Introduction
Apoptosis, also known as programmed cell death, can
be induced by both intrinsic and extrinsic pathways. The intrinsic
pathway is also known as mitochondria pathway and is regulated by
different molecules such as XIAP (inhibitors of apoptosis) and
Bcl-2 family. The extrinsic pathway is activated by tumour necrosis
factors (TNFs) and its down-stream transcription factors are also
involved in intrinsic pathway of apoptosis. The extrinsic pathway
involves the promotion of apoptosis via the ligand-activation of
death receptors (1). For example,
TNF-α binding to its receptors leads to form two complexes; one can
lead to activation of pro-survival NF-κB pathway and another one
activates the apoptosis pathway through Fas associated death domain
(FADD) and activation of caspase-8 (2). Protein tyrosine phosphatases (PTPs)
play crucial roles in regulation of cell functions such as
proliferation and survival. PTPs deficiency leads to several
physiologic abnormalities and dysregulation of apoptosis resulted
by PTP deficiency may play a profound role in these disorders. For
example, PTP-PEST (tyrosine-protein phosphatase non-receptor type
12, PTPN12) makes cells more sensitive to the anti-Fas and TNF-α
induced apoptosis. PTP-PEST is cleaved by caspase-3, which
increases its catalytic activity and changes its protein structure.
Furthermore, the PTP-PEST proteolysis facilitates cellular
detachment during apoptosis (3).
Unlike PTP-PEST, PTP1B (PTPN1) has no caspase cleavage site and it
is not cleaved by caspases during apoptosis. However, there is a
report showing activated PTP1B contributes to STAT3
dephosphorylation and induces apoptosis in human glioma cells
(3,4). PTP1B and its catalytic activity are
required for inositol-requiring kinase 1 (IRE1) signalling which
activates JNK and p38 MAPK. Moreover, endoplasmic reticulum
(ER)-induced apoptosis is decreased in cells lacking PTP1B and
PTP1B null mice are resistant to Fas-induced liver damage because
of absence of PTP1B-mediated suppression of pro-survival NF-κB and
ERK signalling (5,6). In addition, p53 is required for T
cell protein tyrosine phosphatase (TC-PTP, PTPN2) overexpression
induced apoptosis. In breast cancer cells, MCF-7, the accumulation
of p53 in response to TC-PTP overexpression directly increases
expression of Apaf-1 and pro-apoptotic α-isoform of caspase-1
(7,8).
Currently, the role played by PTPRK in cancer
remains largely unknown. The present study examined the expression
of PTPRK in prostate cancer and the impact of this molecule on
prostate cancer cell apoptosis.
Materials and methods
Cell lines and cells culture
PC-3 (human prostate cancer cell line) and MRC-5
(fibroblast cell line) were obtained from the European Collection
of Animal Cell Cultures (ECACC, Salisbury, UK). DU-145, LNCaP,
CA-HPV-10 and PZ-HPV-7 were obtained from the American Type Culture
Collection (ATCC, Rockville, MD, USA) and PNT-1A and PNT-2C2 were
generously given by Professor Norman Maitland (University of York,
York, UK). Cells were routinely cultured with Dulbecco’s modified
Eagle’s medium containing 10% fetal calf serum and antibiotics at
37°C with 5% CO2.
Human prostate specimens
Fresh tissue samples were collected immediately
after surgery and stored at −80°C until use, with approval of the
Bro Taf Health Authority local research ethics committee. All
patients were informed and participated with written consent. All
the specimens were verified by a consultant pathologist.
Immumohistochemical staining
Frozen specimens of mammary tissues were cut at a
thickness of 6 μm using a cryostat (Leica CM 1900, Leica
Microsystems UK Ltd., Buckinghamshire, UK). The sections were
mounted on super frost plus microscope slides, air dried and then
fixed in the mixture of 50% acetone and 50% methanol for 15 min.
After 10 min air-drying, the slides were placed into OptiMax Wash
Buffer (BioGenex, San Ramon, CA, USA) for 5 min to rehydrate. The
slides were incubated in blocking buffer with 10% horse serum for
20 min and probed anti-PTPRK antibody (Santa Cruz Biotechnology,
Santa Cruz, CA, USA) for 1 h. After three times washes, the slides
were incubated with biotinylated secondary antibody (Multilink
swine anti-goat/mouse/rabbit immunoglobulin, Dako Inc.,
Carpinteria, CA, USA). After washing, slides were placed in
avidin-biotin complex (ABC, Vector Labs) for 30 min.
Diaminobenzidine chromogen (Vector Labs) was then added to the
slides and incubated in the dark for 5 min. The slides were
counterstained with Mayer’s haematoxylin for 1 min and dehydrated
in ascending grades of ethanol before clearing in xylene and
mounting with a cover slip.
Reverse transcription-PCR
Total RNA extraction from frozen tissues and culture
cells was performed using Tri Reagent (Sigma-Aldrich Inc., Saint
Louis, MO, USA). Following reverse transcription into cDNA, PCR was
carried out using ReadyMix PCR Reaction Mix (Sigma-Aldrich Inc.).
Primer sequences are shown in Table
I. Reactions were carried out at the following conditions: 94°C
for 5 min, 30 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C
for 30 sec, followed by a final extension of 7 min at 72°C. PCR
products were separated on a 1.5% agarose gel and photographed
after staining with ethidium bromide.
 | Table I.Primer sequences used in the current
study. |
Table I.
Primer sequences used in the current
study.
| Molecular | Forward primers
(5′-3′) | Reverse primers
(5′-3′) |
|---|
| PTPRK |
AATTACAATTGATGGGGAGA |
CCACTTTTCCACCTGAAGTA |
| PTPRK (Q-PCR) |
AATTACAATTGATGGGGAGA |
ACTGAACCTGACCGTACATATTGTGTGACGATGAAAGC |
| GAPDH |
GGCTGCTTTTAACTCTGGTA |
GACTGTGGTCATGAGTCCTT |
| GAPDH (Q-PCR) |
CTGAGTACGTCGTGGAGTC |
ACTGAACCTGACCGTACAGAGATGACCCTTTTG |
| c-Myc |
TGCTCCATGAGGAGACAC |
TTTCATTGTTTTCCAACTCC |
| c-Myc (Q-PCR) |
TGCTCCATGAGGAGACAC |
ACTGAACCTGACCGTACATGATCCAGACTCTGACCTTT |
| ID1 | TCAACGGCGAGATCAG |
ACTGAACCTGACCGTACAGATCGTCCGCAGGAA |
| p53 |
ATCCTCACCATCATCACACT |
ACTGAACCTGACCGTACACAGGACAGGCACAAACAC |
| Caspase-3 |
GGCGTGTCATAAAATACCAG |
ACTGAACCTGACCGTACAACAAAGCGACTGGATGAA |
| Caspase-8 |
AGAAAGGAGGAGATGGAAAG |
ACTGAACCTGACCGTACAGACCTCAATTCTGATCTGCT |
| Caspase-9 |
AAGCCCAAGCTCTTTTTC |
ACTGAACCTGACCGTACAGTTACTGCCAGGGGACTC |
Real-time quantitative PCR
The level of PTPRK transcripts in the breast cancer
cohort was determined using a real-time quantitative PCR, based on
technology which was modified from a method reported previously
(9). Primer sequences are shown in
Table I. The reaction was carried
out on an iCycler iQ™ (Bio-Rad, Hertfordshire, UK) which is
equipped with an optical unit that allows real-time detection of 96
reactions. The reaction conditions were: 94°C for 12 min, 90 cycles
of 94°C for 15 sec, 55°C for 40 sec (the data capture step) and
72°C for 20 sec. The levels of the transcripts were generated from
an internal standard that was simultaneously amplified with the
samples.
Construction of ribozyme transgene
targeting human PTPRK and the establishment of corresponding stable
transfectants
Anti-human PTPRK hammerhead ribozymes were designed
based on the secondary structure of the gene transcript and
generated using the Zuker RNA mFold program (10). The ribozymes were synthesized and
then cloned into a pEF6/V5-His TOPO vector (Invitrogen, Paisley,
UK). The verified ribozyme transgenes and empty plasmids were
transfected into PC-3 (PC-3PTPRKkd and PC-3pEF) cells,
respectively, using an EasyjetPlus electroporator (EquiBio, Kent,
UK). After a period of selection with 5 μg/ml blasticidin
(up to 10 days), the verified transfectants were cultured in
maintenance medium containing 0.5 μg/ml blasticidin. Primer
sequences of the ribozymes were 5′-CTGCAGTTTGCTCTT
TTTTACAATTAATATCTGATGAGTCCGTGAGGA-3′ and
5′-ACTAGTTCATCCTCCTTCTCCTAGTTGTTTCGTCCT CACGGACT-3′.
Cell growth assay
Prostate cancer cells (3,000 cells/well) were plated
into 96-well plates. Cells were fixed in 4% form-aldehyde after 1
and 5 days of culture (11). The
cells were then stained with 0.5% crystal violet. Absorbance was
determined at a wavelength of 540 nm using a spectrophotometer
(BioTek, ELx800). Growth rate at day 5 (%) = absorbance of day
5/absorbance of day 1 x 100.
Analysing apoptosis using flow
cytometer
Cells (1×105) were seeded into flasks and
underwent different treatments with 200 nM p38 inhibitor
(SB203580), 200 nM JNK inhibitor (SP600125) and serum free control
for three days then both the adherent cells and floating cells were
collected. Following cell collection, experiments were carried out
using the Annexin V kit (Santa Cruz Biotechnology) and analysed
using PartecCyFlow® SL flow cytometry and the
accompanying FloMax software package (Partec GmbH, Münster,
Germany).
Immunoprecipitation (IP) and western blot
analysis
Protein was extracted from 75-cm2 flask
which were initially seeded with 4x106 cells and cultured
overnight. The protein samples were incubated with primary
antibodies (Table II) at 4°C for 1
h then incubated for another hour after the addition of conjugated
A/G protein agarose beads (Santa Cruz Biotechnology). The samples
were washed twice with SDS-free lysis buffer before being boiled
with 1X sample buffer (Sigma-Aldrich Inc.).
| Table II.Primary antibodies used in the
current study. |
Table II.
Primary antibodies used in the
current study.
| Protein target | Cat. no. |
|---|
| Mouse
anti-GAPDH | SC-47724 |
| Rabbit
anti-PTPRK | SC-28906 |
| Mouse anti-p38 | SC-7972 |
| Rabbit
anti-JNK | SC-571 |
| Rabbit
anti-caspase-3 | SC-7148 |
| Mouse
anti-caspase-8 | SC-70501 |
| Mouse
anti-caspase-9 | SC-17784 |
| Mouse
anti-phosphotyrosine | SC-508 |
Equal amounts of protein were separated by SDS-PAGE
and blotted onto nitrocellulose membranes. The membrane was then
probed with the respective primary antibodies and corresponding
peroxidase-conjugated secondary antibodies. Protein bands were
visualised using a chemiluminescence detection kit (Luminata,
Millipore) and photographed using UVITech imager (UVITech
Inc.).
Statistical analysis
Statistical analysis was performed using SigmaPlot
11 (SPSS Inc., Chicago, IL, USA). Data were calculated as the mean
± SD and the Student’s t-test was used for normally distributed
data. Fisher’s exact test was used for comparison of two
independent groups. Each assay was performed at least three times
independently. p<0.05 was considered statistically
significant.
Results
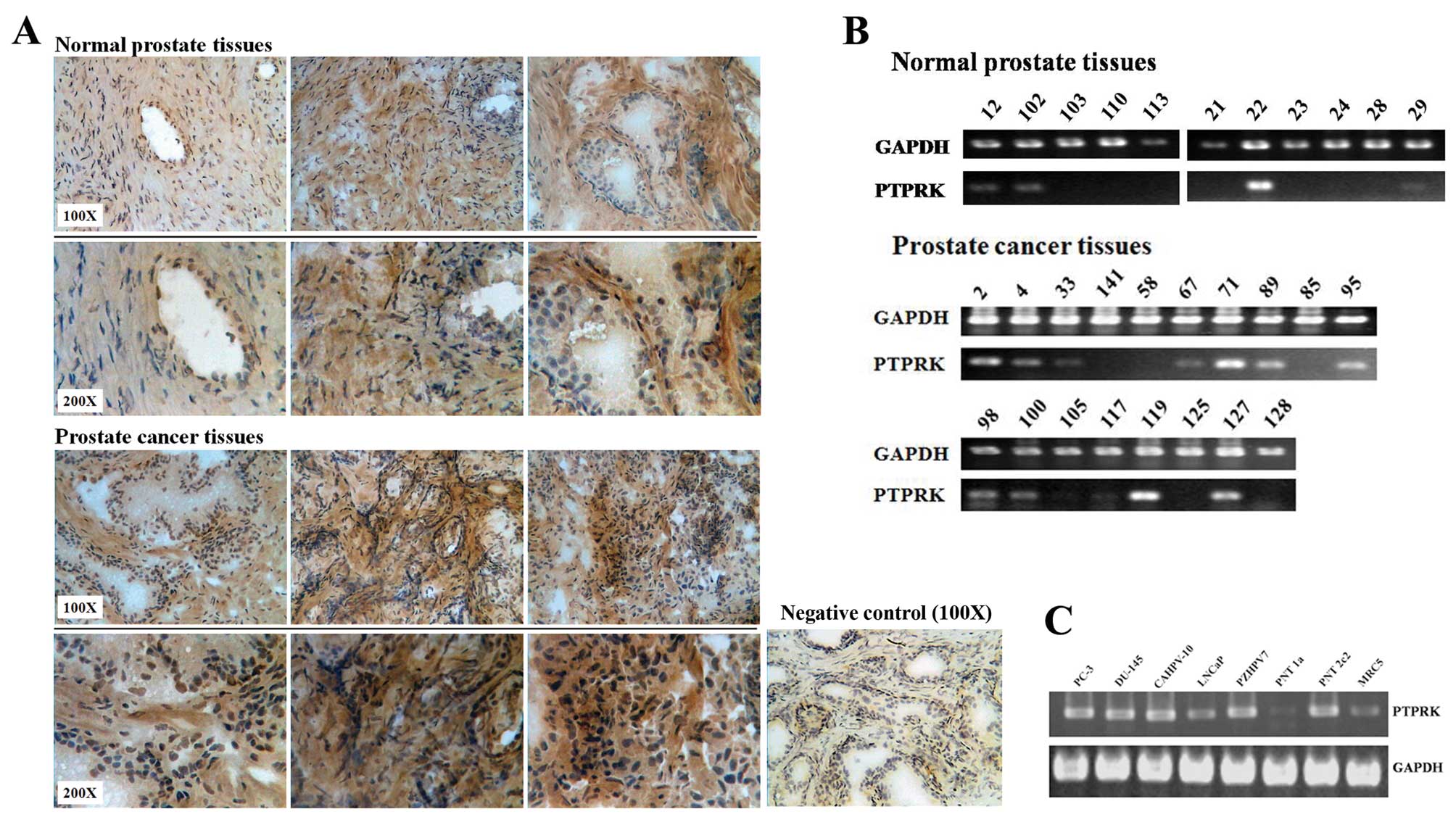
PTPRK expression in prostate tissues and
cell lines
There is a higher PTPRK expression level in prostate
cancer tissues compared with normal prostate tissues using IHC
staining (Fig. 1A). Fig. 1B shows that PTPRK is likely to be
more frequently expressed in prostate cancer tissues (12/18, 66.7%
positive, p= 0.143 vs. normal using Fisher’s exact test) than
normal prostate tissues at mRNA level (4/11, 36.7% positive).
Furthermore, the expression of PTPRK was also examined in the
prostate cell lines PC-3, DU-145, LNCaP, CA-HPV-10, PZ-HPV-7,
PNT-1A, PNT-2C2 and the fibroblast cell line MRC-5. Fig. 1C shows that PTPRK is consistently
expressed in all the prostate cell lines, except PNT-1A, where it
appears to have a lower level of PTPRK mRNA.
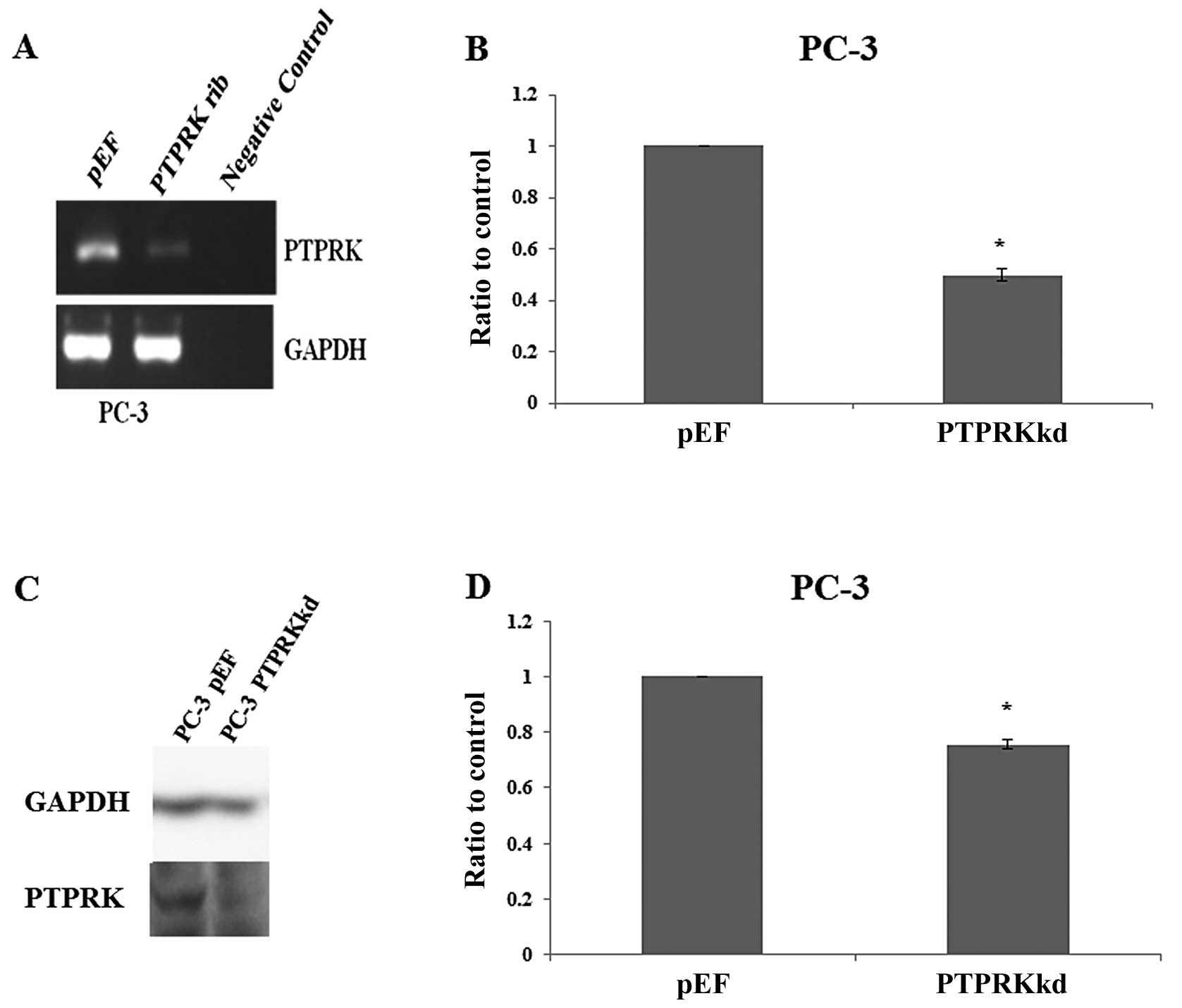
Verification of PTPRK knockdown in PC-3
cells
The expression of PTPRK was knocked down using
ribozyme transgenes targeting human PTPRK mRNA. This was performed
in the prostate cancer cell line PC-3, which expressed PTPRK
(Fig. 1C). The knockdown of PTPRK
was verified in the transfectants using RT-PCR (Fig. 2A), real-time quantitative PCR
(Fig. 2B), western blot analysis
(Fig. 2C) and PTPRK protein band
volume of three repeats which was normalised against corresponding
internal control (Fig. 2D).
Decreased expression of PTPRK was seen in PC-3PTPRKkd
cells which were transfected with ribozyme transgenes compared to
their corresponding empty plasmid control.
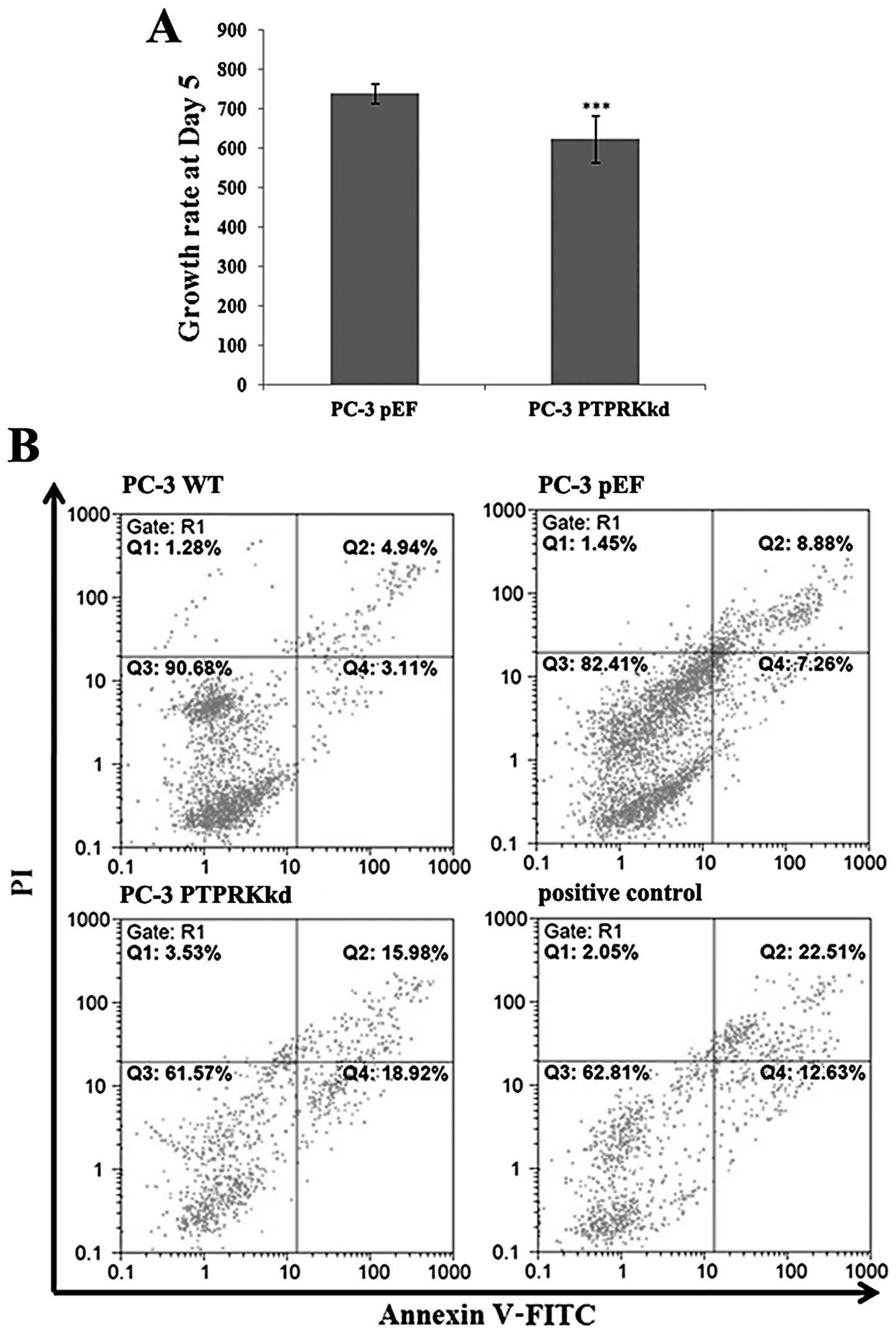
Knockdown of PTPRK reduces in vitro cell
growth assay
Knockdown of PTPRK in PC-3 cells exhibited an impact
on cell growth. The PC-3PTPRKkd cells showed a
decreasing growth rate at day 5 (634.33±58.76, p<0.001) compared
with PC-3pEF (739.35±24.14) (Fig. 3A).
PTPRK knockdown affects apoptosis in
prostate cancer cells
There was an increased proportion of apoptotic cells
(both early and late) in PC-3PTPRKkd (34.90%) compared
to the PC-3pEF control (16.14%) and PC-3 wild-type
(8.05%) cells (Fig. 3B).
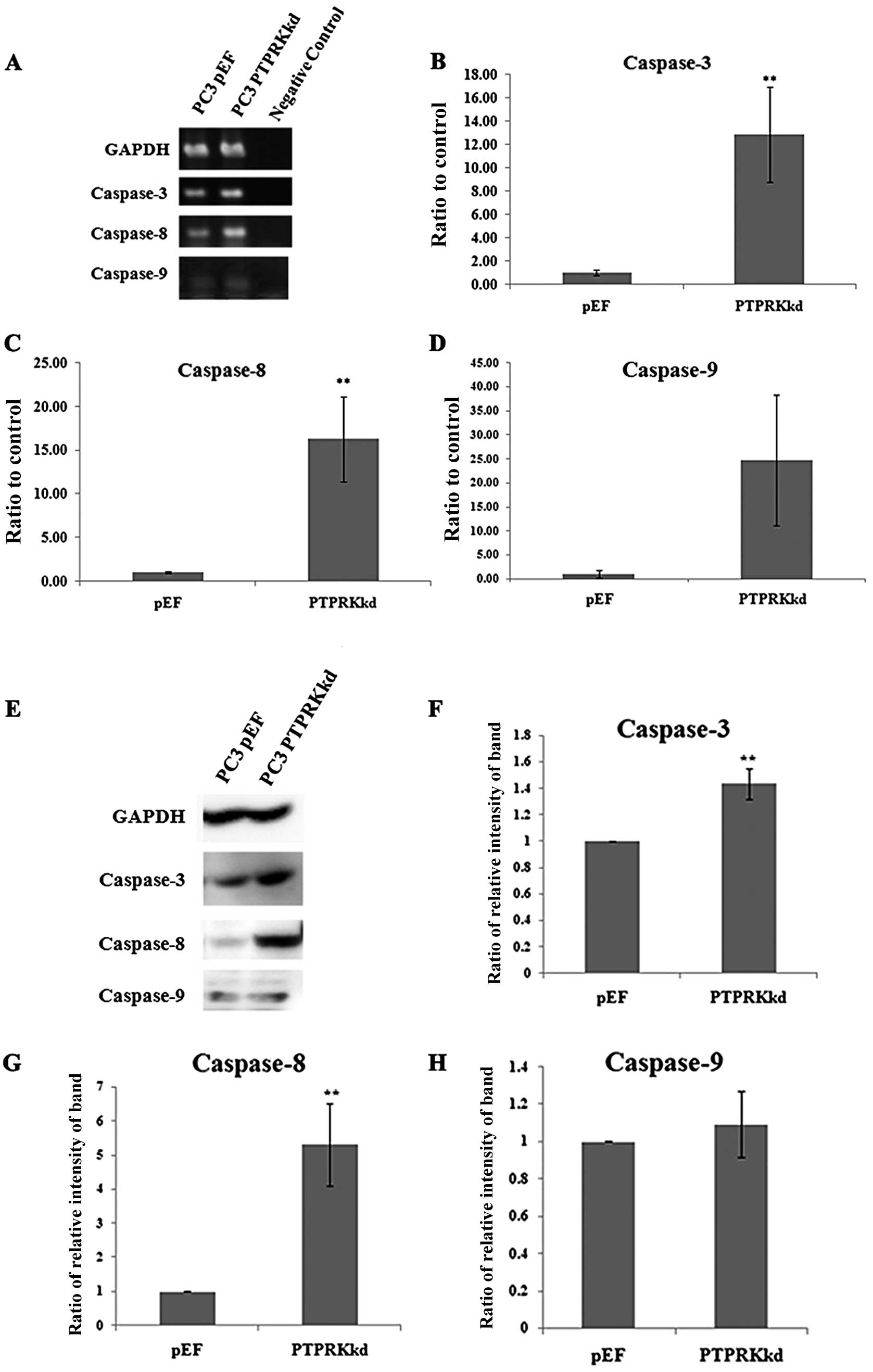
Expression of caspases in the PTPRK
knockdown cells
Caspase-3 is an indicator of apoptosis at the
end-stage. Caspase-8 and caspase-9 are up-stream key factors of
apoptosis; caspase-8 is normally activated by external signalling,
and caspase-9 is activated by internal signalling. Therefore, in
order to further determine whether PTPRK knockdown has an effect on
prostate cancer cell apoptosis, levels of caspase-3, -8 and -9 were
examined in the transfected PC-3 cells using PCR, Q-PCR, and
western blot analysis. PC-3PTPRKkd cells demonstrated
significantly higher expression levels of caspase-3 and caspase-8,
but not caspase-9, compared with PC-3pEF controls cells
in both mRNA and protein levels (Fig.
4).
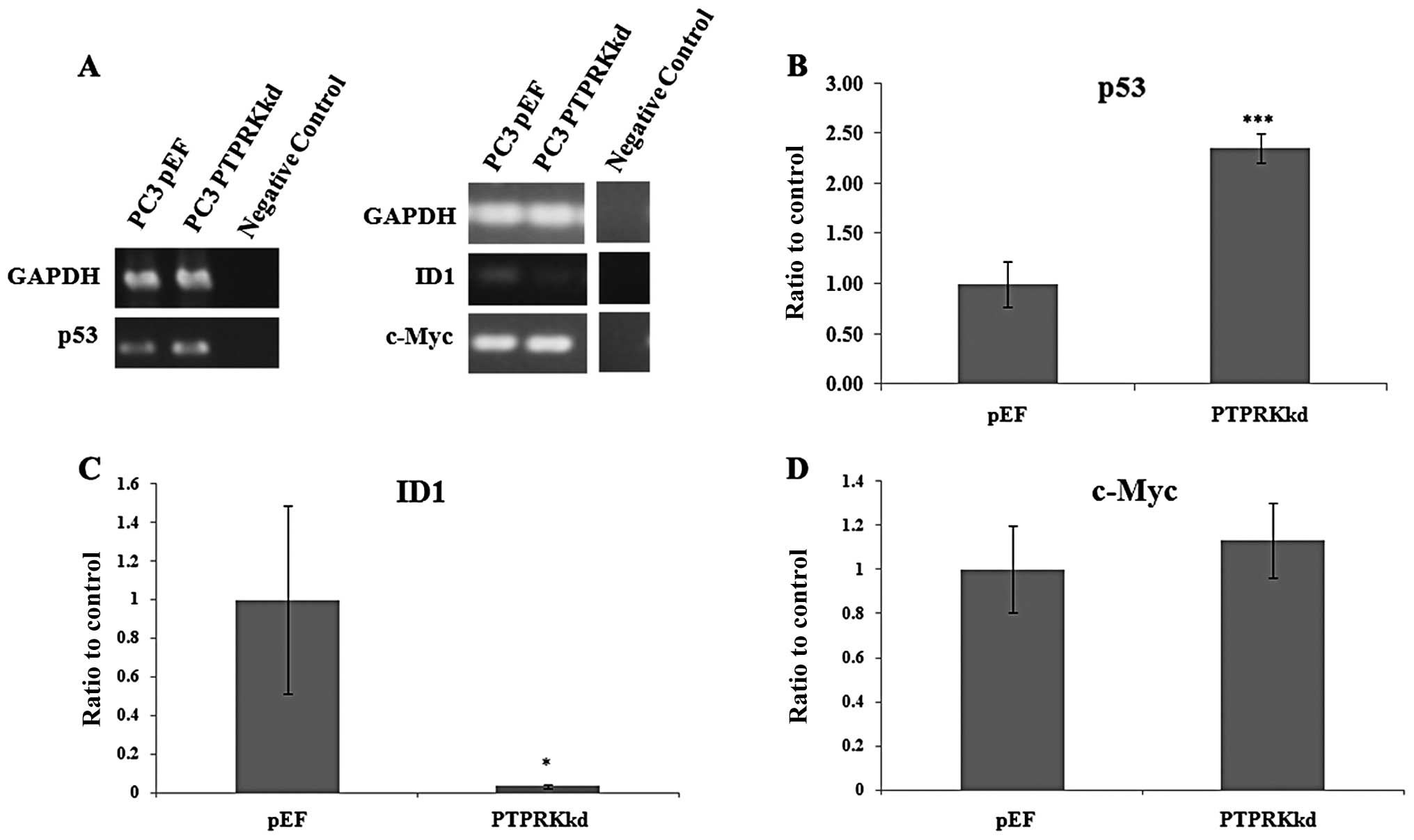
Expression of other genes/molecules
relevant to apoptosis and/or the cell cycle
As PTPRK knockdown was shown to promote the
progression of apoptosis, the expression of a number of relevant
genes was examined using RT-PCR. An upregulation of p53 was also
seen in the PTPRK knockdown cells, whilst a downregulation of ID1
appeared in the same cells (Fig.
5). No effect on the expression of c-Myc was observed in the
PC-3PTPRKkd cells compared with the control.
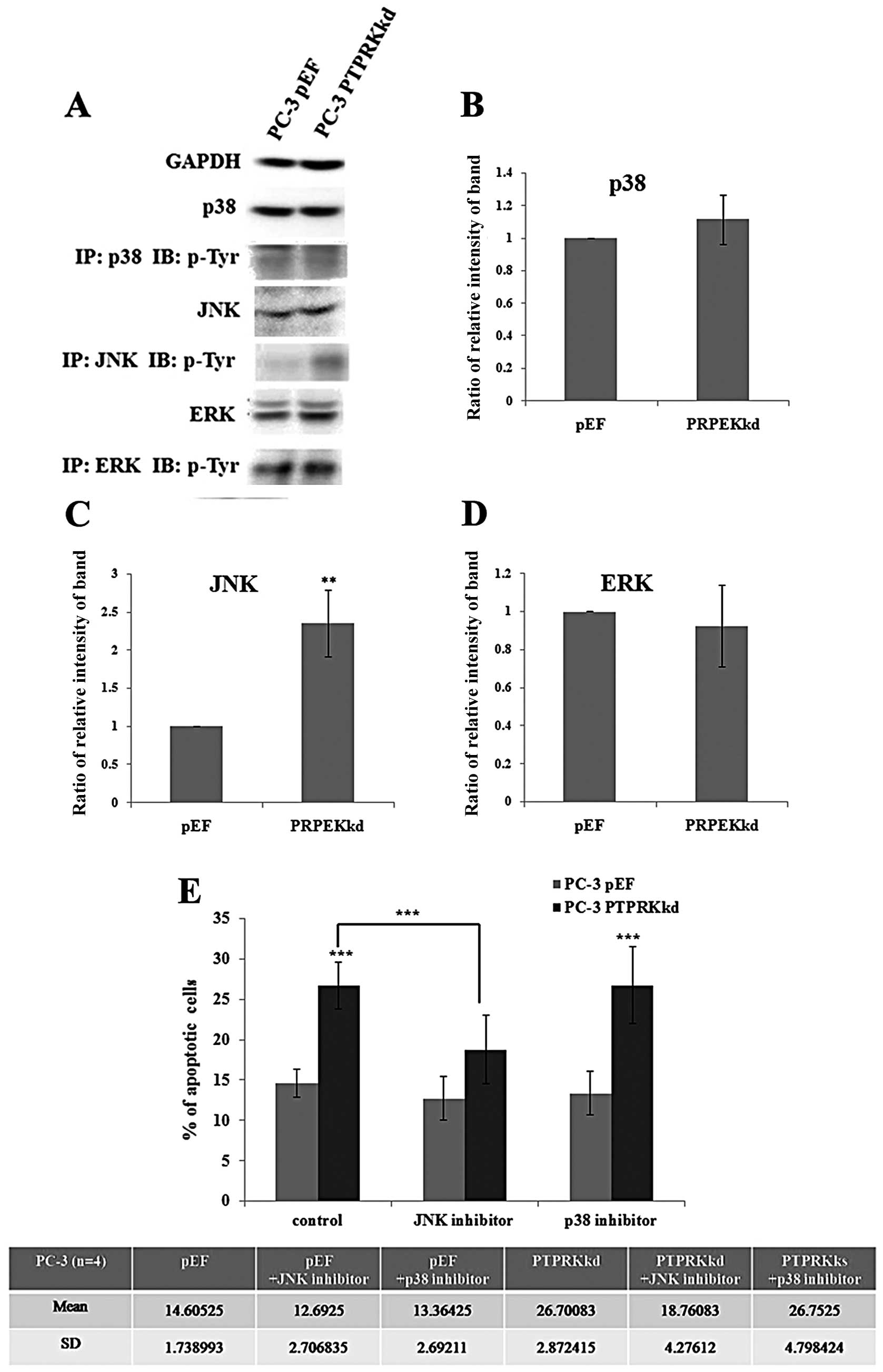
The role of JNK in PTPRK
knockdown-associated apoptosis
The mitogen-activated protein kinase (MAPK) pathway
is the major signalling pathway involved in cellular proliferation
and it affects both apoptosis and the cell cycle. As knockdown of
PTPRK has been shown to impact apoptosis, its effect on the
expression and activations of p38, JNK and ERK in PC-3 cells was
analysed. The PC-3PTPRKkd cell showed a similar level of
protein expression overall in p38, JNK and ERK compared with their
pEF control. Furthermore, levels of phosphorylated p38, JNK and ERK
were analysed using immunoprecipitation and western blot analysis.
A marked increase active p-JNK (Tyr) was seen in the PTPRK
knockdown cells and an increased level of active p38, suggesting
that JNK and p38 may play a role in the regulation of apoptosis in
the PC-3PTPRKkd cells (Fig.
6A).
Additionally, PC-3 cells were treated with p38 and
JNK inhibitors for 72 h and analysed for apoptosis using flow
cytometry. The PC-3pEF control cells showed no effect on
treatment of cells with p38 and JNK inhibitors, but apoptosis of
untreated PC-3PTPRKkd cells (26.70±2.87%) was
dramatically increased compared with PC-3pEF cells
(14.61±1.74%), p<0.001. The PC-3PTPRKkd cells treated
with p38 inhibitor (26.75±4.80%) exhibited similar apoptotic levels
to the untreated cells (13.36±2.69%). However, the
PC-3PTPRKkd cells treated with JNK inhibitor
(18.76±4.28%) exhibited significant reduction of apoptotic cells
compared with untreated PC-3PTPRKkd cells, p<0.001
(Fig. 6B). The addition of the p38
inhibitor did not inhibit the effect on apoptosis, suggesting that
p38 was unlikely to be involved. However, the addition of the JNK
inhibitor diminished the apoptotic effect of PTPRK knockdown. This
suggests that PTPRK knockdown may utilise a signalling pathway via
JNK to impact apoptosis thereby inhibiting cell proliferation.
Discussion
PTPRK is a poorly studied protein phosphatase in the
field of cancer progression. It has been indicated that PTPRK is a
potential tumour suppressor in primary lymphoma of central nervous
system and associated with colorectal cancer and pancreatic islet
tumours (12–15). No study has attempted to define the
function of PTPRK in prostate cells and its potential involvement
in cancer metastasis.
In this study, we demonstrate the presence of PTPRK
expression in 7 prostatic cell lines and 1 human fibroblast cell
line. The cell lines have extensively been used as models for in
vitro studies on prostate cancer. Its expression in tissues
shows that PTPRK is more highly expressed in prostate cancer
tissues compared to normal prostate tissue samples in both mRNA and
protein levels. According to the mRNA levels of PTPRK, its
expression appears at a similar level of that seen in prostate cell
lines. The only exception was PNT-1A which had lower PTPRK
expression. However, the current assessment of PTPRK expression in
prostate cancer is limited and not sufficient to reach any solid
conclusion. Hopefully, the implication of PTPRK in disease
development and progression of prostate cancer can be elucidated by
further investigations using a large clinical cohort of prostate
cancer.
PTPs have also been shown to exhibit different
effects on tumour apoptosis (16).
PTPRK has been shown to be capable of inhibiting the growth of
prostate cancer cells through endogenous alteration of expression.
In general, cell population is controlled by the regulation and
balance between cell proliferation (cell cycle) and cell death
(necrosis and apoptosis). PTPRK knockdown promoted apoptosis in
prostate cancer cells and suppressed in vitro growth.
Moreover, apoptosis was associated with the
activation of caspase-3, -8 and -9, and an increasing Bax:Bcl-2
ratio, followed by release of cytochrome c (17). Caspase-3 is a key molecule in the
late stage of apoptosis; caspase-8 normally is activated by
extrinsic receptor-mediated pathway; and caspase-9 is activated by
intrinsic mitochondrial-mediated pathway (18). In order to analyse how these
molecules were affected in PC-3PTPRKkd cells, the
expression of caspase-3, -8 and -9 was analysed using PCR, Q-PCR
and western blot analysis. There was a significant increase in both
caspase-3 and -8 expressions, but not in caspase-9 following the
knockdown of PTPRK. This result showed that the increased apoptosis
in PC-3PTPRKkd cells was related with extrinsic
signalling rather than mitochondrial signalling.
Most PTPs play a role in promoting apoptosis. For
example, PTP1B has been reported to play a role in the activation
of MAPKs. PTP1B activates JNK and p38 pathways via
inositol-requiring kinase 1 (IRE1) signalling and lack of PTP1B
resulted in decreased levels of ER-induced apoptosis (5,6).
However, SHP-1 (PTPN6) dephosphorylates TrkA which in turn inhibits
NGF-mediated PLCγ1 and Akt phosphorylation, reducing the TrkA
survival signal (19). In
contrast, osteoclastic PTP (PTP-oc) has been reported to promote
c-Src-mediated activation of NF-κB and JNK leading to protection
from apoptosis (20). In this
study, the reduction of PTPRK expression in PC-3 cells shows
promotion of apoptosis and involvement of MAPK signalling pathway.
The tyrosine phosphorylation of JNK was increased in
PC-3PTPRKkd cells and promotion of apoptosis in
PC-3PTPRKkd cells was diminished after treating cells
with the JNK inhibitor (SP600125). These data suggest that PTPRK
downregulates apoptosis in prostate cancer cells by suppressing the
JNK pathway.
We also investigated other apoptosis-related
molecules such as p53, ID1 and c-Myc. These molecules play crucial
roles in the regulation of cell proliferation. Expression of c-Myc
in tumours helps cancerous cells pass through check-points and
progress to the G2/M phase of the cell cycle. In addition,
expression of ID1 activates NF-κB, which then induces Bcl-2 to
inhibit apoptosis. In contrast to these anti-apoptotic factors, p53
acts as a promoter of apoptosis. Furthermore, mRNA expression of
p53 was increased and expression of ID1 was decreased. p53
regulates both cell cycle and apoptosis. There are some reports
that show that expression of p53 is associated with the induction
of apoptosis in different cancer cells (21). p53 silencing was able to suppress
cadmium-induced apoptosis in prostate cells (22). Additionally, activation of JNK can
also upregulate p53 expression, leading to the accumulation of Bax
which induces cell apoptosis in HeLa cells (23). In contrast, ID1 expression
increases NF-κB expression which is associated with the
anti-apoptotic pathway. NF-κB activates Bcl-2 to initiate the
mitochondrial mediated anti-apoptotic effect and also activates
XIAP to inhibit the activities of caspase-3 and -9 (24,25).
These results indicate a complex network affected by PTPRK which
participates in the coordination of cellular functions, making
further investigations into the protein interactions between PTPRK
and the network protein an interesting area to explore in
future.
PTPRK knockdown resulted in increased apoptosis
leading to the inhibition of in vitro growth of prostate
cancer cells. PTPRK is a key factor in coordinating apoptosis via
regulation of the MAPK pathways, in particular the JNK pathway in
prostate cancer cells. Collectively, it is suggested that PTPRK may
a key factor to be included in the signature of diagnosis and
prediction of prostate cancer, and has great potential in the
guidance of personalised treatment of malignancies.
Acknowledgements
The authors would like to thank Cancer
Research Wales for their support.
References
|
1.
|
Wallach D, Kang TB and Kovalenko A: The
extrinsic cell death pathway and the elan mortel. Cell Death
Differ. 15:1533–1541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wajant H and Scheurich P: TNFR1-induced
activation of the classical NF-κB pathway. FEBS J. 278:862–876.
2011.
|
|
3.
|
Halle M, Liu YC, Hardy S, et al: Caspase-3
regulates catalytic activity and scaffolding functions of the
protein tyrosine phosphatase PEST, a novel modulator of the
apoptotic response. Mol Cell Biol. 27:1172–1190. 2007. View Article : Google Scholar
|
|
4.
|
Akasaki Y, Liu G, Matundan HH, et al: A
peroxisome proliferator-activated receptor-gamma agonist,
troglitazone, facilitates caspase-8 and -9 activities by increasing
the enzymatic activity of protein-tyrosine phosphatase-1B on human
glioma cells. J Biol Chem. 281:6165–6174. 2006. View Article : Google Scholar
|
|
5.
|
Gu F, Nguyen DT, Stuible M, Dube N,
Tremblay ML and Chevet E: Protein-tyrosine phosphatase 1B
potentiates IRE1 signaling during endoplasmic reticulum stress. J
Biol Chem. 279:49689–49693. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Sangwan V, Paliouras GN, Cheng A, Dube N,
Tremblay ML and Park M: Protein-tyrosine phosphatase 1B deficiency
protects against Fas-induced hepatic failure. J Biol Chem.
281:221–228. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Gupta S, Radha V, Sudhakar C and Swarup G:
A nuclear protein tyrosine phosphatase activates p53 and induces
caspase-1-dependent apoptosis. FEBS Lett. 532:61–66. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Radha V, Sudhakar C and Swarup G:
Induction of p53 dependent apoptosis upon overexpression of a
nuclear protein tyrosine phosphatase. FEBS Lett. 453:308–312. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Jiang WG, Watkins G, Fodstad O,
Douglas-Jones A, Mokbel K and Mansel RE: Differential expression of
the CCN family members Cyr61, CTGF and Nov in human breast cancer.
Endocr Relat Cancer. 11:781–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zuker M: Mfold web server for nucleic acid
folding and hybridization prediction. Nucleic Acids Res.
31:3406–3415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Jiang WG, Davies G, Martin TA, et al:
Targeting matrilysin and its impact on tumor growth in vivo: the
potential implications in breast cancer therapy. Clin Cancer Res.
11:6012–6019. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Cady FM, O’Neill BP, Law ME, et al:
Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are
indicators of an aggressive clinical course. J Clin Oncol.
26:4814–4819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Nakamura M, Kishi M, Sakaki T, et al:
Novel tumor suppressor loci on 6q22-23 in primary central nervous
system lymphomas. Cancer Res. 63:737–741. 2003.PubMed/NCBI
|
|
14.
|
Starr TK, Allaei R, Silverstein KA, et al:
A transposon-based genetic screen in mice identifies genes altered
in colorectal cancer. Science. 323:1747–1750. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Lu J, Li Q, Donadel G, Notkins AL and Lan
MS: Profile and differential expression of protein tyrosine
phosphatases in mouse pancreatic islet tumor cell lines. Pancreas.
16:515–520. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Julien SG, Dube N, Hardy S and Tremblay
ML: Inside the human cancer tyrosine phosphatome. Nat Rev Cancer.
11:35–49. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Halle M, Tremblay ML and Meng TC: Protein
tyrosine phosphatases: emerging regulators of apoptosis. Cell
Cycle. 6:2773–2781. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Lee ST, Wong PF, Cheah SC and Mustafa MR:
Alpha-tomatine induces apoptosis and inhibits nuclear factor-kappa
B activation on human prostatic adenocarcinoma PC-3 cells. PLoS
One. 6:e189152011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Marsh HN, Dubreuil CI, Quevedo C, et al:
SHP-1 negatively regulates neuronal survival by functioning as a
TrkA phosphatase. J Cell Biol. 163:999–1010. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Amoui M, Sheng MH, Chen ST, Baylink DJ and
Lau KH: A transmembrane osteoclastic protein-tyrosine phosphatase
regulates osteoclast activity in part by promoting osteoclast
survival through c-Src-dependent activation of NFkappaB and JNK2.
Arch Biochem Biophys. 463:47–59. 2007. View Article : Google Scholar
|
|
21.
|
Pietsch EC, Sykes SM, McMahon SB and
Murphy ME: The p53 family and programmed cell death. Oncogene.
27:6507–6521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Aimola P, Carmignani M, Volpe AR, et al:
Cadmium induces p53-dependent apoptosis in human prostate
epithelial cells. PLoS One. 7:e336472012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Cao H, Feng Q, Xu W, et al: Genipin
induced apoptosis associated with activation of the c-Jun
NH2-terminal kinase and p53 protein in HeLa cells. Biol Pharm Bull.
33:1343–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Ling MT, Wang X, Ouyang XS, Xu K, Tsao SW
and Wong YC: Id-1 expression promotes cell survival through
activation of NF-kappaB signalling pathway in prostate cancer
cells. Oncogene. 22:4498–4508. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Peng X, Wang Y, Kolli S, et al: Physical
and functional interaction between the ID1 and p65 for activation
of NF-κB. Am J Physiol Cell Physiol. 303:C267–C277. 2012.PubMed/NCBI
|