Introduction
The incidence of skin cancers, including melanoma
and nonmelanoma, is equivalent to the incidence of malignancies in
all other organs combined (1). It
is estimated that approximately 2 million people are diagnosed with
squamous and basal cell skin cancers annually in the US (2). Epidemiological and clinical studies
indicate that ultraviolet B (UVB) radiation acts as a tumor
initiator, tumor promoter as well as a complete carcinogen for the
development of skin neoplasms (3).
Exposure of the skin to solar UV radiation induces inflammatory
responses, oxidative stress, alterations in cell cycle progression,
DNA damage and suppression of immune responses, all of which have
been implicated in the development of skin cancers (3–7).
UV-induced inflammation is considered as an early event in skin
tumor promotion and the growth of tumors, and chronic and sustained
inflammation plays an important role in all three stages of tumor
development, i.e., initiation, promotion and progression (3). The UV-induced inflammatory responses
are characterized by the development of hyperplastic responses, and
increases in the levels of cyclooxygenase-2 (COX-2) and
prostaglandin (PG) metabolites (3). A link between COX-2/PGE2
and β-catenin signaling has been observed in colon cancer in which
COX-2 is overexpressed and β-catenin signaling contributes to tumor
cell growth (8). The β-catenin
signaling pathway also has been implicated in the growth of
melanomas as well as in the migration of melanoma cells (9–11).
β-catenin, a 90-kDa cytosolic protein, is an
important component of the Wnt pathway. Wnt/β-catenin proteins
regulate expression of various target genes that mediate cellular
processes including proliferation and migration. Activation of
Wnt/β-catenin proteins and mutations in β-catenin are the most
common alterations associated with tumor development and cancer
cell migration or metastasis (9–12)
and the presence of mutated β-catenin is associated with aggressive
tumor growth (9,10). In the canonical model of Wnt
signaling, β-catenin activity is regulated by phosphorylation at
certain key residues by glycogen synthase kinase-3β (GSK-3β) and
casein kinase 1α (CK1α), which leads to its ubiquitination and
subsequent degradation (9,10). Loss of regulation of β-catenin
results in excessive nuclear accumulation and subsequent
stimulation of downstream target genes, which includes the genes
that encode proteins that play key roles in cell proliferation and
tumor growth (13,14).
Smith et al (15) have reported that UVB radiation
induces β-catenin signaling in keratinocytes. While COX-2 and
β-catenin have been linked to colon cancer, it is unclear whether
UVB-induced COX-2-mediated β-catenin expression is linked in skin
cells or whether there is any association between the development
of UVB-induced skin tumors and β-catenin signaling activation. To
establish whether these links occur in UVB-exposed skin, we have
performed short-term and long-term in vivo animal
experiments, in which mice were exposed to acute and chronic UVB
exposures and their effect on β-catenin signaling investigated. We
also examined the status of β-catenin signaling in UVB-induced skin
tumors using SKH-1 hairless mice. We show that exposure of mouse to
UVB radiation induced dose- and time-dependent activation of
β-catenin in the skin, and that this effect of UVB radiation on
β-catenin activation was dependent on UVB-induced inflammatory
mediators or COX-2/PGE2 expression. To verify this link,
we also used COX-2-deficient mice and a high-fat diet
(HF-diet)-induced inflammation-mediated mouse skin tumor model.
Materials and methods
Animals
The 6- to 7-week-old female C3H/HeN and SKH-1
hairless mice used in this study were purchased from Charles River
Laboratory (Wilmington, MA). The breeding pairs of COX-2-deficient
mice were kindly provided by Dr R. Langenbach of National
Institutes of Environmental Health Sciences (NIEHS/NIH), and a
colony of COX-2-deficient mice was maintained in our animal
resource facility, as described (16). COX-2-deficient mice are
heterozygous and can survive for several weeks and lack of any
clear phenotypical differences with their wild-types. Homozygous
mice are resistant in breeding. All mice were maintained under
standard housing conditions of a 12-h dark/12-h light cycle,
relative humidity of 50±10% and a temperature of 24±2°C. The animal
protocol used in this study was approved by the Institutional
Animal Care and Use Committee of the University of Alabama at
Birmingham.
Reagents and antibodies
Antibodies specific for COX-2, EP2, EP4,
proliferating cell nuclear antigen (PCNA), β-catenin (specific for
immunostaining) and associated secondary antibodies (Alexafluor
488-conjugated) were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA). The PGE2 immunoassay kit, indomethacin and
AH6809 were purchased from Cayman Chemical (Ann Arbor, MI). The
antibodies for β-catenin, cyclin D1, cyclin D2, CK1α, GSK-3β,
matrix metalloproteinase (MMP)-2, MMP-9 and c-Myc were obtained
from Cell Signaling Technology (Beverly, MA).
UVB irradiation of mice
The dorsal hair of C3H/HeN and COX-2-deficient mice
was shaved with electric clippers at least 24 h before UVB
exposure. The backs of the mice were UVB irradiated as described
earlier (6) using a band of four
FS20 UVB lamps (Daavlin, UVA/UVB Research Irradiation Unit, Bryan,
OH) equipped with an electronic controller to regulate UV dosage.
The UV lamps emit UVB (280–320 nm; ∼80% of total energy) and UVA
(320–375 nm; ∼20% of total energy). The peak emission of UV
radiation is at 314 nm. This UV unit enables us to enter the UV
dose in millijoules and variations in energy output are compensated
automatically so that the desired UV dose is delivered.
Treatment of mice with indomethacin and
EP2 receptor antagonist
To assess the effect of COX-2 inhibitor
(indomethacin) or EP2 antagonist (AH6809) on UVB-induced
inflammatory responses and the expression levels of β-catenin
proteins, the mice were treated topically with indomethacin (50
μg in 0.2 ml acetone) (17,18)
or AH6809 (25 μg in 0.2 ml acetone) (17,19),
which were applied to the shaved dorsal skin of C3H/HeN mice 30 min
before UVB exposure. Control animals were treated topically with
acetone (0.2 ml acetone). Animals were sacrificed 24 h after the
last UVB exposure, and skin samples were collected for the analysis
of protein biomarkers. Each group had 4–5 animals.
Immunoassay analysis of
PGE2
Skin samples were homogenized in 100 mM phosphate
buffer, pH 7.4, as described earlier using a polytron homogenizer
(PT3100, Fisher Scientific, Rockford, IL). The supernatants were
collected after centrifugation for analysis of the levels of
PGE2 using the Cayman PGE2 Enzyme Immunoassay
Kit following the manufacturer’s protocol (Cayman Chemical).
Western blot analysis
The skin or skin tumor samples were pooled from two
or three mice in each group (n=4–5 per group), and at least 2 sets
of samples were prepared for western blot analysis. Skin or tumor
lysates were prepared for western blot analysis as described
previously (6). Proteins (30–60
μg) were resolved by 8–10% sodium dodecyl
sulfate/polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes. The membranes were incubated in blocking
buffer for 1 h at room temperature and then incubated with the
primary antibodies overnight at 4°C. The membrane was then washed
with phosphate buffered saline (PBS) and incubated with
HRP-conjugated secondary antibody. Protein bands were visualized
using the enhanced chemiluminescence detection reagents. Equal
loading of proteins on the gel was verified by re-probing the
membrane with anti-β-actin antibody.
Immunofluorescent detection of
β-catenin
Immunohistochemical detection of β-catenin in skin
tumor samples was performed as detailed previously (11). Briefly, sections were
deparaffinized and rehydrated in a graded series of alcohols.
Following the antigen retrieval process, cells were permeabilized
with 0.2% Triton X-100 (Sigma, St. Louis, MO) in PBS and then
incubated with β-catenin-specific antibody for 2 h at room
temperature. The cells were washed with PBS buffer and β-catenin
was detected by an Alexafluor 488-conjugated secondary antibody.
Sections were mounted with Vectashield mounting medium for
fluorescence and stained with DAPI (Vector Laboratories,
Burlingame, CA). Sections were observed using a fluorescence
detection-equipped microscope and photographed.
Statistical analysis
The statistical significance of difference for
PGE2 between the values of control and treatment groups
was determined using analysis of variance (ANOVA). P<0.05 was
considered to indicate a statistically significant difference.
Results
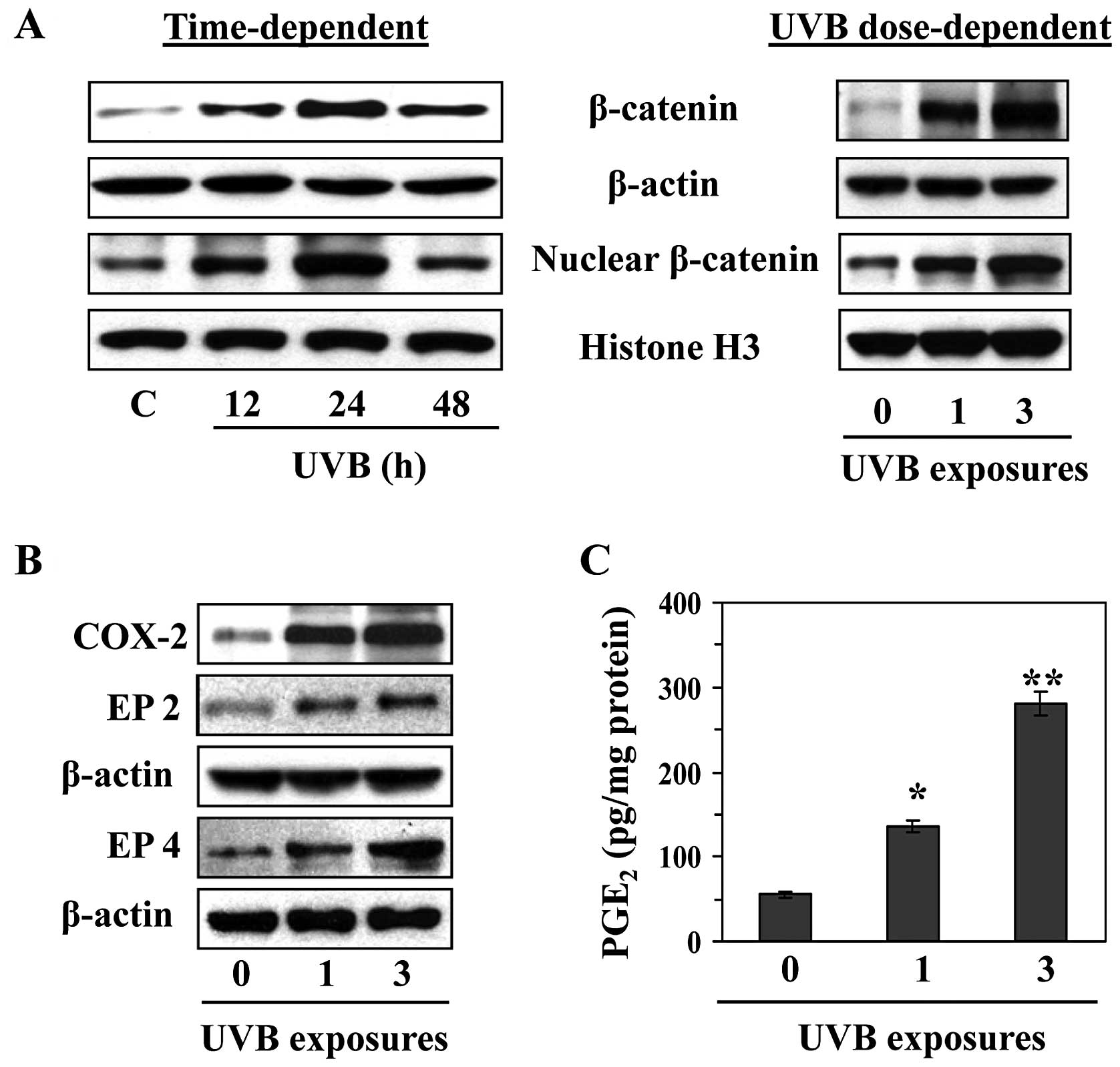
UVB irradiation enhances the levels of
β-catenin in mouse skin
C3H/HeN mice were exposed to a single dose of UVB
(180 mJ/cm2), and mice were sacrificed at different time
points after UVB irradiation to determine the kinetics of
UVB-induced β-catenin expression. Skin samples were collected and
lysates were subjected to western blot analysis to determine the
levels of β-catenin. As shown in Fig.
1A (left panel), the expression of β-catenin in the mouse skin
was upregulated at 12 and 24 h after UVB exposure and then declined
at or after 48 h compared to the expression level of β-catenin at
24 h after UV irradiation. The levels of nuclear β-catenin followed
the same time course. We also compared the effect of single and
multiple (3X) exposures of UVB irradiation on the expression of
β-catenin in the skin. As shown in Fig. 1A (right panel), the effects of UVB
radiation on the levels of cytosolic and nuclear β-catenin
expression were exacerbated when the skin was exposed to UVB
radiation three times on three consecutive days as compared with
the levels of β-catenin in the skin after a single UVB
exposure.
UVB-induced activation of β-catenin is
associated with enhanced expression of COX-2, PGE2 and
PGE2 receptors
To determine whether induction of β-catenin after
UVB irradiation is linked with UVB-induced inflammation and
inflammatory mediators, we analyzed the levels of COX-2,
PGE2 and the receptors of PGE2 (EP2 and EP4)
in the mouse skin. For this purpose mice were exposed either once
or three times on three consecutive days. Skin samples were
collected 24 h after the last UVB exposure. Western blot analysis
revealed that exposure of the skin to UVB resulted in higher
expression of COX-2 (Fig. 1B) and
PGE2 (Fig. 1C) as
compared to non-UVB-exposed mouse skin and this higher COX-2 and
PGE2 expression was dependent on the number of UVB
exposures. As most of the biological functions of PGE2
are mediated through its receptors, we examined the levels of two
PGE2 receptors, EP2 and EP4, which are the more
prominent receptors in the skin. As shown in Fig. 1B, the levels of EP2 and EP4 were
higher in UVB-irradiated skin compared to non-UVB-exposed skin and
the levels of these receptors were higher in skin which had been
irradiated three times with UVB as compared with skin that had been
subject to only one UVB exposure.
COX-2-deficient mice are resistant to
upregulation of β-catenin in UVB-exposed skin
If COX-2 stimulates UVB-induced expression of
β-catenin in mice, then COX-2-deficiency in mice should block or
inhibit UVB-induced upregulation of β-catenin. Thus, to confirm the
role of COX-2 or PGE2 in UVB-induced expression of
β-catenin in mouse skin, we used COX-2-deficient mice
(COX-2+/−). It is established that these mice do not
undergo significant enhancement of UVB-mediated inflammation or
inflammatory mediators in the skin, including PGE2. The
COX-2-deficient mice were exposed to acute UVB (180
mJ/cm2) radiation and sacrificed at different time
points (12, 24 and 48 h) after UVB exposure. Skin samples were
collected and lysates prepared for analysis of β-catenin by western
blot analysis. As shown in Fig. 2,
there was no marked increase in the levels of β-catenin in the
UVB-exposed skin of the COX-2-deficient mice at 12 h after UVB
irradiation as compared with non-UVB-exposed skin of
COX-2-deficient mice. However, the presence of COX-2 protein was
noted at 24 and 48 h after UVB-irradiation of mice. As the mice
used for this study were heterozygous COX-2-deficient, the residual
presence of β-catenin is not surprising. However, the levels of
β-catenin were clearly higher in the UVB-exposed skin of wild-type
mice at 12 and 24 h after UVB exposure. The level of β-catenin was
declined at/or after 48 h of UVB exposure compared to 24 h time
point. These results indicate that COX-2-deficient mice are
resistant to UVB-induced activation of β-catenin signaling.
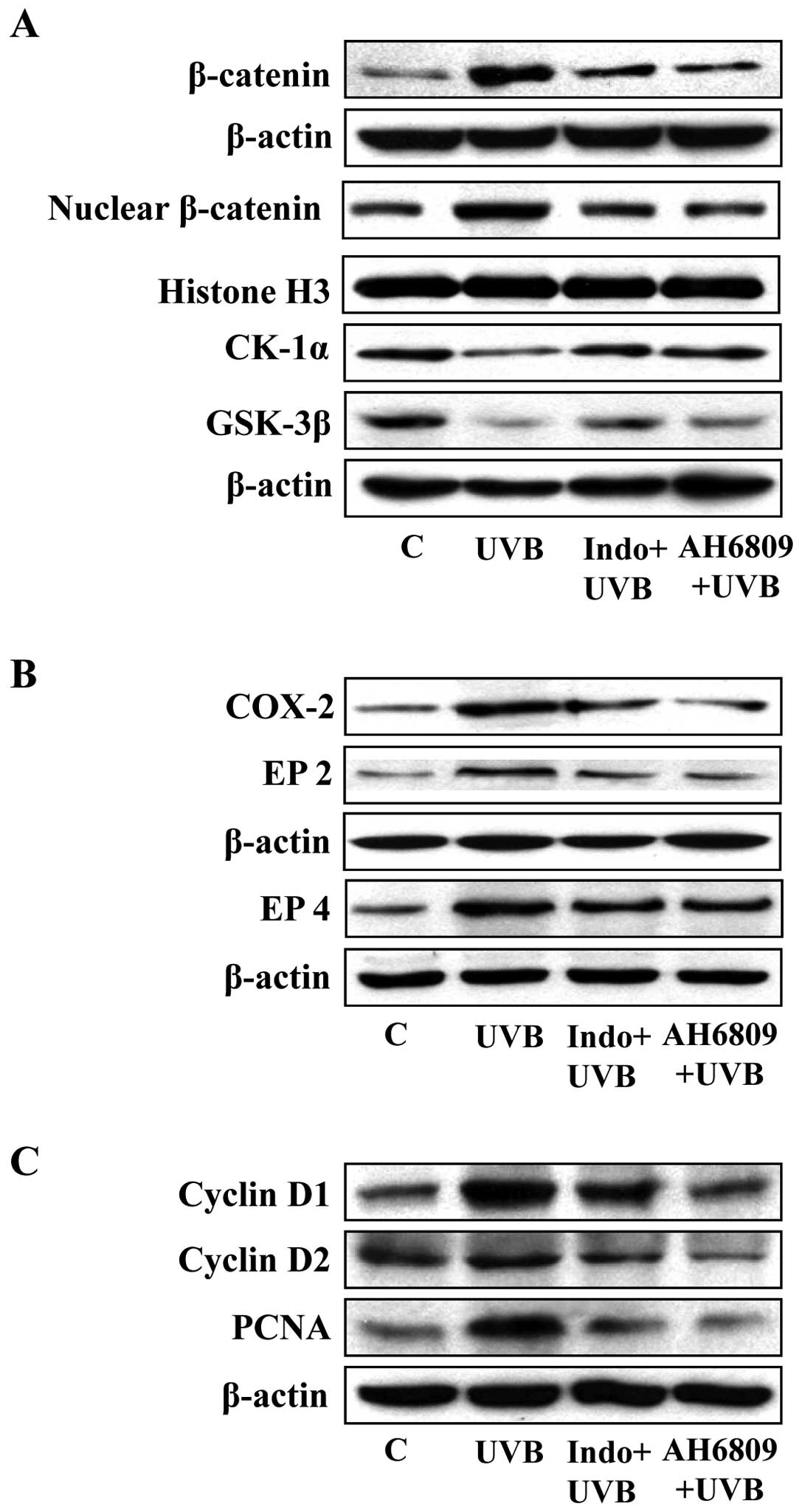
Treatment of mice with COX-2 inhibitor or
EP2 antagonist inhibits UVB-induced activation of β-catenin
signaling in mice
To further identify whether the induction of COX-2
in UVB-exposed skin is responsible for the activation of β-catenin
signaling, the effects of UVB radiation on the activation of
β-catenin was determined in mice that had been administered the
COX-2 inhibitor, indomethacin. As most of the biologic functions of
PGE2 are mediated through the PGE2 receptor
EP2, we also tested the effects of PGE2 on the
activation of β-catenin signaling by using an EP2 antagonist
(AH6809) to block the action of PGE2 in UVB-exposed
skin. In these experiments, mice were UVB irradiated on three
consecutive days with or without topical application of
indomethacin or the AH6809. The mice were sacrificed 24 h after the
last UVB exposure and skin samples processed for protein analysis.
As shown in Fig. 3A, western blot
analysis data confirmed the levels of cytosolic and nuclear
β-catenin were higher in the UVB-exposed skin than in the
non-UVB-irradiated mouse skin. Topical application of indomethacin
or EP2 antagonist before each exposure to UVB resulted in lower
accumulation of β-catenin in the mouse skin compared to the levels
in the mouse skin that was irradiated to UVB but was not treated
with indomethacin or EP2 antagonist. Additionally, we found that
the levels of CK1α and GSK-3β were elevated in the skin of mice
which were UVB-exposed and were treated with either indomethacin or
EP2 antagonist compared to the UVB-irradiated skin of mice which
were not treated with indomethacin or EP2 antagonist.
Effect of indomethacin and EP2 antagonist
on UVB-induced inflammatory responses
As overexpression of COX-2 and PGE2 in
UVB-exposed skin are prominent biomarkers of inflammation and have
a role in activation of β-catenin signaling, we determined the
effects of indomethacin and the EP2 antagonist on UVB-induced
expression of COX-2 in parallel to the analysis of the effects on
the expression of β-catenin. As shown in Fig. 3B, the results confirmed that the
levels of COX-2, EP2 and EP4 were higher in the UVB-exposed skin of
mice than the non-UVB-exposed mouse skin. As would be anticipated,
the levels of COX-2 were markedly lower in the indomethacin-treated
UVB-exposed skin than the UVB-exposed untreated mouse skin and
there was a similar reduction in the levels of EP2 and EP4. Similar
effects on COX-2, EP2 and EP4 proteins were found when the mouse
skin was treated with EP2 antagonist prior to UVB exposure
(Fig. 3B). In the same set of
experiments, we determined the effects of indomethacin and EP2
antagonist on biomarkers of proliferation, including PCNA, cyclin
D1 and cyclin D2. As shown in Fig.
3C, treatment of mice with indomethacin or EP2 antagonist
inhibited UVB-enhanced expression of PCNA, cyclin D1 and cyclin D2
as compared to the expression of these biomarkers of proliferation
in the skin of UVB-irradiated mice which were not treated with
indomethacin or EP2 antagonist.
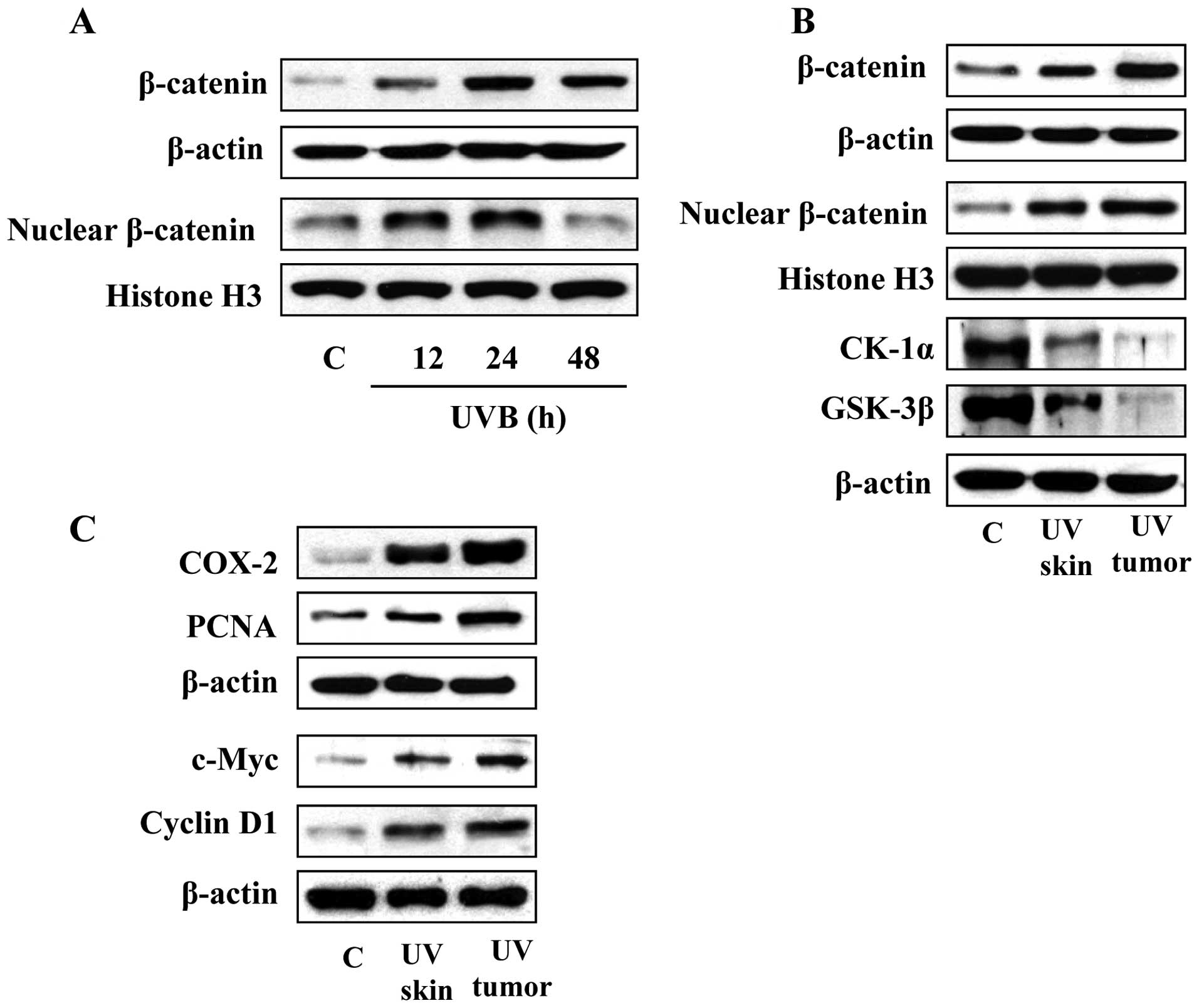
Analysis of UVB-induced activation of
β-catenin signaling in the SKH-1 hairless mouse model
To further verify our results obtained in C3H/HeN
mouse model with respect to UVB-induced β-catenin, we conducted
experiments using the SKH-1 hairless mouse model. In this mouse
model, the animals were exposed to acute UVB irradiation (180
mJ/cm2) and sacrificed at 12, 24 and 48 h after UVB
irradiation. Skin lysates were subjected to the analysis of the
levels of β-catenin using western blot analysis. As shown in
Fig. 4A, the levels of β-catenin
and nuclear β-catenin were upregulated at the 12 and 24 h time
points and thereafter the levels declined, indicating a time course
response similar to that observed in the C3H/HeN mice (Fig. 1A). The SKH-1 hairless mouse is a
well-established model for analysis of UVB-induced skin tumor
formation. We therefore exposed the SKH-1 hairless mice to UVB
radiation 3 times per week for total of 24 weeks to determine the
long-term effects of UV radiation on β-catenin signaling and its
association with tumor formation. After 24 weeks of UVB
irradiation, mice were sacrificed and tumors and tumor uninvolved
skin samples collected for the analysis of β-catenin-associated
signaling proteins. As shown in Fig.
4B, western blot analysis revealed that long-term exposure of
the mice to UVB irradiation resulted in upregulation of the levels
of β-catenin compared with the skin samples obtained from the
non-UVB-irradiated control mice. The levels of β-catenin in tumor
samples were higher than the UVB-irradiated skin samples obtained
from the mice in the same group. Additionally, the levels of CK1α
and GSK-3β were reduced in UVB-exposed skin as well as in tumor
samples as compared to non-UVB-exposed control skin.
To further verify the potential link between the
expression of UVB-induced β-catenin and the expression of
biomarkers of UVB-induced inflammation/proliferation, the skin and
tumor samples from the same experiment were analyzed for expression
of various biomarkers of these responses. The results of western
blot analysis revealed that the expression levels of COX-2, PCNA,
c-Myc and cyclin D1 were higher in UVB-induced skin tumors and
UVB-exposed skin (tumor uninvolved) as compared with the skin of
non-UVB-exposed control animals (Fig.
4C).
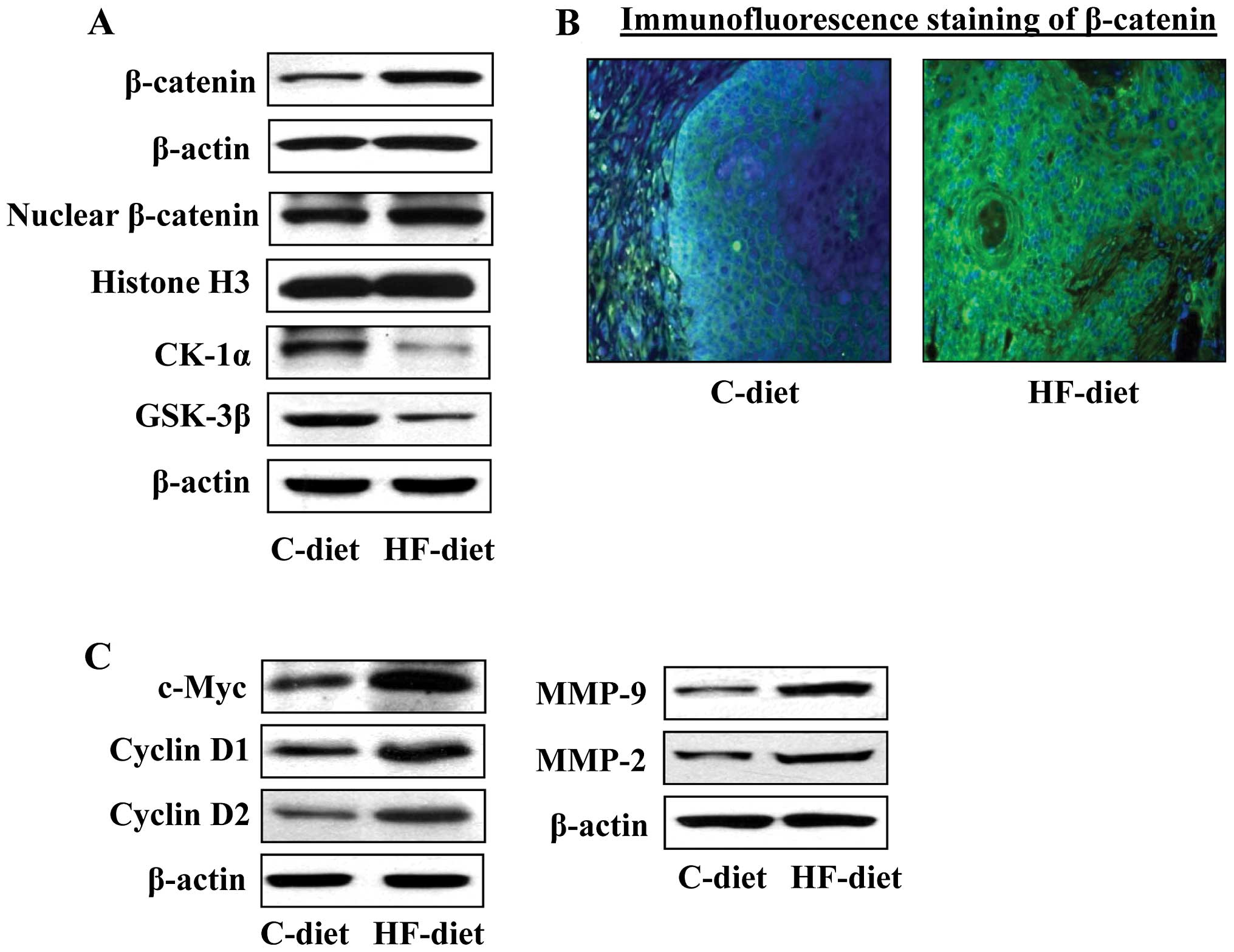
Status of β-catenin and β-catenin
signaling molecules in high-fat diet-induced skin tumors in
UVB-irradiated skin
In our recent studies, we found that administration
of the HF-diet enhances UVB-induced skin carcinogenesis in SKH-1
hairless mice in terms of tumor multiplicity and tumor growth/size
compared to the skin carcinogenesis in control-diet-fed mice
(20). In these experiments, we
also found that the enhancement of photocarcinogenesis observed on
administration of the HF-diet to mice was associated with the
higher levels of inflammation and inflammatory mediators than were
observed in skin of the mice fed a control diet. Based on these
observations, we used this tumor model in our present study to
determine whether HF-diet-induced inflammation in UVB-induced skin
tumor is associated with enhancement in the levels or activation of
β-catenin. Skin tumor samples from experiments described above were
analyzed for biomarkers related to β-catenin signaling. As shown in
Fig. 5A, western blot analysis
revealed that the levels of total β-catenin as well as nuclear
β-catenin were higher in the skin tumor samples from HF-diet-fed
mice as compared with the skin tumors of mice fed a control diet.
These results were further verified by intracellular staining of
β-catenin in tumor sections. The intensity of intracellular
staining of β-catenin was higher in the skin tumors of HF-diet-fed
mice compared with the intracellular staining in the skin tumors of
C-diet-fed mice (Fig. 5B).
Concomitantly, the levels of CK1α and GSK-3β were lower in the skin
tumors of HF-diet-fed mice than the skin tumors of mice fed the
control diet (Fig. 5A). The
downstream signaling targets of β-catenin signaling also were
analyzed in the skin tumors from HF-diet and C-diet-fed mice. As
shown in Fig. 5C, we found that
the levels of c-Myc, cyclin D1, cyclin D2, and MMP-2 and MMP-9 were
higher in the skin tumors of HF-diet-fed mice as compared to the
skin tumors of mice fed the control diet. These data support our
concept that activation of β-catenin signaling in UVB-exposed skin
is mediated through enhanced inflammation.
Discussion
Epidemiological, clinical and experimental studies
clearly indicate a link between chronic UVB exposure and greater
risk of non-melanoma skin cancer in humans. Although many genetic,
epigenetic and environmental factors play a role in the risk of
skin cancers, the chronic and sustained generation of inflammation
and inflammatory mediators in UVB-exposed skin are considered to be
potent regulators of tumor promotion and progression. In
particular, UVB-induced overexpression of COX-2 and PGE2
in the skin have been associated with multiple signaling pathways
responsible for tumor cell survival, resistance to apoptotic cell
death and suppression of immune system (3,21).
Despite this, little is known about the potential link between the
COX-2/PGE2 axis and β-catenin signaling in UVB-exposed
skin and UVB-induced skin tumors. It has been shown that
PGE2 can promote the growth of colon cancer cells by
activating β-catenin signaling (8). The study by Smith et al
(15) demonstrated that UVB
radiation-induced β-catenin signaling is increased by COX-2
expression in human and mouse keratinocytes, and also in
UVB-irradiated mouse skin. Thus, it was interesting to extend the
studies on the effect of UVB radiation on β-catenin signaling and
also to check the levels of β-catenin in UVB-induced skin tumors.
In this report, we provide evidence that UVB-induced upregulation
of COX-2/PGE2 results in activation of β-catenin
signaling in the mouse skin and contributes to skin tumor
development by using different mouse models.
The data presented herein provide evidence that
UVB-induced COX-2 expression and PGE2 production
promotes β-catenin signaling in UVB-exposed mouse skin. The lack of
upregulation and activation of β-catenin signaling in UVB-exposed
COX-2-deficient mice supports the evidence that UVB-induced COX-2
overexpression may be necessary for the activation of β-catenin.
The activation of β-catenin signaling in UVB-exposed skin under the
influence of inflammatory mediators, such as COX-2 and
PGE2, was verified using two strains of mice, C3H/HeN
and SKH-1 hairless mice (Figs. 1
and 4). Additionally, we found
that treatment of mice with indomethacin (an inhibitor of COX-2)
and the EP2 antagonist suppressed the accumulation as well as
activation of β-catenin signaling in UVB-exposed skin, and
established their effects on CK1α, GSK-3β and downstream targets of
cell proliferation (Fig. 3).
As β-catenin has been shown to have a role in tumor
growth (8), we subjected SKH-1
hairless mice to a standard photocarcinogenesis protocol (20,22).
At the termination of the photocarcinogenesis experiment, mice were
sacrificed and UVB-induced skin tumors and tumor uninvolved skin
samples were collected and analyzed for the biomarkers of
inflammation and the molecules of β-catenin signaling. The levels
of β-catenin were markedly higher in chronically UVB-exposed skin
and UVB-induced skin tumors compared with the skin samples from
non-UVB-exposed control mice. The levels of COX-2 expression and
downstream targets of β-catenin signaling were also upregulated in
skin tumors suggesting the role of COX-2 overexpression in
activation of β-catenin signaling in UVB-induced skin tumors
(Fig. 4).
Finally, to further support the evidence that
UVB-induced inflammation, specifically overexpression of COX-2 and
PGE2, has a role in the activation of β-catenin
signaling, we used a high fat-diet-induced inflammation model
(20). We have found that
administration of high-fat diet induces higher expression of COX-2
and PGE2 in UVB-induced skin tumors than the skin tumors
in control-diet-fed mice (20). We
used this tumor model, and determined the relationship between
COX-2/PGE2 and activation of β-catenin signaling. As
shown in Fig. 5, we found that the
levels of β-catenin and their downstream targets, such as c-Myc,
cyclin D1, cyclin D2, MMP-2 and MMP-9, were higher in skin tumors
of high-fat diet-fed mice compared with the skin tumors of
control-diet fed mice. These data provide additional evidence that
overexpression of COX-2 and PGE2 have a role in
activation of β-catenin signaling in UVB-exposed skin and
UVB-induced skin tumorigenesis. In addition to
COX-2/PGE2, UVB-induced expression of epidermal growth
factor receptor (EGFR) and Akt also has been shown to be involved
in activation of β-catenin signaling. EGFR activation promotes
β-catenin signaling by promoting disassociation of β-catenin from
α-catenin (23). There is evidence
that β-catenin signaling contributes to chemically-induced skin
cancer. In the two-stage chemical skin carcinogenesis in mice, an
increased accumulation of nuclear β-catenin was observed (24). These findings in combination with
our data suggest that UVB-induced β-catenin signaling contributes
to skin carcinogenesis and that it is stimulated by UVB-induced
inflammation. Thus, this study suggests that UVB-induced skin
cancer can be inhibited by targeting β-catenin signaling.
Acknowledgements
Authors thank Dr Fiona Hunter for
editorial assistance.
References
|
1.
|
Housman TS, Feldman SR, Williford PM,
Fleischer AB Jr, Goldman ND, Acostamadiedo JM and Chen GJ: Skin
cancer is among the most costly of all cancers to treat for the
medicare population. J Am Acad Dermatol. 48:425–429. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
American Cancer Society: Cancer Facts and
Figures, 2010. American Cancer Society; Atlanta, GA: 2010
|
|
3.
|
Mukhtar H and Elmets CA:
Photocarcinogenesis: Mechanisms, models and human health
implications. Photochem Photobiol. 63:355–447. 1996. View Article : Google Scholar
|
|
4.
|
Katiyar SK: Oxidative stress and
photocarcinogenesis: Strategies for prevention. Oxidative Stress,
Disease and Cancer. Singh KK: Imperial College Press; London: pp.
933–964. 2006, View Article : Google Scholar
|
|
5.
|
Rivas JM and Ullrich SE: The role of IL-4,
IL-10, and TNF-alpha in the immune suppression induced by
ultraviolet radiation. J Leukoc Biol. 56:769–775. 1994.PubMed/NCBI
|
|
6.
|
Meeran SM, Akhtar S and Katiyar SK:
Inhibition of UVB-induced skin tumor development by drinking green
tea polyphenols is mediated through DNA repair and subsequent
inhibition of inflammation. J Invest Dermatol. 129:1258–1270. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Katiyar SK, Matsui MS and Mukhtar H:
Kinetics of UV light-induced cyclobutane pyrimidine dimers in human
skin in vivo: An immunohistochemical analysis of both epidermis and
dermis. Photochem Photobiol. 72:788–793. 2000. View Article : Google Scholar
|
|
8.
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a Gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Gavert N and Ben-Ze’ev A: β-catenin
signaling in biological control and cancer. J Cell Biochem.
102:820–828. 2007.
|
|
10.
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Vaid M, Prasad R, Sun Q and Katiyar SK:
Silymarin targets β-catenin signaling in blocking
migration/invasion of human melanoma cells. PLoS One.
6:e230002011.
|
|
12.
|
Rimm DL, Caca K, Hu G, Harrison FB and
Fearon ER: Frequent nuclear/cytoplasmic localization of
beta-catenin without exon 3 mutations in malignant melanoma. Am J
Pathol. 154:325–329. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Li YJ, Wei ZM, Meng YX and Ji XR:
Beta-catenin up-regulates the expression of cyclinD1, c-myc and
MMP-7 in human pancreatic cancer: relationships with carcinogenesis
and metastasis. World J Gastroenterol. 11:2117–2123. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lowy AM, Clements WM, Bishop J, Kong L,
Bonney T, Sisco K, Aronow B, Fenoglio-Preiser C and Groden J:
β-catenin/Wnt signaling regulates expression of the membrane type 3
matrix metalloproteinase in gastric cancer. Cancer Res.
66:4734–4741. 2006.
|
|
15.
|
Smith KA, Tong X, Abu-Yousif AO, Mikulec
CC, Gottardi CJ, Fischer SM and Pelling JC: UVB radiation-induced
β-catenin signaling is enhanced by COX-2 expression in
keratinocytes. Mol Carcinog. 51:734–745. 2012.
|
|
16.
|
Langenbach R, Loftin C, Lee C and Tiano H:
Cyclooxygenase knockout mice: models for elucidating
isoform-specific functions. Biochem Pharmacol. 58:1237–1246. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chun KS and Langenbach R: The
prostaglandin E2 receptor, EP2, regulates survivin expression via
an EGFR/STAT3 pathway in UVB-exposed mouse skin. Mol Carcinog.
50:439–448. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Chun KS, Akunda JK and Langenbach R:
Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by
activating the prostaglandin E2 receptors, EP2 and EP4. Cancer Res.
67:2015–2021. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Chun KS, Lao HC, Trempus CS, Okada M and
Langenbach R: The prostaglandin receptor EP2 activates multiple
signaling pathways and beta-arrestin1 complex formation during
mouse skin papilloma development. Carcinogenesis. 30:1620–1627.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Vaid M, Singh T, Prasad R and Katiyar SK:
Intake of high-fat diet stimulates the risk of ultraviolet
radiation-induced skin tumors and malignant progression of
papillomas to carcinoma in SKH-1 hairless mice. Toxicol Appl
Pharmacol. 274:147–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Prasad R and Katiyar SK: Prostaglandin E2
promotes ultra-violet radiation-induced immune suppression through
DNA hypermethylation. Neoplasia. 15:795–804. 2013.PubMed/NCBI
|
|
22.
|
Vaid M, Sharma SD and Katiyar SK:
Honokiol, a phytochemical from the Magnolia plant, inhibits
photocarcinogenesis by targeting UVB-induced inflammatory mediators
and cell cycle regulators: Development of topical formulation.
Carcinogenesis. 31:2004–2011. 2010.
|
|
23.
|
Ji H, Wang J, Nika H, Hawke D, Keezer S,
Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K and Lu Z:
EGF-induced ERK activation promotes CK2-mediated disassociation of
alpha-catenin from beta-catenin and transactivation of
beta-catenin. Mol Cell. 36:547–559. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bhatia N and Spiegelman VS: Activation of
Wnt/beta-catenin/Tcf signaling in mouse skin carcinogenesis. Mol
Carcinog. 42:213–221. 2005. View
Article : Google Scholar
|