Introduction
Cancer upregulated gene 2 (CUG2) was identified as a
candidate gene that is commonly upregulated in various tumor
tissues, such as ovarian, liver, colon, and lung tissues, and is
known to play a crucial role in tumorigenesis. CUG2 was mapped to
chromosome 6q22.32; it spans ∼8.5 kb with a three-exon structure
and encodes an 88-amino-acid polypeptide (1). Further study revealed that CUG2 is a
new centromere component that is required for proper kinetochore
function during cell division (2).
CUG2 has been shown to exert an oncogenic effect in a transplanted
model using NIH3T3 cells expressing CUG2, in a manner similar to
Ras (1). Whereas CUG2
overexpression has been shown to activate Ras and MAPKs including
p38 MAPK, which eventually facilitates oncolytic reoviral
replication (3), our previous
studies showed that CUG2 confers resistance to oncolytic vesicular
stomatitis virus (VSV) infection (4), as well as induces cell migration and
drug resistance (4), through
activation of Stat1 (5).
Autophagy is an ancient catabolic process necessary
in maintaining homeostasis in eukaryotic cells, whereby long-lived
cytoplasmic proteins and organelles are degraded and nutrients are
provided under starvation or stress conditions (6). It is a programmed, sequential process
mainly involving autophage-related gene (Atg) products and has
roles in biological processes including development, aging, and
degeneration (7). Aberrant
regulation of autophagy is associated with many diseases, such as
cancer and neurodegenerative diseases (8,9).
Initial reports associating autophagy with cancer showed that
allelic loss of the essential autophagy gene Beclin 1
(BECN1) is prevalent in human breast, ovarian, and prostate
cancers (10) and that
Becn1+/− mice develop mammary gland hyperplasias,
lymphomas, and lung and liver tumors (11). Subsequent studies demonstrated that
Atg5−/− and Atg7−/− mice
develop liver adenomas (12). In
addition, autophagy has been reported to be involved in virus
replication (13–15). Autophagy induction requires the
antiviral eIF2α kinase signaling pathway (including PKR and eIF2α),
and eIF2α kinase signaling is antagonized by the herpes simplex
virus (HSV-1) neurovirulence gene product ICP34.5 (16). Autophagy plays a key role in the
recognition of certain RNA viruses by delivering viral replication
intermediates through Toll-like receptor-7 (TLR-7) activation in
plasmacytoid dendritic cells (pDCs) (17) and other cells via retinoic
acid-inducible gene I (RIG-I) and melanoma
differentiation-associated gene 5 (MDA-5) (18–20).
This study aimed to determine whether autophagy is
also involved in resistance to oncolytic VSV in A549-CUG2 cells. We
show that autophagy impairment induced reactive oxygen species
(ROS) formation, which led to decreased ISG15 production due to
overexpression of CUG2, eventually sensitizing the cells to
VSV-induced apoptosis. We propose that autophagy impairment is a
potential strategy for successful VSV virotherapy of
CUG2-overexpressing tumors.
Materials and methods
Cell cultures and virus
amplification
Human lung cancer A549 cells stably transfected with
CUG2 expression plasmid (A549-CUG2) were used. The cells were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS), 1% penicillin, 1% streptomycin, and puromycin (0.5
μg/ml) at 5% CO2 and 37°C. VSV (Indiana strain),
purchased from the ATCC (Manassas, VA, USA), was propagated in L929
cells, and viral titer was measured in plaque-forming units.
Reagents and antibodies
For immunoblotting, antibodies against Atg5, ISG15,
and poly-ADP-ribose polymerase (PARP) were acquired from Cell
Signaling Biotechnology (Danvers, MA, USA). Beclin 1 and β-actin
antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). VSV glycoprotein (G) antibody was obtained from Abcam
(Cambridge, MA, USA).
Western blotting assays
Cells were harvested and lysed in lysis buffer [150
mM NaCl, 1% NP-40, 50 mM Tris-HCl (pH 7.5)] containing 0.1 mM
Na2VO3, 1 mM NaF and protease inhibitors
(Sigma, St. Louis, MO, USA). For immunoblotting, proteins from
whole-cell lysates were resolved by 10 or 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then
transferred onto nitrocellulose membranes. Primary antibodies were
used at 1:1,000 or 1:2,000 dilution, and secondary antibodies
conjugated to horseradish peroxidase were used at 1:2,000 dilution
in 5% non-fat dry milk. After the final washing, the nitrocellulose
membranes were analyzed by enhanced chemiluminescence assay using
the LAS 4000 mini system (Fuji, Tokyo, Japan).
Reverse transcription-polymerase chain
reaction
Total RNA was extracted from cells using the RNeasy
mini kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions. Three micrograms of total RNA was
converted to cDNA using Superscript II reverse transcriptase
(Invitrogen, Carlsbad, CA, USA), and polymerase chain reaction
(PCR) was performed using the following specific primers: human
ISG15, 5′-ATG GGC TGG GAC CTG ACG GTG AAG AT-3′ (sense) and
5′-TTA GCT CCG CCC GCC AGG CTC TGT-3′; human IFIT3, 5′-ATG
AGT GAG GTC ACC AAG AAT-3′ (sense) and 5′-TCA GTT CAG TTG CTC TGA
GTT A-3′ (antisense). The cDNA for each reaction was diluted, and
PCR was run at the optimized cycle number. β-actin mRNA was also
analyzed as an internal standard. After amplification, the products
were subjected to electrophoresis on 2.0% agarose and detected by
ethidium bromide staining.
Transfection with short interference
RNA
Cells were trypsinized and incubated overnight to
achieve 60–70% confluence before short interference RNA (siRNA)
transfection. Atg5 siRNA [500 nM; Bioneer, Daejeon, Korea; sense,
5′-ACU UUG CUG UAA CCC UGU A(dTdT)-3′; antisense, 5′-UAC AGG GUU
ACA GCA AAG U(dTdT)-3′], Beclin 1 siRNA [500 nM; Bioneer; sense,
5′-ACU UUG CUG UAA CCC UGU A(dTdT)-3′; antisense, 5′-UAC AGG GUU
ACA GCA AAG U(dTdT)-3′], ISG15 siRNA [500 nM; Bioneer; sense,
5′-ACU UUG CUG UAA CCC UGU A(dTdT)-3′; antisense, 5′-UAC AGG GUU
ACA GCA AAG U(dTdT)-3′], or negative control siRNA (Bioneer) was
mixed with Lipofectamine 2000 (Invitrogen). The cells were
incubated with the transfection mixture for 6 h and then rinsed
with RPMI-1640 containing 10% FBS. The cells were incubated for 48
h before harvest.
Measurement of ROS
The intracellular ROS levels were determined by
using an oxidative-sensitive fluorescence dye,
2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA; Molecular
Probes, Eugene, OR, USA). Cells were treated with 20 μM
DCF-DA for 30 min and then observed under a fluorescence microscope
(Carl Zeiss, Axio-D1, Oberkochen, Germany).
Results
Suppression of Atg5 or Beclin 1
expression sensitizes A549-CUG2 cells to VSV-induced apoptosis
Our previous study showed that CUG2 overexpression
induces resistance to VSV infection by activation of the
Stat1-2′-5′ oligoadenylate synthetase-like 2 (OASL 2) signaling
pathway (5). Several lines of
evidence have shown that autophagy is implicated with viral
replication (13–15), and moreover other studies showed
that Atg5-deficient mouse embryonic fibroblast (MEF) cells
exhibit resistance to VSV through hyperproduction of type I IFN
(20,21). Based on these studies, we tested
what role Atg5 protein plays in VSV infection in our cell model. We
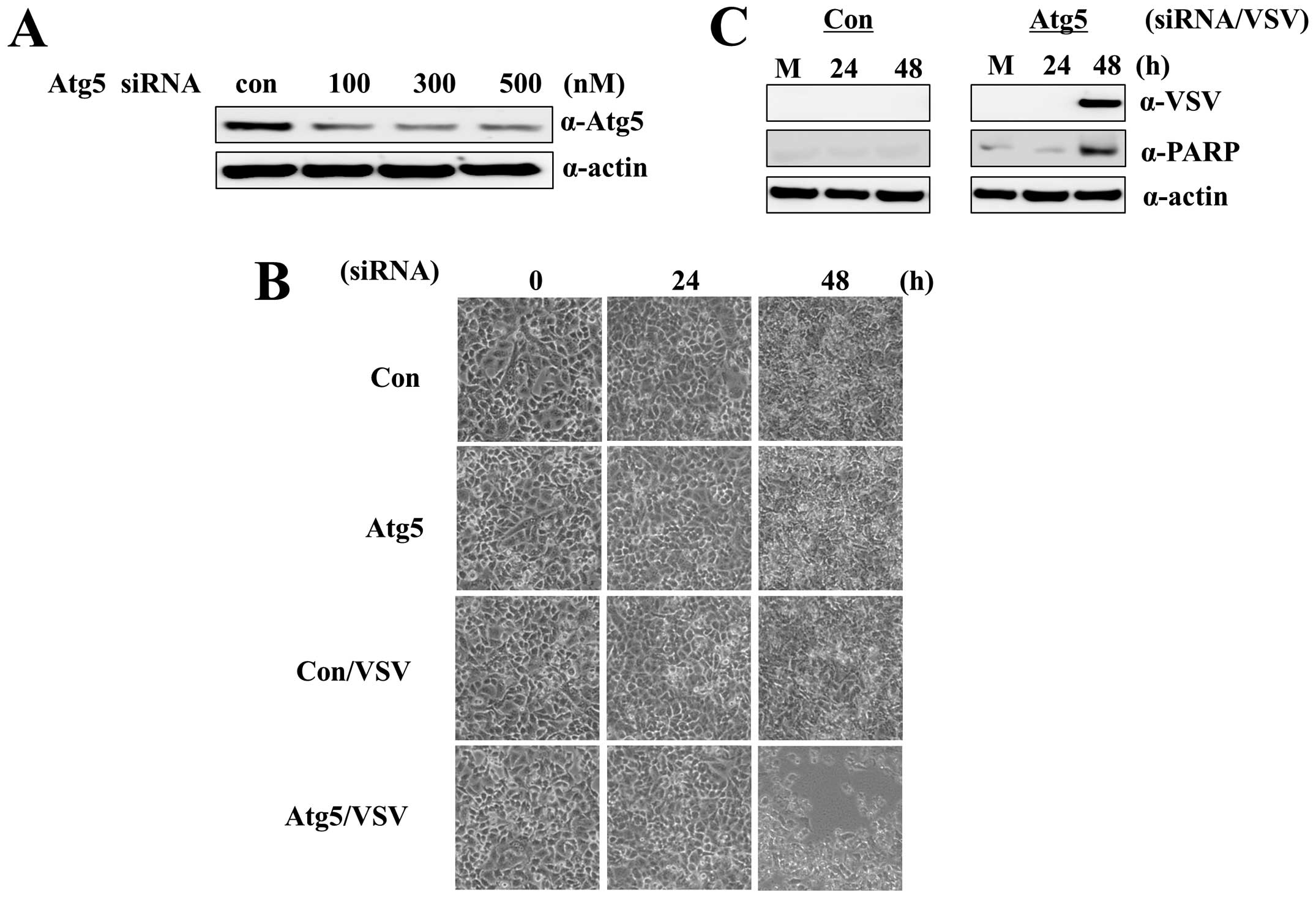
first optimized the concentration of Atg5 siRNA (500 nM) to
significantly reduce Atg5 protein levels in A549-CUG2 cells
(Fig. 1A). VSV infection alone did
not induce cytolysis in A549-CUG2 cells, as seen in mouse colon
cancer cells overexpressing human CUG2 (Fig. 1B). In contrast to previous reports
(20,21), A549-CUG2 cells treated with VSV and
Atg5 siRNA clearly exhibited cytolysis at 60 h after infection
(Fig. 1B). Control A549 cells
stably expressing an empty vector showed acceleration of cytolysis
after treatment with VSV and Atg5 siRNA (data not shown). A549-CUG2
cells cotreated with VSV and Atg5 siRNA underwent apoptosis caused
by VSV replication (Fig. 1C).
These results suggest that autophagy protects A549-CUG2 cells from
VSV-induced apoptosis.
To confirm our observation that suppression of
autophagic gene expression sensitizes A549-CUG2 cells to
VSV-induced apoptosis, we suppressed another autophagic gene,
Beclin 1, also known as Atg6. Treatment with Beclin 1
siRNA (500 nM) clearly suppressed Beclin 1 protein expression in
A549-CUG2 cells at 48 h after treatment (Fig. 2A). Cotreatment with VSV and Beclin
1 siRNA induced apoptosis in A549-CUG2 cells (Fig. 2B); the apoptotic cell death was
attributed to VSV replication (Fig.
2C). This result also confirmed our hypothesis that autophagy
protects A549-CUG2 cells from VSV-induced apoptosis.
Suppression of Atg5 or Beclin 1
expression induces ROS formation in A549-CUG2 cells
Next, we investigated the mechanism by which
suppression of Atg5 or Beclin 1 expression sensitizes cells to
VSV-induced apoptosis. Recent studies have reported that impaired
autophagy in pathogenic diseases including cancer shows accumulated
damaged mitochondria and deregulated ROS levels (22). We therefore asked whether
suppression of Atg5 or Beclin 1 expression is related to ROS
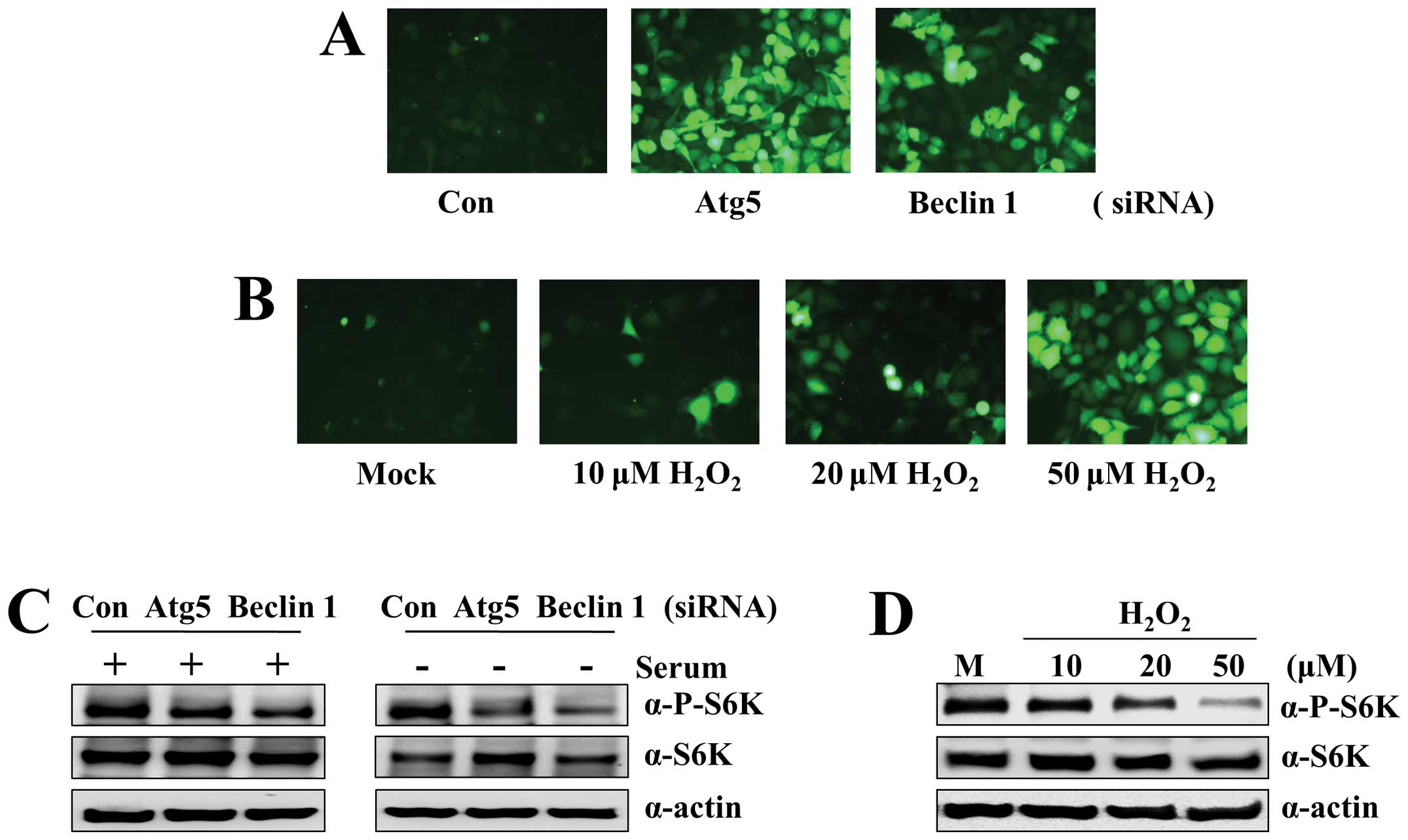
formation in A549-CUG2 cells. To answer this question, we measured,
using DCF-DA fluorescence, the ROS levels in A549 cells treated
with Atg5 or Beclin 1 siRNA. We found that the cells treated with
Atg5 or Beclin 1 siRNA showed enhanced fluorescence compared with
control siRNA-treated cells at 48 h after transfection (Fig. 3A). As a control, A549-CUG2 cells
were treated with H2O2 at different
concentrations. We observed that H2O2 at
20–50 μM induced fluorescence intensity similar to that in
cells treated with Atg5 or Beclin 1 siRNA (Fig. 3B). These results indicate that the
accumulation of damaged mitochondria induced ROS formation in
autophagy-deficient cells. Because it was previously reported that
the kinase mammalian target of rapamycin (mTOR) stimulates type I
IFN production via phosphorylation of its effector proteins
including S6 kinase (23), we also
examined the activation status of S6 kinase during autophagic
suppression in the presence or absence of serum. We found that Atg5
or Beclin 1 suppression decreased the levels of phosphorylated S6
kinase in the presence of serum and more drastic reduction of
phosphorylated S6 kinase levels under serum-free conditions
(Fig. 3C). The addition of
H2O2 into the medium during culture of
A549-CUG2 cells led to an oxidative stress-mediated reduction of S6
kinase phosphorylation, similar to the pattern seen in Atg5 or
Beclin 1 suppression (Fig. 3D).
This result indicates that oxidative stress caused by autophagy
impairment inhibits the activation of S6 kinase, which is an
essential step in type I IFN production.
Suppression of ISG15 caused by treatment
with Atg5 or Beclin 1 siRNA in A549-CUG2 cells confers
susceptibility to VSV infection
Next, we analyzed the association between ROS
formation due to suppression of autophagic gene expression and
reduction of antiviral activity. Our previous study revealed an
upregulation of antiviral genes such as OASL2 and ISG15. OASL2
expression showed more dependence on Stat1 activation than did
ISG15 expression (5). We thus
examined the transcript levels of ISG15 after treatment with Atg5
or Beclin 1 siRNA. We found that the ISG15 transcript levels were
reduced after treatment with Atg5 or Beclin 1 siRNA compared to
those in the cells treated with control siRNA (Fig. 4A and B).The decreased ISG15
transcript level led to suppression of ISG15 protein expression
under Atg5- or Beclin 1-deficient conditions. Therefore, we
investigated whether the reduced expression of ISG15 transcripts
sensitized A549-CUG2 cells to VSV infection. We determined the
optimum concentration of ISG15 siRNA to be 500 nM for the
suppression of ISG15 transcript expression (Fig. 4C). Treatment with VSV and ISG15
siRNA sensitized A549-CUG2 cells to VSV-induced apoptosis (Fig. 4D and E) caused by VSV replication;
on the other hand, VSV infection failed to induce apoptosis in
control siRNA-treated A549-CUG2 cells, as shown in Figs. 1B and 2B. This result indicates that suppression
of ISG15 by Atg5 or Beclin 1 siRNA renders A549-CUG2 cells
susceptible to VSV infection. Taken together, these results suggest
that suppression of autophagy leads to excessive ROS, which impairs
the cell’s innate immunity, thus rendering it susceptible to VSV
infection.
ROS sensitizes A549-CUG2 cells to
VSV-induced apoptosis
On the basis of our previous results (Figs. 1, 2 and 4),
we hypothesized that ROS formation, caused by the accumulation of
damaged mitochondria due to deficiency in autophagic genes, reduces
ISG15 protein levels in A549-CUG2 cells, leading to VSV-induced
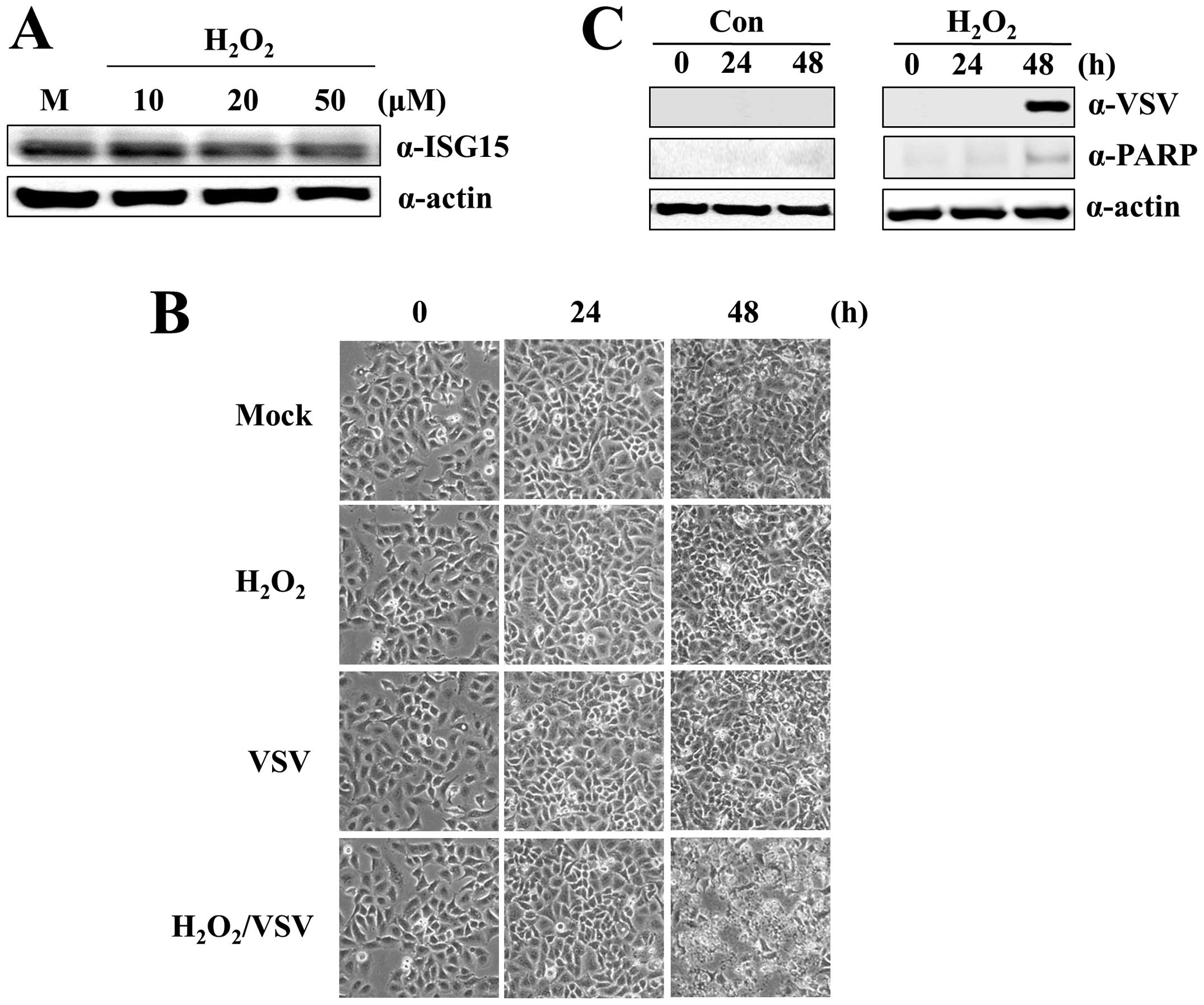
apoptosis. To test this hypothesis, we treated A549-CUG2 cells with
different concentrations of H2O2.
H2O2 at 50 μM significantly decreased
ISG15 protein levels (Fig. 5A).
A549-CUG2 cells treated with VSV and H2O2
were susceptible to VSV-induced apoptosis (Fig. 5B and C) caused by VSV replication.
However, VSV infection or H2O2 treatment
alone did not induce apoptosis. This result confirms that ROS
formation sensitizes A549-CUG2 cells to VSV-induced apoptosis.
Discussion
Autophagy has been considered as an intrinsic
antiviral mechanism against viral infection (13–15).
The autophagic process allows sequestration of the virus inside the
autophagosome, leading to their destruction in the lysosome
(24). Autophagy is also involved
in the digestion of endogenously synthesized viral proteins for MHC
II presentation of viral antigens, reflecting the role of autophagy
in adaptive immunity (25). Recent
evidence also shows that autophagy is linked to type I IFN
induction through TLR-7 in the cell’s antiviral response (17). However, viruses also have evolved
strategies to escape antiviral autophagy. For example, the
α-herpesvirus HSV-1 ICP34.5 inhibits the induction of autophagy by
targeting Beclin 1 and PKR-mediated phosphorylation of eIF2α
(26,27). KSHV and γ-HV68 viral Bcl-2 proteins
attenuate autophagy through direct interaction with Beclin 1
(28,29). Additionally, influenza A virus
matrix protein 2 (M2), a proton-selective ion channel, has been
identified to be necessary and sufficient for the inhibition of
autophagosomal maturation (30).
Mouse hepatitis virus (MHV) uses autophagosome-like structures for
its production (31).
In the case of VSV, autophagy stimulates IFNα
secretion by pDCs in response to VSV recognition by TLR-7 in mice
(17). In vivo, autophagy
protects against lethal infection in Drosophila and inhibits
viral replication in Drosophila cells (32). However, autophagy negatively
regulates RIG-I-mediated induction of type I IFN in VSV-infected
MEFs. In particular, autophagy-defective Atg5−/− MEF
cells exhibit enhanced RLR signaling, increased IFN secretion and
resistance to VSV infection (20,21).
In contrast, we observed VSV susceptibility in A549-CUG2 cells with
suppressed Atg5 expression. ROS formation is a common phenomenon
observed in Atg5−/− MEF cells and A549-CUG2 cells
treated with Atg5 siRNA; however, differential results were
observed for VSV infection. We assume that this discrepancy might
be attributed to cellular context, but we are investigating in more
detail the mechanism(s) whereby autophagy impairment induces
sensitization of A549-CUG2 cells to VSV.
Autophagy impairment could induce premature
senescence in human primary fibroblasts through p53 activation due
to increased ROS formation resulting from accumulation of
dysfunctional mitochondria. Further investigation revealed that
phosphorylation of S6 kinase and ribosomal S6 protein was decreased
due to ROS-caused autophagy impairment (33). Consistent with these results, we
observed a decrease in phospho-S6 kinase levels due to autophagy
impairment. Moreover, one study reported that mTOR signaling
stimulates type I IFN production via phosphorylation of its target
proteins 4E-BPs and S6 kinase 1/2 (34). MEF cells and mice lacking S6 kinase
were more susceptible to VSV infection than their WT counterparts
as a result of an impaired type I IFN response (23). On the basis of these results, we
speculate that decreased levels of phospho-S6 kinase leads to
reduced IFN production, which eventually results in the
sensitization to VSV of A549-CUG2 cells treated with Atg5 or Beclin
1 siRNA.
In addition, we observed that ROS caused by
autophagy impairment led to downregulatiion of ISG15 expression
levels in A549-CUG2 cells. The antiviral role of ISG15 has been
supported by the observations that the overexpression of ISG15
represses Sindbis virus replication in multiple organs of IFN
receptor-deficient mice and protects the host against virus-induced
lethality (35). Several
IFN-induced antiviral proteins were recently identified as cellular
targets of ISG15, including PKR, MxA, and RIG-I, suggesting that
ISG15 conjugation may play an important regulatory role in the
IFN-mediated antiviral response (36,37).
However, ISG15-deficient mice were initially reported to exhibit
normal IFN signaling and resistance to VSV infection (38). In this study, we showed that
suppression of ISG15 by siRNA sensitized A549-CUG2 cells to VSV
infection. This discrepancy in the ISG15 antiviral ability may be
attributed to the experimental setting. We found only a few
antiviral genes such as OASL2, ISG15, IFIT3, IP-10 and GBP3 to be
upregulated in the microarray analysis. Among them, OASL2 (strongly
dependent on Stat1 activation) and ISG15 (less dependent on Stat1
activation) were the most abundant (5) so that suppression of ISG15, a major
component of antiviral protein in A549-CUG2 cells, may reduce the
threshold against viral infectivity. We thus propose that
CUG2-mediated antiviral activity can be reduced by ISG15 deficiency
in A549 cells, which eventually enhances sensitivity of the cells
to VSV infection. This study revealed that autophagy with intrinsic
antiviral activity affects the efficiency of VSV treatment on
tumors overexpressing CUG2. Not only activation of Stat1 but also
autophagy as an antiviral mechanism can cause difficulties in
virotherapy. Therefore, we suggest that autophagy impairment can be
a potential strategy for VSV treatment of tumor cells
overexpressing CUG2.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry of
Health & Welfare, Republic of Korea (1120140).
References
|
1.
|
Lee S, Gang J, Jeon SB, et al: Molecular
cloning and functional analysis of a novel oncogene,
cancer-upregulated gene 2 (CUG2). Biochem Biophys Res Commun.
360:633–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kim H, Lee M, Lee S, et al:
Cancer-upregulated gene 2 (CUG2), a new component of centromere
complex, is required for kinetochore function. Mol Cells.
27:697–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Park EH, Park EH, Cho IR, et al: CUG2, a
novel oncogene confers reoviral replication through Ras and p38
signaling pathway. Cancer Gene Ther. 17:307–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Malilas W, Koh SS, Kim S, et al: Cancer
upregulated gene 2, a novel oncogene, enhances migration and drug
resistance of colon cancer cells via STAT1 activation. Int J Oncol.
43:1111–1116. 2013.PubMed/NCBI
|
|
5.
|
Malilas W, Koh SS, Srisuttee R, et al:
Cancer upregulated gene 2, a novel oncogene, confers resistance to
oncolytic vesicular stomatitis virus through STAT1-OASL2 signaling.
Cancer Gene Ther. 20:125–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Qu X, Yu J, Bhagat G, et al: Promotion of
tumorigenesis by heterozygous disruption of the Beclin 1 autophagy
gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Levine B: Cell biology: autophagy and
cancer. Nature. 446:745–747. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Takamura A, Komatsu M, Hara T, et al:
Autophagy-deficient mice develop multiple liver tumors. Genes Dev.
25:795–800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Espert L, Codogno P and Biard-Piechaczyk
M: Involvement of autophagy in viral infections: antiviral function
and subversion by viruses. J Mol Med. 85:811–823. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Shoji-Kawata S and Levine B: Autophagy,
antiviral immunity, and viral countermeasures. Biochim Biophys
Acta. 1793:1478–1484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Levine B and Deretic V: Unveiling the
roles of autophagy in innate and adaptive immunity. Nat Rev
Immunol. 7:767–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Mulvey M, Poppers J, Sternberg D and Mohr
I: Regulation of eIF2alpha phosphorylation by different functions
that act during discrete phases in the herpes simplex virus type 1
life cycle. J Virol. 77:10917–10928. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lee HK, Lund JM, Ramanathan B, Mizushima N
and Iwasaki A: Autophagy-dependent viral recognition by
plasmacytoid dendritic cells. Science. 315:1398–1401. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Yoneyama M, Kikuchi M, Natsukawa T, et al:
The RNA helicase RIG-I has an essential function in double-stranded
RNA-induced innate antiviral responses. Nat Immunol. 5:730–737.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kato H, Takeuchi O, Sato S, et al:
Differential roles of MDA5 and RIG-I helicases in the recognition
of RNA viruses. Nature. 441:101–105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Jounai N, Takeshita F, Kobiyama K, et al:
The Atg5 Atg12 conjugate associates with innate antiviral immune
responses. Proc Natl Acad Sci USA. 104:14050–14055. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tal MC, Sasai M, Lee HK, Yordy B, Shadel
GS and Iwasaki A: Absence of autophagy results in reactive oxygen
species-dependent amplification of RLR signaling. Proc Natl Acad
Sci USA. 106:2770–2775. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kongara S and Karantza V: The interplay
between autophagy and ROS in tumorigenesis. Front Oncol. 2:1712012.
View Article : Google Scholar
|
|
23.
|
Alain T, Lun X, Martineau Y, et al:
Vesicular stomatitis virus oncolysis is potentiated by impairing
mTORC1-dependent type I IFN production. Proc Natl Acad Sci USA.
107:1576–1581. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Schmid D, Dengjel J, Schoor O, Stevanovic
S and Munz C: Autophagy in innate and adaptive immunity against
intracellular pathogens. J Mol Med. 84:194–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Dengjel J, Schoor O, Fischer R, et al:
Autophagy promotes MHC class II presentation of peptides from
intracellular source proteins. Proc Natl Acad Sci USA.
102:7922–7927. 2005.PubMed/NCBI
|
|
26.
|
Talloczy Z, Virgin HW IV and Levine B:
PKR-dependent autophagic degradation of herpes simplex virus type
1. Autophagy. 2:24–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Talloczy Z, Jiang W, Virgin HW IV, et al:
Regulation of starvation- and virus-induced autophagy by the
eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA.
99:190–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Sinha S, Colbert CL, Becker N, Wei Y and
Levine B: Molecular basis of the regulation of Beclin 1-dependent
autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy.
4:989–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Pattingre S, Tassa A, Qu X, et al: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Gannage M, Dormann D, Albrecht R, et al:
Matrix protein 2 of influenza A virus blocks autophagosome fusion
with lysosomes. Cell Host Microbe. 6:367–380. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Prentice E, Jerome WG, Yoshimori T,
Mizushima N and Denison MR: Coronavirus replication complex
formation utilizes components of cellular autophagy. J Biol Chem.
279:10136–10141. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Dreux M and Chisari FV: Viruses and the
autophagy machinery. Cell Cycle. 9:1295–1307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kang HT, Lee KB, Kim SY, Choi HR and Park
SC: Autophagy impairment induces premature senescence in primary
human fibroblasts. PLoS One. 6:e233672011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Cao W, Manicassamy S, Tang H, et al:
Toll-like receptor-mediated induction of type I interferon in
plasmacytoid dendritic cells requires the rapamycin-sensitive
PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 9:1157–1164. 2008.
View Article : Google Scholar
|
|
35.
|
Lenschow DJ, Giannakopoulos NV, Gunn LJ,
et al: Identification of interferon-stimulated gene 15 as an
antiviral molecule during Sindbis virus infection in vivo. J Virol.
79:13974–13983. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Malakhov MP, Kim KI, Malakhova OA, Jacobs
BS, Borden EC and Zhang DE: High-throughput immunoblotting.
Ubiquitiin-like protein ISG15 modifies key regulators of signal
transduction. J Biol Chem. 278:16608–16613. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Zhao C, Denison C, Huibregtse JM, Gygi S
and Krug RM: Human ISG15 conjugation targets both IFN-induced and
constitutively expressed proteins functioning in diverse cellular
pathways. Proc Natl Acad Sci USA. 102:10200–10205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Osiak A, Utermohlen O, Niendorf S, Horak I
and Knobeloch KP: ISG15, an interferon-stimulated ubiquitin-like
protein, is not essential for STAT1 signaling and responses against
vesicular stomatitis and lymphocytic choriomeningitis virus. Mol
Cell Biol. 25:6338–6345. 2005. View Article : Google Scholar
|