Introduction
Pancreatic cancer is a devastating disease with an
overall median survival time of 4–6 months, making it the fourth
major cause of cancer-related deaths in the US (1). Contributing to the lethality of the
disease is its ability to grow undetected until it reaches a
metastatic state, where surgery, the only curative option, has
little effect (2,3). Despite advances in chemotherapy, its
impact on long-term survival has been minimal. Thus, there remains
a compelling need to develop new and efficacious chemotherapeutic
agents for pancreatic cancer.
Non-steroidal anti-inflammatory drugs (NSAIDs) have
demonstrated antineoplastic properties in pancreatic cancer;
however, their efficacy and safety as anticancer agents are limited
(4). For example, sulindac, alone
or in combination with other drugs, modestly inhibited the growth
of pancreatic cancer in pre-clinical models (5–8).
Chronic use of sulindac, however, is associated with significant
gastrointestinal and renal toxicity. Prompted by these
considerations, our group has synthesized phospho-sulindac (P-S). A
key structural feature of P-S is the covalent modification of the
-COOH moiety, a major culprit for its gastrointestinal toxicity,
with a diethylphosphate group via a linker. Phospho-sulindac (P-S)
has demonstrated significant efficacy against colon cancer,
substantially exceeding that of sulindac (9). P-S is also much safer than sulindac
(9,10), especially regarding its ability to
spare the gastroduodenal mucosa (11). Hence, we hypothesized that P-S
would be efficacious against pancreatic cancer.
Nuclear factor of activated T-cells (NFAT) are a
family of nuclear transcription factors primarily involved in the
regulation of T-cell activation and differentiation (12). Recent studies, on the other hand,
revealed non-canonical functions of NFAT in several malignancies,
most notably in pancreatic cancer, where it drives cancer
progression via the promotion of cell proliferation, invasion and
angiogenesis (13). Aberrant
regulation of NFAT thus contributes to drug resistance to diverse
therapeutic agents (14,15).
In the current study, we demonstrate that P-S
inhibits pancreatic cancer growth in vitro and in
vivo. We identified NFATc1-dependent signaling as a mechanism
of drug resistance in pancreatic cancer; and showed that the
activation of NFATc1 is an unfavorable prognostic factor that
predicts poor tumor response to P-S, which can be overcome by
pharmacological intervention.
Materials and methods
Reagents
Phospho-sulindac (P-S) was synthesized as reported
(16). Sulindac was purchased from
Sigma (St. Louis, MO, USA). Annexin V was purchased from Invitrogen
(Grand Island, NY, USA). Propidium iodide (PI) and
5-bromo-2’-deoxyuridine (BrdU) were obtained from BD Bioscience
(San Jose, CA, USA). All general solvents and reagents were of HPLC
grade or the highest grade commercially available.
Cell lines
Human pancreatic cancer cell lines BxPC-3, Mia
PaCa-2, Panc-1 and HPAF II were obtained from American Type Tissue
Collection (ATCC, Manassas, VA, USA). Cells were grown at 37°C in
5% CO2 in the specific medium suggested by ATCC and
supplemented with 10% fetal calf serum (Mediatech, Herndon, VA,
USA), penicillin 50 U/ml and streptomycin 50 μg/ml (Life
Technologies, Grand Island, NY, USA).
NFATc1 knockdown BxPC-3 and Mia PaCa-2 cell lines
were generated using MISSION Lentiviral Packaging Mix
(Sigma-Aldrich, St. Louis, MO, USA). Cells were infected with the
lentivirus and then subjected to 3 μg/ml of puromycin to
generate stable cell lines. Decreased levels of NFATc1 were
confirmed by western blotting.
The NFATc1 Mia PaCa-2 overexpression cell line was
generated using NFATc1 human cDNA clone from Origene (Rockville,
MD, USA). Cells were transfected using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA) and selected with 1 mg/ml of
G418.
Cytokinetic analyses
Cell viability was measured by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide assay
following the protocol of the manufacturer (Roche Diagnostics,
Indianapolis, IN, USA). Apoptosis was assessed by Annexin
V/propidium iodide (PI) staining (Life Technologies). Cell
proliferation was determined by the bromodeoxyuridine (BrdU)
incorporation method (BD Biosciences, San Jose, CA, USA).
Gene expression microarray
Mia PaCa-2 cells were treated with vehicle or
1.5×IC50 P-S for 30 min or 2 h. Total RNA was isolated
from cell lines using an RNA Extraction kit (Qiagen Inc., Valencia,
CA, USA). Samples were submitted to Genome Explorations (Memphis,
TN, USA) and analyzed on the Affymetrix Human Genome U133 gene chip
(Cleveland, OH, USA).
Immunofluorescence
Cells were seeded in an 8-well glass chamber slide
(Lab-Tek, Rochester, NY, USA). The following day, cells were
treated with vehicle or P-S for 24 h. Cells were fixed in 4%
paraformaldehyde (15 min). Block and NFATc1 antibody (Abcam,
Cambridge, MA, USA) incubations were performed in PBS with 2% BSA
and 0.3% Triton. Cells were mounted with Vectashield mounting media
with diamidinophenylindole (DAPI; Vector Laboratories Inc.,
Burlingame, CA, USA) and imaged using a Zeiss Axioplan inverted
fluorescence microscope (Thornwood, NY, USA).
Western blotting
After treatment, cells were lysed on ice with 1%
Triton lysis buffer containing 2.5 mmol/l 4-nitrophenylphosphate,
1% SDS and 0.25% sodium deoxycholate for 30 min. The cell lysate
(25 μg) was fractionated by SDS-electrophoresis gel and
transferred onto a PVDF membrane. This was followed by
immunoblotting with NFATc1 (Abcam), pERK, pAKT (Cell Signaling,
Danvers, MA, USA), COX-2 (Cayman Chemical, Ann Arbor, MI, USA) and
c-Myc (Santa Cruz, Santa Cruz, CA, USA) antibodies. Secondary
antibodies conjugated with horseradish peroxidase (HRP) (Santa Cruz
Biotechnology) were applied to the membrane, followed by
development on X-ray film.
qRT-PCR
Total RNA was isolated from cell lines using TRIzol
reagent as per the instructions of the manufacturer (Life
Technologies). Ten micrograms of RNA was used in
reverse-transcription using random primers and the M-MLV Reverse
Transcriptase kit (Sigma). The following primer pairs were used for
mouse NFATc1: forward 5′-CCAGTCATC GGCGGGAAGAAGA-3′; reverse
5′-TATACACCCCCAG ACCGCATCAGC-3′.
Pancreatic cancer xenografts
All animal experiments were approved by the
Institutional Animal Care and Use Committee at the Stony Brook
University. Six-week-old female BALB/c nude mice (Charles River,
Wilmington, MA, USA) were xenografted subcutaneously with Mia
PaCa-2 cells (1.5×106 in 100 μl PBS) in the right
and left flank. The animals were then treated for 4 weeks with
vehicle or P-S (100 mg/kg/day, p.o.) (n=6/group).
To study the effect of NFAT expression on drug
response, animals were separated into 6 groups (n=6/group) and
treated by with vehicle (corn oil, p.o.) or P-S (100 mg/kg/day,
p.o.) for one week prior to the inoculation of wild-type, NFAT Kd
or NFAT overexpressing Mia PaCa-2 cells into the right and left
flank. Treatment continued for 4 weeks. For all studies, tumor size
was measured using a digital microcaliper. Upon study completion,
animals were euthanized and tumors collected, measured and embedded
in OCT.
Immunohistochemistry
Mia PaCa-2 xenograft tissue was fixed in 10%
phosphate-buffered formalin for 16 h, dehydrated and embedded into
paraffin blocks as previously described (17). Slides were prepared and stained for
Ki-67 (Santa Cruz Biotechnology), TUNEL (Roche Applied Science,
Indianapolis, IN, USA), COX-2 (Cayman Chemicals), or NFATc1 (Abcam)
positive cells. A pathologist blinded to sample identity scored the
number of positive cancer cells and the number of all (positive and
negative) cancer cells to calculate the percentage of positive
cells.
Statistical analysis
Results from at least 3 independent experiments and
expressed as the mean ± SD were analyzed by the Student’s t-test.
p<0.05 was considered significant.
Results
P-S inhibits the growth of pancreatic
cancer cells in vitro and in human pancreatic xenografts
Table I summarizes
the 24, 48 and 72 h IC50 values of P-S as well as those
of sulindac in a panel of 4 human pancreatic cancer cell lines:
BxPC-3, Mia PaCa-2, Panc-1 and HPAF-II. In all four cell lines, P-S
showed an enhanced potency when compared to sulindac that was
time-dependent, becoming higher the longer the incubation period.
At 24 h, P-S showed an average potency enhancement of 22.5-fold
over sulindac; at 48 h, 36.0-fold; and at 72 h, 60.8-fold.
 | Table I.The 24-h IC50 values of P-S
and sulindac in a panel of pancreatic cancer cell lines. |
Table I.
The 24-h IC50 values of P-S
and sulindac in a panel of pancreatic cancer cell lines.
| Cell line | P-S IC50,
μM/potency enhancementa |
|---|
|
|---|
| 24 h | 48 h | 72 h |
|---|
| BxPC-3 | 106±4 >19 | 42±2 >48 | 15±2 >133 |
| Mia-PaCa-2 | 79±2 >25 | 60±2 >33 | 55±1 >36 |
| Panc-1 | 99±2 >20 | 83±4 >32 | 51±2 >39 |
| HPAF-II | 76±2 >26 | 65±2 >31 | 57±1 >35 |
We next evaluated the efficacy of P-S in nude mice
bearing human pancreatic cancer cell xenografts. As shown in
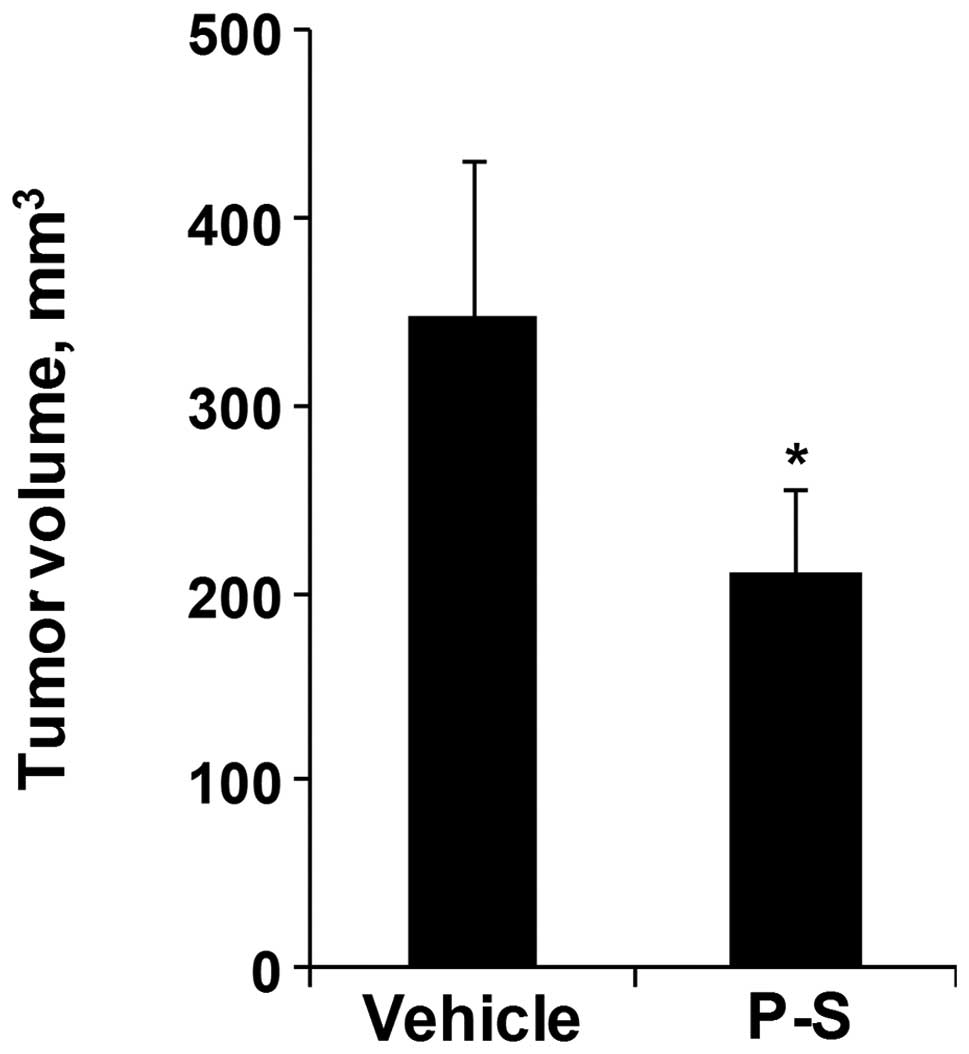
Fig. 1, tumor volume was decreased
by 45.3% in P-S (p.o.) treated group in comparison to vehicle
(p<0.05). Moreover, the P-S treated groups exhibit no changes in
body weight or signs of toxicity. Thus, P-S is an efficacious drug
in inhibiting pancreatic cancer growth in vitro and in
vivo.
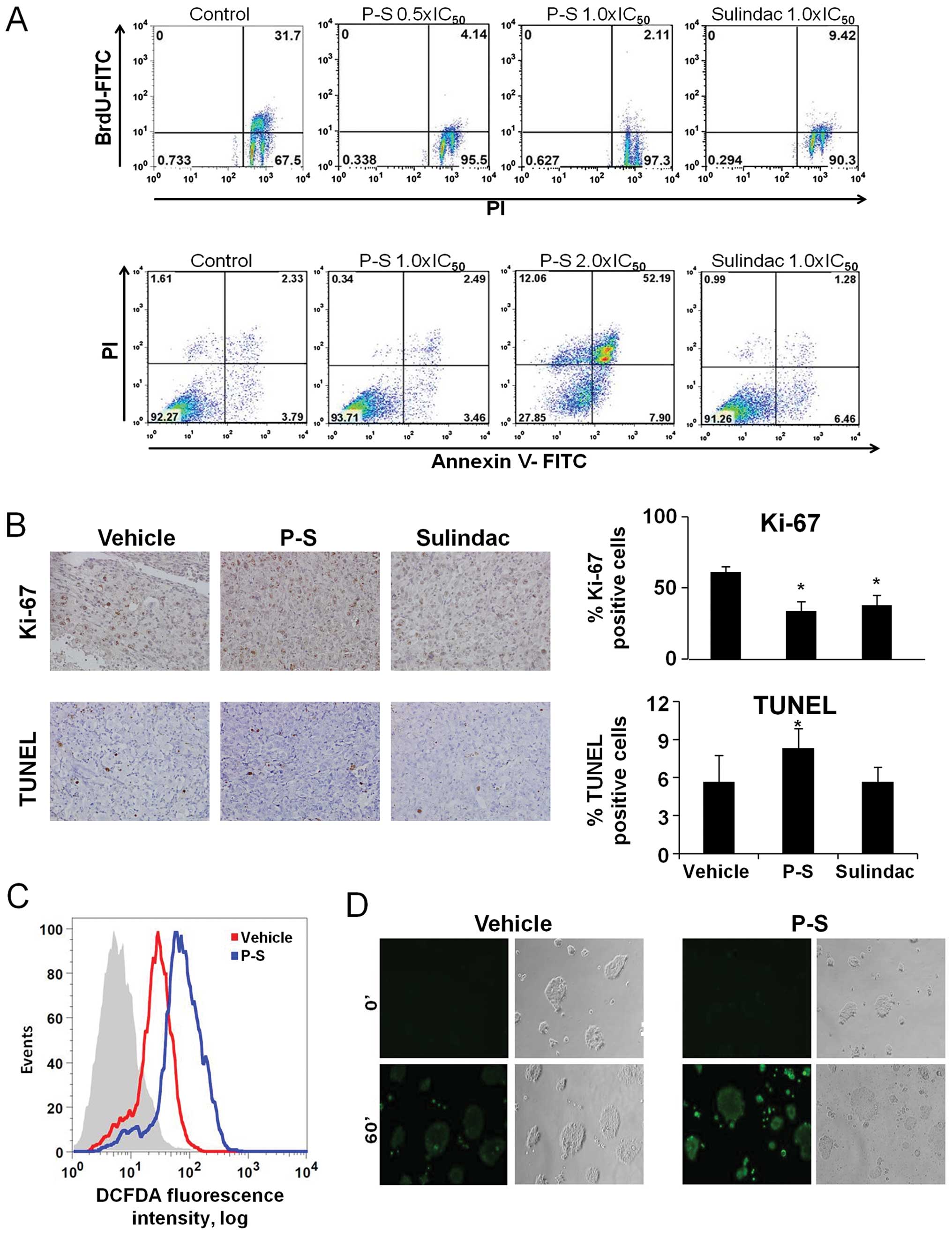
The cytokinetic effect of P-S
Given that P-S suppresses the growth of pancreatic
cancer cells, we determined the cytokinetic effect of P-S in BxPC-3
cells. P-S had a significant inhibitory effect on cell
proliferation (Fig. 2A), reducing
BrdUFITC(+) cells by 87.0% at 0.5×IC50, and 93.3% at
1.0×IC50. Sulindac 1.0×IC50 reduced BrdU(+)
cells by 70.3%, 20% less in comparison to P-S 1.0×IC50.
These results show that while both P-S and sulindac are able to
reduce pancreatic cancer cell proliferation, P-S was more potent
than its parent compound.
The effect of P-S on apoptosis was analyzed in
BxPC-3 cells (Fig. 2A). There was
no significant induction of apoptosis at P-S 1.0×IC50 or
sulindac 1.0×IC50. However, P-S at 2.0×IC50
induced a dramatic increase in Annexin V-positive cells, with
apoptotic cells comprising 60% of the total. These results show
that P-S induces cell death in BxPC-3 cells in a
concentration-dependent manner. Similar results were observed in
Mia PaCa-2 cells (data not shown).
The cytokinetic effect of P-S on proliferation and
apoptosis was analyzed in vivo (Fig. 2B). P-S inhibited cell
proliferation, as indicated by the reduction in Ki-67 positive
cells by 44.9% (p= 0.006) in Mia PaCa-2 xenografts. P-S also
increased apoptosis in these xenografts. TUNEL positive cells were
increased by 31.9% in comparison to vehicle.
P-S induces ROS in BxPC-3 cells
Previous studies have shown that P-S induces
reactive oxygen species (ROS) in various types of cancer cells
(10,16). BxPC-3 cells were treated with P-S
1.0×IC50 for 1 h and ROS levels were determined using
the DCFDA general ROS probe (Fig.
2C). There was a 3-fold increase in ROS in cells treated with
P-S (p<0.05) compared to the vehicle. Live cell imaging for ROS
levels was also performed in BxPC-3 cells treated either with
vehicle or P-S for 1 h (Fig. 2D),
using the DCFDA general ROS probe. Similarly, there was a notable
increase in ROS in cells treated with P-S in comparison to
vehicle.
Microarray analysis of gene expression in
Mia PaCa-2 cells treated with P-S
To further understand the signaling effects of Mia
PaCa-2 cells, we analyzed gene expression of vehicle or P-S
(1.5×IC50) treated cells using the Affymetrix Human
Genome U133 gene chip. Gene expression of NFATc1 and AP1, members
of the B- and T-cell receptor pathways, was upregulated >2-fold
in response to treatment with P-S. NFATc1 is a member of the NFAT
(nuclear factor of activated T-cells) family of transcription
factors initially identified as regulators of T-lymphocyte
activation (12,13). NFATc1 has been shown to be
overexpressed in pancreatic cancer and contributing to the
aggressive nature of the disease (12,13,20).
Effect of P-S on the expression of NFATc1
and its downstream targets COX-2 and c-Myc in BxPC-3 and Mia-PaCa-2
cells
Sustained activation of calcineurin-NFAT
transcription pathway has a pro-proliferative effect in pancreatic
cancer cells through the transcription activation of oncogenic
c-myc (20) and cyclooxygenase-2
(COX-2) (21). To investigate the
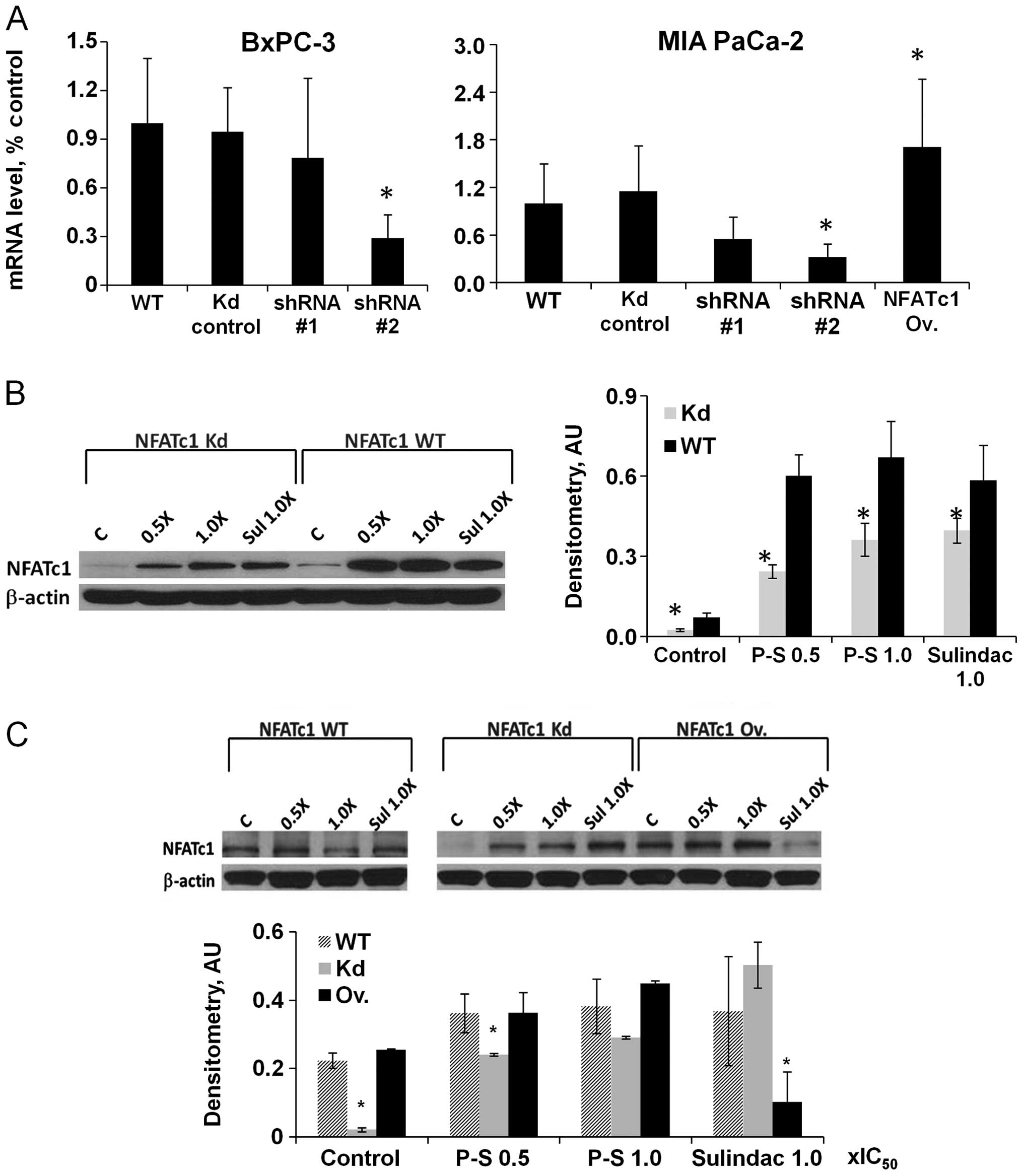
effect of P-S on NFATc1 signaling, we generated NFATc1-knockdown
cell lines from BxPC-3 and Mia PaCa-2 pancreatic cancer cells; and
overexpressed NFATc1 in Mia PaCa-2 cells. Knockdown or
overexpression of NFATc1 gene and protein expression was confirmed
by quantitative real-time PCR and western blotting (Fig. 3A).
P-S (0.5×IC50 and 1×IC50) or
sulindac (1×IC50) treatment in BxPC-3 and Mia PaCa-2
(NFATc1 WT, knockdown or over-expressing) cells induced the
expression of NFATc1 protein (Fig. 3B
and C). In addition, nuclear accumulation of NFATc1, indicative
of NFATc1 activation, was increased dose-dependently upon treatment
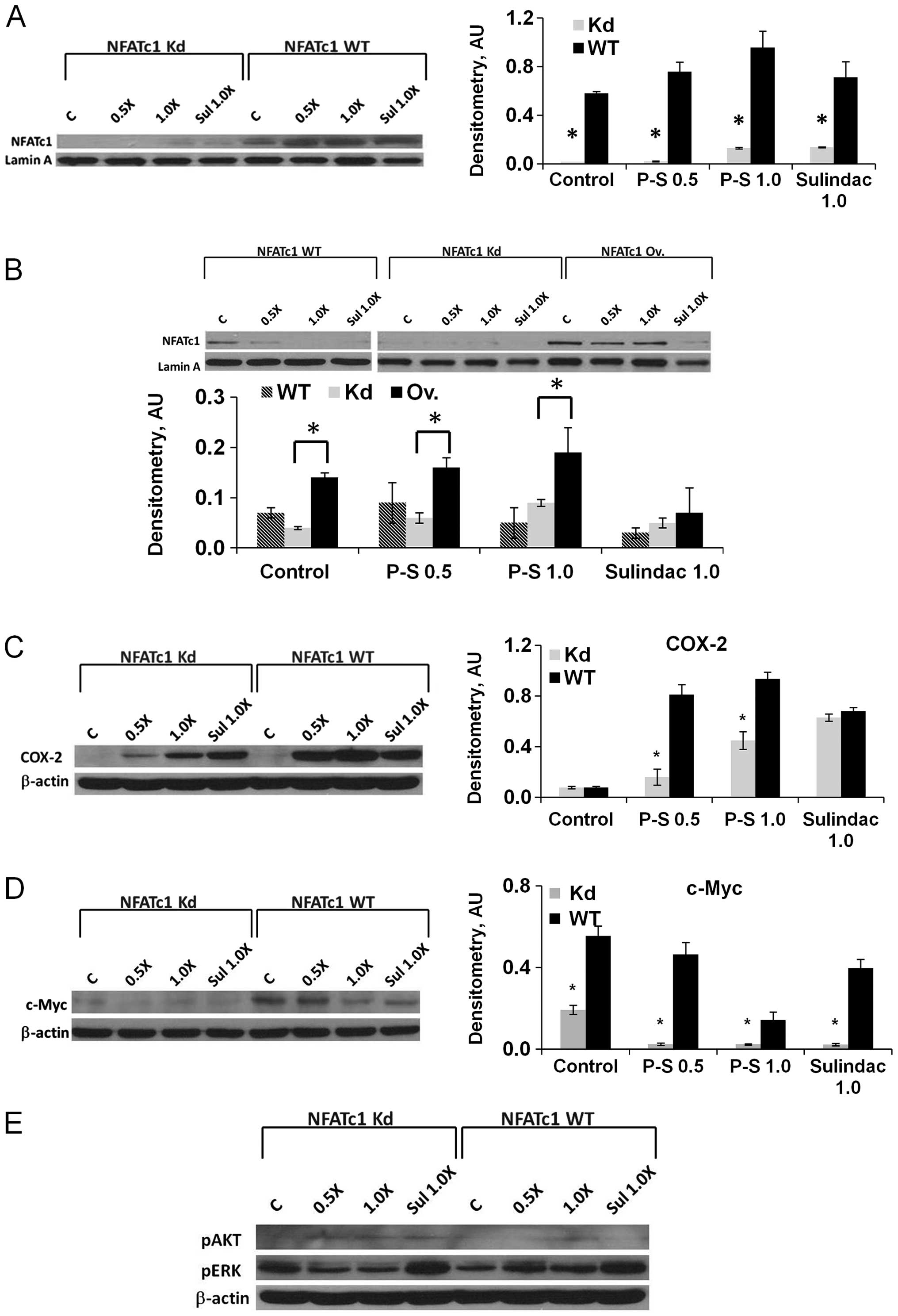
with P-S in BxPC-3 and Mia PaCa-2 cells (Fig. 4A and B). In all cases, the protein
levels of NFATc1 were lower in NFATc1-knockdown cells compared to
wild-type cells, suggesting that shRNA effectively abrogates the
induction of NFATc1 by P-S (p<0.05). In summary, P-S induced the
expression of NFATc1 in pancreatic cancer cells in
vitro.
Next, we investigated the expression of c-myc and
COX-2, target genes of NFATc1, in pancreatic cancer cells in
response to P-S treatment. While no difference in COX-2 protein
expression (Fig. 4C) was observed
in untreated wild-type and NFATc1-knockdown BxPC-3 cells, P-S
treatment induced COX-2 expression in both cell lines, with a more
dramatic effect in the wild-type cells. A similar effect was also
observed after treatment with sulindac. The expression of c-Myc was
suppressed in NFATc1-knockdown cells compared to wild-type
(Fig. 4D). In contrast to COX-2,
however, c-Myc expression decreased in response to P-S in a
concentration-dependent manner; and the effect was greater in
NFATc1-knockdown cells. While it is apparent that NFATc1 positively
regulates c-Myc expression, the c-Myc suppressing effects of P-S
appears to be NFAT-independent.
NFATc1 expression modulates the
anticancer effect of P-S in vitro
Given the importance of NFATc1 in pancreatic
carcinogenesis and the effect of P-S in its expression, we examined
its role as a modulator of the therapeutic efficacy of P-S in
pancreatic cancer. NFATc1-knockdown BxPC-3 and Mia PaCa-2 cells
were both sensitized to the cytotoxic effects of P-S, as indicated
by the 3- and 2.4-fold reduction in 24 h IC50 values
compared to the control shRNA-expressing cells (Table II). On the other hand, the
cytotoxic effect of P-S is reduced in NFATc1 overexpressing Mia
PaCa-2 cells (Table II). These
results suggest that NFATc1 expression levels negatively regulate
cellular response to P-S. We additionally examined the effect of
non-NSAID anticancer agents on BxPC-3 WT and NFATc1-knockdown
cells, including: fluorouracil (a pyrimidine analogue), valproic
acid (histone deacetylase inhibitor), and irinotecan (topoisomerase
poison). Table III shows that for
the three drugs tested, an enhanced potency was observed in
NFATc1-knockdown cells in comparison to WT (1.6- to 2.2-fold).
These findings indicate that NFATc1 protein expression in
pancreatic cancer cells decreases the potency of diverse anticancer
agents, suggesting that NFATc1 plays a key role in modulating drug
response in pancreatic cancer.
| Table II.Effect of NFATc1 knockdown or
overexpression on 24-h IC50 values of P-S in BxPC-3 and
MIA PaCa-2 cells. |
Table II.
Effect of NFATc1 knockdown or
overexpression on 24-h IC50 values of P-S in BxPC-3 and
MIA PaCa-2 cells.
| Cell line | P-S
IC50, μM |
|---|
|
|---|
| Mock
transfected | NFATc1
knockdown | NFATc1
overexpression |
|---|
| BxPC-3 | 104±2 | 35±2 | ND |
| Mia-PaCa-2 | 93±1 | 38±2 | 111±2 |
| Table III.Effect of NFATc1 knockdown on 24-h
IC50 values of chemotherapeutic drugs in BxPC-3
cells. |
Table III.
Effect of NFATc1 knockdown on 24-h
IC50 values of chemotherapeutic drugs in BxPC-3
cells.
| Drug | P-S
IC50, μM |
|---|
|
|---|
| Mock
transfected | NFATc1
knockdown | Potency
enhancement |
|---|
| Fluorouracil | 586±2 | 263±2 | 2.2 |
| Valproic acid | 543±1 | 338±3 | 1.6 |
| Irinotecan | 58.5±4 | 26.8±1 | 2.2 |
NFATc1 expression modulates the
anticancer effect of P-S in vivo
We determined the effect of NFATc1 knockdown or
overexpression on the tumor response to P-S in vivo.
Following a prevention protocol, animals were first treated with
P-S for one week prior to subcutaneous injection of wild-type,
NFATc1-knockdown and NFATc1-overexpressing Mia PaCa-2 cells.
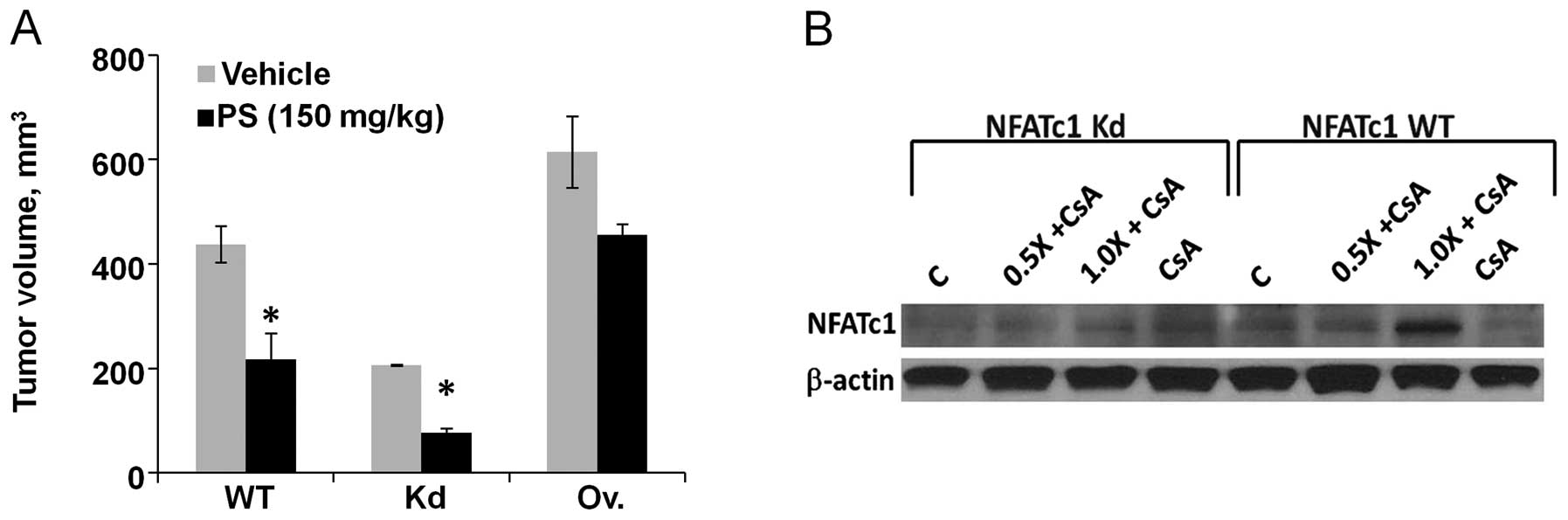
Animals were then treated for 4 more weeks. As shown in Fig. 5A, tumor volume was significantly
reduced by 50.4% in P-S-treated wild-type compared to the vehicle
(p=0.013). In NFATc1 knockdown tumors, P-S treatment caused a 62.8%
reduction in tumor volume compared to the vehicle (p=0.0005). In
contrast, P-S alone had a weak inhibitory effect (25.9% inhibition)
on tumors overexpressing NFATc1 and the effect was not
statistically significant. Consistent with in vitro
findings, NFATc1 expression is an important factor that negatively
regulates the tumor responsiveness to P-S in vivo.
Pharmacological targeting of NFATc1
sensitizes pancreatic cancer cells to the cytotoxic effect of
P-S
The immunosuppressive drug cyclosporin A (CsA) is an
inhibitor of NFAT-mediated transcription activity. CsA prevents
dephosphorylation of NFAT by calcineurin, leading to sequestration
of phosphorylated NFAT in the cytoplasm. To analyze the effect of
NFATc1 inhibition on cellular response to P-S, BxPC-3 wild-type and
NFATc1-knockdown cells were pre-treated with CsA (1 μM for 1
h), followed by either 0.5× or 1.0×IC50 P-S for 24 h. A
slight (1.2-fold) enhancement of potency of P-S was observed in
combination with CsA in wild-type cells, while no effect was
observed in NFATc1-knockdown cells (data not shown). Western blot
analysis showed a significant reduction in the protein levels of
NFATc1 in both the wild-type and NFATc1 knockdown cells treated
with CsA alone or P-S plus CsA (Fig.
5B). P-S increased the expression of NFATc1, while CsA reduced
it. These results suggest that CsA antagonizes the induction of
NFATc1 by P-S, thereby potentiating the cytotoxic effect of
P-S.
Discussion
Pancreatic cancer is among the most lethal of human
cancers and it is highly resistant to many chemotherapeutic drugs.
Our findings demonstrate that P-S possesses considerable efficacy
in the pre-clinical models of pancreatic cancer while being
apparently safe, establish that NFATc1 is a critical factor in
mediating drug resistance in pancreatic cancer, and that targeting
NFATc1 improves tumor response to chemotherapeutic drugs, including
P-S.
In a panel of four human pancreatic cancer cell
lines (Kras wild-type or mutant), P-S was consistently more
potent (19- to >100-fold) than sulindac in inhibiting their
growth. The antitumor efficacy of P-S was also established in a
pancreatic cancer xenograft model which encompass Kras
wild-type (BxPC-3) and mutant (Mia PaCa-2) human pancreatic cancer
cell lines. In both cases, P-S significantly inhibited the growth
of the xenografts compared to control, irrespective of the
Kras mutation status. Apart from efficacy, P-S also exhibits
a favorable safety profile, evidenced by the apparent lack of organ
toxicity, and more importantly, minimal gastrointestinal side
effects, a dose-limiting toxicity of its parent NSAID sulindac
(10).
Underlying the growth inhibitory potency of PS was
its combined cytokinetic effect consisting of suppressed
proliferation and enhanced apoptosis. Such an effect was observed
both in vitro and in vivo. Induction of oxidative
stress is a key mechanism of action for several anticancer agents,
including P-S. As cancer cells have elevated ROS generation and are
under increased intrinsic oxidative stress, these cells are more
vulnerable to further oxidative insults induced by ROS-generating
agents. Consistent with previous reports (10,16,18,19,22),
there is a significant increase in ROS in response to P-S in
pancreatic cancer cells. The cytokinetic effect and induction of
ROS contributes to the antitumor effect of P-S in pancreatic
cancer.
Our study also unravels a novel role of NFATc1 in
mediating drug resistance in pancreatic cancer. P-S triggered
profound upregulation of NFATc1 and its nuclear translocation in
pancreatic cancer cells, leading to robust induction of its
transcriptional targets, including COX-2. It is conceivable that
such an induction of pro-proliferative and pro-survival factors by
NFATc1 has important implications for drug response and resistance.
Indeed, genetic silencing of NFATc1 in pancreatic cancer cells
enhanced the potency of P-S; while its ectopic expression conferred
drug resistance. Accordingly, NFATc1-knockdown Mia PaCa-2
xenografts were notably more responsive to P-S compared to
wild-type, whereas NFATc1-overexpressing xenografts were
insensitive to P-S. This observation was extended to
mechanistically diverse anti-cancer agent (5-FU, valproic acid and
irinotecan); suggesting that NFATc1 promotes a broad resistance to
chemotherapeutic drugs in pancreatic cancer.
Given the important role of NFATc1 in drug
resistance, it represents a novel prognostic factor for predicting
drug response and a potential therapeutic target for improving
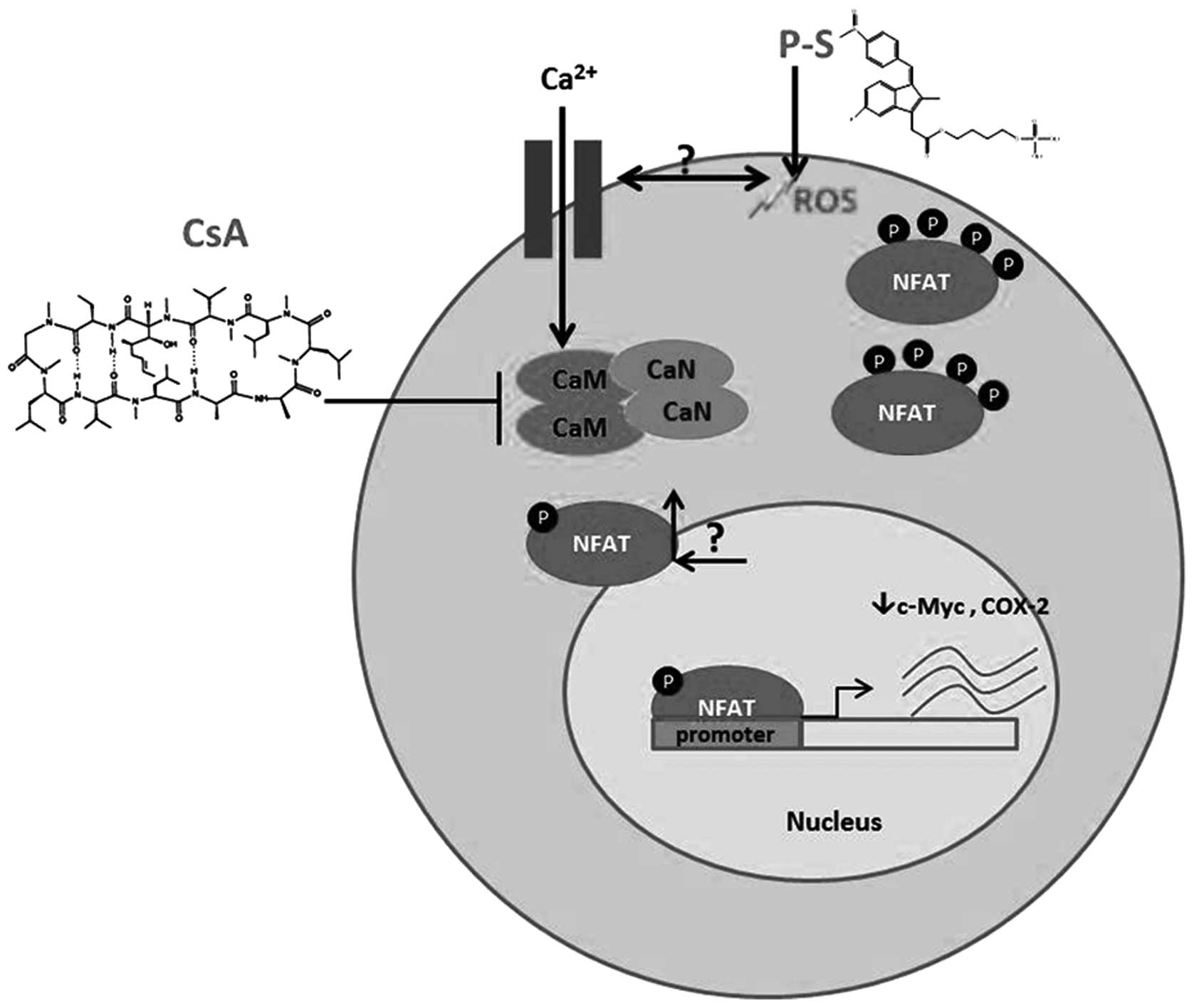
tumor responsiveness to chemo-therapeutic drugs. NFATc1 activation
is regulated by Ca2+/calcineurin signaling pathway
(23). Fluctuation in
Ca2+ levels simulates calcineurin to dephosphorylate
NFATc1, which then translocates to the nucleus to activate gene
expression. Pharmacological inhibition of calcineurin by CsA blocks
NFATc1 activation as well as its nuclear translocation; and more
importantly, CsA treatment sensitizes pancreatic cancer cells to
P-S. Our findings thus provide a biochemical basis for synergism
between P-S and CsA in pancreatic cancer, and suggest that the
combination therapy with P-S and NFATc1 inhibitors may be a
promising chemotherapeutic approach to improve treatment
outcomes.
In conclusion, our study demonstrates that P-S is a
promising agent that can effectively inhibit Kras wild-type
and Kras-mutant pancreatic cancer in vitro and in
vivo. We uncovered a novel role of NFATc1 in modulating drug
response in pancreatic cancer, and proposed a pharmacological
approach to overcome the drug resistance associated with NFATc1
activation (outlined in Fig. 6).
Overall, the effectiveness and safety of P-S in the treatment of
pancreatic cancer suggest that this compound merits further
evaluation.
Acknowledgements
We thank Gerardo Mackenzie, Yu Sun,
Liqun Huang, Jennie L. Williams, Ping Ji and Ninche Alston for all
their helpful comments and suggestions. This study was supported by
the National Institute of Health Grants R01-CA139453; N01-CN-43302
WA#7, RCA153662A. This study was also supported in part by the
3MT-NSF-IGERT. B.R. has an equity position in Medicon
Pharmaceuticals, Inc.
References
|
1.
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
2.
|
Hezel AF, Kimmelman AC, Stanger BZ,
Bardeesy N and Depinho RA: Genetics and biology of pancreatic
ductal adeno-carcinoma. Genes Dev. 20:1218–1249. 2006. View Article : Google Scholar
|
|
3.
|
Bardeesy N and DePinho RA: Pancreatic
cancer biology and genetics. Nat Rev Cancer. 2:897–909. 2002.
View Article : Google Scholar
|
|
4.
|
Wolfe MM, Lichtenstein DR and Singh G:
Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N
Engl J Med. 340:1888–1899. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Molina MA, Sitja-Arnau M, Lemoine MG,
Frazier ML and Sinicrope FA: Increased cyclooxygenase-2 expression
in human pancreatic carcinomas and cell lines: growth inhibition by
nonsteroidal anti-inflammatory drugs. Cancer Res. 59:4356–4362.
1999.PubMed/NCBI
|
|
6.
|
Yip-Schneider MT, Wu H, Ralstin M,
Yiannoutsos C, Crooks PA, Neelakantan S, Noble S, Nakshatri H,
Sweeney CJ and Schmidt CM: Suppression of pancreatic tumor growth
by combination chemotherapy with sulindac and LC-1 is associated
with cyclin D1 inhibition in vivo. Mol Cancer Ther. 6:1736–1744.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Yip-Schneider MT, Nakshatri H, Sweeney CJ,
Marshall MS, Wiebke EA and Schmidt CM: Parthenolide and sulindac
cooperate to mediate growth suppression and inhibit the nuclear
factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer
Ther. 4:587–594. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yip-Schneider MT and Schmidt CM: MEK
inhibition of pancreatic carcinoma cells by U0126 and its effect in
combination with sulindac. Pancreas. 7:337–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Huang L, Zhu C, Sun Y, Xie G, Mackenzie
GG, Qiao G, Komninou D and Rigas B: Phospho-sulindac (OXT-922)
inhibits the growth of human colon cancer cell lines: a
redox/polyamine-dependent effect. Carcinogenesis. 31:1982–1990.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Mackenzie GG, Sun Y, Huang L, Xie G,
Ouyang N, Gupta RC, Johnson F, Komninou D, Kopelovich L and Rigas
B: Phosphosulindac (OXT-328), a novel sulindac derivative, is safe
and effective in colon cancer prevention in mice. Gastroenterology.
139:1320–1332. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Xie G, Nie T, Mackenzie GG, Sun Y, Huang
L, Ouyang N, Alston N, Zhu C, Murray OT, Constantinides PP,
Kopelovich L and Rigas B: The metabolism and pharmacokinetics of
phosphosulindac (OXT-328) and the effect of
difluoromethylornithine. Br J Pharmacol. 165:2152–2166. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Muller MR and Rao A: NFAT, immunity and
cancer: a transcription factor comes of age. Nat Rev Immunol.
10:645–656. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Mancini M and Toker A: NFAT proteins:
emerging roles in cancer progression. Nat Rev Cancer. 9:810–820.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Cippa PE, Kraus AK, Lindenmeyer MT, Chen
J, Guimezanes A, Bardwell PD, Wekerle T, Wuthrich RP and Fehr T:
Resistance to ABT-737 in activated T lymphocytes: molecular
mechanisms and reversibility by inhibition of the calcineurin-NFAT
pathway. Cell Death Dis. 3:e2992012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Gregory MA, Phang TL, Neviani P,
Alvarez-Calderon F, Eide CA, O’Hare T, Zaberezhnyy V, Williams RT,
Druker BJ, Perrotti D and Degregori J: Wnt/Ca2+/NFAT
signaling maintains survival of Ph+leukemia cells upon
inhibition of Bcr-Abl. Cancer Cell. 18:74–87. 2010.
|
|
16.
|
Sun Y and Rigas B: The thioredoxin system
mediates redox-induced cell death in human colon cancer cells:
implications for the mechanism of action of anticancer agents.
Cancer Res. 68:8269–8277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Ouyang N, Williams JL, Tsioulias GJ, Gao
J, Iatropoulos MJ, Kopelovich L, Kashfi K and Rigas B: Nitric
oxide-donating aspirin prevents pancreatic cancer in a hamster
tumor model. Cancer Res. 66:4503–4511. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sun Y, Huang L, Mackenzie GG and Rigas B:
Oxidative stress mediates through apoptosis the anticancer effect
of phosphononsteroidal anti-inflammatory drugs: implications for
the role of oxidative stress in the action of anticancer agents. J
Pharmacol Exp Ther. 338:775–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nie T, Wong CC, Alston N, Aro P,
Constantinides PP and Rigas B: Phospho-ibuprofen (MDC-917)
incorporated in nano-carriers: Anti-cancer activity in vitro and in
vivo. Br J Pharmacol. 166:991–1001. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Buchholz M, Schatz A, Wagner M, Michl P,
Linhart T, Adler G, Gress TM and Ellenrieder V: Overexpression of
c-myc in pancreatic cancer caused by ectopic activation of NFATc1
and the Ca2+/calcineurin signaling pathway. EMBO J.
25:3714–3724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Duque J, Fresno M and Iniguez MA:
Expression and function of the nuclear factor of activated T cells
in colon carcinoma cells: involvement in the regulation of
cyclooxygenase-2. J Biol Chem. 280:8686–8693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Cao Z and Li Y: Chemical induction of
cellular antioxidants affords marked protection against oxidative
injury in vascular smooth muscle cells. Biochem Biophys Res Commun.
292:50–57. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Hogan PG, Chen L, Nardone J and Rao A:
Transcriptional regulation by calcium, calcineurin, and NFAT. Genes
Dev. 17:2205–2232. 2003. View Article : Google Scholar : PubMed/NCBI
|