Introduction
Renal cell carcinoma (RCC) accounts for 3% of adult
solid tumours and ∼30–40% of patients present with metastatic
disease which has a poor prognosis, with a 5-year survival of
<10%. Identification of novel therapeutic targets or biomarkers
for prognostic, diagnostic or predictive use remains a priority.
Cell surface proteins are involved in a number of vital cellular
processes which are altered in tumourigenesis and constitute ideal
targets for small molecule or antibody based therapies. Soluble
shed forms may also act as circulating biomarkers, making this a
subcellular compartment of particular interest in proteomic-based
biomarker identification studies.
Investigation of the most common form of inherited
RCC led to identification of the von Hippel-Lindau (VHL) tumour
suppressor gene (1) and it is now
clear that loss of VHL function also occurs in a large proportion
of sporadic RCCs of the conventional (clear cell) subtype (2). VHL has been implicated in numerous
biological processes and has a well established role in regulation
of the transcription factor hypoxia-inducible factor (HIF), acting
as the substrate recognition component of an E3 ubiquitin ligase
complex that targets HIF-α subunits for polyubiquitination and
proteasomal degradation in an oxygen-dependent manner. In cells
exposed to hypoxia or lacking functional VHL, HIF-α is stabilised,
resulting in a number of gene expression changes including the
upregulation of vascular endothelial growth factor (VEGF),
platelet-derived growth factor (PDGF) and carbonic anhydrase IX
(CAIX). It is clear that although the HIF pathway is central to VHL
function and tumourigenesis, HIF-independent functions, some of
which involve other substrates of VHL ubiquitin ligase, contribute
to its role (3,4).
Targeting cell surface proteins that are downstream
of VHL is already being exploited, as illustrated by the receptor
tyrosine kinase inhibitors sunitinib and sorafenib (5). Similarly CAIX, one of the most
consistently upregulated proteins in conventional RCC, has been
investigated in many studies including assessment of soluble forms
in serum and urine (6). The plasma
membrane was therefore chosen as a subcellular fraction to focus on
for biomarker identification. During optimisation of a plasma
membrane protein enrichment strategy based on cell surface
biotinylation and avidin affinity chromatography for a comparative
study (7) using the VHL-defective
UMRC2- renal cancer cell line as a model system, purified proteins
were catalogued using 1D PAGE followed by in-gel tryptic digestion
and LC-MS/MS (GeLC-MS/MS). This identified several plasma membrane
proteins previously associated with RCC and a number of proteins of
interest as potential biomarkers due to their dysregulation in
other cancers and/or their known cellular functions. These included
β-dystroglycan, the transmembrane subunit of the dystroglycan 1
protein, which was selected for further study and shown to exhibit
a VHL-dependent change in glycoform in UMRC2 cells. Using an
oligonucelotide array targeting genes involved in glycosylation,
changes in several key enzymes were found in UMRC2−/+VHL cells
supporting a role for VHL-mediated changes in glycosylation in
tumourigenesis. Altered expression of bifunctional
UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase
(GNE) was confirmed in UMRC2−/+VHL cells and also found to occur in
conventional RCC.
Materials and methods
Cell culture and human tissue
samples
VHL-deficient RCC cells (UMRC2, RCC4 and 786-0)
transfected with vector control (−) and VHL (+VHL) were cultured as
previously described (8). For
validation studies, samples of macroscopically viable conventional
RCC representing a range of grades (1–4) and
stages (I–IV) of disease and matched distant normal renal cortical
tissue were selected from a bank of fresh frozen samples collected
and processed as previously described (8) following ethics committee approval and
with informed consent.
Preparation of plasma membrane fractions
and whole cell lysates
A modified protocol of the method described by Zhao
and co-workers (9) was adopted to
isolate cell surface exposed plasma membrane proteins, using
biotin-labelling of cells with EZ-link Sulfo-NHS-S-S-biotin (Perbio
Science UK Limited, Cramlinghton, UK) and subsequent purification
of biotinylated proteins with streptavidin sepharose™ high
performance beads (GE Healthcare, Little Chalfont, UK). Each step
of the protocol was optimised to maximise yield and enrichment of
plasma membrane proteins; details of the final protocol have been
described elsewhere (7). Whole
tissue and cell lysates were prepared in RIPA buffer containing
Complete™ mini protease inhibitor cocktail tablet (1 per 2.5 ml;
Roche, Burgess Hill, UK) or Laemmli sample buffer as previously
described (7).
Western blotting
Western blotting was carried out using the
EnvisionTM+-based detection system (Dako, Ely, UK)
(8). Primary antibodies against
the following proteins were used: rabbit polyclonal antibodies to
GLUT-1 (Abcam plc, Cambridge, UK; 1:8,000), GNE (Sigma-Aldrich,
Poole, UK; 1:250) mouse monoclonal antibodies to β-actin
(Sigma-Aldrich, clone AC15, 1:400,000), β-dystroglycan (BD
Biosciences, San Jose, CA, USA; clone 56, 0.5 μg/ml),
β-dystroglycan (Novocastra, Milton Keynes, UK; clone 43DAG/8D5, 0.1
μg/ml), Golgin-84 (Abcam plc; clone 26, 0.5 μg/ml),
glucose regulated protein (GRP) 94 (Bioquote Limited, York, UK;
clone 9610, 0.5 μg/ ml), heat shock protein (HSP) 70
(Bioquote Limited; clone C92F3A-5, 0.05 μg/ml), lamin A/C
(BD Biosciences; clone 14, 0.75 μg/ml), NADH ubiquinol
oxidoreductase 39 kDa (Invitrogen; clone 20C11, 0.5 μg/ml),
Na/K-ATPase α1 (Novus Biologicals Inc., Littleton, USA; clone
464.4, 0.4 μg/ ml). Western blots were normalised using
parallel Coomassie-stained gels and additionally by probing with
antibodies to β-actin. The optimal concentration of primary
antibodies was pre-determined by titrations using whole UMRC2- cell
lysates and linearity was confirmed by probing serial dilutions of
protein load. Negative control blots were probed with irrelevant
antibodies. Western blots were scanned as 12-bit images using a
Personal Densitometer SI (GE Healthcare) and analysed using
ImageQuant software.
GeLC-MS/MS analysis of the enriched
plasma membrane fraction
Purified plasma membrane proteins (40 μg)
from UMRC2- cells were resolved by SDS-PAGE (10% T). Gels were
stained with colloidal Coomassie and lanes divided into 2-mm gel
slices which were subjected to in-gel tryptic digestion as
previously described (7). Online
nano-LC/MS/ MS was performed on an Agilent 1100 nano-HPLC system
(Agilent Technologies, South Queensferry, UK) coupled with a QSTAR
XL (Applied Biosystems, Warrington, UK).
Protein Pilot (version 1.0, Applied Biosystems) and
Analyst (version 2.0, Applied Biosystems) were used to extract and
process the MS/MS spectra. Data were searched against the Celera
mammalian protein database (KBM55.0.20050302. fasta) restricted to
human (187835 entries) with the Paragon algorithm (10) using the following parameters:
digestion: trypsin, search effort: rapid, Instrument: QSTAR ESI
(mass tolerance 0.2 Da for MS and MS/MS ions), cysteine alkylation:
iodoacetamide. Protein identification required at least two
peptides with 95% confidence. The ProGroup algorithm was used to
generate a minimal set of protein identifications. False discovery
rates at the protein level (that is, requiring two significant
peptides) were estimated to be 0.0024% by searching a decoy version
of the database generated by EMBOSS (11).
Protein deglycosylation of whole cell
lysates
Removal of N-linked glycans from glycoproteins was
carried out using the GlycoProfile™ II Enzymatic In-solution
N-Deglycosylation kit (Sigma-Aldrich), according to the
manufacturer’s protocol. Briefly, protein from whole UMRC2−/+ cell
extracts prepared in RIPA buffer was adjusted to 1X reaction buffer
(20 mM NH4HCO3) and 2 μl of denaturing
solution [2% (w/v) OCG, 100 mM β-mercaptoethanol] was added and
samples incubated for 10 min at room temperature. Deglycosylation
was carried out for 17 h at 37°C with the addition of 10 μl
of peptide-N-glycosidase (PNGase) F (500 U/ml). The reaction was
terminated by freezing the samples at −80°C. Complete removal of N-
and O-linked glycans was performed using the Glycoprotein
Deglycosylation kit (Merck, Nottingham, UK), according to the
manufacturer’s protocol. Protein from whole UMRC2−/+VHL cell
extracts prepared in RIPA buffer was diluted in 5X reaction buffer
(250 mM sodium phosphate buffer, pH 7.0), followed by the addition
of 2.5 μl of denaturing solution [2% (w/v) SDS, 1 M
β-mercaptoethanol] and 3.75 μl of 15% (v/v) Triton X-100.
Enzymatic deglycosylation was carried out by the addition of 1
μl of each enzyme (Table I)
and samples were incubated at 37°C for 24 h. The reaction was
terminated by freezing the samples at −80°C. In both cases mock
reactions where deglycosylation enzymes were substituted with an
equivalent volume of H2O were carried out in
parallel.
 | Table I.Exoglycosidases used to remove N- and
O-linked glycans from proteins. |
Table I.
Exoglycosidases used to remove N- and
O-linked glycans from proteins.
| Enzyme | Substrate |
|---|
| N-Glycosidase F
(5,000 U/ml) | All
asparagine-linked complex, hybrid, or high mannose oligosaccharides
unless α1,3-core fucosylated |
|
Endo-α-N-acetylgalactosaminidase (1.25
U/ml) | Serine- or
threonine-linked unsubstituted Galβ1,3GalNAcα |
|
α2–3,6,8,9-neuraminidase (5 U/ml) | Non-reducing
terminal branched and unbranched sialic acids |
| β-1,4-galactosidase
(3 U/ml) | Only β1,4-linked,
non-reducing terminal galactose |
|
β-N-acetylglucosaminidase (45 U/ml) | All non-reducing
terminal β-linked N-acetylglucosamine residues |
Deglycosylation was examined by Western blotting
with analysis of GLUT-1 being used as an internal control. In
addition, as an independent deglycosylation control, bovine fetuin
(Sigma-Aldrich or provided in the Glycoprotein Deglycosylation kit)
was deglycosylated under the same conditions as the cell lysates
and the glycan removal and subsequent shift in molecular weight was
observed by silver staining.
Glycoarray analysis
RNA was extracted from three independent replicates
of each of the UMRC2, RCC4 and 786-0 cell lines (all −/+VHL) using
the Qiagen RNeasy Mini kit and used to probe the GLYCOv3
oligonucleotide array (https://www.functionalglycomics.org), a custom
Affymetrix GeneChip (Affymetrix, Santa Clara, CA, USA) designed for
the Consortium for Functional Glycomics and including probes for
1,188 human probe-ids encoding a number of classes of protein
including glycosyltransferases, glycan degradation proteins,
nucleotide sugar synthesis and transporter proteins and glycan
binding proteins (https://www.functionalglycomics.org). Total RNA sample
quality was checked with an Agilent Bioanalyzer (Agilent
Technologies, Palo Alto, CA, USA). RNA from each preparation was
labelled using the MessageAmp II-Biotin Enhanced Amplification kit
(Ambion Inc., Austin, TX, USA). Hybridization and scanning of the
GLYCOv3 chip were performed according to the Affymetrix recommended
protocols (12). The chips were
scanned using the Affymetrix GeneChip Scanner 3000 using default
settings and a target intensity of 250 for scaling. Chips had a
background <100 intensity units and a GAPDH 3′/5′ ratio <1.8.
Robust Multichip Average (RMA) was used to convert the intensity
values to expression values (13,14).
RMA consists of a three step approach which uses a background
correction, a quantile normalization and summarizes the probe set
information by using Tukey’s median polish algorithm. All
processing of the data was performed within the Bioconductor
project and the R program software (R is available as Free Software
under the terms of the Free Software Foundation’s GNU General
Public License). The-fold changes and standard errors were
estimated by fitting a linear model for each gene and empirical
Bayes smoothing was applied to the standard errors for all the
samples at the same time. The linear modeling approach and the
empirical Bayes statistics as implemented in the Limma package in
the R software were employed for differential expression analysis
(15,16). Statistics were obtained for
transcripts with the multiple testing adjusted (Benjamini-Hochberg)
p-value level of 0.05. Filtering was performed so that probe-sets
with a fold change of <1.3 were eliminated from the results.
Results
A method for enrichment of plasma membrane proteins
using cell surface biotinylation and avidin affinity chromatography
was optimised using the VHL-defective renal cancer cell line
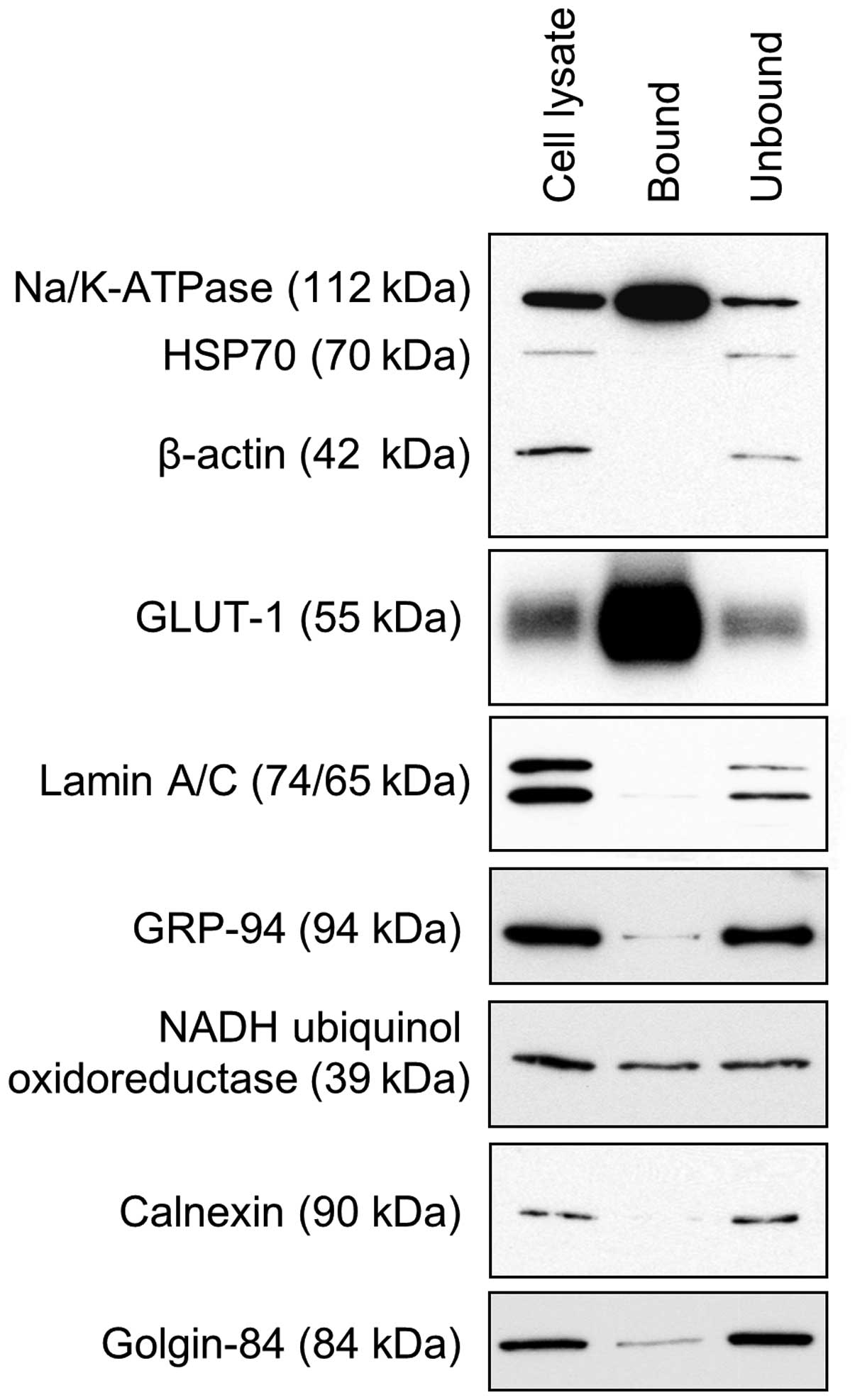
UMRC2-. Western blot analysis (Fig.
1) showed significant enrichment of the plasma membrane
proteins Na/K-ATPase α1 and GLUT-1 compared to the unbound fraction
or a whole cell lysate. The abundant cytosolic proteins HSP70 and
β-actin were almost undetectable as were proteins specific to
endoplasmic reticulum (GRP94 and calnexin) and the nucleus (lamin
A/C). Low levels of golgin-84 were found whilst the mitochondrial
protein NADH-ubiquinol oxidoreductase 39 kDa was present at more
significant levels, indicating that some contamination may be
present.
Using a GeLC-MS/MS approach, a total of 3,991
peptides were identified corresponding to 814 unique proteins with
at least two significant peptides (the complete date set is
available at www.proteomics.leeds.ac.uk), of which 183 (22%) were
known plasma membrane proteins (a selection of these are shown in
Table II); this represents a
significant enrichment compared with 5% of the 956 proteins
identified in a parallel analysis of a whole cell lysate. The
identified proteins included several of interest in the context of
VHL/RCC including integrin α3, transferrin receptor 1, epidermal
growth factor receptor (EGFR) and CAIX.
| Table II.Selected proteins identified by
GeLC-MS/MS. |
Table II.
Selected proteins identified by
GeLC-MS/MS.
| Accession no. | Protein name | Unused protein
score | Percent
coverage | Significant
peptides (>95%) |
|---|
| Q16790 | Carbonic anhydrase
IX | 10.00 | 20.92 | 5 |
| O43570 | Carbonic anhydrase
XII | 6.00 | 20.06 | 3 |
| P00533 | Epidermal growth
factor receptor | 53.81 | 42.81 | 25 |
| P08183 | Multidrug
resistance protein 1 | 11.04 | 19.14 | 3 |
| Q969J9 | Dystroglycan 1 | 5.40 | 7.26 | 3 |
| Q13740 | MEMD protein
(CD166) | 24.71 | 41.24 | 12 |
| P02786 | Transferrin
receptor protein 1 | 47.05 | 52.24 | 23 |
| Q8WUM6 | Integrin β-1 | 30.64 | 38.47 | 14 |
| P05106 | Integrin β-3 | 12.07 | 13.83 | 6 |
| P06756 | Integrin α-V | 50.17 | 46.95 | 21 |
| P18084 | Integrin β-5
precursor | 16.30 | 25.28 | 7 |
| P23229 | Integrin α-6
precursor | 4.53 | 3.19 | 2 |
| P26006 | Integrin α-3 | 19.99 | 18.29 | 9 |
| P21796 | Voltage-dependent
anion-selective channel protein 1 | 15.46 | 50.00 | 7 |
| P45880 | Voltage-dependent
anion-selective channel protein 2 | 11.05 | 37.76 | 5 |
| Q9Y277 | Voltage-dependent
anion-selective channel protein 3 | 16.16 | 54.77 | 8 |
| P05023 |
Sodium/potassium-transporting ATPase α-1
chain | 54.27 | 40.66 | 25 |
| P54709 |
Sodium/potassium-transporting ATPase β-3
chain | 4.18 | 23.30 | 2 |
| O15153 | Sodium bicarbonate
cotransporter | 30.25 | 34.40 | 13 |
| P53985 | Monocarboxylate
transporter 1 | 11.82 | 14.80 | 6 |
| P13987 | CD59 | 3.99 | 35.94 | 2 |
| P55285 | K-cadherin | 6.19 | 9.62 | 3 |
| P19022 | N-cadherin | 9.64 | 14.68 | 4 |
| Q6PHR3 | Melanoma cell
adhesion molecule | 23.42 | 37.31 | 12 |
| P27487 | Dipeptidyl
peptidase IV | 14.37 | 22.19 | 7 |
One protein selected for further study was the
DAG1 gene product dystroglycan-1, a protein that has been
previously implicated in carcinogenesis (17). Dystroglycan-1 comprises two
subunits, α and β, which are generated by proteolytic cleavage of
the α/β precursor polypeptide. The peptides identified in this
study (corresponding to amino acids 702–714, 783–793 and 795–823)
were all from β-dystroglycan, a 43-kDa type I transmembrane protein
consisting of amino acids 654–895 of the molecule (18) that anchors the extracellular
α-subunit to the cell surface.
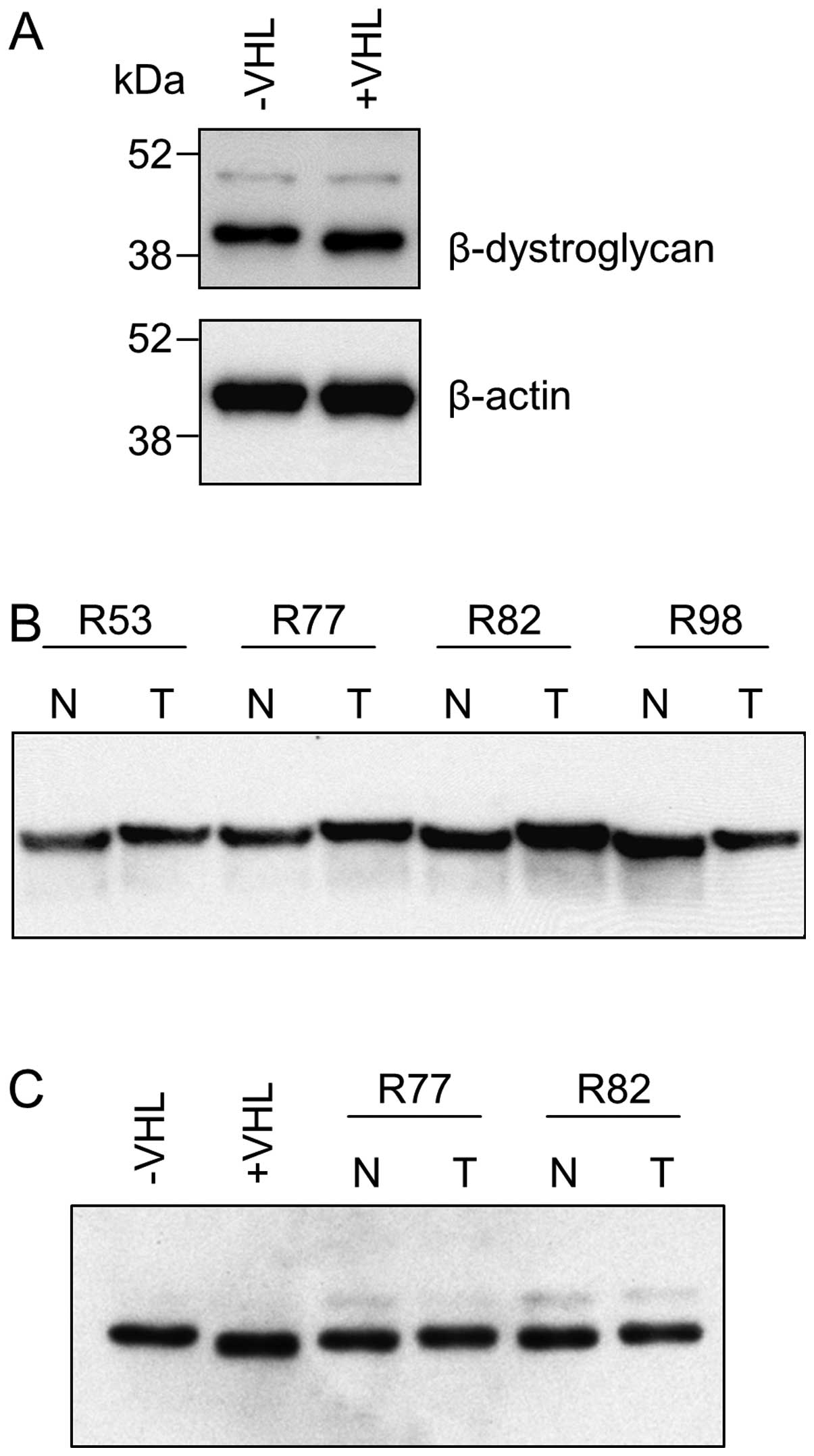
Comparative analysis of UMRC2−/+VHL whole cell
lysates by Western blotting using an antibody raised against amino
acids 655–767 (clone 56) towards the N-terminus of the
β-dystroglycan molecule that recognises the full length protein (43
kDa) but not the fragment reported to migrate at ∼31 kDa (19,20)
showed no difference in expression level but a slightly increased
electrophoretic mobility (corresponding to a difference of <5
kDa) in UMRC2+VHL cells (Fig. 2A).
This was confirmed using an alternative anti-β-dystroglycan
antibody (clone 43DAG/8D5) recognising a C-terminal epitope. This
alteration was not seen in two other renal cancer cell lines (RCC4
and 786-0 −/+ VHL) but a similar change was seen in vivo in
12/15 matched normal and RCC tissue samples (Fig. 2B). The form of β-dystroglycan seen
in tumour tissue was found to co-migrate with that in UMRC2− cells
but the form in normal renal tissue migrated more slowly than that
in UMRC2+VHL cells, thus the overall difference was smaller in
magnitude (Fig. 2C).
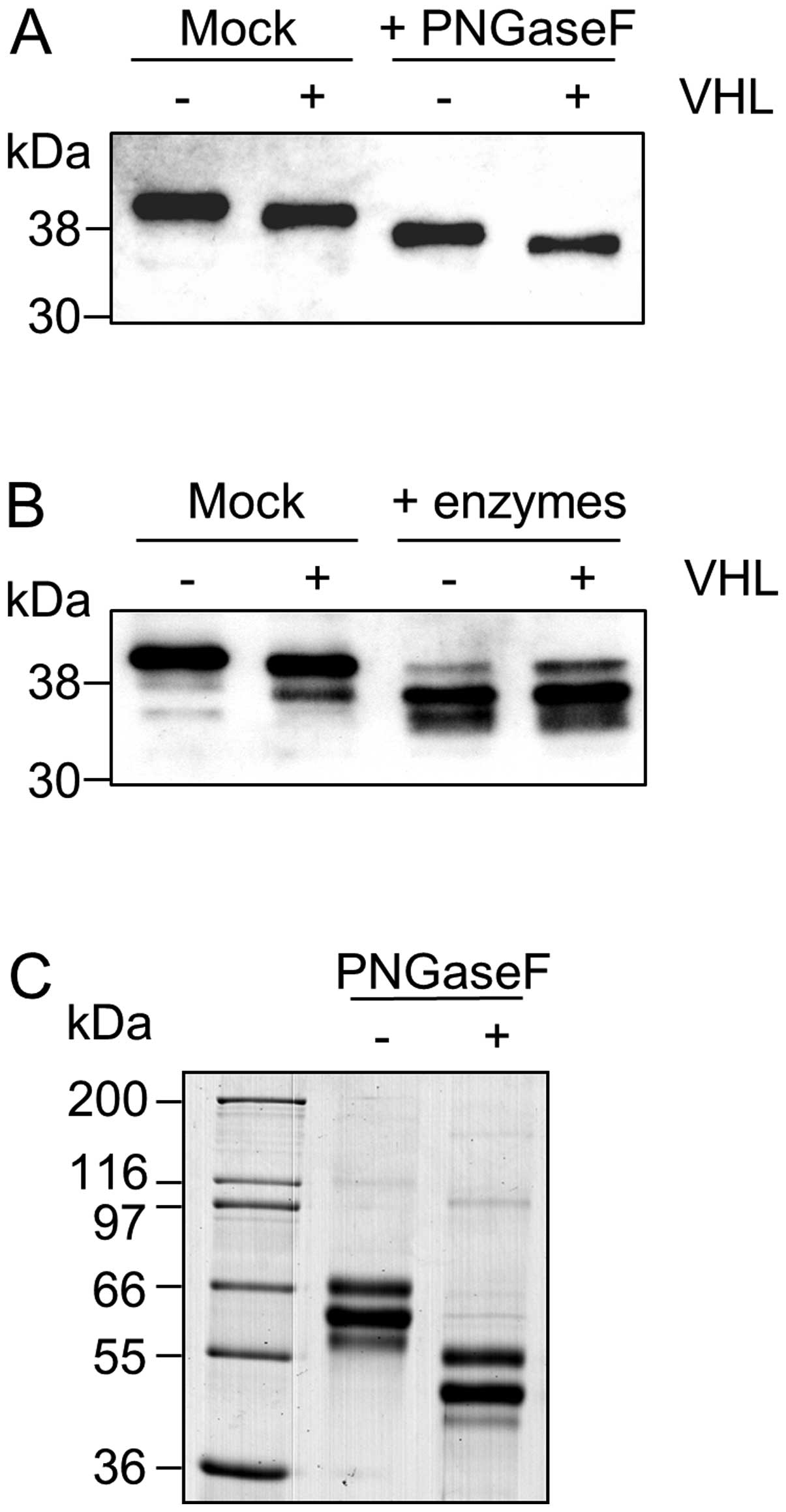
Removal of N-glycans with PNGase F resulted in a
shift in molecular weight of both β-dystroglycan isoforms with the
difference in size between the UMRC2− and +VHL cells still apparent
(Fig. 3A). Using a combination of
exoglycosidases together with PNGase F to remove both N- and
O-linked glycans eliminated the difference in size seen between
UMRC2- and +VHL cells (Fig. 3B)
strongly supporting differential glycosylation as being the cause
of the difference, with O-linked glycans contributing at least part
of the change. For deglycosylation experiments, GLUT-1 was
monitored as an internal control and bovine fetuin as an external
control (for an example see Fig.
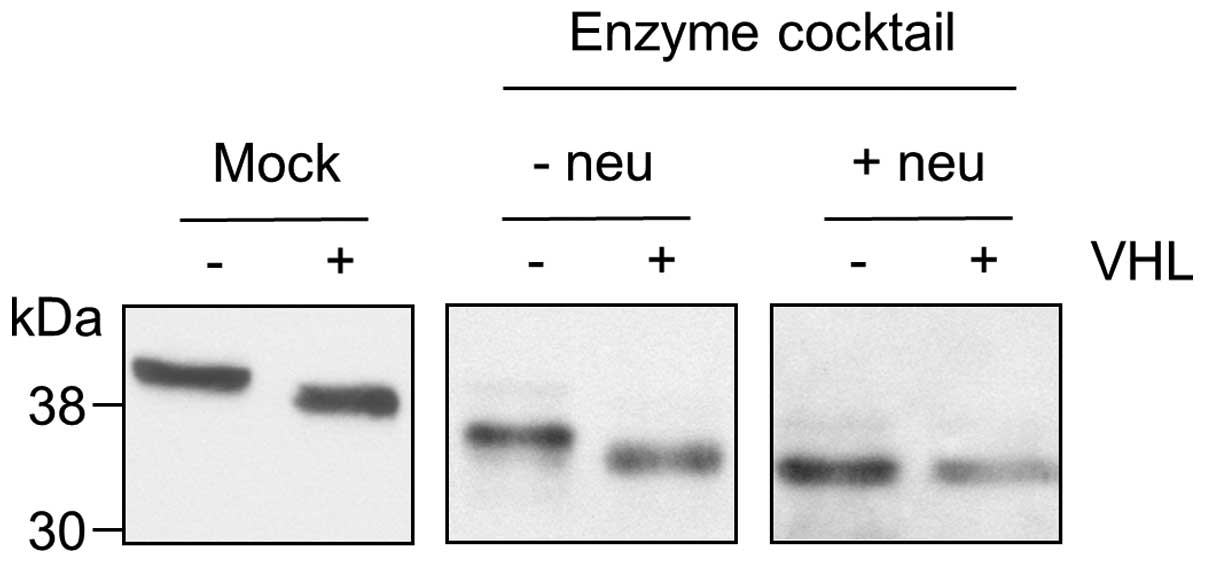
3C). When α2–3,6,8,9-neuraminidase, which specifically removes
all non-reducing terminal branched and unbranched sialic acid
residues, was omitted from the deglycosylation reaction, the
difference in size of β-dystroglycan between UMRC2− and +VHL cells
was still apparent (Fig. 4).
Conversely, if deglycosylation was carried out using only
α2–3,6,8,9-neuraminidase, the difference in size was eliminated
(data not shown). Taken together these data indicate that the
difference between β-dystroglycan isoforms is due predominantly to
a change in the level of sialylation. The presence of protease
inhibitors in the extracts used for the experiment, together with
the absence of a change in mobility in the mock reactions carried
out without enzymes strongly suggest that proteolytic activity did
not contribute to the reactions (additional bands were present in
both the mock reactions and the deglycosylated samples in the
example shown in Fig. 3B, but
these were of significantly lower intensity than the major forms
and not present in every reaction as illustrated in Fig. 4). The smaller magnitude of change
in tissue made resolution of forms of β-dystroglycan more difficult
thus confident interpretation of deglycosylation experiments with
tissues was not possible.
Examination of the expression profiles of genes
involved in glycosylation using a glycosylation focussed microarray
showed changes in several molecules (the complete date set is
available at www.proteomics.leeds.ac.uk), several of which related
to possible alterations in sialylation (Table III). These included GNE, which
encodes bifunctional UDP-N-acetylglucosamine
2-epimerase/N-acetylmannosamine kinase, which was upregulated in
UMRC2− cells and NPL, which encodes a sialic acid lyase, which was
downregulated. As a positive control, changes in other gene classes
on the chip which were found in UMRC2 cells included upregulation
of TGF-β and downregulation of clusterin in -VHL cells which are
both known VHL-associated changes (21,22).
| Table III.Genes involved in sialylation with
altered expression in UMRC2−/+VHL cells. |
Table III.
Genes involved in sialylation with
altered expression in UMRC2−/+VHL cells.
| Gene name | Accession no.
(NCBI) | Protein name | Fold change in
UMRC2a |
|---|
| NPL | AF338436 | Sialic acid
lyase | 1.8-fold ↓ |
| GNE | NM_005476.2 |
UDP-GlcNAc-2-epimerase/ManAc kinase | 5.2-fold ↑ |
| ST3GAL6 | NM_006100.2 | Sialyl transferase
10 | 2.1-fold ↑ |
| ST6GAL1 | NM_173216.1 | Sialyltransferase
1 | 1.7-fold ↑ |
| NEU1 | BC000722 | Sialidase-1 | 1.3-fold ↓ |
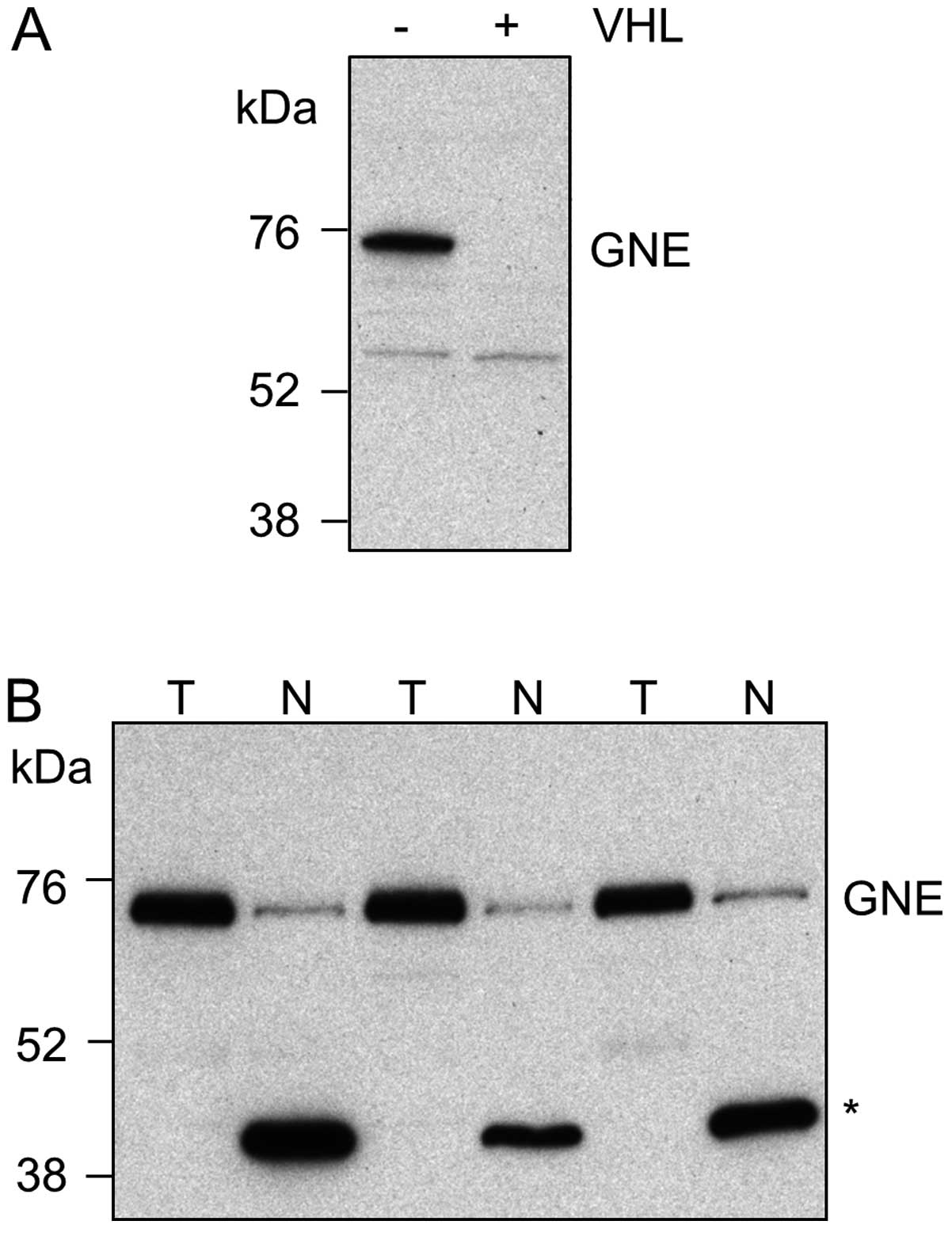
Expression of GNE, the gene of interest which showed
the greatest magnitude of change at the mRNA level between
UMRC2−/+VHL cells, was investigated further. Western blot analysis
showed that GNE protein levels changed in UMRC2−/+VHL cells in a
manner that paralleled the changes seen at the mRNA level (Fig. 5A). Furthermore, in matched normal
kidney cortex and conventional RCC tissues, GNE was found to be
upregulated in 6/8 tumours (Fig.
5B). In normal kidney tissue a dominant lower molecular weight
band was also seen; unlike the higher molecular weight band, this
was not recognised by an alternative antibody to GNE raised against
a different epitope and it remains to be determined whether this
represents a smaller form of GNE or a cross reacting protein
species.
Discussion
The tumour suppressor gene VHL plays a
central role in development of conventional RCC and the
characterisation of VHL-regulated proteins and pathways
offers promise in identifying new biomarkers and therapeutic
targets. In RCC both global mRNA expression profiling of VHL cell
line pairs (23–25) and complementary proteomic
approaches (7,8,22,26)
have succeeded in identifying changes that are relevant in
tumourigenesis.
Previous analysis of the membrane proteome of renal
cancer cells fractionated from a post-nuclear supernatant using a
60% sucrose cushion followed by a 15–60% sucrose gradient
identified high expression of CD70 and showed that this protein
could act as a target in antibody-targeted cytotoxic therapy
(27). A further study used cell
surface capturing (CSC) technology, where glycans are labelled with
biocytin hydrazide and following digestion, labelled
N-glycopeptides purified by avidin affinity chromatography,
combined with stable isotope labelling with amino acids in cell
culture (SILAC) to compare the cell surface of −/+VHL cells
(26). In the present study, cell
surface biotinylation and avidin affinity chromatography was used
to enrich plasma membrane proteins as part of method development
for a quantitative comparative proteomic study. The presence of
NADH ubiquinol oxidoreductase 39 kDa in the enriched fraction
suggested some contamination from other organelles, but this
protein is a subunit of a respiratory chain complex demonstrated to
localise to the plasma membrane (28). Similarly, a number of proteomic
studies have reported intracellular proteins on the cell surface,
such as cytoplasmic and ER lumenal chaperones (29–31).
The enrichment strategy described here was used in combination with
SILAC in a study which identified upregulation of the adhesion
molecules CD166 and CD147 in VHL defective cells and some RCC
tissues (7).
Whilst examining the enriched fraction to assess the
extent of profiling of proteins of relevance in RCC, β-dystroglycan
was selected for further analysis due to the known involvement of
dystroglycan in cancer. Dystroglycan is a transmembrane
glycoprotein encoded by the DAG1 gene that is processed into
two subunits − the transmembrane β domain and the extracellular α
domain. Loss of α-dystroglycan expression and correlations with
prognosis have been reported in a number of tumour types (32–36).
In RCC, loss of α-dystroglycan correlated with high grade disease
and was an independent predictor of shorter disease-free and
overall survival (37). Combined
loss of α-dystroglycan and p27kip1 defined a group of
patients with particularly poor outcome (38). Many studies analysing
α-dystroglycan used antibodies recognising glycosylation-dependent
epitopes and changes in glycosylation have been suggested to
account for loss of α-dystroglycan staining (39,40).
Changes in expression of both LARGE and
β3-N-acetylglucosaminyltransferase-1 restored glycosylation of
α-dystroglycan and altered tumour cell behaviour (41,42).
Changes in expression of β-dystroglycan in cancer
are less consistent with some studies finding no change in
expression. Loss of β-dystroglycan was found in some cancers
including prostate, breast, colon and oesophageal (32,43–45)
and relationships with progression were reported for breast and
colon cancers (32). In oral SCC
loss of β-dystroglycan was reported in poorly differentiated
tumours (19) whilst in a separate
study the presence of the 31 kDa β-dystroglycan fragment correlated
with lymph node metastasis and tumour differentiation (46).
Characterisation of β-dystroglycan showed that its
form changed in UMRC2 cells in a VHL-dependent manner. No evidence
was found to suggest that this was due to changes in
phosphorylation or alternative splicing (using dephosphorylation
with lambda protein phosphatase and by RT-PCR respectively, data
not shown). However, deglycosylation experiments indicated that
this change was due, at least in part, to differential sialylation.
A similar but smaller change was also seen in the majority of RCC
samples compared with matched normal kidney cortex. Previous
studies in RCC did not report this change in glycosylation in
β-dystroglycan (38) which may be
due to the gel systems used as resolution of the forms, which
differ by <5 kDa, is difficult, especially in tissues.
As mentioned above, a truncated ∼31-kDa fragment of
β-dystroglycan lacking the extracellular domain has been identified
in cell lines and tissues (20,47)
with processing by matrix metalloproteinases being implicated in
its formation (46–48); an MMP-9 cleavage site has recently
been defined (49). Tyrosine
phosphorylated forms have also been described (50,51).
In a study examining the role of dystroglycan in prostate cancer
cell lines, β-dystroglycan was shown to exhibit reversible cell
density dependent changes in form, with lower molecular weight
forms of 38–43 kDa due to mis-glycosylation and bands at 31 and 26
kDa resulting from proteolysis being seen in supra-confluent cells
(52). The altered glycosylation
in our study seems to be distinct from this effect. Treatment with
PNGase F resulted in a similar increase in gel mobility of
β-dystroglycan in UMRC2− and +VHL cells thus N-glycosylation was
present irrespective of VHL status. The cells used in our study
were all harvested at the same growth state (that is, approaching
confluence), but it is possible that VHL alters the point at which
a density-dependent change in form is induced.
Possible mechanisms underlying the VHL-dependent
changes in sialylation were investigated using a glycoarray.
Overall, the glycoarray results for the three −/+VHL cell line
pairs analysed did not show obvious patterns, which may reflect the
complexity of glycosylation, the potential for differences between
differing genetic backgrounds and the limitations of cell line
models. Altered expression of glycogenes involved in sialylation
that were seen in UMRC2 cells, like the change in form of
β-dystroglycan, were restricted to this cell line, with the
exception of NPL, expression of which was found to be VHL-dependent
in all three cell lines. Previous studies have also found
differences in VHL-dependent gene expression in different cell
lines and this is apparent here not just for genes involved in
glycosylation but also known VHL-regulated genes present on the
array, with no one cell line pair behaving as an outlier.
In UMRC2−/+VHL cells, lower expression of NEU1 which
encodes sialidase-1, which localises to the lysosome and the cell
surface and increased expression of the sialyltransferases ST3GAL6
and ST6GAL1, in UMRC2− cells may contribute to altered sialylation.
Similarly, upregulation of GNE, which is a key enzyme in sialic
acid biosynthesis, together with downregulation of NPL, which is
involved in sialic acid turnover, in UMRC2− cells may alter the
availability of sialic acid and thereby affect sialylation. Indeed
there is mounting evidence in the literature that substrate
availability and levels of GNE do impact on sialylation. In an
analysis of N-linked sialoglycopeptides in human pancreatic
carcinoma (SW1990) cells, increased metabolic flux through the
sialic acid pathway by exogenously supplied substrate was found to
selectively increase the sialylation of individual glycoproteins
(53). In hematopoietic cell
lines, GNE was found to be an important regulator of cell surface
sialylation (54) and knockdown of
GNE in HEK293 cells reduced total cell surface sialic acid content
(55). Expression of GNE in
UMRC2−/+VHL cells validated the changes found using the microarray,
with reduced expression of GNE being seen in VHL transfectants and
GNE levels were also upregulated in a significant proportion of
tumours compared to normal renal tissue. Changes in tumour tissue
at the mRNA level reported in microarray data sets correlate with
this result (56,57).
A similar finding of involvement of a tumour
suppressor gene in glycosylation has been previously reported, with
altered expression of glycosyltransferases and decreased
sialylation of N- and O-glycans being seen in Capan-1 pancreatic
carcinoma cells following transfection with p16INK4a
(58). In an extension of this
study, decreased levels of GNE were found to be an important
consequence of p16INK4a transfection, correlating with
loss of membrane bound sialic acid and hyposialylation of α5 and β1
integrin (56). The changes in
these integrin subunits were very similar to the change seen in
β-dystroglycan.
The results described here raise the question of the
functional impact of changes in glycosylation of β-dystroglycan in
renal cancer and there is clearly enormous potential for
glycosylation-dependent biomarkers (59). Alterations in glycosylation
associated with cancer progression are well documented and include
changes in sialylation (reviewed in ref. 60). Changes in glycosylation in RCC have
been shown using lectin staining (61) and we have previously described
VHL-dependent changes in glycosylation of CD166 (7). The extent to which glycosylation of
other proteins can also be regulated by VHL remains to be
determined. Further work is now required to build on the
preliminary findings presented here and establish the extent of
changes in glycosylation in RCC that may be attributable to VHL and
their functional consequences.
Acknowledgements
This study was carried out through
grant funding from Cancer Research UK and a collaborative
studentship from the University of Leeds and AstraZeneca. We also
thank the patients and staff of the Urology and Oncology
Departments at Leeds.
References
|
1.
|
Latif F, Tory K, Gnarra J, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Young AC, Craven RA, Cohen D, et al:
Analysis of VHL gene alterations and their relationship to clinical
parameters in sporadic conventional renal cell carcinoma. Clin
Cancer Res. 15:7582–7592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Nyhan MJ, O’Sullivan GC and McKenna SL:
Role of the VHL (von Hippel-Lindau) gene in renal cancer: a
multifunctional tumour suppressor. Biochem Soc Trans. 36:472–478.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Li L and Kaelin WG Jr: New insights into
the biology of renal cell carcinoma. Hematol Oncol Clin North Am.
25:667–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Flaherty KT and Puzanov I: Building on a
foundation of VEGF and mTOR targeted agents in renal cell
carcinoma. Biochem Pharmacol. 80:638–646. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Závada J, Závadová Z, Zat’ovičová M, Hyrsl
L and Kawaciuk I: Soluble form of carbonic anhydrase IX (CA IX) in
the serum and urine of renal carcinoma patients. Br J Cancer.
89:1067–1071. 2003.
|
|
7.
|
Aggelis V, Craven RA, Peng J, et al:
Proteomic identification of differentially expressed plasma
membrane proteins in renal cell carcinoma by stable isotope
labelling of a von Hippel-Lindau transfectant cell line model.
Proteomics. 9:2118–2130. 2009. View Article : Google Scholar
|
|
8.
|
Craven RA, Hanrahan S, Totty N, et al:
Proteomic identification of a role for the von Hippel Lindau tumour
suppressor in changes in the expression of mitochondrial proteins
and septin 2 in renal cell carcinoma. Proteomics. 6:3880–3893.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Zhao Y, Zhang W, Kho Y and Zhao Y:
Proteomic analysis of integral plasma membrane proteins. Anal Chem.
76:1817–1823. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Shilov IV, Seymour SL, Patel AA, et al:
The Paragon Algorithm, a next generation search engine that uses
sequence temperature values and feature probabilities to identify
peptides from tandem mass spectra. Mol Cell Proteomics.
6:1638–1655. 2007. View Article : Google Scholar
|
|
11.
|
Rice P, Longden I and Bleasby A: EMBOSS:
the European Molecular Biology Open Software Suite. Trends Genet.
16:276–277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lockhart DJ, Dong H, Byrne MC, et al:
Expression monitoring by hybridization to high-density
oligonucleotide arrays. Nat Biotechnol. 14:1675–1680. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Bolstad BM, Irizarry RA, Åstrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e152003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Smyth GK: Linear models and empirical
Bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article 3. 2004 View Article : Google Scholar
|
|
16.
|
Smyth GK: Limma: linear models for
microarray data. Bioinformatics and Computational Biology Solutions
Using R and Bioconductor. Gentleman R, Carey VJ, Dudoit S, Irizarry
RA and Huber W: Springer; New York, NY: pp. 397–420. 2005,
View Article : Google Scholar
|
|
17.
|
Sgambato A and Brancaccio A: The
dystroglycan complex: from biology to cancer. J Cell Physiol.
205:163–169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Holt KH, Crosbie RH, Venzke DP and
Campbell KP: Biosynthesis of dystroglycan: processing of a
precursor propeptide. FEBS Lett. 468:79–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jing J, Lien CF, Sharma S, Rice J, Brennan
PA and Górecki DC: Aberrant expression, processing and degradation
of dystroglycan in squamous cell carcinomas. Eur J Cancer.
40:2143–2151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Losasso C, Di Tommaso F, Sgambato A, et
al: Anomalous dystroglycan in carcinoma cell lines. FEBS Lett.
484:194–198. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ananth S, Knebelmann B, Grüning W, et al:
Transforming growth factor β1 is a target for the von Hippel-Lindau
tumor suppressor and a critical growth factor for clear cell renal
carcinoma. Cancer Res. 59:2210–2216. 1999.
|
|
22.
|
Nakamura E, Abreu-e-Lima P, Awakura Y, et
al: Clusterin is a secreted marker for a hypoxia-inducible
factor-independent function of the von Hippel-Lindau tumor
suppressor protein. Am J Pathol. 168:574–584. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Zatyka M, da Silva NF, Clifford SC, et al:
Identification of cyclin D1 and other novel targets for the von
Hippel-Lindau tumor suppressor gene by expression array analysis
and investigation of cyclin D1 genotype as a modifier in von
Hippel-Lindau disease. Cancer Res. 62:3803–3811. 2002.PubMed/NCBI
|
|
24.
|
Wykoff CC, Sotiriou C, Cockman ME, et al:
Gene array of VHL mutation and hypoxia shows novel hypoxia-induced
genes and that cyclin D1 is a VHL target gene. Br J Cancer.
90:1235–1243. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Jiang Y, Zhang W, Kondo K, et al: Gene
expression profiling in a renal cell carcinoma cell line:
dissecting VHL and hypoxia-dependent pathways. Mol Cancer Res.
1:453–462. 2003.PubMed/NCBI
|
|
26.
|
Boysen G, Bausch-Fluck D, Thoma CR, et al:
Identification and functional characterization of pVHL-dependent
cell surface proteins in renal cell carcinoma. Neoplasia.
14:535–546. 2012.PubMed/NCBI
|
|
27.
|
Adam PJ, Terrett JA, Steers G, et al: CD70
(TNFSF7) is expressed at high prevalence in renal cell carcinomas
and is rapidly internalised on antibody binding. Br J Cancer.
95:298–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Yonally SK and Capaldi RA: The F(1)F(0)
ATP synthase and mitochondrial respiratory chain complexes are
present on the plasma membrane of an osteosarcoma cell line: an
immunocytochemical study. Mitochondrion. 6:305–314. 2006.
View Article : Google Scholar
|
|
29.
|
Shin BK, Wang H, Yim AM, et al: Global
profiling of the cell surface proteome of cancer cells uncovers an
abundance of proteins with chaperone function. J Biol Chem.
278:7607–7616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Jang JH and Hanash S: Profiling of the
cell surface proteome. Proteomics. 3:1947–1954. 2003. View Article : Google Scholar
|
|
31.
|
Tjalsma H, Pluk W, van den Heuvel LP,
Peters WH, Roelofs R and Swinkels DW: Proteomic inventory of
‘anchorless’ proteins on the colon adenocarcinoma cell surface.
Biochim Biophys Acta. 1764:1607–1617. 2006.
|
|
32.
|
Sgambato A, Migaldi M, Montanari M, et al:
Dystroglycan expression is frequently reduced in human breast and
colon cancers and is associated with tumor progression. Am J
Pathol. 162:849–860. 2003. View Article : Google Scholar
|
|
33.
|
Sgambato A, Caredda E, Leocata P, et al:
Expression of alpha-dystroglycan correlates with tumour grade and
predicts survival in oral squamous cell carcinoma. Pathology.
42:248–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Jiang X, Rieder S, Giese NA, Friess H,
Michalski CW and Kleeff J: Reduced alpha-dystroglycan expression
correlates with shortened patient survival in pancreatic cancer. J
Surg Res. 171:120–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Moon YW, Rha SY, Zhang X, et al:
Increments of alpha-dystroglycan expression in liver metastasis
correlate with poor survival in gastric cancer. J Surg Oncol.
100:459–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Sgambato A, De Paola B, Migaldi M, et al:
Dystroglycan expression is reduced during prostate tumorigenesis
and is regulated by androgens in prostate cancer cells. J Cell
Physiol. 213:528–539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Sgambato A, Camerini A, Amoroso D, et al:
Expression of dystroglycan correlates with tumor grade and predicts
survival in renal cell carcinoma. Cancer Biol Ther. 6:1840–1846.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Sgambato A, Camerini A, Genovese G, et al:
Loss of nuclear p27kip1 and alpha-dystroglycan is a
frequent event and is a strong predictor of poor outcome in renal
cell carcinoma. Cancer Sci. 101:2080–2086. 2010.
|
|
39.
|
Calogero A, Pavoni E, Gramaglia T, et al:
Altered expression of alpha-dystroglycan subunit in human gliomas.
Cancer Biol Ther. 5:441–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Shimojo H, Kobayashi M, Kamigaito T,
Shimojo Y, Fukuda M and Nakayama J: Reduced glycosylation of
alpha-dystroglycans on carcinoma cells contributes to formation of
highly infiltrative histological patterns in prostate cancer.
Prostate. 71:1151–1157. 2011. View Article : Google Scholar
|
|
41.
|
de Bernabé DB, Inamori K,
Yoshida-Moriguchi T, et al: Loss of alpha-dystroglycan laminin
binding in epithelium-derived cancers is caused by silencing of
LARGE. J Biol Chem. 284:11279–11284. 2009.PubMed/NCBI
|
|
42.
|
Bao X, Kobayashi M, Hatakeyama S, et al:
Tumor suppressor function of laminin-binding alpha-dystroglycan
requires a distinct beta3-N-acetylglucosaminyltransferase. Proc
Natl Acad Sci USA. 106:12109–12114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Henry MD, Cohen MB and Campbell KP:
Reduced expression of dystroglycan in breast and prostate cancer.
Hum Pathol. 32:791–795. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Cross SS, Lippitt J, Mitchell A, et al:
Expression of beta-dystroglycan is reduced or absent in many human
carcinomas. Histopathology. 53:561–566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Parberry-Clark C, Bury JP, Cross SS and
Winder SJ: Loss of dystroglycan function in oesophageal cancer.
Histopathology. 59:180–187. 2011.PubMed/NCBI
|
|
46.
|
Shang ZJ, Ethunandan M, Górecki DC and
Brennan PA: Aberrant expression of beta-dystroglycan may be due to
processing by matrix metalloproteinases-2 and -9 in oral squamous
cell carcinoma. Oral Oncol. 44:1139–1146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Yamada H, Saito F, Fukuta-Ohi H, et al:
Processing of beta-dystroglycan by matrix metalloproteinase
disrupts the link between the extracellular matrix and cell
membrane via the dystroglycan complex. Hum Mol Genet. 10:1563–1569.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Zhong D, Saito F, Saito Y, Nakamura A,
Shimizu T and Matsumura K: Characterization of the protease
activity that cleaves the extracellular domain of
beta-dystroglycan. Biochem Biophys Res Commun. 345:867–871. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Bozzi M, Inzitari R, Sbardell D, et al:
Enzymatic processing of beta-dystroglycan recombinant ectodomain by
MMP-9: identification of the main cleavage site. IUBMB Life.
61:1143–1152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Sotgia F, Bonuccelli G, Bedford M, et al:
Localization of phospho-beta-dystroglycan (pY892) to an
intracellular vesicular compartment in cultured cells and skeletal
muscle fibers in vivo. Biochemistry. 42:7110–7123. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
James M, Nuttall A, Ilsley JL, et al:
Adhesion-dependent tyrosine phosphorylation of beta-dystroglycan
regulates its interaction with utrophin. J Cell Sci. 113:1717–1726.
2000.PubMed/NCBI
|
|
52.
|
Mitchell A, Mathew G, Jiang T, et al:
Dystroglycan function is a novel determinant of tumor growth and
behavior in prostate cancer. Prostate. 73:398–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Almaraz RT, Tian Y, Bhattarcharya R, et
al: Metabolic flux increases glycoprotein sialylation: implications
for cell adhesion and cancer metastasis. Mol Cell Proteomics.
11:M112.0175582012. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Keppler OT, Hinderlich S, Langner J,
Schwartz-Albiez R, Reutter W and Pawlita M: UDP-GlcNAc 2-epimerase:
a regulator of cell surface sialylation. Science. 284:1372–1376.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Möller H, Böhrsch V, Lucka L, Hackenberger
CP and Hinderlich S: Efficient metabolic oligosaccharide
engineering of glycoproteins by UDP-N-acetylglucosamine
2-epimerase/N-acetylmannosamine kinase (GNE) knock-down. Mol
Biosyst. 7:2245–2251. 2011.PubMed/NCBI
|
|
56.
|
Kemmner W, Kessel P, Sanchez-Ruderisch H,
et al: Loss of UDP-N-acetylglucosamine
2-epimerase/N-acetylmannosamine kinase (GNE) induces apoptotic
processes in pancreatic carcinoma cells. FASEB J. 26:938–946. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, et al: Oncomine 3.0: genes, pathways, and networks in a
collection of 18,000 cancer gene expression profiles. Neoplasia.
9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
André S, Sanchez-Ruderisch H, Nakagawa H,
et al: Tumor suppressor p16INK4a - modulator of glycomic
profile and galectin-1 expression to increase susceptibility to
carbohydrate-dependent induction of anoikis in pancreatic carcinoma
cells. FEBS J. 274:3233–3256. 2007.
|
|
59.
|
Meany DL and Chan DW: Aberrant
glycosylation associated with enzymes as cancer biomarkers. Clin
Proteomics. 8:72011. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Brooks SA, Carter TM, Royle L, et al:
Altered glycosylation of proteins in cancer: what is the potential
for new anti-tumour strategies. Anticancer Agents Med Chem. 8:2–21.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Ferguson RE, Vasudev NS, Selby PJ and
Banks RE: Protein glycosylation and renal cancer. Encyclopedia of
Genetics, Genomics, Proteomics and Bioinformatics. Jorde LB, Little
P, Dunn M and Subramaniam S: John Wiley and Sons; 2006, View Article : Google Scholar
|