) (Wako) CHOP-KO-DR and DR-wild-type cells were
maintained in Dulbecco’s modified Eagle’s medium (Sigma)
supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin
(100 μg/ml). All cell lines were cultured in a humidified
incubator containing 5% CO2 and 95% air at 37°C.
Assessment of the viable number of cells
among cultured cells
The number of viable cells was assessed by CellTiter
Blue, a cell viability assay kit (Promega Co., Madison, WI), with
fluorescence measurements at 570 nm for excitation and 590 nm for
fluorescence emission.
Immunoblotting
Immunoblotting was performed as previously described
(30). In brief, cells were lysed
with RIPA lysis buffer (Nacalai Tesque, Kyoto, Japan) containing 1
mM PMSF, 0.15 U/ml aprotinin, 10 mM EDTA, 10 mg/ml sodium fluoride
and 2 mM sodium orthovanadate. Cellular proteins were quantified
using a DC Protein Assay kit of Bio-Rad (Richmond, CA). Equal
amounts of proteins were loaded onto the gels, separated by
SDS-PAGE and transferred onto Immobilon-P membrane (Millipore
Corp., Bedford, MA). The membranes were probed with first
antibodies (Abs) such as anti-LC3B antibody (Ab) (Novus Biological,
Inc., Littleton, CO), anti-p62 monoclonal (m) Ab (sequestsome-1),
anti-ubiquitin mAb and anti-GAPDH mAb (Santa Cruz, CA),
anti-cleaved-caspase-3 Ab, anti-PARP Ab, anti-CHOP mAb (Cell
Signaling Technology, MA). Immunoreactive proteins were detected
with horseradish peroxidase-conjugated second Abs and an enhanced
chemiluminescence reagent (ECL) (Millipore). Densitometry was
performed using a Molecular Imager, ChemiDoc XRS System
(Bio-Rad).
Gene expression analysis
Total RNA was isolated from cell pellets using
Isogen (Nippon Gene, Tokyo, Japan) and genomic DNA was removed
using RQ1 RNase-Free DNase (Promega) at 37°C for 30 min, followed
by extraction with phenol chloroform and ethanol precipitation.
Reverse-transcription using a PrimeScript RT Master Mix (Takara Bio
Inc. Ohtsu, Japan) was performed according to the manufacturer’s
instructions. Real-time PCR was performed on 3 ng of cDNA using
validated SYBR Green gene expression assays for human and mouse
ER-stess related genes (CHOP, BAX, BIM, DR5, GADD34 and
TRB3) in combination with SYBR Premix Ex Taq II Tli RNase H
Plus (Takara Bio Inc.). The sequences of primers are listed in
Table I. Quantitative real-time
PCR was performed in duplicates in a Thermal Cycler Dice Real-Time
System TP800 (Takara) under the following conditions: initial cDNA
denaturation at 95°C for 30 sec, followed by 45 cycles of the
sequence of denaturation at 95°C for 5 sec and simultaneous
annealing and extension at 60°C for 30 sec. The data were analyzed
using Thermal Cycler Dice Real-Time System Software (Takara) and
the comparative Ct method
(2−ΔΔCt) was used for relative
quantification of gene expression. The data of real-time PCR
products were standardized to GAPDH as an internal control.
To confirm the specific amplification of target genes, each gene
product was further separated by 1.5% agarose gel after real-time
PCR to detect a single band at the theoretical product size, as
well as analysis of the dissociation curve for detecting a single
peak.
 | Table ISequence of primers for real-time
PCR. |
Table I
Sequence of primers for real-time
PCR.
| Symbol | Species | Accession no. | Forward
(5′-3′) | Reverse
(5′-3′) | Products size
(bp) |
|---|
| CHOP | h | NM_004083.5 |
AAATCAGAGCTGGAACCTGAGGA |
CCATCTCTGCAGTTGGATCAGTC | 112 |
| m | NM_007837.3 |
AATAACAGCCGGAACCTGAGGA |
CCCAATTTCATCTGAGGACAGGA | 200 |
| BAX | h | NM_138761.3 |
GAACCATCATGGGCTGGACA |
CCACAAAGATGGTCACGGTCTG | 132 |
| m | NM_007527.3 |
CAGGATGCGTCCACCAAGAA |
GTTGAAGTTGCCATCAGCAAACA | 165 |
| BIM | h | NM_207002.2 |
CATCATCGCGGTATTCGGTTC |
AAGGTTGCTTTGCCATTTGGTC | 141 |
| m | NM_207680.2 |
TCCTGTGCAATCCGTATCTCC |
CGCAAGCTTCCATACGACAGT | 70 |
| DR5 | h | NM_003842.4 |
AAGTGCCGCACAGGGTGTCC |
GCTGGGACTTCCCCACTGTGC | 116 |
| m | NM_020275.4 |
GTCCAGCTGGCCTACAGC |
GCTTGCAGTTCCCTTCTGAC | 87 |
| GADD34 | h | NM_014330.3 |
AACCAGCAGTTCCCTTCCTG |
TTGCCTCTCGCTCACCATAC | 74 |
| m | NM_008654.2 |
AGGAGAAGCTGGGTCCCTAC |
GGTCACATCTTGGGTCAAGG | 131 |
| TRB3 | h | NM_021158.3 |
CGCTGACCGTGAGAGGAAGAAGC |
TCGGCTGCCTTGCCCGAGTA | 159 |
| m | NM_175093.2 |
CGCTTTGTCTTCAGCAACTGT |
TCATCTGATCCAGTCATCACG | 83 |
| GAPDH | h | NM_002046.3 |
GCACCGTCAAGGCTGAGAAC |
TGGTGAAGACGCCAGTGGA | 138 |
| m | NM_008084.2 |
TGTGTCCGTCGTGGATCTGA |
TTGCTGTTGAAGTCGCAGGAG | 150 |
Assessment of aggresome formation
Assessment of aggresome formation was performed
using a ProteoStat® Aggresome Detection kit according to
the manufacturer’s instructions (Enzo Life Sciences, Farmingdale,
NY) (31). Cells were fixed with
4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and
incubated with ProteoStat aggresome dye. Aggresome was analyzed by
flow cytometry using a Partec PAS I Flow Cytometer (Partec,
Münster, Germany) with a 488-nm laser with fluorescence detection
in the FL3 channel. After staining with ProteoStat aggresome dye,
cells were further stained with 4′,6-diamidino-2-phenylindole
(DAPI) and cell suspensions were sedimented and fixed on slide
glasses using Shandon Cytospin III (Shandon Southern Products Ltd.,
Cheshire, UK) to make slide glass preparations. Analysis by
fluorescence microcopy was performed using a Texas Red filter for
imaging the cell aggresome signal and a DAPI filter for imaging the
nuclear signal using a digital microscope BZ-9000 (Keyence Co.,
Osaka, Japan).
Assessment of apoptosis
Cells were stained with Annexin V and propidium
iodide (PI) using an Annexin V-FITC Apoptosis Detection kit (Wako)
according to the manufacturer’s protocol. Fluorescent intensities
were detected by flow cytometry using a Partec PAS I flow cytometer
(Partec). Annexin V-FITC binding was monitored using an FITC signal
detector (FL1, 520 nm) and PI staining was monitored phycoerythrin
emission signal detector (FL3, 590–650 nm). We also performed
morphological observation for assessment of apoptosis. Cell
suspensions were sedimented and fixed on slide glasses using
Shandon Cytospin III (Shandon Southern Products Ltd.); preparations
were then stained with May-Grünwald-Giemsa and examined using a
digital microscope BZ-9000 (Keyence Co.).
Statistics
All data are given as the mean ± SD. Statistical
analysis was performed by using Mann-Whitney’s U test
(two-tailed).
Results
Apoptosis and autophagy induction after
treatment with BZ in MM cell lines
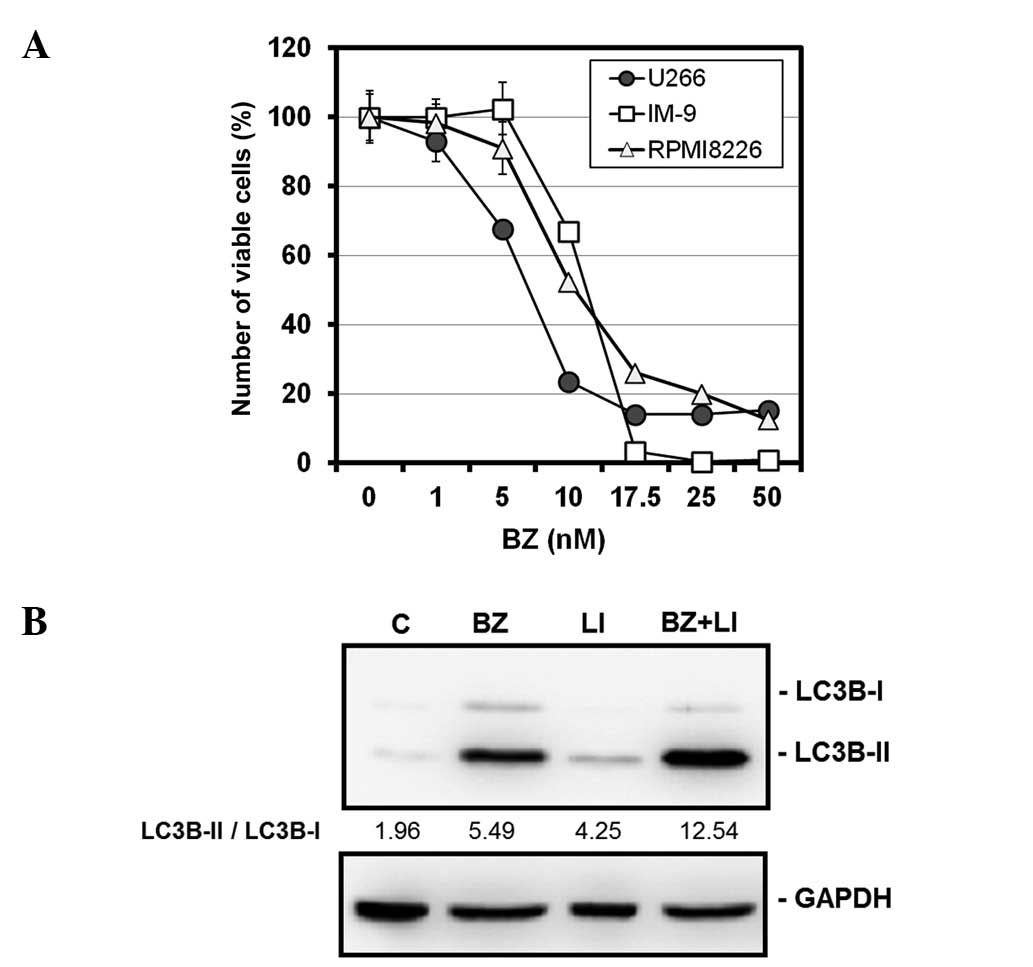
BZ induced cell growth inhibition in a
dose-dependent manner in all three MM cell lines tested.
IC50 (50% inhibitory concentrations) of each cell line
was 7.2 nM for U266, 10.5 nM for RPMI8226 and 12.2 nM for IM-9,
respectively (Fig. 1A).
Morphological features and immunoblotting with anti-cleaved
caspase-3 and anti-PARP Abs all revealed apoptosis induction after
treatment with BZ, as previously reported elsewhere (10). Immunoblottings with anti-LC3B and
anti-p62 Abs demonstrated that treatment with myeloma cells with BZ
resulted in increased expression ratios of LC3B-II to LC3B-I, along
with decreased expression levels of p62 (10). Combined treatment with BZ and
lysosomal inhibitors such as pepstatin A and E64d further increased
the ratio of LC3II-B to LC3B-I, compared with those after treatment
with either BZ or lysosomal inhibitors alone in U266 cells
(Fig. 1B). This result indicated
that increased ratios of LC3B-II to LC3B-I in response to BZ are
due to autophagy induction rather than blocking autophagic flux as
previously reported (27,32).
Macrolide antibiotics blocked autophagy
flux and sensitized to BZ in MM cells
We previously reported that combined treatment with
BZ and bafilomycin A1, which is an autophagy inhibitor,
synergistically enhanced ER-stress-mediated apoptosis in MM cells
(10). Recent reports demonstrated
that CAM attenuated the late stage of autophagy, although its
mechanism still remains unclear (28). Additionally, AZM has been reported
to block autophagy in macrophage (29). Since bafilomycin A1 is a
macrolide, we speculated that macrolide antibiotics might share the
same target(s) for blocking autophagy and might induce the same
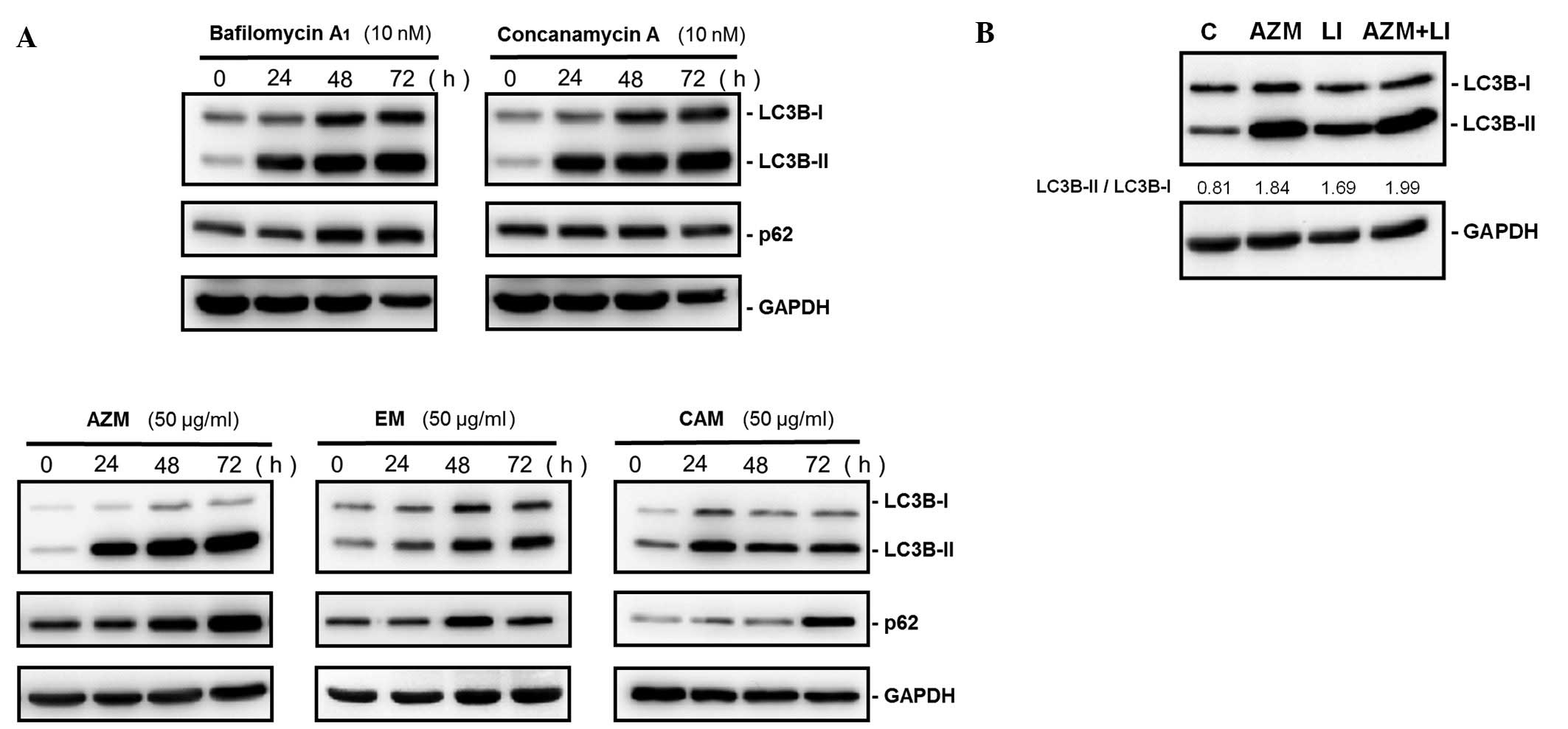
effect in MM cell growth. As indicated in Fig. 2A, immunoblotting with anti-LC3B Ab
demonstrated that treatment of U266 cells with bafilomycin
A1, concanamycin A, AZM, CAM or EM increased the
expression ratios of LC3B-I to LC3B-II. However, p62, which is a
substrate of autophagylysosomal proteolysis, increased after
treatment with these macrolides. Unlike BZ, combined treatment with
lysosomal inhibitors and AZM did not indicate any further increase
of the LC3B-II/LC3B-I ratios, compared with those by treatment with
lysosomal inhibitors or AZM alone (Fig. 2B). These data indicate that all
macrolide antibiotics tested block autophagy flux.
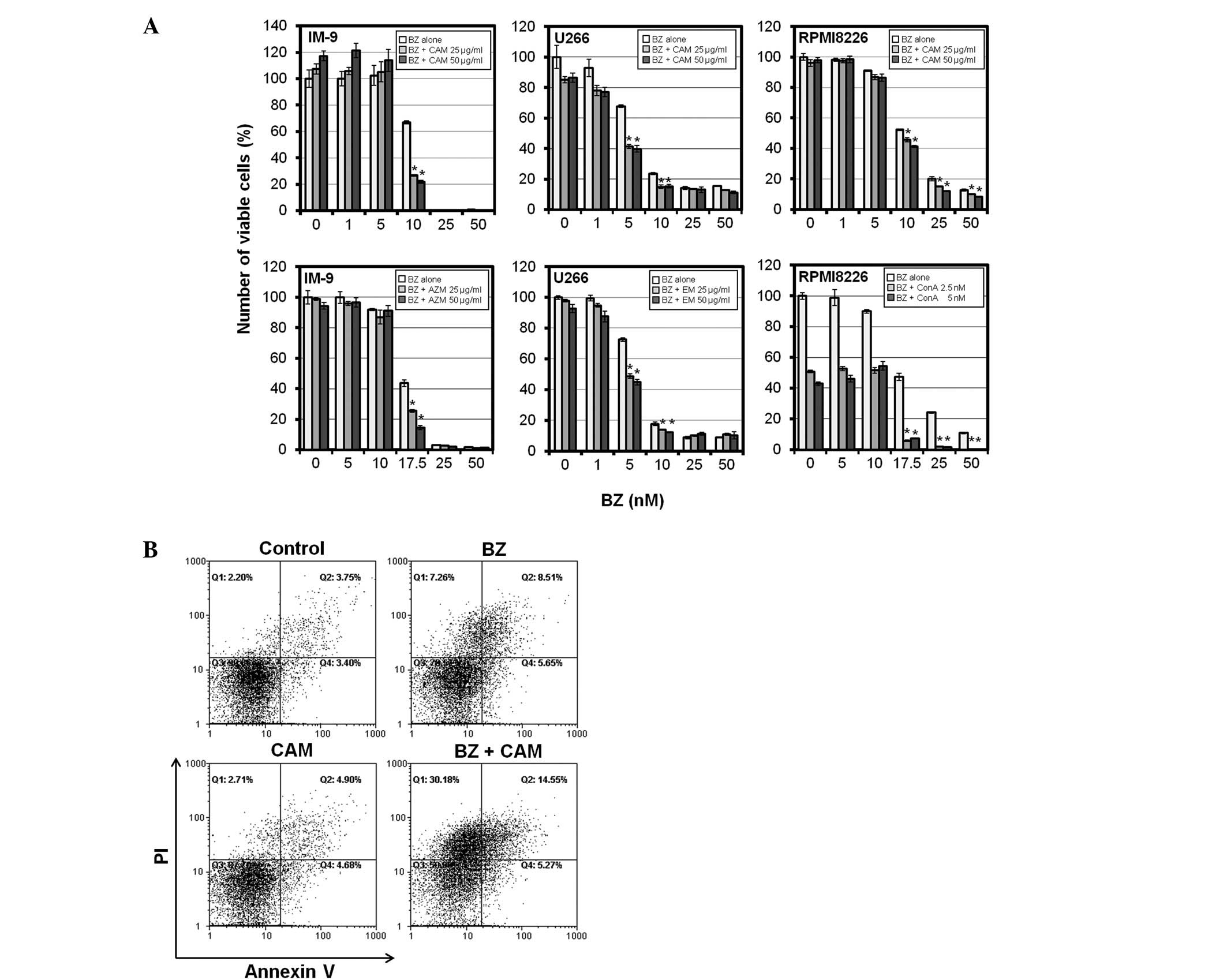
We next investigated whether a macrolide antibiotic
increases the sensitivity of BZ in MM cells as well as bafilomycin
A1(10). Treatment with
AZM, CAM, or EM alone indicated little or almost no cytotoxicity at
up to 100 μg/ml in MM cell lines (data not shown). However,
a combination of AZM, CAM, or EM (at 25 and 50 μg/ml) with
BZ enhanced BZ-induced cytotoxicity in MM cell lines including
IM-9, U266 and RPMI8226 (Fig. 3A).
In addition, flow cytometry of PI/Annexin V double staining
revealed that CAM enhanced BZ-induced apoptosis in IM-9 cells,
although treatment with CAM alone indicated no apoptosis induction
(Fig. 3B).
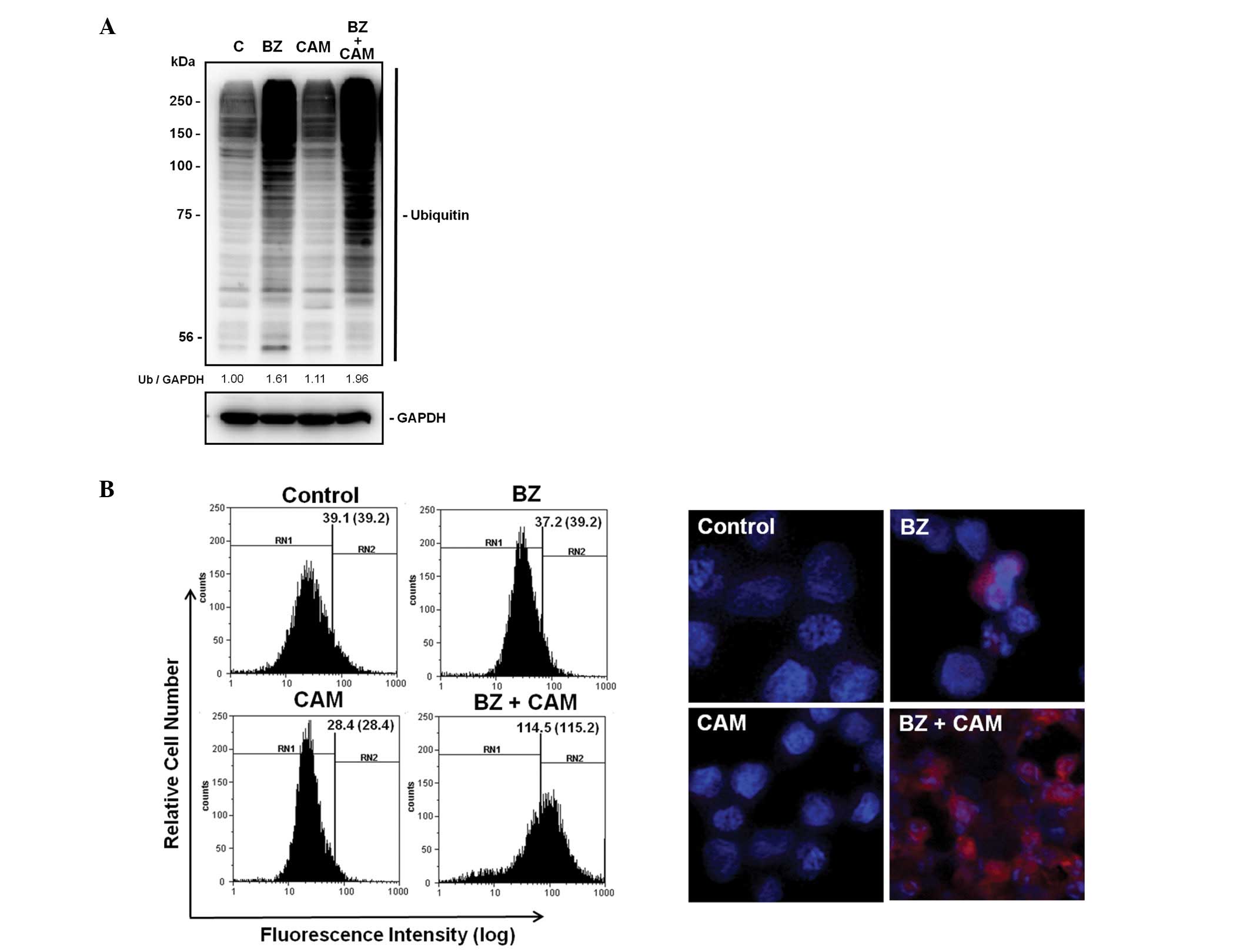
Accumulation of ubiquitinated proteins
and aggresome formation after combined treatment with CAM plus BZ
in MM cells
All data presented above suggest that two major
intracellular proteolytic systems (e.g., the ubiquitin-proteasome
system and the autophagy-lysosome system) could be simultaneously
blocked by combined treatment with BZ and a macrolide antibiotic.
It was reported that, in addition to proteasome-mediated protein
degradation, polyubiquitinated proteins are also degraded by the
autophagy-lysosome pathway via docking protein p62 which has both a
ubiquitin-associated domain and an LC3-interacting lesion (25,26).
Immunoblotting with anti-ubiquitin Ab indicate that treatment of
U266 cells with BZ plus CAM further increased intracellular
ubiquitinated proteins, compared with that by BZ treatment, while
treatment with CAM alone had no effect on protein ubiquitination.
Furthermore, aggresome formation was dramatically increased after
combined treatment with BZ plus CAM in IM-9 cells (Fig. 4).
Involvement of CHOP induction for
enhanced cytotoxicity by combined treatment with BZ and CAM against
MM cells
It has been reported that ER-stress-mediated CHOP
induction is involved in the cytotoxicity of BZ in various kinds of
cells (10,33,34).
This was also supported by data indicating that translational
inhibition using cycloheximide attenuated BZ-induced cytotoxicity
in U266 cells (data not shown). Therefore, we next examined whether
combined treatment with BZ and CAM increases ER stress-loading on
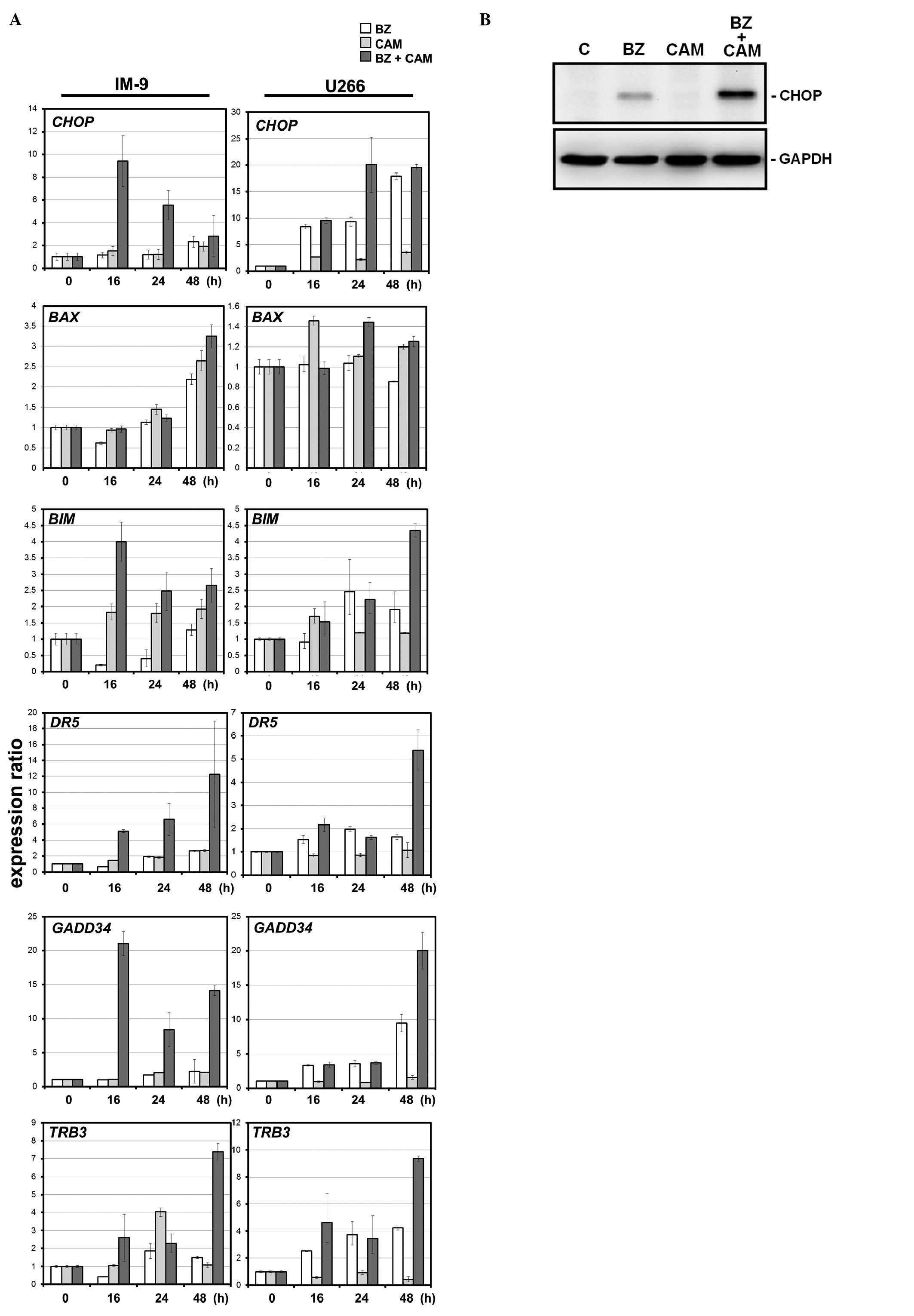
MM cells. Real-time PCR indicated that the levels of
ER-stress-related genes were more pronounced by combined treatment
with BZ and CAM than with either BZ or CAM alone (Fig. 5). Treatment with CAM alone
indicated little effect on gene expression. In addition,
proapoptotic genes (BIM, BAX, DR5 and TRB3) that are
transcriptionally regulated by CHOP (35) were more pronounced with combined
treatment than with treatment with BZ alone. These data strongly
suggested that the simultaneous inhibition of two major protein
degradation systems resulted in the enhancement of ER stress and
appeared to lead to CHOP activation and subsequent apoptosis
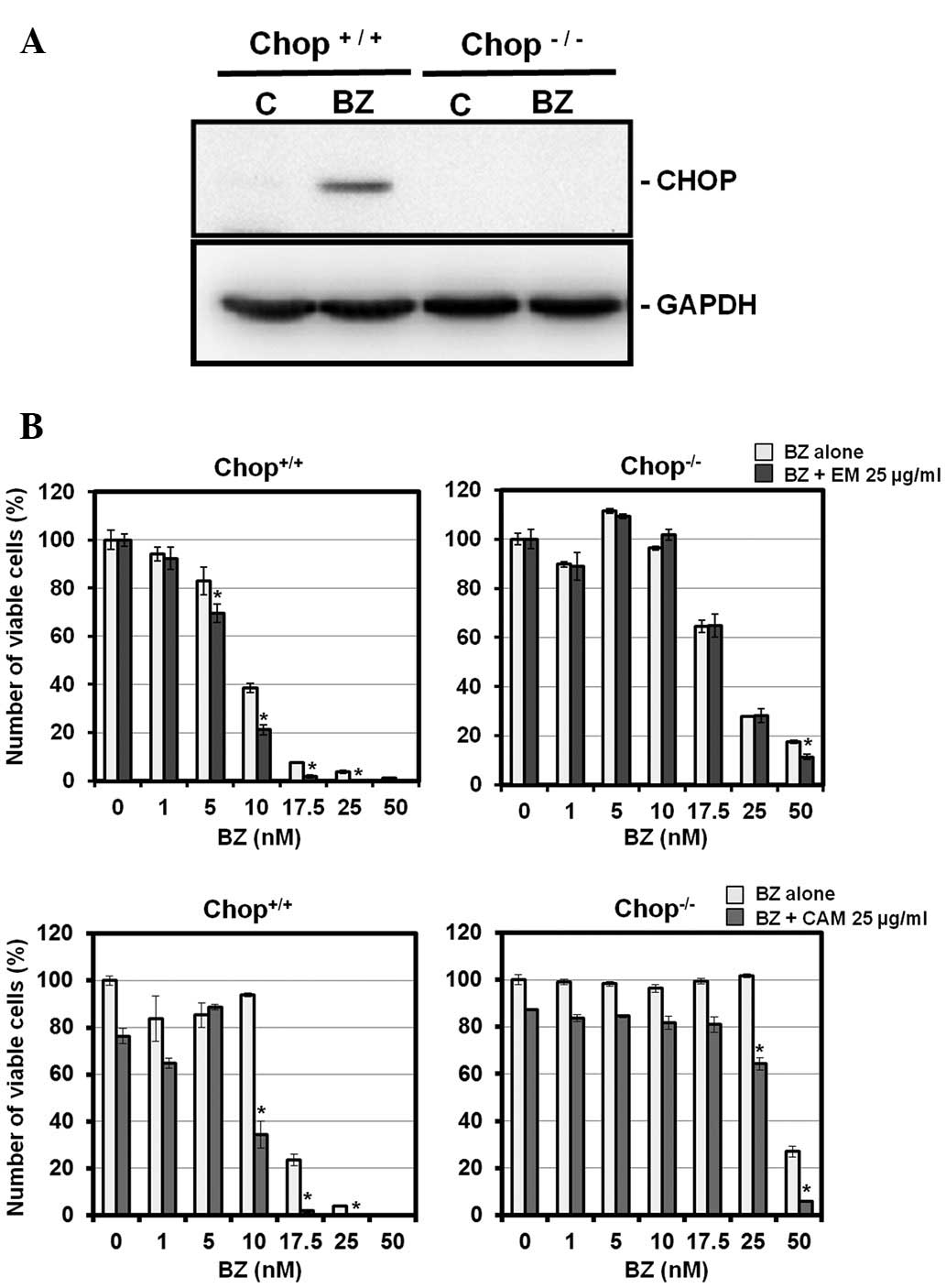
induction. To prove this hypothesis, we used a CHOP knockout MEF
cell line. Fig. 6 illustrates that
CHOP−/− MEF cells were more resistant to BZ than
wild-type MEF cells. Pronounced cytotoxicity was detected with
combined treatment with BZ and EM or CAM in wild-type MEF cells as
well as MM cell lines. It is noteworthy that this enhancement was
almost completely canceled in CHOP−/− MEF cells. This
result indicates that cytotoxicity enhanced by a combination of BZ
and EM or CAM is mediated through CHOP induction. Like MM cell
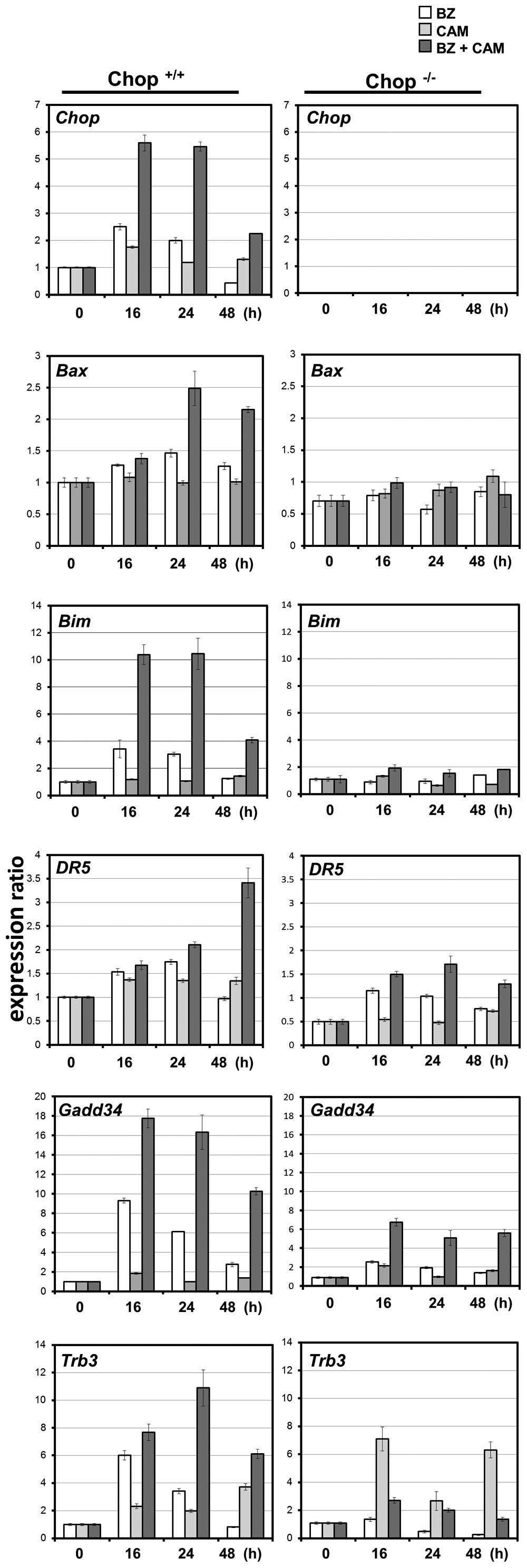
lines, the expression profiles of CHOP-regulated proapoptotic genes
were all pronounced with a combination of BZ and CAM in wild-type
MEF cells, but not in CHOP−/− MEF cells (Fig. 7).
Discussion
In the present study, we demonstrated that treatment
with AZM, CAM, or EM, all of which are widely used macrolide
antibiotics in routine medical care, enhanced BZ-induced
cytotoxicity in MM cells, although these macrolides themselves
exhibited almost no cytotoxicity (Fig.
3). Furthermore, we clearly demonstrated that combined
treatment with BZ and one of the macrolides enhances CHOP induction
and the expression levels of the proapoptotic genes
transcriptionally regulated by CHOP (Figs. 5 and 7). Since CHOP knockout MEF cells
completely canceled the enhanced cytotoxicity (Fig. 6), ER-stress-mediated CHOP induction
appears to be involved in this phenomenon. In addition to the
ubiquitinproteasome system, it was reported that polyubiquitinated
proteins are engulfed into autophagosome and are degraded by the
autophagy-lysosome system via binding to p62 docking protein, which
has both an LC3-interacting region and a ubiquitin-associated
domain (25,26). Thus, by binding ubiquitin via their
C-terminal ubiquitin-associated domains, p62-mediated degradation
of ubiquitinated cargo occurs by selective autophagy. First, we
demonstrated that macrolide antibiotics suppressed autophagy flux,
as previously reported with CAM and AZM (Fig. 2) (28,29).
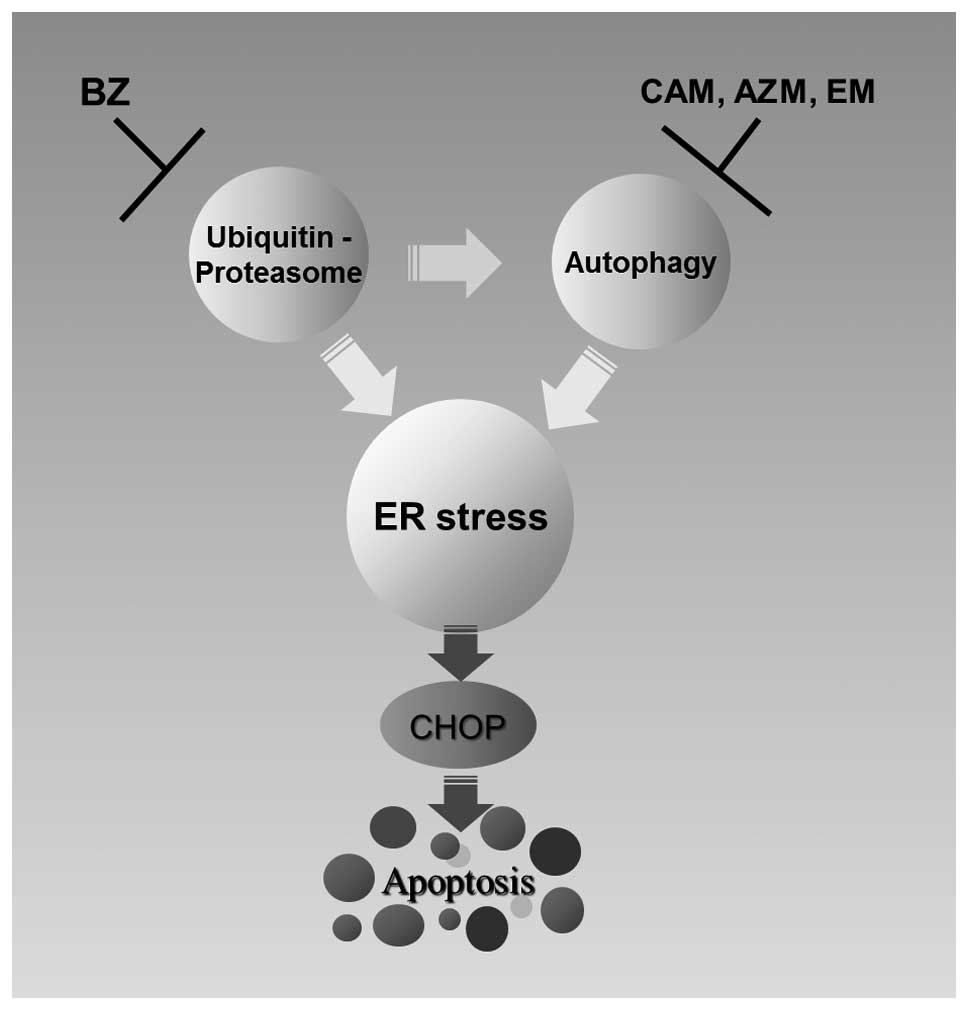
Therefore, blocking the two major protein degradation systems
appears to result in loading excess ER stress, due to complete
inhibition of ERAD (36,37). This was supported by our
observation that intercellular ubiquitinated proteins were
increased by BZ plus CAM, compared with that by BZ alone (Fig. 4A). Second, aggresome formation was
dramatically increased by combining two reagents (Fig. 4B). Third, the expressions of
ER-stress-related genes, including CHOP, were increased by combined
treatment (Figs. 5 and 7). Therefore, simultaneous inhibition of
the ubiquitin-proteasome system and the autophagy-lysosome system
enhances ER-stress-mediated apoptosis in MM cells (Fig. 8).
A similar phenomenon was previously reported
regarding enhanced cytotoxicity by the combination of BZ and an
inhibitor for histone deacetylase 6 (HDAC6) (38). Unfolded proteins are transported to
microtubule-organizing center (MTOC), where the lysosomes are
enriched and degraded through the autophagy-lysosome pathway. HDAC6
deacetylates α-tubulin, which is thought to be a component of the
MTOC; and knockdown of HDAC6 resulted in reducing autophagy
(38). Tubacin, a small molecule
inhibitor of HDAC6, prevented deacetylation of α-tubulin and
produced accumulation of polyubiquitinated proteins and apoptosis
and further acts synergistically with BZ to induce cytotoxicity in
MM cells (39). Based on our
results presented here, these data also can be explained by
enhanced loading of ER-stress by simultaneously targeting the
autophagy-lysosome pathway by tubacin and the ubiquitinprotease
pathway by BZ (Fig. 8). In our
system, dramatic enhancement of aggresome formation was detected
(Fig. 4B). Aggresome formation
therefore appears to provide another system for delivery of
aggregated protein from cytoplasm to lysosomes for degradation and
may reduce ER stress (40).
The molecular mechanism of autophagy induction in
response to BZ is still unclear. A recent report demonstrated that
BZ treatment induced autophagy in the breast cancer cell line MCF-7
by the proteasomal stabilization of ATF4 and ATF4-dependent
upregulation of LC3B (41). ATF4,
which is a transcription factor and a component of the PERK pathway
in UPR (22), facilitated
autophagy through direct binding to a cyclic AMP response
element-binding site in the LC3B promoter, following upregulation
of LC3B and autophagy induction in response to severe hypoxia
(22,42,43).
Therefore, crosstalk between the autophagy-lysosome system and ER
stress was suggested (10,22). Although macrolides blocked the
autophagy flux, combined treatment with BZ and one macrolide for
>48 h resulted in enhanced autophagy induction, compared with BZ
alone in U266 cells (data not shown).
The unexpected effects of macrolide antibiotics on
MM cells discussed here are supported by several previous reports
(28,29,44,45).
CAM was reported to attenuate autophagy and to induce cell growth
inhibition in MM cells (28),
although our data indicated almost no cell growth inhibition. AZM
also reportedly blocked autophagy in macrophage and was thus
assumed to increase the risk of Mycobacterium abcessus
infection in cystic fibrosis patients who need to take AMZ for a
long period (29). Furthermore,
CAM enhanced tyrosine kinase inhibitor (TKI)-induced cell death in
chronic myelogenous leukemia (CML) cells by inhibiting late-stage
autophagy (44). Combining CAM
plus TKI achieved remarkable molecular responses in four
consecutive advanced-CML patients who were resistant to TKI alone
(45). We have observed that CML
cells are constitutively exposed to ER-stress with high expression
levels of ER-stress-related genes, including GRP78 and CHOP, which
may be due to abnormal BCR-ABL fusion protein synthesis (Miyazawa
et al, unpublished data). Therefore, enhanced cytotoxicity
could be explained in terms of loading excess ER-stress in CML
cells. Interestingly, BiRD therapy consisting of Baixin (CAM),
Revlimid (lenalidomide) and dexamethasone resulted in high complete
and overall response rates in MM, although the molecular mechanism
for using CAM was not clarified (46,47).
CAM has immunomodulatory properties, partially mediated by the
suppression of interleukin-6 and other inflammatory cytokines.
There might also be a direct anti-neoplastic effect mediated by CAM
(48). Certain macrolide
antibiotics have been reported to exert some antitumor activities
in non-small lung cancer and melanoma (49–52).
Although the underlying molecular mechanism has not yet been
clarified, ER-stress-mediated apoptosis might be involved in some
of these antitumor activities.
It is also important to consider the target(s) of
macrolides for blocking autophagy flux. Our previous report
demonstrated that combined treatment with BZ and bafilomycin
A1 (BAF), often used as an autophagy inhibitor,
synergistically induces MM cell death in vitro (10). BAF is a macrolide antibiotic that
was initially characterized for its selective inhibition of a
proton-pumping V-ATPase (53). At
nanomoler concentrations, BAF disrupts the vesicular proton
gradients and ultimately increases the pH of acidic vesicles
(53). This disruption of
vesicular acidification in response to BAF appears to prevent the
fusion of autophagosomes with lysosomes, resulting in inhibition of
autophagy (54). It was reported
that treatment with AZM increased lysosomal pH in macrophage, which
may lead to inhibition of the lysosomal hydrolases having an
optimal low pH for their enzymatic activities (29). Therefore, V-ATPase is a strong
candidate for the target of macrolides. Further study is required
to identify the target molecule(s) involved in this phenomenon.
Our study confirms that inhibiting the
autophagy-lysosome system with a macrolide antibiotic can strongly
enhance the efficacy of BZ and provides the foundation for clinical
trials of BZ in combination with AZM, CAM, or EM for treating MM
patients.
Acknowledgements
This study was supported in part by
funds from the Private University Strategic Research-Based Support
Project (Molecular Information-based Intractable Disease Research
Project) from the Ministry of Education, Culture, Sports, Science
and Technology of Japan to K.M. (2008–2013) and a Grant-in-Aid for
Scientific Research (C) from the Ministry of Education, Culture,
Sports, Science and Technology of Japan to K.M. (no. 22591050).
References
|
1.
|
Richardson PG, Barlogie B, Berenson J, et
al: A phase 2 study of bortezomib in relapsed, refractory myeloma.
N Engl J Med. 348:2609–2617. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Moreau P, Richardson PG, Cavo M, Orlowski
RZ, San Miguel JF, Palumbo A and Harousseau JL: Proteasome
inhibitors in multiple myeloma: 10 years later. Blood. 120:947–959.
2012.PubMed/NCBI
|
|
3.
|
Anderson KC: New insights into therapeutic
targets in myeloma. Hematology Am Soc Hematol Educ Program.
2011:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bird JM, Owen RG, D’Sa S, et al:
Guidelines for the diagnosis and management of multiple myeloma
2011. Br J Haematol. 154:32–75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Laubach J, Richardson P and Anderson K:
Multiple myeloma. Annu Rev Med. 62:249–264. 2011. View Article : Google Scholar
|
|
6.
|
Feinman R, Siegel DS and Berenson J:
Regulation of NF-kB in multiple myeloma: therapeutic implications.
Clin Adv Hematol Oncol. 2:162–166. 2004.PubMed/NCBI
|
|
7.
|
Li ZW, Chen H, Campbell RA, Bonavida B and
Berenson JR: NF-kappaB in the pathogenesis and treatment of
multiple myeloma. Curr Opin Hematol. 15:391–399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Hideshima T, Chauhan D, Richardson P, et
al: NF-kappa B as a therapeutic target in multiple myeloma. J Biol
Chem. 277:16639–16647. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Chauhan D, Hideshima T, Mitsiades C,
Richardson P and Anderson KC: Proteasome inhibitor therapy in
multiple myeloma. Mol Cancer Ther. 4:686–692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kawaguchi T, Miyazawa K, Moriya S, et al:
Combined treatment with bortezomib plus bafilomycin A1 enhances the
cytocidal effect and induces endoplasmic reticulum stress in U266
myeloma cells: crosstalk among proteasome, autophagy-lysosome and
ER stress. Int J Oncol. 38:643–654. 2011.
|
|
11.
|
Hideshima T, Ikeda H, Chauhan D, et al:
Bortezomib induces canonical nuclear factor-kappaB activation in
multiple myeloma cells. Blood. 114:1046–1052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Li C, Chen S, Yue P, Deng X, Lonial S,
Khuri FR and Sun SY: Proteasome inhibitor PS-341 (bortezomib)
induces calpain-dependent IkappaB(alpha) degradation. J Biol Chem.
285:16096–16104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jia L, Gopinathan G, Sukumar JT and
Gribben JG: Blocking autophagy prevents bortezomib-induced
NF-kappaB activation by reducing I-kappaBalpha degradation in
lymphoma cells. PLoS One. 7:e325842012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Conticello C, Giuffrida R, Adamo L, et al:
NF-kappaB localization in multiple myeloma plasma cells and
mesenchymal cells. Leuk Res. 35:52–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Obeng EA, Carlson LM, Gutman DM,
Harrington WJ Jr, Lee KP and Boise LH: Proteasome inhibitors induce
a terminal unfolded protein response in multiple myeloma cells.
Blood. 107:4907–4916. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Meister S, Schubert U, Neubert K, et al:
Extensive immunoglobulin production sensitizes myeloma cells for
proteasome inhibition. Cancer Res. 67:1783–1792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Periyasamy Thandavan S, Jackson WH,
Samaddar JS, et al: Bortezomib blocks the catabolic process of
autophagy via a cathepsin-dependent mechanism, affects endoplasmic
reticulum stress and induces caspase-dependent cell death in
antiestrogen-sensitive and resistant ER+ breast cancer
cells. Autophagy. 6:19–35. 2010.
|
|
18.
|
Fels DR, Ye J, Segan AT, et al:
Preferential cytotoxicity of bortezomib toward hypoxic tumor cells
via overactivation of endoplasmic reticulum stress pathways. Cancer
Res. 68:9323–9330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ri M, Iida S, Nakashima T, et al:
Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5
in preventing the accumulation of unfolded proteins and fatal ER
stress. Leukemia. 24:1506–1512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Herr I and Debatin KM: Cellular stress
response and apoptosis in cancer therapy. Blood. 98:2603–2614.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Verfaillie T, Salazar M, Velasco G and
Agostinis P: Linking ER stress to autophagy: potential implications
for cancer therapy. Int J Cell Biol. 2010:9305092010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kirkin V, McEwan DG, Novak I and Dikic I:
A role for ubiquitin in selective autophagy. Mol Cell. 34:259–269.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Korolchuk VI, Menzies FM and Rubinsztein
DC: Mechanisms of cross-talk between the ubiquitin-proteasome and
autophagylysosome systems. FEBS Lett. 584:1393–1398. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Nakamura M, Kikukawa Y, Takeya M, Mitsuya
H and Hata H: Clarithromycin attenuates autophagy in myeloma cells.
Int J Oncol. 37:815–820. 2010.PubMed/NCBI
|
|
29.
|
Renna M, Schaffner C, Brown K, et al:
Azithromycin blocks autophagy and may predispose cystic fibrosis
patients to mycobacterial infection. J Clin Invest. 121:3554–3563.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Moriya S, Miyazawa K, Kawaguchi T, Che XF
and Tomoda A: Involvement of endoplasmic reticulum stress-mediated
CHOP (GADD153) induction in the cytotoxicity of
2-aminophenoxazine-3-one in cancer cells. Int J Oncol. 39:981–988.
2011.PubMed/NCBI
|
|
31.
|
Shen D, Coleman J, Chan E, Nicholson TP,
Dai L, Sheppard PW and Patton WF: Novel cell- and tissue-based
assays for detecting misfolded and aggregated protein accumulation
within aggresomes and inclusion bodies. Cell Biochem Biophys.
60:173–185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar
|
|
33.
|
Komatsu S, Miyazawa K, Moriya S, et al:
Clarithromycin enhances bortezomib-induced cytotoxicity via
endoplasmic reticulum stress-mediated CHOP (GADD153) induction and
autophagy in breast cancer cells. Int J Oncol. 40:1029–1039.
2012.
|
|
34.
|
Nawrocki ST, Carew JS, Maclean KH, et al:
Myc regulates aggresome formation, the induction of Noxa, and
apoptosis in response to the combination of bortezomib and SAHA.
Blood. 112:2917–2926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Bays NW, Gardner RG, Seelig LP, Joazeiro
CA and Hampton RY: Hrd1p/Der3p is a membrane-anchored ubiquitin
ligase required for ER-associated degradation. Nat Cell Biol.
3:24–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Tsai YC and Weissman AM: Ubiquitylation in
ERAD: reversing to go forward? PLoS Biol. 9:e10010382011.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Lee JY, Koga H, Kawaguchi Y, et al: HDAC6
controls auto-phagosome maturation essential for
ubiquitin-selective quality-control autophagy. EMBO J. 29:969–980.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Hideshima T, Bradner JE, Wong J, Chauhan
D, Richardson P, Schreiber SL and Anderson KC: Small-molecule
inhibition of proteasome and aggresome function induces synergistic
antitumor activity in multiple myeloma. Proc Natl Acad Sci USA.
102:8567–8572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Rodriguez Gonzalez A, Lin T, Ikeda AK,
Simms Waldrip T, Fu C and Sakamoto KM: Role of the aggresome
pathway in cancer: targeting histone deacetylase 6-dependent
protein degradation. Cancer Res. 68:2557–2560. 2008.PubMed/NCBI
|
|
41.
|
Milani M, Rzymski T, Mellor HR, Pike L,
Bottini A, Generali D and Harris AL: The role of ATF4 stabilization
and autophagy in resistance of breast cancer cells treated with
Bortezomib. Cancer Res. 69:4415–4423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Rzymski T, Milani M, Singleton DC and
Harris AL: Role of ATF4 in regulation of autophagy and resistance
to drugs and hypoxia. Cell Cycle. 8:3838–3847. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Rzymski T, Milani M, Pike L, et al:
Regulation of autophagy by ATF4 in response to severe hypoxia.
Oncogene. 29:4424–4435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Schafranek L, Leclercq TM, White DL and
Hughes TP: Clarithromycin enhances dasatinib-induced cell death in
chronic myeloid leukemia cells, by inhibition of late stage
autophagy. Leuk Lymphoma. 54:198–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Carella AM, Beltrami G, Pica G, Carella A
and Catania G: Clarithromycin potentiates tyrosine kinase inhibitor
treatment in patients with resistant chronic myeloid leukemia. Leuk
Lymphoma. 53:1409–1411. 2012. View Article : Google Scholar
|
|
46.
|
Gay F, Rajkumar SV, Coleman M, et al:
Clarithromycin (Biaxin)-lenalidomide-low-dose dexamethasone (BiRd)
versus lenalidomide-low-dose dexamethasone (Rd) for newly diagnosed
myeloma. Am J Hematol. 85:664–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Niesvizky R, Jayabalan DS, Christos PJ, et
al: BiRD (Biaxin [clarithromycin]/Revlimid
[lenalidomide]/dexamethasone) combination therapy results in high
complete- and overall-response rates in treatment-naive symptomatic
multiple myeloma. Blood. 111:1101–1109. 2008.
|
|
48.
|
Mikasa K, Sawaki M, Kita E, et al:
Significant survival benefit to patients with advanced
non-small-cell lung cancer from treatment with clarithromycin.
Chemotherapy. 43:288–296. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Ohara T, Morishita T, Suzuki H, Masaoka T,
Ishii H and Hibi T: Antibiotics directly induce apoptosis in B cell
lymphoma cells derived from BALB/c mice. Anticancer Res.
24:3723–3730. 2004.PubMed/NCBI
|
|
50.
|
Wada T, Sata M, Sato J, et al:
Clarithromycin suppresses invasiveness of human lung adenocarcinoma
cells. Chemotherapy. 53:77–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Yatsunami J, Fukuno Y, Nagata M, et al:
Roxithromycin and clarithromycin, 14-membered ring macrolides,
potentiate the antitumor activity of cytotoxic agents against mouse
B16 melanoma cells. Cancer Lett. 147:17–24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Hamada K, Kita E, Sawaki M, Mikasa K and
Narita N: Antitumor effect of erythromycin in mice. Chemotherapy.
41:59–69. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar
|
|
54.
|
Klionsky DJ, Elazar Z, Seglen PO and
Rubinsztein DC: Does bafilomycin A1 block the fusion of
autophagosomes with lysosomes? Autophagy. 4:849–950. 2008.
View Article : Google Scholar : PubMed/NCBI
|