Introduction
Breast cancer is the most common female cancer and
∼70–75% of cases express oestrogen receptor α (ERα) (1). Tamoxifen (TAM), a selective estrogen
receptor (ER) modulator, has been used extensively in the clinical
management of primary and advanced breast cancer and is also widely
employed as a preventive agent after surgery for breast cancer
(2). A larger number of clinical
studies over the past 30 years have shown that TAM can reduce the
incidence and regression of breast carcinoma among women worldwide
(3). Despite the relative safety
and significant anti-neoplastic activities of tamoxifen, most
initially responsive breast tumors develop resistance to its
(4). In addition, TAM is also
known to increase the expression of vascular endothelial growth
factor (VEGF), which is an undesirable effect in breast cancer
treatment (5,6). Extensive studies have shown that
VEGF, a receptor tyrosine kinase, plays an important role in
tumorigenesis, and blocking the VEGF signaling pathway can reduce
tumor-associated angiogenesis and blood vessel-dependent metastasis
(7,8). Therefore, combination therapies of
tamoxifen with an anti-VEGF signaling agent aimed at inhibition of
both ER-mediated signaling and VEGF stimulated stromal activation,
may be a potential means of delaying the arrival of resistance.
Cyclooxygenase-2 (COX-2) is an inducible gene, whose
expression is undetectable in most normal tissues. Accumulating
evidence shows that overexpression of COX-2 was commonly related to
different types of carcinomas, including breast carcinoma (9–12).
COX-2 has been implicated in breast tumorigenesis based on its
increased expression in a significant fraction of breast carcinomas
and the protective effects of nonsteroidal anti-inflammatory drugs
(NSAIDs) against breast cancer (13). Hoeben et al (14) have indicated that COX-2
overexpression is correlated with induction of VEGF expression and
therefore tumor angiogenesis in human breast cancers. Inhibition of
COX-2 by non-steroidal anti-inflammatory drugs (NSAD) leads to
restricted angiogenesis and downregulates production of VEGF
(15). Although accumulating
evidence suggests that NSAIDs, such as celecoxib sensitize cancer
cells to ATM-induced apoptosis, it remains unclear whether it is
COX-2 dependent or independent (16). Furthermore, NSAIDs could result in
liver injury, which would limit their clinical applications
(17,18).
Silencing of COX-2 by RNAi would avoid the side
effects induced by NSAIDs. Accumulating evidence shows that
down-regulation of COX-2 expression by RNAi inhibited tumor cell
proliferation and colony formation in vitro in different
types of cancer cells (12,19,20).
COX-2 silencing abolished the metastatic potential of highly
malignant breast cancer cells (10). In addition, Lin et al
documented that combining gene therapy with various drugs enhances
transduction efficiency resulting in enhanced tumor cell killing
(21). Therefore, in this study,
we knocked out the COX-2 gene via a replication-incompetent
lentivirus (LV-COX-2), then LV-COX-2 combination with TAM for
suppressing VEGF expression and simultaneously reducing doses of
ATM.
The objective of the present study was to evaluate
the potency of LV-COX-2 in combination with TAM in inhibiting
breast cancer cell growth, proliferation, and angiogenesis in
vitro and reveal the underlying molecular mechanisms involved
in TAM-induced apoptosis. In addition, tumor growth ability in nude
mice was detected to define the combination treatment effect in
tumorigenesis in vivo.
Materials and methods
Cell culture
Human breast cancer cell lines MCF-7 and HEK293
(human embryonic kidney cells) were bought from the Shanghai Cell
Collection (Shanghai, China). Cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL, Grand Island, NY, USA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Gibco-BRL) at 37°C in a 5% CO2 atmosphere and at 95%
humidity.
Reagents
Stock solutions of 10 mM TAM (Sigma-Aldrich, St.
Louis, MO, USA) were dissolved in dimethylsulfoxide (DMSO,
Sigma-Aldrich), stored at −20°C, and diluted in fresh medium just
before use. TRIzol reagent kit and Coomassie Blue R-250 from
Gibco-BRL, Invitrogen Corp. (Carlsbad, CA, USA); Nonidet P-40 lysis
buffer, chemiluminescent peroxidase substrate, propidium iodide
(PI), 4′,6-diamidino-2-phenylindole (DAPI),
3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT),
and sense and antisense VEGFR2 oligo primers from Sigma-Aldrich;
and pyrogallol and H2O2 from Merck
(Whitehouse Station, NJ, USA). Stock solutions of PI, DAPI, and MTT
were prepared by dissolving 1 mg of each compound in 1 ml of
phosphate buffered saline (PBS). The solution was protected from
light, stored at 4°C, and used within 1 month.
Animals
Female BALB/c nude mice aged 4–5 weeks were obtained
from the Experimental Animal Center of the Jilin University
(Changchun, China). All procedures were performed according to
institutional guidelines and conformed to the National Institutes
of Health guidelines on the ethical use of animals.
Generation of plasmids and recombinant
lentivirus
To inhibit the expression of COX-2, a short hairpin
RNA (shRNA) targeting the COX-2 transcript was designed. The
synthesized oligonucleotides containing specific target sequence, a
loop, the reverse complement of the target sequence, a stop codon
for U6 promoter and two sticky ends were cloned into pGCSIL-GFP
lentivirus vector according to the instructions (Shanghai Gene Chem
Co. Ltd., China). The targeting sequences corresponding to the
siRNAs for COX-2 (GeneBank accession no. NM_000963.2) was as
followed: bases on 290–310 (sense 5′-AAACTGCTCAACACCGGAATT-3′).
Sequences for the scrambled control (NC) for siRNA are
AATTCTCCGAACGTGTCACGT (sense). This sequence does not target any
gene product and have no significant sequence similarity to human
gene sequences, being essential for determining the effects of
siRNA delivery.
To produce the lentivirus, the HEK293 cells were
transfected with 20 μg of LV-COX-2, and LV-NC plasmid
together with 15 μg of pHelper-1.0 and 10 μg of
pHelper-2.0 packaging plasmids, respectively (22). The culture medium was collected
within 48 h after transfection, concentrated by
ultracentrifugation, aliquoted, and stored at −80°C until used. The
titer of lentivirus was determined by hole-by-dilution titer assay
as described (23). Four days
after a single exposure of HEK293 cells to the lentivirus, strong
green fluorescence was observed in >90% of cells, indicating a
high and stable transduction of the lentiviral vector system. The
final titer of LV-COX-2, and LV-NC were 5×108 TU/ml and
4×108 TU/ml, respectively.
RNA isolation and real-time RT-PCR
Total RNA was extracted from cultured cells using
TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. RNA was reverse-transcribed into cDNA by a
Primescript™ RT reagent kit according to the manufacturer’s
protocols (Takara, Japan). Quantitative real-time polymerase chain
reaction (RT-PCR) assays were carried out using SYBR Green
Real-Time PCR Master Mix (Toyobo, Osaka, Japan) and RT-PCR
amplification equipment using specific primers: COX-2, sense strand
5′-CCCTTGGGTGTCAAAGGTAAA-3′, antisense strand
5′-AAACTGATGCGTGAAGTGCTG-3′, β-actin, sense strand
5′-GCGAGCACAGAGCCTCGCCTTTG-3′, antisense strand
5′-GATGCCGTGCTCGATGGGGTAC-3′. The PCR conditions were as follows: a
pre-denaturing at 95°C for 3 min, followed by 40 cycles of
denaturation at 95°C for 10 sec, annealing/extension at 58°C for 20
sec, final extension of 72°C for 10 min. The amplification
specificity was checked by melting curve analysis. The
2−ΔΔCT method was used to calculate the relative
abundance of target gene expression generated by Rotor-Gene
Real-Time Analysis Software 6.1.81. The expression of interest
genes were determined by normalization of the threshold cycle (Ct)
of these genes to that of the control β-actin.
Western blot analysis
Cultured cells were washed twice with
phosphate-buffered saline (PBS, pH 7.2), then cells were lysed with
Triton X-100 in HEPES buffer (150 mM NaCl, 50 mM HEPES, 1.5 mM
MgCl2, 1% Triton X-100, 0.1% SDS, protease inhibitor
cocktail (Sigma), 100 mM NaF and 100 mM
Na3VO4) for 30 min. Cell lysates were
clarified by centrifugation (10,000 g, 15 min), and protein
concentrations were determined using the Bradford reagent (Sigma,
Germany). Protein samples were separated on an 8–15%
SDS-polyacrylamide gel (SDS-Page) and transferred onto
nitrocellulose membranes (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), followed by blocking in TBS containing 5% skim milk
and 0.1% Tween-20 for 2 h at room temperature, and then
immunoblotted with specific primary antibodies and incubated with
corresponding horseradish peroxidase-conjugated secondary antibody.
The other primary antibodies used in the western blot analyses
were: anti-VEGF, anti-VEGFR, anti-COX-2 (all Cell Signaling
Technology, Beverly, MA, USA), mouse monoclonal anti-β-actin
(Sigma-Aldrich), and mouse monoclonal anti-caspase-3, mouse
monoclonal anti-caspase-8 (Santa Cruz Biotechnology). Secondary Abs
used for immunodetection were: HRP-conjugated goat anti-mouse IgG
and goat anti-rabbit IgG (Amersham Biosciences, Uppsala, Sweden).
All immunoblots were visualized by enhanced chemiluminescence
(Pierce).
Proliferation assays
To measure the effect of LV-COX-2 and ATM alone or
both on cell proliferation,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was used. MCF-7 cells grown in monolayers were harvested and
dispensed in 96-well culture plates in 100 μl of DMEM at a
concentration of 5×103 cells per well. After 24 h, cells
without any treatment, cells treated only with LV-NC, and cells
transfected with LV-COX-2, cells treated only with ATM, cells
treated with ATM combination with LV-COX-2 were seeded in 96-well
plates and left at 37°C for 48 h. The cells were then washed with
PBS and incubated in 50 μl of 0.5 mg/ml MTT in culture
medium at 37°C for 4 h. Following the addition of 100 μl of
isopropanol, the absorbance was read at 595 nm in an ELISA plate
reader (Molecular Devices Corp., Sunnyvale, CA, USA). The mean
proliferation of cells without any treatment was expressed as
100%.
Detection of apoptosis
MCF-7 cells were cultured in 6-well plates in DMEM
with 10% FBS medium and were treated with LV-NC, LV-COX-2, ATM and
ATM combination with LV-COX-2, respectively, for 48 h. The cover
slips were washed three times with phosphate-buffered saline (PBS)
and single cell suspensions were fixed in 1% PBS. Cells were
stained with 100 μg/ml acridine orange (AO) and 100
μg/ml ethidium bromide (EB) for 1 min. Then cells were
observed under a fluorescence microscope. At least 200 cells were
counted and the percentage of apoptotic cells was determined. In
addition, we also detected caspase-3 and -8 protein expression by
western blotting as an additional indicator of apoptosis.
Cell cycle analysis
To determine the cell cycle distribution,
5×105 MCF-7 cells were plated in 60-mm dishes and
treated with LV-NC, LV-COX-2, ATM and ATM combination with
LV-COX-2, respectively, for 48 h. After treatment, the cells were
collected by trypsinization, fixed in 70% ethanol, and kept at
−20°C overnight for fixation. Cells were washed in PBS, resuspended
in 1 ml of PBS containing 100 μg/ml RNase and 40
μg/ml PI and incubated in the dark for 30 min at room
temperature. The distribution of cells in the cell cycle phases
were analyzed from the DNA histogram with a FACSCaliber flow
cytometer (Becton-Dickinson, San Jose, CA, USA) and CellQuest
software (CA, USA).
Cell migration assay
The migration assay was performed using a 12-well
Boyden chamber (Neuro Probe) with an 8-μm pore size.
Approximately 1×105 cells were seeded into upper wells
of the Boyden chamber and incubated for 6 h at 37°C in medium
containing 1% FBS. Medium containing 10% FBS was used as a
chemoattractant in the bottom wells. Cells that did not migrate
through the pores of the Boyden chamber were manually removed with
a rubber swab. Cells that migrated to the lower side of the
membrane were stained with hematoxylin and eosin and photographed
using an inverted microscope.
Wound-healing assay
To assess the effect of TAM and LV-COX-2 on cell
migration, wound-healing assay was performed. MCF-7 cells
(1×105) were plated in 12-well plates in complete growth
medium. After 24 h of growth, a scratch was made through the
confluent cell monolayer, and then the cells were treated with
LV-COX-2, LV-NC, TAM, TAM combination with LV-COX-2, respectively,
in 3 ml of complete medium. At 48 h post-treatment, cells were
stained with hematoxylin and eosin (HE). Cells invading the wound
line were observed under an inverted phase-contrast microscope
using 20×, Leica DMR, Germany.
Cell invasion assay
Cell invasion was determined using trans-well
chambers made from polycarbonate membrane filters with a pore size
of 8 μm. Transwell filters in 6-well plates were coated with
Matrigel, hydrated for ∼2 h in the tissue culture incubator with
500 μl serum-free culture media in the bottom and 500
μl in the top of the chamber. After hydration of the
Matrigel, 5×105 MCF-7 cells were plated in 500 μl
serum-free medium on top of chamber, while 2 ml medium 10% FCS were
placed in the lower chambers. TAM, LV-NC, LV-COX-2, LV-COX-2
combination with ATM were added to the upper chambers,
respectively. Cells without any drug were used as control. After 48
h of incubation, the filters were removed, washed twice in PBS and
fixed in 10% formalin for 15 min. After fixing at room temperature,
the chambers are rinsed in PBS and stained with 0.2% crystal violet
staining solution for 30 min. After washing the chambers by PBS,
the cells at the top of the Matrigel membrane were carefully
removed by cotton swabs. At this time all cells that remain had
invaded to the bottom side of the membrane. Cell invasion was
observed with an immunofluorescence microscope by counting the
cells that had invaded into the bottom of the Cell Culture Insert.
All experiments were performed in triplicate.
Measurement of prostaglandin-E2 (PGE2)
production
PGE2 synthesis was determined by competitive
enzyme-linked immunosorbent assay (ELISA) as previously described
(24). In brief, MCF-7 cells were
treated with LV-NC, LV-COX-2, ATM and ATM combination with LV-COX-2
respectively for 48 h in 12-well plates, and then these culture
media were centrifuged to remove cell debris. Cell-free culture
media were collected at indicated time, and then PGE2 levels were
determined by competitive ELISA as described using the kit
manufacturer (Cayman Chemical, Ann Arbor, MI, USA).
Measurement of VEGF levels
To measure VEGF levels, 5×105 MCF-7 cells
were plated in 6-well plates and incubated under culture conditions
overnight, and the medium was replaced by serum-free culture
conditioned medium. LV-NC, LV-COX-2, ATM and ATM combination with
LV-COX-2, respectively, were added to the culture, and the medium
was collected at 72 h. VEGF levels were measured using a VEGF
enzyme linked immunosorbent assay (ELISA) kit (DVE00, R&D
Systems, Minneapolis, MN, USA) according to the manufacturer’s
instructions. The optical density at 570 nm of each well was
measured using an ELISA reader (μQuant; Biotek Instruments,
Inc., Winooski, VT, USA). In addition, we also detected VEGF and
VEGFR protein expression level by western blotting.
Tumor xenograft in nude mice
Exponentially growing MCF-7 cells were harvested and
a tumorigenic dose of 2×106 cells was injected
intraperitoneally into 4–5-week-old female BALB mice. When tumors
reached 100–200 mm3, mice were divided randomly into
five groups (10 mice per group). The control group received 1%
polysorbate resuspended in deionized water. The other four groups
were treated with LV-NC (1×108 PFU/dose), ATM (2 mg/kg
body weight), LV-COX-2 (1×108 PFU/dose), or ATM plus
LV-COX-2 (ATM, 1 mg/kg body weight; LV-COX-2, 1×108
PFU/dose respectively) intraperitoneally on alternative days for 3
weeks. The tumor size was measured using caliper before the
treatment injections were given, and on day 7, 14 and 21 of
treatment. On day 21, the animals were euthanized using chloroform
and their spleen tissues were collected and cultured for a
splenocyte surveillance study.
Assay of splenocyte proliferation
Spleens from treated mice were collected, and
single-cell spleen suspensions were pooled in serum-free RPMI-1640
by filtering the suspension through a sieve mesh with the aid of a
glass homogenizer to exert gentle pressure on the spleen fragments.
The detail assay of splenocyte proliferation was previously
described (16).
Statistical analysis
Data from at least three independent experiments are
expressed as mean ± SD. Statistical comparison of more than two
groups was performed using one-way ANOVA followed by a Tukey
post hoc test. Statistical analyses were undertaken using
GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA, USA)
and the SPSS® statistical package, version 16.0 (SPSS
Inc., Chicago, IL, USA) for Windows®. P<0.05 was
considered statistically significant.
Results
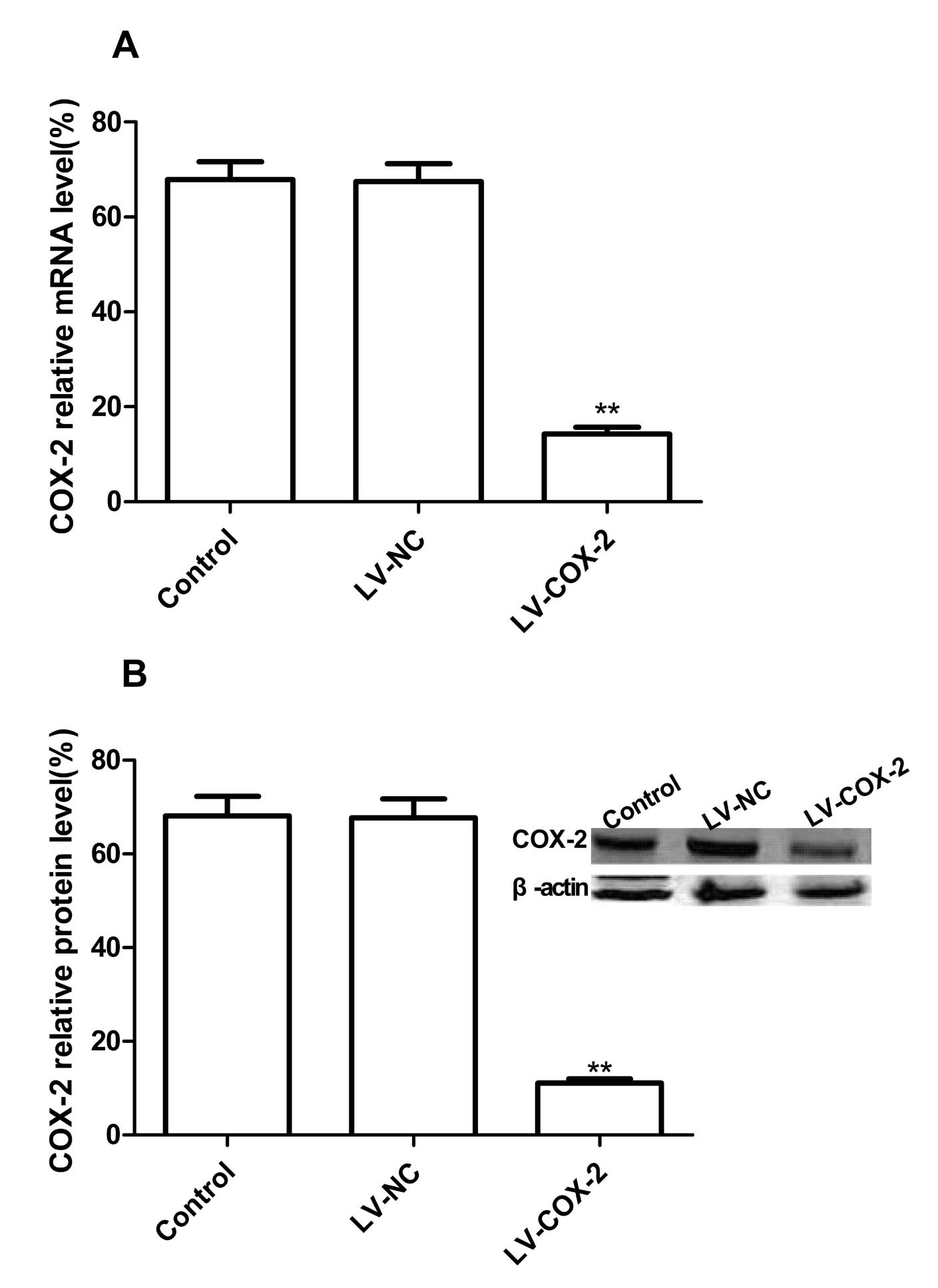
Downregulation of COX-2 expression by
LV-COX-2
To silence COX-2 expression, we constructed a
recombinant lentiviral carrying a based shRNA against COX-2,
LV-COX-2, to inhibit COX-2 expression in breast carcinoma cell
lines. To evaluate the silencing capacity of LV-COX-2, MCF-7 were
treated with LV-COX-2, LV-NC as negative controls. Real-time RT-PCR
and western blotting were performed to detect the COX-2 mRNA
expression and protein expression levels at 3 days
posttransfection. Our results showed no significant inhibition in
COX-2 mRNA expression in LV-NC group, there is no significantly
difference in LV-NC group and control group (PBS group)
(P>0.05). On the other hand, COX-2 mRNA expression in LV-COX-2
group was significantly decreased compared to LV-NC group and
control group after injection (Fig.
1A, p<0.01). On protein level, there was no significant
inhibition in COX-2 protein expression found in LV-NC group and
control group (P>0.05), while the band density decreased
dramatically in the LV-COX-2 group as compared with the LV-NC and
control group (P<0.01) (Fig.
1B). These results demonstrated that LV-COX-2 significantly
decreased COX-2 protein expression in the breast cancer cell line
(P<0.01).
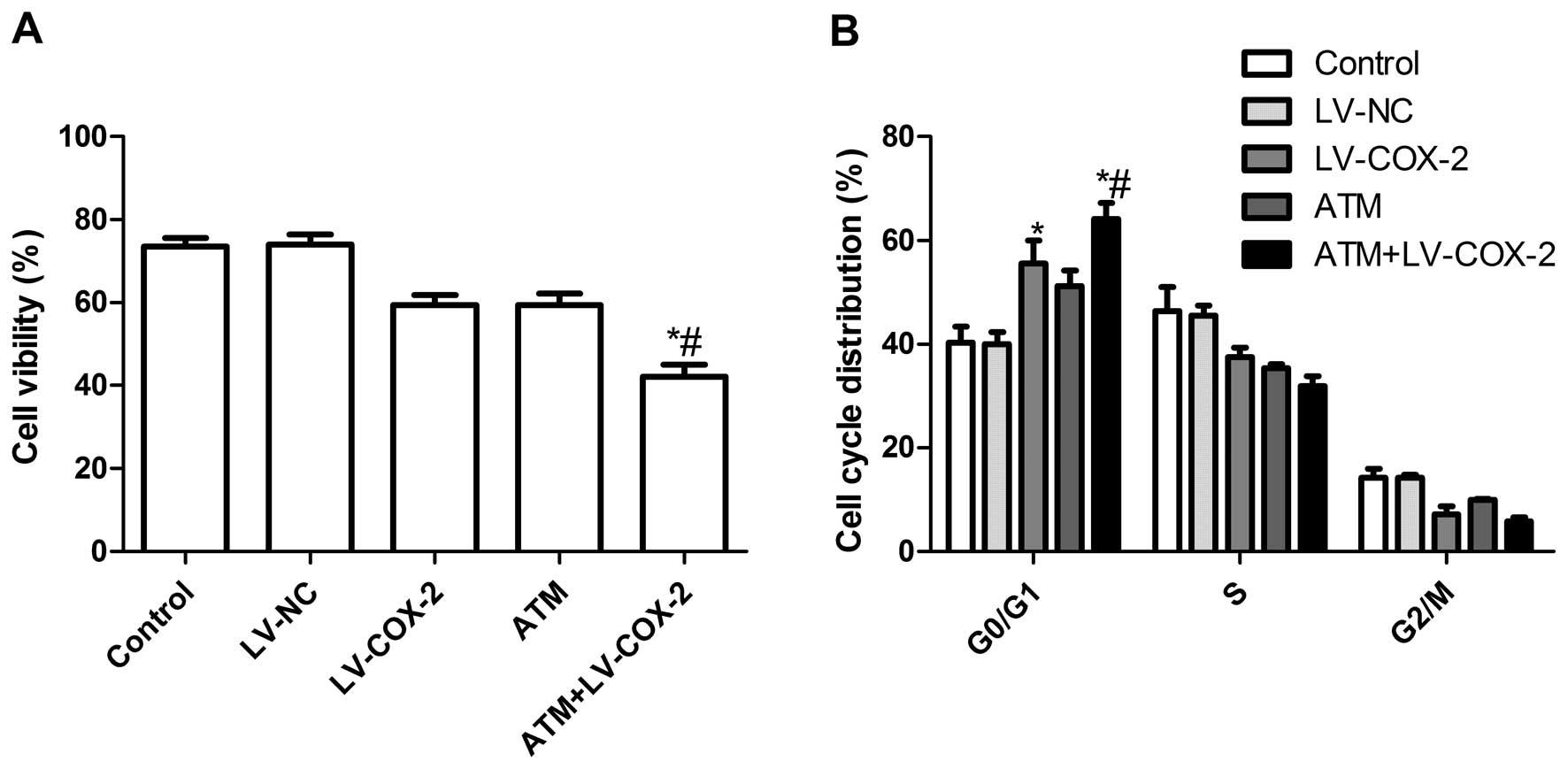
Effects of ATM and LV-COX-2 alone or
combination on MCF-7 cell proliferation and the cell cycle
To evaluate the effect of LV-COX-2 and ATM alone and
both on the cell viability of breast cancer cells in vitro,
MTT assay was performed for 48 h when MCF-7 were treated with
LV-COX-2 and ATM alone or combination. It was found that the
inhibitory rates of LV-COX-2 and ATM alone or combination treatment
were higher than control group and LV-NC treatment group
(P<0.01). There is no significance different between LV-NC group
and control group (P>0.05). In addition, the inhibitory rates of
LV-COX-2 in combination with ATM group were higher than LV-COX-2
group and ATM treatment group (Fig.
2A).
The effects of LV-COX-2 and ATM on the cell cycles
of MCF-7 cells were then analyzed by flow cytometry. MCF-7 cells
treated with LV-COX-2 or ATM had an increased percentage of arrest
at the G0/G1 phase compared with untreated cells and LV-NC control
(Fig. 2B). The LV-COX-2
combination with ATM resulted in an even greater percentage of
arrest at the G0/G1 phase than the higher doses of either drug
alone (P<0.01).
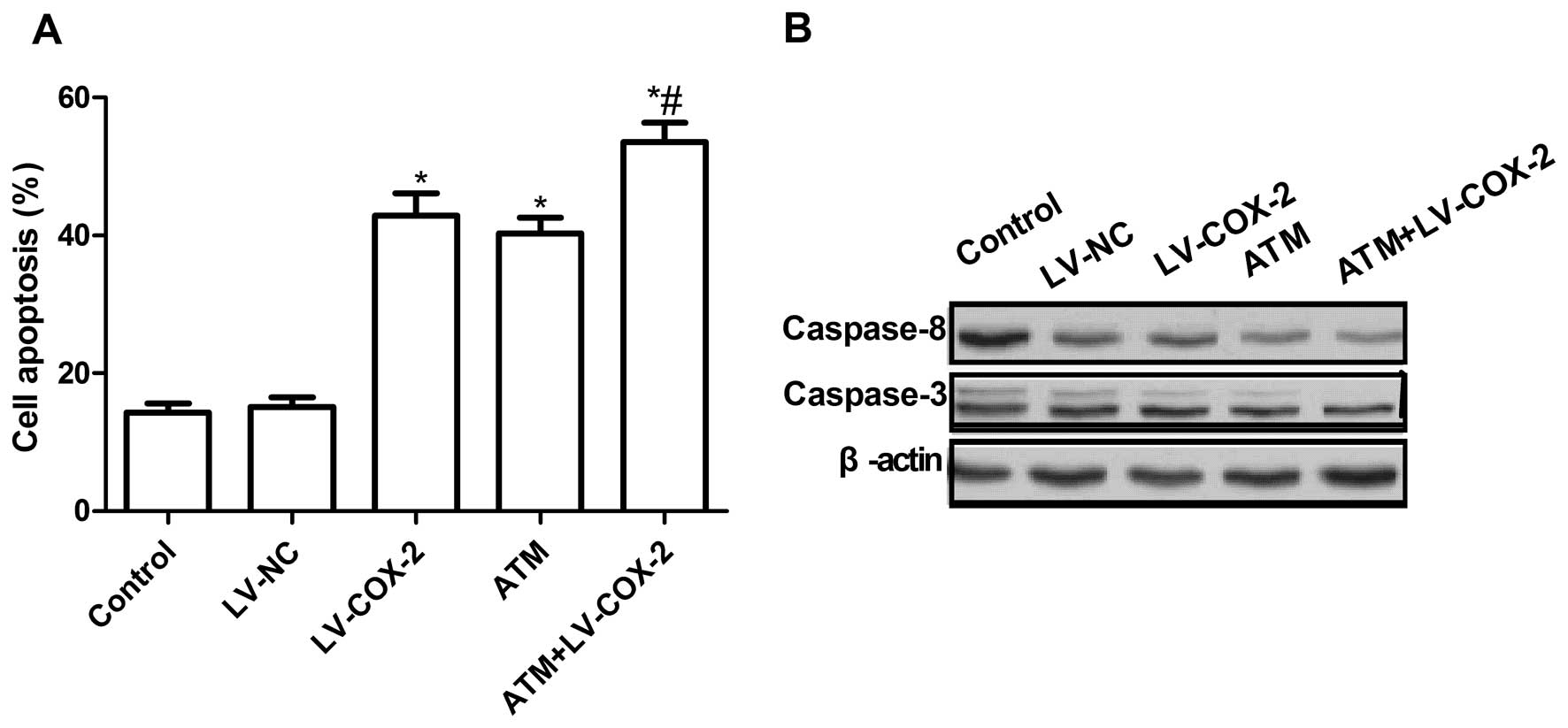
Effects of ATM and LV-COX-2 alone or
combination on MCF-7 cell apoptosis
To investigate whether the LV-COX-2 and ATM alone or
combination could induce of apoptosis, we analyzed apoptosis after
treatment with LV-COX-2 and ATM. It was found that MCF-7 cells
treated with LV-COX-2 or ATM could significantly induce cell
apoptosis compared with untreated cells and LV-NC control (Fig. 3A). Treatment with combination of
LV-COX-2 and ATM led to a dramatic increase in apoptotic cells
compare to single LV-COX-2 group and control group (P<0.01)
(Fig. 3A).
To explore the possible mechanism of induction of
cell apoptosis of combination with LV-COX-2 and ATM, expression
patterns of caspase-3 and -8 were determined by western blotting.
The results showed that combination with LV-COX-2 and ATM could
significantly decrease the expression of apoptosis inhibiting
genes, caspase-3 and -8, in MCF-7 cells, compared to LV-COX-2 or
ATM alone (P<0.01) (Fig.
3B).
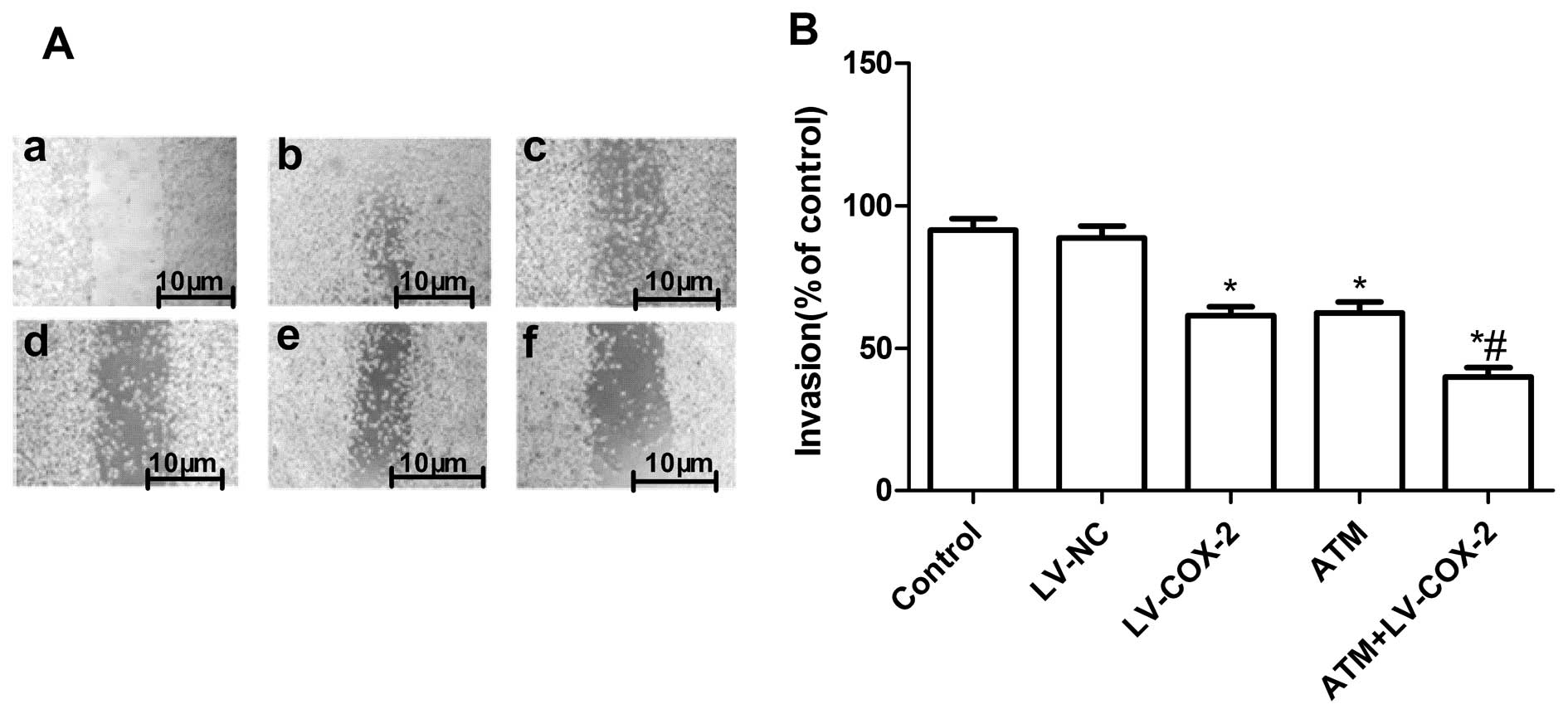
Effects of ATM and LV-COX-2 on MCF-7 cell
migration and cell invasion
To ascertain the inhibitory effect of TAM and
LV-COX-2 as a single or combined treatment on breast cancer
migration, wound-healing assay was performed to investigate their
effects on the migration potential of MCF-7 cells. After 48-h
treatment, cells in the control group and LV-NC group efficiently
spread into the wound area to such an extent that the wound
boundary was not apparent, while only some cells in TAM or LV-COX-2
treated group spread forward in MCF-7 cells. The cell migration in
combination group was less than either drug alone (Fig. 4A).
The ability of TAM and LV-COX-2 to reduce the
invasiveness of MCF7 cells was further investigated by the
transwell system assay. It was found that invasion was also
decreased significantly with TAM or LV-COX-2 treatment compared to
control and LV-NC (P<0.01) (Fig.
4B). Compared with the results with either agent alone, the
combination of TAM and LV-COX-2 greatly inhibited MCF-7 cell
invasion.
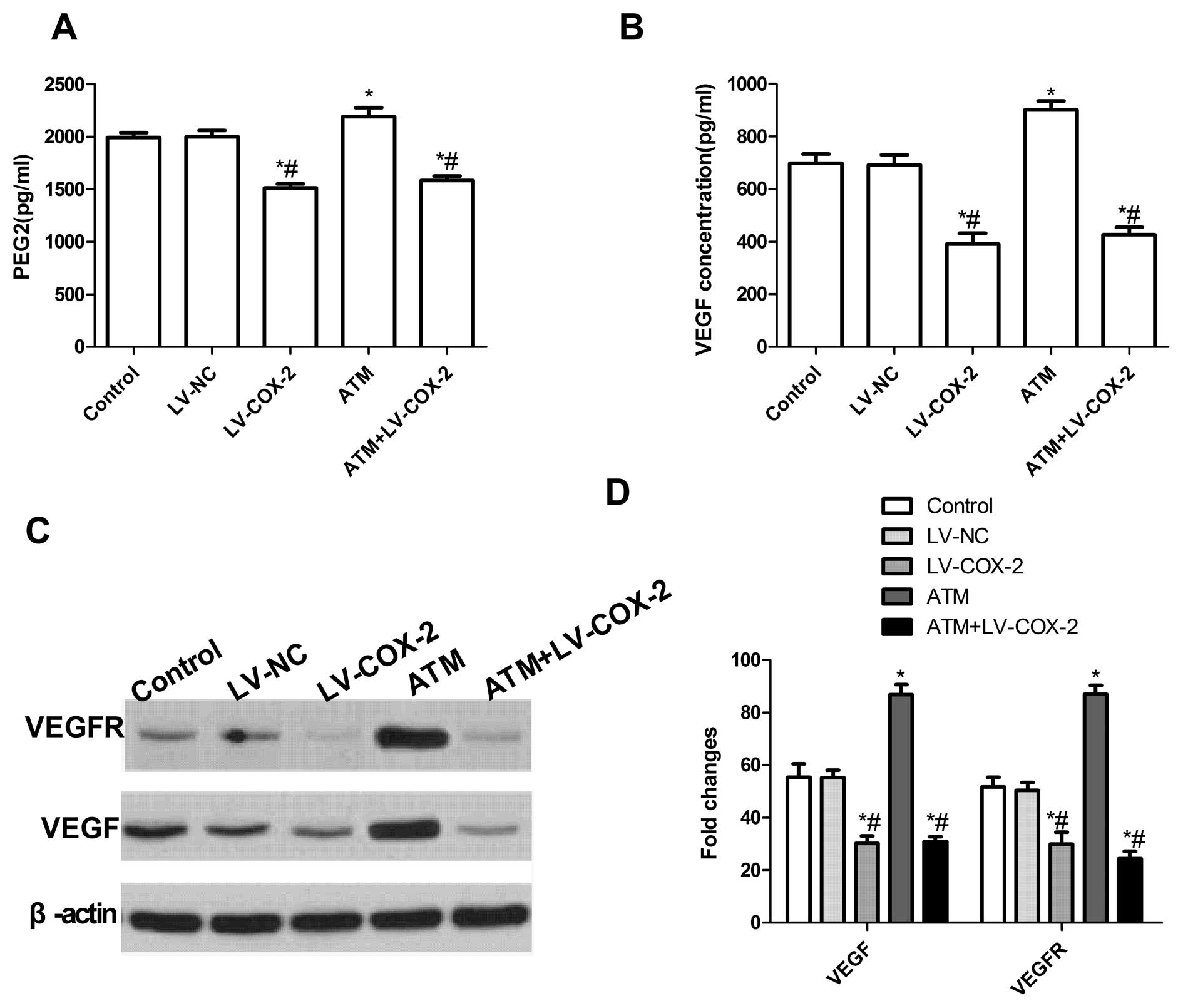
Effects of LV-COX-2 and ATM on PGE2
production and VEGF expression level in MCF-7 cells
To examine the effect of ATM and LV-COX-2 alone or
combination on PGE2 production in MCF-7 cells, ELISA was performed.
As shown in Fig. 5A, LV-COX-2
single or combination with ATM inhibited PGE2 production. However,
ATM increased PGE2 production compared to the other groups.
We also investigated the role of TAM and LV-COX-2 in
the inhibition of secretory VEGF, a pro-angiogenic factor
responsible for the migration and invasion of breast cancer cells.
VEGF secretion in serum-free culture conditioned medium was
assessed in MCF-7 cells by ELISA 24 h post-treatment. The results
showed that TAM alone considerably upregulated VEGF secretion and
the LV-COX-2 or LV-COX-2 combination TAM significantly decreased
VEGF secretion compared with no treatment and LV-NC treatment
(Fig. 5B).
To further study the possible mechanism of induction
VEGF expression with LV-COX-2 and ATM, VEGF and VEGFR expression
were determined by western blotting. The results showed that
combination with LV-COX-2 and ATM or single LV-COX-2 could
significantly decrease the expression of VEGF, VEGFR expression
compared to control group and LV-NC group (P<0.01) (Fig. 5C and D). On the other hand, ATM
dramatically increased the expression of VEGF, VEGFR expression
compared to control group and LV-NC group (P<0.01) (Fig. 5C and D). These results implied that
combination with LV-COX-2 and ATM could inhibit PGE2 production and
VEGF secretion by inhibition VEGF and VEGFR expression.
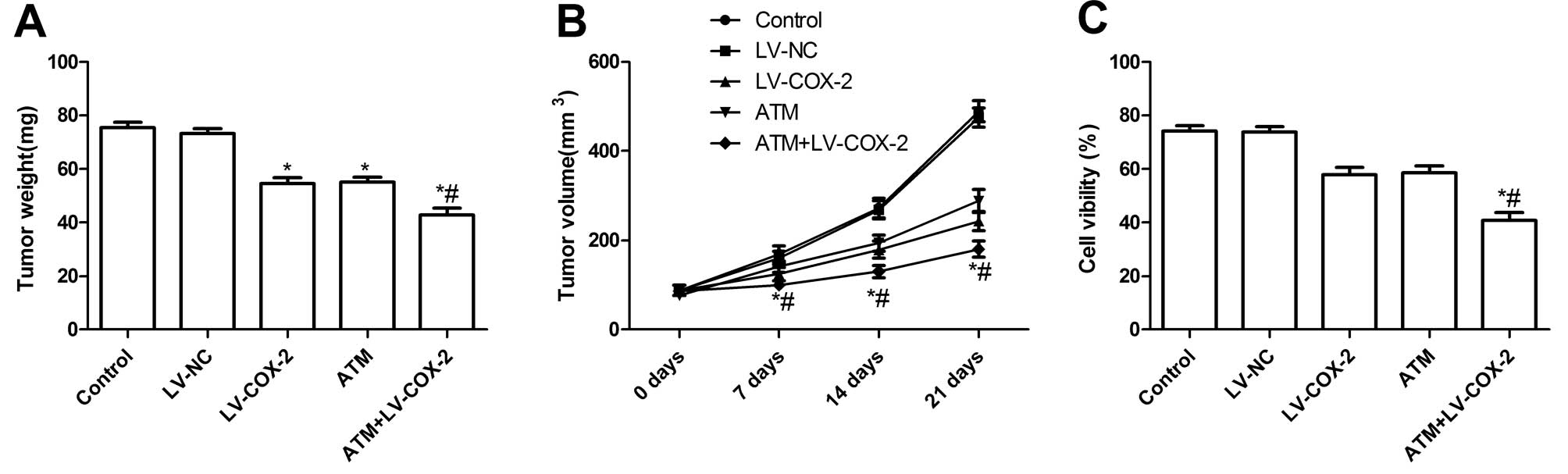
Antitumor activity of TAM and LV-COX-2 in
nude mice bearing MCF-7 tumors
We assessed the in vivo therapeutic efficacy
of TAM and LV-COX-2 in female BALB mice bearing MCF-7 breast tumors
cells. Mice were sacrificed and tumor tissue harvested 21 days
after treatment. Then tumor weight of the animals was measured.
Tumor weight of LV-COX-2 and ATM alone or combination group was
lower than those of control group and LV-NC group (Fig. 6A). Compared with the results with
either agent alone, the combination of TAM and LV-COX-2 greatly
inhibited tumor growth. In addition, we found that tumor volume
after treatment with LV-COX-2 and ATM was significantly slower for
MCF-7 tumor cells compared with control group and LV-NC group
(Fig. 6B). Treatment with
combination of LV-COX-2 and ATM led to a dramatic inhibition of
tumor growth compared to single LV-COX-2 group and control group
(P<0.01) (Fig. 6B). These
results indicate that LV-COX-2 combination with ATM treatment in
breast cancer cells markedly suppresses their tumorigenicity in
mice.
We also assess the efficacy of LV-COX-2 combination
with ATM in modulating splenocyte proliferation using MTT assay. As
shown in Fig. 6C, the inhibitory
rates of LV-COX-2 and ATM alone or combination significantly
increased compared to control group and LV-NC group (P<0.01).
The inhibitory rates of LV-COX-2 combination with ATM treatment
group were higher than single drug groups, which demonstrated that
combination treatment could inhibit MCF-7 cell proliferation.
Discussion
Estrogen can cause a general upregulation of genes
regulating cell proliferation and survival and the downregulation
of genes with anti-proliferative or pro-apoptotic activity
resulting in growth stimulation and apoptosis suppression (25). Therefore, anti-estrogens are able
to decrease cancer cell proliferation and induce cell death
signaling pathways (26).
Consequently, tamoxifen (TAM), a selective estrogen receptor (ER)
modulator, has been described as ‘the most important drug developed
in the history of breast cancer’ (27) and can induce cell cycle arrest
leads to an accumulation of cancer cells in G0/G1 phase of the cell
cycle (28) and induce apoptosis
of breast cancer cells (29).
However, a large number of initial clinical studies of TAM
displayed its anti-angiogenic and VEGF reducing ability in various
tumor models (30–33). In addition, an undesirable response
leading to enhanced metastasis and angiogenesis and resulting in
inferior outcomes, when prolonged administration of TAM causes
intracellular VEGF levels to rise in patients (34). In this context, we made an attempt
to decrease intracellular VEGF levels by decreasing ATM dose in
breast cancer cells. Furthermore, recently studies showed that
siRNA-mediated knockdown of COX-2 expression inhibited VEGF
expression (35). Therefore, in
this study, we knocked out COX-2 gene via a replication-incompetent
lentivirus (LV-COX-2), then LV-COX-2 combination with TAM for
reducing doses of ATM. The result showed that this novel
combination inhibited COX-2 expression, downregulated the level of
PGE2 and decreased the production of vascular endothelial growth
factor (VEGF) in tumors.
A recent study showed that the combination of TAM
and CXB at nontoxic levels exerts potent anti-angiogenic effects by
decrease in VEGF expression (16),
which was in agreement with our results. However, the celecoxib
analgesia also faces the gastrointestinal side effects (36) and tolerance as observed in a rat
model of inflammatory pain (37).
Furthermore, celecoxib could result in liver injury (17,18).
These side effects would limit their clinical applications. In this
study, we selected LV-COX-2 combiantion with ATM avoiding the side
effects induced by celecoxib.
Extensive studies showed that shRNA and transgene
carried by lentiviral vector can be transcribed into precursor mRNA
where shRNA is embedded in the 3′UTR or in the intron of transgene
(38–40). It has been shown that RNA
combination anticancer drug could achieve better antitumor activity
(21). Luni et al
demonstrated that when melanoma differentiation associated gene-7
(mda-7) via a replication-incompetent adenovirus (Ad.mda-7) and
gefitinib are used in combination they might provide an effective
therapeutic approach for NSCLC since these agents target multiple
cell survival pathways and are equally effective against NSCLC
cells (41). In the present study,
we used a novel strategy of combining a gene therapy approach,
LV-COX-2, with ATM, and evaluated its antitumor effects against
breast cancer cells. The result showed that LV-COX-2 combination
with TAM in breast cancer cells significantly suppressed the
proliferation and metastasis, and induced tumor apoptosis in
vitro, and tumor growth also was suppressed in vivo.
In conclusion, our in vitro studies
demonstrate that when LV-COX-2 and ATM are used in combination they
might provide an effective therapeutic approach for breast cancer
since these agents target induction breast cell apoptosis and are
equally effective against breast cancer cells. Additionally,
further studies with in vivo tumor models also confirmed
that LV-COX-2 combination with ATM could suppress breast tumor
growth. Therefore, it may be worthwhile to consider the combination
treatment as novel therapeutic strategy for further evaluation in
clinical trials.
Acknowledgements
This study was supported by Science
and Technology Research and Innovation Team Fund of Jilin province
(JL2011038).
References
|
1.
|
Jordan VC and O’Malley BW: Selective
estrogen-receptor modulators and antihormonal resistance in breast
cancer. J Clin Oncol. 25:5815–5824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Brauch H and Jordan VC: Targeting of
tamoxifen to enhance antitumour action for the treatment and
prevention of breast cancer: the ‘personalised’ approach? Eur J
Cancer. 45:2274–2283. 2009.PubMed/NCBI
|
|
3.
|
Cuzick J, Sestak I, Pinder SE, et al:
Effect of tamoxifen and radiotherapy in women with locally excised
ductal carcinoma in situ: long-term results from the UK/ANZ DCIS
trial. Lancet Oncol. 12:21–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Roberts CG, Millar EK, O’Toole SA, et al:
Identification of PUMA as an estrogen target gene that mediates the
apoptotic response to tamoxifen in human breast cancer cells and
predicts patient outcome and tamoxifen responsiveness in breast
cancer. Oncogene. 30:3186–3197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Garvin S, Nilsson UW and Dabrosin C:
Effects of oestradiol and tamoxifen on VEGF, soluble VEGFR-1, and
VEGFR-2 in breast cancer and endothelial cells. Br J Cancer.
93:1005–1010. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Qu Z, Van Ginkel S, Roy AM, et al:
Vascular endothelial growth factor reduces tamoxifen efficacy and
promotes metastatic colonization and desmoplasia in breast tumors.
Cancer Res. 68:6232–6240. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Majeti BK, Lee JH, Simmons BH and Shojaei
F: VEGF is an important mediator of tumor angiogenesis in malignant
lesions in a genetically engineered mouse model of lung
adenocarcinoma. BMC Cancer. 13:2132013. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Chen CT and Hung MC: Beyond anti-VEGF:
dual-targeting antiangiogenic and antiproliferative therapy. Am J
Transl Res. 5:393–403. 2013.PubMed/NCBI
|
|
9.
|
Breinig M, Schirmacher P and Kern MA:
Cyclooxygenase-2 (COX-2) - a therapeutic target in liver cancer?
Curr Pharm Des. 13:3305–3315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Stasinopoulos I, O’Brien DR, Wildes F,
Glunde K and Bhujwalla ZM: Silencing of cyclooxygenase-2 inhibits
metastasis and delays tumor onset of poorly differentiated
metastatic breast cancer cells. Mol Cancer Res. 5:435–442. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
de Heer P, Sandel MH, Guertens G, et al:
Celecoxib inhibits growth of tumors in a syngeneic rat liver
metastases model for colorectal cancer. Cancer Chemother Pharmacol.
62:811–819. 2008.PubMed/NCBI
|
|
12.
|
Narayanan BA, Narayanan NK, Davis L and
Nargi D: RNA interference-mediated cyclooxygenase-2 inhibition
prevents prostate cancer cell growth and induces differentiation:
modulation of neuronal protein synaptophysin, cyclin D1, and
androgen receptor. Mol Cancer Ther. 5:1117–1125. 2006. View Article : Google Scholar
|
|
13.
|
Singh-Ranger G, Salhab M and Mokbel K: The
role of cyclooxygenase-2 in breast cancer: review. Breast Cancer
Res Treat. 109:189–198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angiogenesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Hasegawa K, Ichikawa W, Fujita T, et al:
Expression of cyclooxygenase-2 (COX-2) mRNA in human colorectal
adenomas. Eur J Cancer. 37:1469–1474. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kumar BN, Rajput S, Dey KK, et al:
Celecoxib alleviates tamoxifen-instigated angiogenic effects by
ROS-dependent VEGF/VEGFR2 autocrine signaling. BMC Cancer.
13:2732013. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lacroix I, Lapeyre-Mestre M, Bagheri H, et
al: Nonsteroidal anti-inflammatory drug-induced liver injury: a
case-control study in primary care. Fundam Clin Pharmacol.
18:201–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rudnick DA, Shikapwashya O, Blomenkamp K
and Teckman JH: Indomethacin increases liver damage in a murine
model of liver injury from alpha-1-antitrypsin deficiency.
Hepatology. 44:976–982. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Park JW, Park JE, Lee JA, Lee CW and Kim
CM: Cyclooxygenase-2 (COX-2) is directly involved but not decisive
in proliferation of human hepatocellular carcinoma cells. J Cancer
Res Clin Oncol. 132:184–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Wang R, Wang X, Lin F, Gao P, Dong K and
Zhang HZ: shRNA-targeted cyclooxygenase (COX)-2 inhibits
proliferation, reduces invasion and enhances chemosensitivity in
laryngeal carcinoma cells. Mol Cell Biochem. 317:179–188. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lin T, Gu J, Zhang L, et al: Enhancing
adenovirus-mediated gene transfer in vitro and in vivo by addition
of protamine and hydrocortisone. J Gene Med. 5:868–875. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Coleman JE, Huentelman MJ, Kasparov S, et
al: Efficient large-scale production and concentration of
HIV-1-based lentiviral vectors for use in vivo. Physiol Genomics.
12:221–228. 2003.PubMed/NCBI
|
|
23.
|
Deglon N, Tseng JL, Bensadoun JC, et al:
Self-inactivating lentiviral vectors with enhanced transgene
expression as potential gene transfer system in Parkinson’s
disease. Hum Gene Ther. 11:179–190. 2000.PubMed/NCBI
|
|
24.
|
Tai MH, Weng CH, Mon DP, Hu CY and Wu MH:
Ultraviolet C irradiation induces different expression of
cyclooxygenase 2 in NIH 3T3 cells and A431 cells: the roles of
COX-2 are different in various cell lines. Int J Mol Sci.
13:4351–4366. 2012. View Article : Google Scholar
|
|
25.
|
Frasor J, Danes JM, Komm B, Chang KC,
Lyttle CR and Katzenellenbogen BS: Profiling of estrogen up- and
down- regulated gene expression in human breast cancer cells:
insights into gene networks and pathways underlying estrogenic
control of proliferation and cell phenotype. Endocrinology.
144:4562–4574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Renoir JM, Bouclier C, Seguin A, Marsaud V
and Sola B: Antioestrogen-mediated cell cycle arrest and apoptosis
induction in breast cancer and multiple myeloma cells. J Mol
Endocrinol. 40:101–112. 2008. View Article : Google Scholar
|
|
27.
|
Zheng J and Yao Z: Effect of tamoxifen on
apoptosis and drug resistance of breast cancer cells in vitro.
Zhonghua Zhong Liu Za Zhi. 22:55–57. 2000.In Chinese.
|
|
28.
|
Thiantanawat A, Long BJ and Brodie AM:
Signaling pathways of apoptosis activated by aromatase inhibitors
and antiestrogens. Cancer Res. 63:8037–8050. 2003.
|
|
29.
|
Rajput S, Kumar BN, Sarkar S, et al:
Targeted apoptotic effects of thymoquinone and tamoxifen on XIAP
mediated Akt regulation in breast cancer. PLoS One. 8:e613422013.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Blackwell KL, Haroon ZA, Shan S, et al:
Tamoxifen inhibits angiogenesis in estrogen receptor-negative
animal models. Clin Cancer Res. 6:4359–4364. 2000.PubMed/NCBI
|
|
31.
|
Marson LP, Kurian KM, Miller WR and Dixon
JM: The effect of tamoxifen on breast tumour vascularity. Breast
Cancer Res Treat. 66:9–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Garvin S and Dabrosin C: Tamoxifen
inhibits secretion of vascular endothelial growth factor in breast
cancer in vivo. Cancer Res. 63:8742–8748. 2003.
|
|
33.
|
McNamara DA, Harmey J, Wang JH, Kay E,
Walsh TN and Bouchier-Hayes DJ: Tamoxifen inhibits endothelial cell
proliferation and attenuates VEGF-mediated angiogenesis and
migration in vivo. Eur J Surg Oncol. 27:714–718. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ruohola JK, Valve EM, Karkkainen MJ,
Joukov V, Alitalo K and Harkonen PL: Vascular endothelial growth
factors are differentially regulated by steroid hormones and
antiestrogens in breast cancer cells. Mol Cell Endocrinol.
149:29–40. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Liu H, Xiao J, Yang Y, et al: COX-2
expression is correlated with VEGF-C, lymphangiogenesis and lymph
node metastasis in human cervical cancer. Microvasc Res.
82:131–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Mallen SR, Essex MN and Zhang R:
Gastrointestinal tolerability of NSAIDs in elderly patients: a
pooled analysis of 21 randomized clinical trials with celecoxib and
nonselective NSAIDs. Curr Med Res Opin. 27:1359–1366. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Rezende RM, Paiva-Lima P, Dos Reis WG,
Camelo VM, Bakhle YS and de Francischi JN: Celecoxib induces
tolerance in a model of peripheral inflammatory pain in rats.
Neuropharmacology. 59:551–557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Du G, Yonekubo J, Zeng Y, Osisami M and
Frohman MA: Design of expression vectors for RNA interference based
on miRNAs and RNA splicing. FEBS J. 273:5421–5427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Stegmeier F, Hu G, Rickles RJ, Hannon GJ
and Elledge SJ: A lentiviral microRNA-based system for single-copy
polymerase II-regulated RNA interference in mammalian cells. Proc
Natl Acad Sci USA. 102:13212–13217. 2005. View Article : Google Scholar
|
|
40.
|
Samakoglu S, Lisowski L, Budak-Alpdogan T,
et al: A genetic strategy to treat sickle cell anemia by
coregulating globin transgene expression and RNA interference. Nat
Biotechnol. 24:89–94. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Emdad L, Lebedeva IV, Su ZZ, et al:
Combinatorial treatment of non-small-cell lung cancers with
gefitinib and Ad.mda-7 enhances apoptosis-induction and reverses
resistance to a single therapy. J Cell Physiol. 210:549–559. 2007.
View Article : Google Scholar : PubMed/NCBI
|