Introduction
Chronic myeloid leukemia (CML) is a hematological
malignancy characterized by the presence of the Philadelphia
chromosome (1). This chimeric
chromosome generates the fusion gene BCR-ABL, produces fusion
proteins and leads to constitutive activation of the tyrosine
kinase (TK). Deregulated TK activity results in activation of
several downstream signaling pathways, including PI3K/AKT, Ras/MAPK
and STAT pathways, which are implicated in mitogenic signaling and
cell survival (2,3).
Treatment of CML was revolutionized by the advent of
imatinib mesylate (IM), a targeted agent which inhibits the
activity of BCR-ABL (4). IM has
dramatically improved the prognosis of CML patients in chronic
phase. However, patients in advanced stage (blast crisis) manifest
drug resistance against IM, leading to relapse, due to
amplification of BCR-ABL and the acquisition of BCR-ABL-independent
mechanisms (5).
Autophagy is an important intracellular catabolic
pathway. Autophagy helps cells to maintain cellular homeostasis. It
is associated with pro-survival functions in cancer cells during
cytotoxic insults such as starvation, hypoxia and chemo-therapy
(6). Recent studies suggest that
targeting autophagy may present a novel strategy for modulating IM
resistance in CML cells including CML stem cells (7). However, the mechanism of
autophagy-mediated IM-resistance remains largely unclear and
strategies for targeting autophagy remain challenging.
Studies (7–9) have shown that IM induced autophagy
and autophagy inhibitors boosted the therapeutic efficacy in CML
cell lines such as 32D-P210 and K562. However, lack of effective
small molecular inhibitors has constrained further research and
clinical applications. We therefore identified a novel autophagy
inhibitor named specific and potent autophagy inhibitor-1
(spautin-1), which inhibits starvation-induced autophagy in solid
tumor cells (10). Whether
spautin-1 affects IM-induced autophagy and cytotoxicity in CML
cells is unknown. Therefore, we sought to investigate the
anti-leukemic activity of spautin-1 in CML and the underlying
molecular mechanisms.
We report that spautin-1 increased the cytotoxicity
of IM in both K562 cell line and primary cells. Co-treatment of
spautin-1 and IM enhanced cell apoptosis by inhibiting IM-induced
autophagy as well as inactivating PI3K/AKT and activating
downstream GSK3β, which downregulates the expression of
anti-apoptotic proteins Mcl-1 and Bcl-2. Taken together, our
findings suggest that when combined with spautin-1, the efficacy of
IM was greatly enhanced. The combination strategy may be an
interesting option to treat CML cases in the future.
Materials and methods
Reagents
IM was purchased from Selleckchem, USA. Spautin-1
was kindly provided by Shanghai Institute of Organic Chemistry,
China Academy of Science. Both were dissolved in DMSO
(Sigma-Aldrich) and stored in the dark at −20°C. LC3B (anti-rabbit,
1:2,000) was purchased from Sigma-Aldrich. Beclin-1 (anti-rabbit,
1:2,000), Bcl-2 (anti-rabbit, 1:1,000), Mcl-1 (anti-rabbit,
1:1,000) and Bim (anti-rabbit, 1:1,000) were obtained from Abcam.
PARP (anti-rabbit, 1:1,000), caspase-3 (anti-rabbit, 1:1,000), AKT
(anti-rabbit, 1:1,000), p-AKTser473 (anti-rabbit,
1:1,000), GSK3β (anti-rabbit, 1:1,000), p-GSK3βser9
(anti-rabbit, 1:1,000) and tubulin (anti-rabbit, 1:2,000) were
derived from Cell Signaling Technology. ULK1 (anti-rabbit,
1:1,000), ATG5 (anti-rabbit, 1:1,000) and ATG7 (anti-rabbit,
1:1,000) were sourced from Epitomics.
Cell culture and patients
K562 cells were generously provided by Shanghai
Institute of Hematology and maintained in RPMI-1640 medium (Gibco,
Life Technologies, USA) with 10% heat-inactivated fetal bovine
serum (FBS) (Gibco, Life Technologies) and 1%
penicillin-streptomycin (Gibco, Life Technologies) at 37°C in a
humidified atmosphere containing 5% CO2.
The diagnosis of CML was based on clinical features,
hematological characteristics and the presence of the Ph
chromosome, as described previously (11). The trial was approved by
Institutional Review Boards and Ethics Committees. Patients signed
written informed consent, in accordance with the Declaration of
Helsinki. Bone marrow mononuclear cells from CML patients were
isolated by Ficoll density centrifugation. Briefly, after bone
marrow aspiration, citrate-anticoagulated blood was mixed with
Ficoll reagent (Cedarlane) in PBS (1:1). The leukocyte-rich plasma
was subjected to density gradient centrifugation (2,000 rpm, 30
min). The cell pellet containing neutrophils was recovered and the
contaminating erythrocytes were removed by hypotonic lysis with
H2O. Cells were washed twice with PBS and kept in
RPMI-1640 with 10% FBS. Cell viability, as determined by trypan
blue exclusion, was consistently >95%.
Cell proliferation assays by Cell
Counting Kit-8 (CCK-8)
Cell proliferation was evaluated using CCK-8
(Beyotime). Cells (1×105/ml) were seeded into 96-well
plates in triplicate and then treated with 125 to 4,000 nM IM alone
or in combination with spautin-1 (10 μM). After 48 h of
incubation, 10 μl of CCK-8 reagent was added to each well.
Four hours later, the absorbance was read at 450 nm using a
microplate reader (Bio-Rad). The background absorbance was measured
in wells containing only dye solution and culture medium. Data
presented are the values subtracting the background absorbance
values from the total absorbance values. The mean of the
triplicates were calculated.
Fluorescence microscopy
Apoptotic morphology was studied by staining the
cells with Hoechst 33258 (KeyGen Biotech) fluorescent stain. Cells
(1×105/ml) were seeded into a 12-well plate with
indicated concentration of IM (500 nM) for 12 h. Then spautin-1 (10
μM) or DMSO was added to K562 medium for further 36 h. After
incubation, cells were stained with 20 mg/ml of Hoechst 33258 for
10 min and observed under a fluorescence microscope (Olympus).
Flow cytometry
The percentage of apoptotic cells was analyzed by
flow cytometry using Annexin V-FITC Apoptosis Detection assay kit I
(BD Pharmingen, Lot 25015). Cells (1×105/ml) were washed
with ice-cold PBS and resuspended in cold Annexin V-binding buffer
containing Annexin V-FITC and propidium iodide (PI). The samples
were incubated at room temperature in the dark for 10 min and the
total volume was adjusted to 500 μl with Annexin V-binding
buffer. The number of stained cells was assessed by flow cytometer
(BD FACScan). Early apoptotic cells were defined as positive for
Annexin V-FITC but negative for PI staining and late apoptotic
cells were positive for both Annexin V-FITC and PI staining.
Cell cycle analysis was performed by fixing cells in
70% ethanol for 12 h at 4°C, followed by incubation with 1 mg/ml
RNase A for 30 min at 37°C. Subsequently, cells were stained with
PI (50 μg/ml) (Becton-Dickinson, San Jose, CA, USA) in
phosphate-buffered saline (PBS), 0.5% Tween-20, and analyzed using
a flow cytometer (BD FACScan).
Western blot analysis
Briefly, K562 cells (1×105/ml) were
cultured in 6-well plates, harvested at specific intervals and
lysed in lysis buffer (Beyotime). Protein concentration was
measured by the BCA protein assay (Beyotime). Equal amounts of
protein were resolved on 8 or 12% SDS-PAGE gel and transferred to a
PVDF membrane. After blocking with phosphate-buffered saline (PBS)
containing 5% non-fat milk and 0.1% Tween-20 for 2 h, membranes
were incubated with primary antibodies at 4°C overnight and
secondary antibodies for 2 h at room temperature. Protein-antibody
complexes were detected by an enhanced chemiluminescence
immunoblotting ECL (Beyotime). Immunoblots were quantified using
ImageJ2x, and the levels of protein were normalized to tubulin
levels.
Statistical analysis
All assays were performed in triplicate and data
expressed as mean values ± SD. Statistical significance of
differences between groups was determined using Student’s t-test.
Probability value ≤0.05 was considered significant and marked by
asterisks in the figures. All statistical analysis was conducted
using SPSS 13.0.
Results
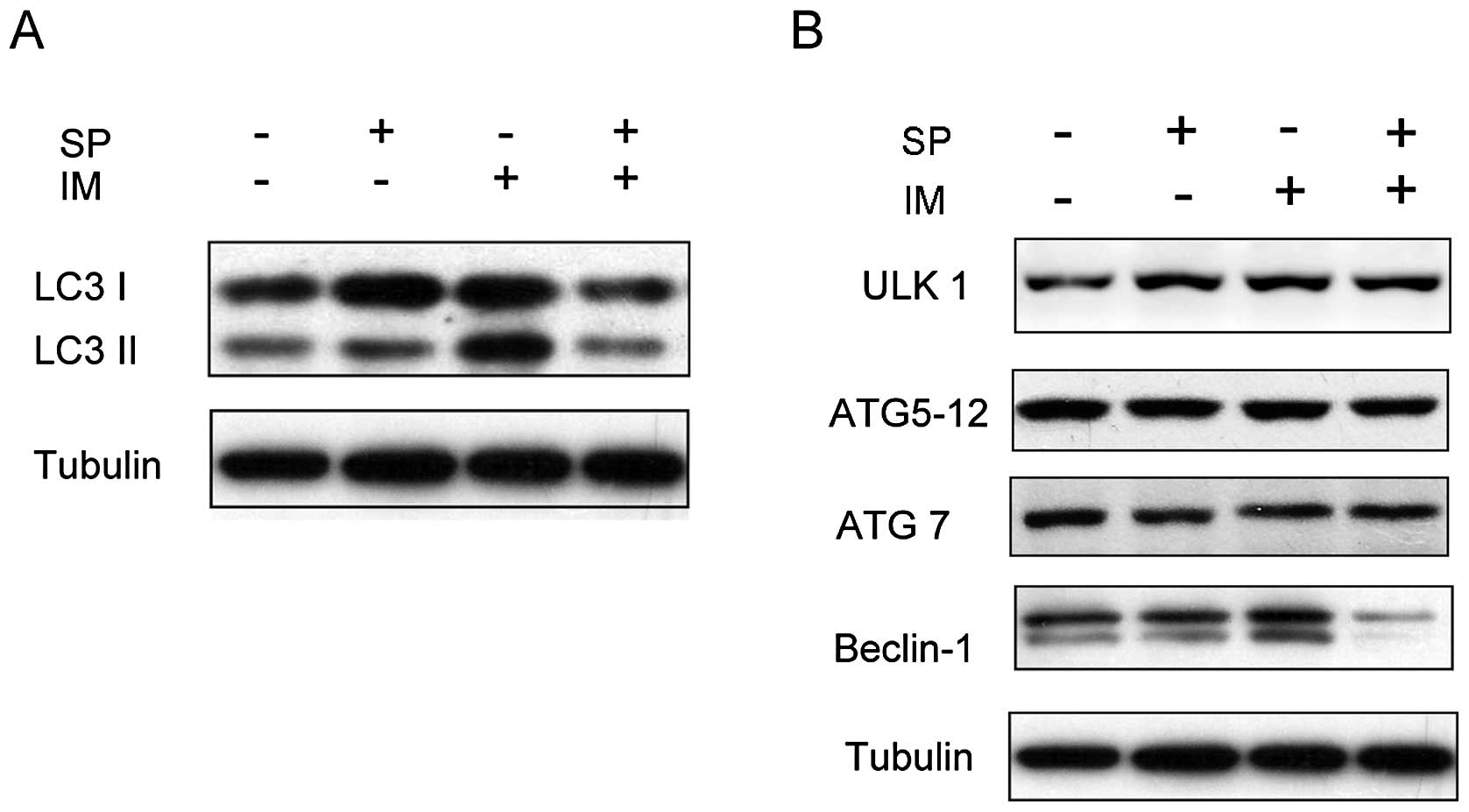
Spautin-1 inhibits IM-induced autophagy
in K562 cells
Previous studies reported autophagy underlying IM
resistance in CML cells (8,12,13).
To investigate whether spautin-1 affected IM-induced autophagy, we
determined microtubule-associated protein light chain 3 (LC3)
conversions (LC3-I to LC3-II) by immunoblot analysis. LC3 is now
widely used to monitor autophagy. The amount of LC3-II correlates
with the number of autophagosomes. Autophagy was activated in K562
cells after treatment with 0.5 μM IM for 48 h, which was
consistent with previous reports (9). Spautin-1 showed strong inhibition of
IM-induced autophagy in K562 cells (Fig. 1A and C). We also observed the
expression of a few autophagy-related factors, such as Beclin-1,
ULK1, ATG5-12 complex and ATG7 in K562 cells. The results showed
that spautin-1 markedly attenuated the protein levels of Beclin-1
induced by IM, but had no significant effect on other autophagy
factors (Fig. 1B and C). These
findings suggested that spautin-1 inhibited IM-induced autophagy in
a Beclin-1-dependent manner.
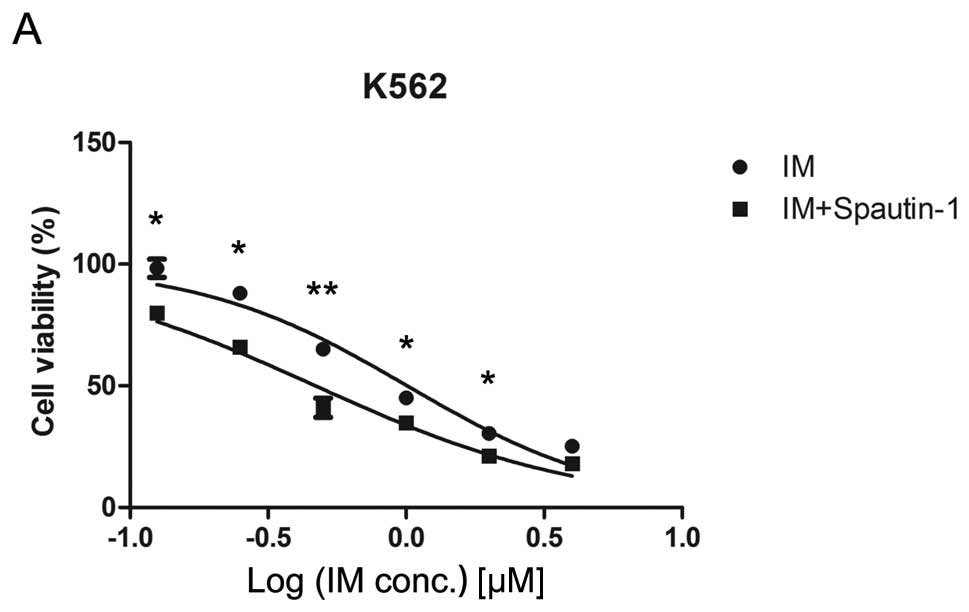
Spautin-1 enhances IM-induced
cytotoxicity in K562 cells
Several studies demonstrated that IM-induced
autophagy was a protective response preventing CML cells from
therapy-induced cell death (7,14).
The observed effect of spautin-1 in autophagy suggested that it
rendered CML cells increasingly susceptible to IM. To verify this
idea, we compared the growth inhibitory effect of IM on K562 cells
before and after inhibition of autophagy by spautin-1. K562 cells
were treated with varying concentrations of IM in the presence or
absence of 10 μM spautin-1 for 48 h. It was followed by CCK8
assay and morphological examination of the treated cells. As shown
in Fig. 2A, IM inhibited the
growth of K562 cells with 50% inhibition (IC50) of 1.03
μM at 48 h. In contrast, co-treatment with spautin-1
increased IM-induced inhibition of cell viability with
IC50 of 0.45 μM. Spautin-1 alone showed no
significant impact on cell viability after 48 h of incubation
(Fig. 2B). Microscopic evidence
indicated that IM combined with spautin-1 significantly boosted
IM-induced cell death (Fig. 2C).
These results were confirmed by fluorescent microscopy of Hoechst
33258 staining (Fig. 2D and E).
The data suggested that autophagy inhibitor spautin-1 enhanced
IM-induced cytotoxicity in K562 cells.
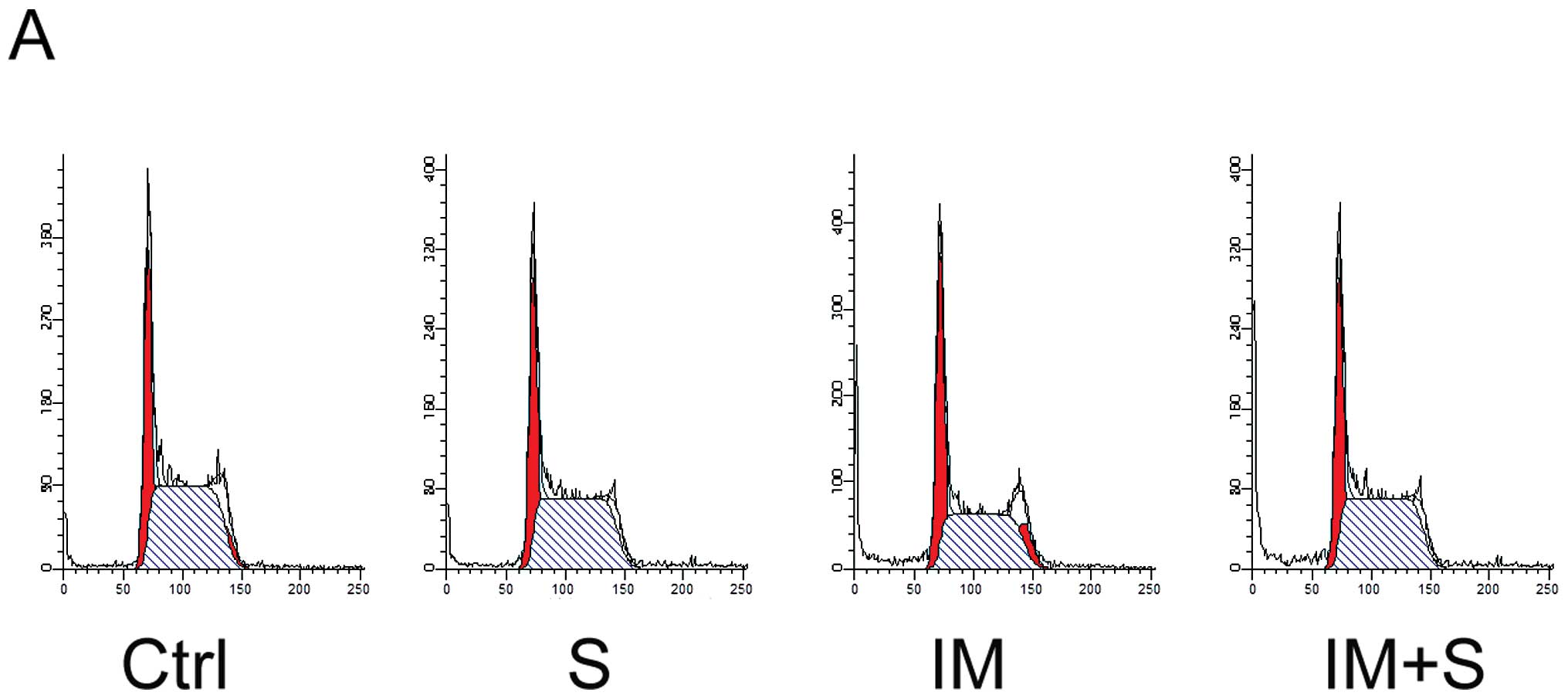
Spautin-1 enhances IM-induced apoptosis
in K562 cells
We further investigated whether spautin-1 affected
cell cycle or cell death in IM-treated K562 cells. Cell cycle and
apoptosis were analyzed by flow cytometry after staining with PI
and PI/Annexin V, respectively. No sign of obvious cell cycle
arrest was found in any group (Fig.
3A). However, after treated with 250 or 500 nM IM for 48 h,
with or without spautin-1, co-treatment with spautin-1 enhanced
IM-induced apoptosis from 8.4 and 23.9% to 15.5 and 36.4%,
respectively (Fig. 3B and C).
Additionally, spautin-1 alone showed no significant impact on
apoptosis. We also found that spautin-1 increased IM-induced
caspase-3 cleavage, which was an indicator of cell apoptosis.
Consistently, the level of cleaved product of caspase substrate
poly(ADP-ribose) polymerase (PARP) correlated with the activation
of caspase (Fig. 3D and E). To
investigate the mechanism of spautin-1 in promoting IM-induced
apoptosis of K562 cells, we focused on Bcl-2 family proteins, which
played a key role in cell apoptosis. Compared with IM alone,
co-treatment of spautin-1 and IM remarkably downregulated
anti-apoptotic proteins including Bcl-2 and myeloid cell leukemia-1
(Mcl-1) (Fig. 3F). These data
collectively illustrated that spautin-1 promoted IM-induced CML
cell apoptosis by reducing the expression of anti-apoptotic
proteins Bcl-2 and Mcl-1.
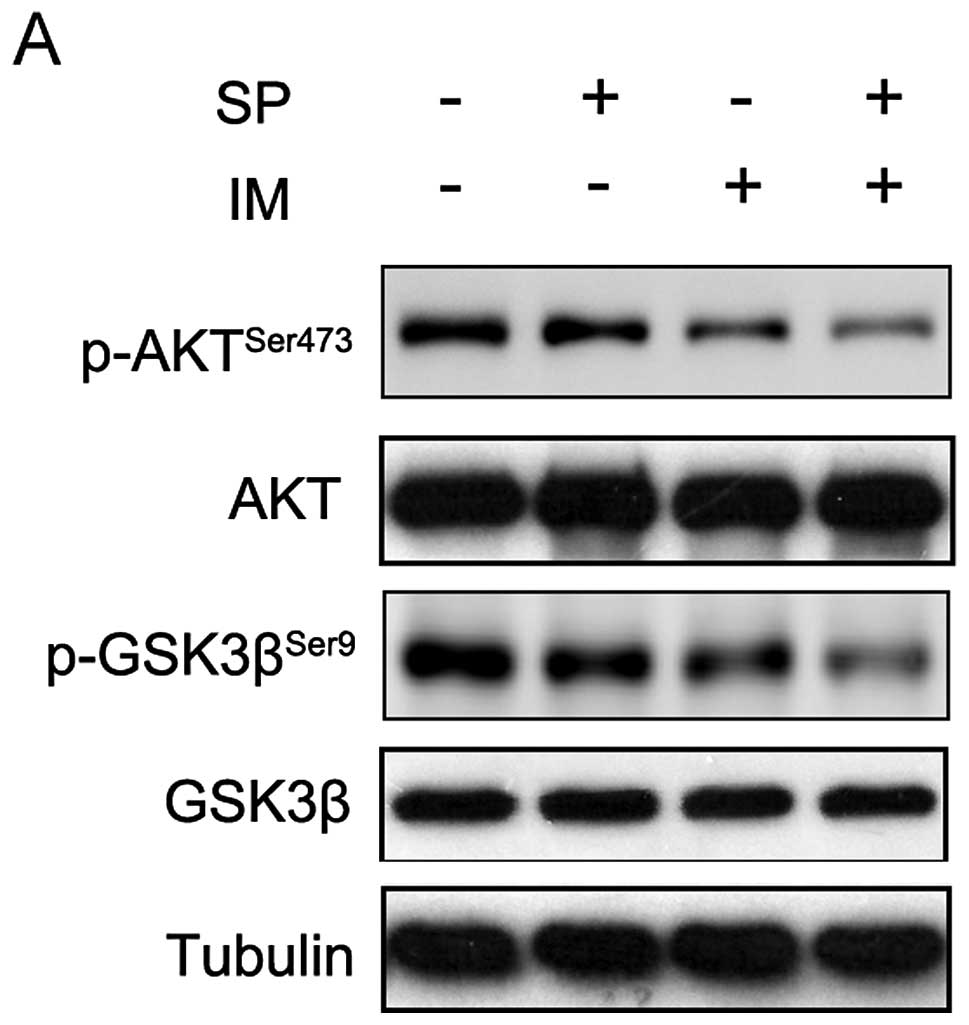
AKT/GSK3β is involved in spautin-1
pro-apoptotic activity in CML cells
We determined the mechanism underlying regulation of
IM-induced CML cell apoptosis by spautin-1. Accumulating evidence
supports that PI3K/AKT/mTOR signaling pathway was involved in
autophagy and apoptosis (15–17).
We investigated whether this pathway functioned in inhibiting
autophagy and promoting the apoptosis by spautin-1 in IM-treated
K562 cells. As shown in Fig. 4A,
the combination of IM and spautin-1 sharply suppressed
AKTser473 phosphorylation compared with IM alone. The
total AKT level was unchanged. AKT comprises many kinase substrates
with diverse functionalities. We detected some identified ATK
substrates, such as p27, FOXO and GSK3β. Interestingly,
GSK3βser9 phosphorylation was also markedly reduced in
IM/spautin-1 co-treated K562 cells compared with cells treated with
IM alone (Fig. 4). In each group,
total GSK3β was unaffected. Phosphorylation at Ser9 is a major
mechanism for inhibiting GSK3β enzymatic activity (18). These results indicated that the
suppression of AKT and activating downstream GSK3β were involved in
autophagy inhibition and promotion of apoptosis in spautin-1/IM
co-treated CML cells.
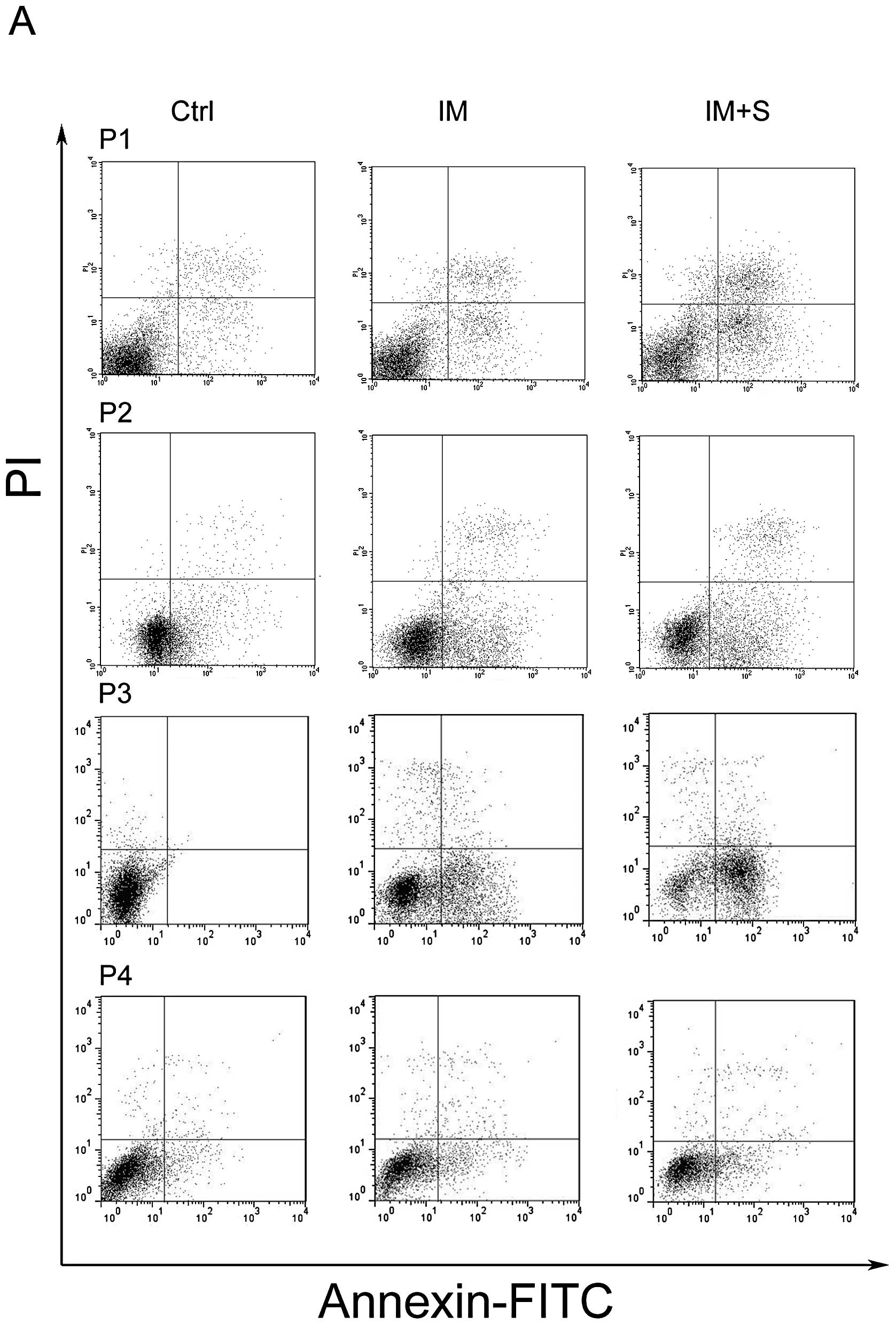
Spautin-1 potentiates the efficacy of IM
in primary CML cells
The aforementioned results indicated that spautin-1
enhanced the cytotoxic sensitivity of IM in CML cell line K562. We
therefore, wondered whether spautin-1 affected primary CML cells
similarly. Isolated primary cells from four CML patients were used
to assess the pro-apoptotic activity of spautin-1. Cell apoptosis
was analyzed by flow cytometry with PI/Annexin V staining.
Consistently with findings in K562 cells, combination of IM and
spautin-1 significantly promoted cell death in primary cells. The
mean apoptotic rate was 40.2±11.4% and 24.8±6.9% in the combination
group and IM alone group, respectively (P<0.05) (Fig. 5). The above data indicated that
spautin-1 enhanced IM-induced cell death both in K562 and primary
CML cells.
Discussion
IM is associated with a substantial therapeutic
effect in CML and has been endorsed as the first line therapy in
CML by international guidelines (19). However, CML cells may respond to IM
in a variety of ways ranging from initiation of cell death to the
activation of survival pathways such as autophagy (7,20,21).
In our study, the novel autophagy inhibitor, spautin-1, exhibited
significant efficacy in enhancing IM-induced cytotoxicity in both
K562 and CML cells.
Several studies have shown that the blockage of
autophagy by pharmacologic inhibitors or genetic knockdown of
critical autophagy-related genes enhanced the cytotoxicity of IM on
CML cells (12). Considering the
crucial role played by autophagy in the treatment of CML and the
shortage of effective autophagy inhibitors, we studied the efficacy
of spautin-1, which has been identified to inhibit
starvation-induced autophagy. We observed that spautin-1 markedly
inhibited IM-induced autophagy by downregulating the autophagy
protein Beclin-1. The Beclin1/VPS34 complex plays a crucial role in
autophagosome formation. In fact, Beclin-1 has been implicated in
the progression of solid tumors and leukemia (22). Earlier studies reported that
inhibition of autophagy by knockdown of Beclin-1 sensitized CML
cells to chemo-therapy drugs (7,23).
Spautin-1 specifically targeted Beclin-1 and inhibited IM-induced
autophagy, which may contribute to enhanced effect of IM in CML. We
found that spautin-1 enhanced IM-induced cytotoxicity in CML cell
line K562, decreasing the IC50 from 1 to 0.5 μM.
This result was also confirmed in primary cells from CML patients.
Therefore, spautin-1 sensitized the CML cells to IM cytotoxicity,
which correlated with autophagy inhibition.
We investigated the cell cycle and apoptosis in K562
cells and found that spautin-1 significantly promoted the
IM-induced apoptosis of K562 cells in a caspase-dependent manner
but did not change cell cycle distribution compared with IM alone.
Previous studies have shown that IM induced CML cell apoptosis
mainly by activating Bcl-2 family proapoptotic proteins: Bim and
Bad (24). However, Bim was not
affected either by IM alone or combined with spautin-1 in our
experiments. This phenomenon may be attributed to a low
concentration of IM (0.5 μM). Interestingly, the combination
of IM and spautin-1 significantly downregulated the anti-apoptotic
protein Bcl-2 and Mcl-1 compared with IM alone. This indicated that
spautin-1 promoted IM-induced CML cell apoptosis by downregulating
anti-apoptotic protein Bcl-2 and Mcl-1.
To further elucidate the mechanism of the
synergistic effect of spautin-1 and IM, we focused on the PI3K/AKT
signaling pathway, which regulated apoptosis and autophagy. Our
results showed that IM partially reduced the phosphorylation level
of AKTSer437, leading to decreased activity of AKT.
Interestingly, co-treatment of IM and spautin-1 further inhibited
the phosphorylation of AKT compared with IM alone. We found
spautin-1 alone inhibited the phosphorylation of AKT. The PI3K/AKT
pathway was frequently activated in leukemia, promoting survival
and preventing apoptosis in CML cells, especially in IM-resistant
cells (25). It has been reported
that treatment with PI3K/ATK pathway inhibitor effectively
inhibited the resistance to IM of CML cells (17). Our results suggested that spautin-1
might promote IM-induced apoptosis in CML cells by inactivating
AKT.
Among numerous AKT downstream substrates, we focused
on mTOR, FOXO1, Bad, p27 and GSK3β, which correlated with cell
apoptosis and proliferation. It has been established that Ser9
dephosphorylation of GSK-3β results in its activation and is
associated with cell apoptosis (26). Our results showed that
GSK3βSer9 phosphorylation level was significantly
downregulated by co-treatment of spautin-1 and IM, indicating that
GSK3β was actually activated. The active GSK3β triggers
phosphorylation-mediated proteasomal degradation of the
anti-apoptotic protein Mcl-1 (27–29).
It has been reported that downregulation of Mcl-1 through GSK-3β
activation contributes to chemically-induced apoptosis in acute
myeloid leukemia cells (30). We
also found that Mcl-1 protein level in K562 cells was markedly
reduced when co-treated with spautin-1 and IM, but not when treated
with IM alone. Therefore, it is suggested that spautin-1 activated
GSK3βSer9 by inactivating AKT, which ultimately resulted
in downregulation of the anti-apoptotic protein Mcl-1.
In conclusion, spautin-1 enhanced IM-induced
cytotoxicity in K562 cell line. Spautin-1 inhibited IM-induced
autophagy and restrained the pathway, which led to CML cell
survival. On the other hand, spautin-1 promoted cell apoptosis by
activating GSK3β through PI3K/AKT, which reduced the anti-apoptotic
protein Mcl-1. Spautin-1 was also effective in primary CML
cells.
Altogether, our study indicates that spautin-1 is a
promising new approach for CML treatment in combination with IM.
Additional studies of the signaling pathways related to autophagy
and cell death, especially the AKT/GSK3β should provide effective
therapeutic strategies against CML.
Acknowledgements
The authors wish to thank Dr Dawei Ma
(Shanghai Institute of Organic Chemistry, China Academy of Science)
for kindly providing spautin-1 and technical assistance.
References
|
1.
|
Elzinga BM, Nyhan MJ, Crowley LC,
O’Donovan TR, Cahill MR and McKenna SL: Induction of autophagy by
Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates
Bcr-Abl protein. Am J Hematol. 88:455–462. 2013. View Article : Google Scholar
|
|
2.
|
Cortez D, Reuther G and Pendergast AM: The
Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and
stimulates G1-to-S phase transition in hematopoietic cells.
Oncogene. 15:2333–2342. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Yang X, Lin J, Gong Y, et al:
Antileukaemia effect of rapamycin alone or in combination with
daunorubicin on ph+ acute lymphoblastic leukaemia cell line.
Hematol Oncol. 30:123–130. 2012.
|
|
4.
|
Carew JS, Nawrocki ST, Giles FJ and
Cleveland JL: Targeting autophagy: a novel anticancer strategy with
therapeutic implications for imatinib resistance. Biologics.
2:201–204. 2008.
|
|
5.
|
Burchert A: Roots of imatinib resistance:
a question of self-renewal? Drug Resist Updat. 10:152–161. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Yu Y, Yang L, Zhao M, et al: Targeting
microRNA-30a-mediated autophagy enhances imatinib activity against
human chronic myeloid leukemia cells. Leukemia. 26:1752–1760. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bellodi C, Lidonnici MR, Hamilton A, et
al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View
Article : Google Scholar
|
|
9.
|
Crowley LC, Elzinga BM, O’Sullivan GC and
McKenna SL: Autophagy induction by Bcr-Abl-expressing cells
facilitates their recovery from a targeted or nontargeted
treatment. Am J Hematol. 86:38–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Liu J, Xia H, Kim M, et al: Beclin1
controls the levels of p53 by regulating the deubiquitination
activity of USP10 and USP13. Cell. 147:223–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhao M, Yang M, Yang L, et al: HMGB1
regulates autophagy through increasing transcriptional activities
of JNK and ERK in human myeloid leukemia cells. BMB Rep.
44:601–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Salomoni P and Calabretta B: Targeted
therapies and autophagy: new insights from chronic myeloid
leukemia. Autophagy. 5:1050–1051. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Mishima Y, Terui Y, Taniyama A, et al:
Autophagy and autophagic cell death are next targets for
elimination of the resistance to tyrosine kinase inhibitors. Cancer
Sci. 99:2200–2208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yu Y, Cao L, Yang L, Kang R, Lotze M and
Tang D: microRNA 30A promotes autophagy in response to cancer
therapy. Autophagy. 8:853–855. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Sheng Z, Ma L, Sun JE, Zhu LJ and Green
MR: BCR-ABL suppresses autophagy through ATF5-mediated regulation
of mTOR transcription. Blood. 118:2840–2848. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Tong Y, Liu YY, You LS and Qian WB:
Perifosine induces protective autophagy and upregulation of ATG5 in
human chronic myelogenous leukemia cells in vitro. Acta Pharmacol
Sin. 33:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Burchert A, Wang Y, Cai D, et al:
Compensatory PI3-kinase/Akt/mTor activation regulates imatinib
resistance development. Leukemia. 19:1774–1782. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Frame S, Cohen P and Biondi RM: A common
phosphate binding site explains the unique substrate specificity of
GSK3 and its inactivation by phosphorylation. Mol Cell.
7:1321–1327. 2001. View Article : Google Scholar
|
|
19.
|
Linke R and Dempke W: Management of
imatinib-resistant CML patients. Onkologie. 30:574–580. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Drullion C, Tregoat C, Lagarde V, et al:
Apoptosis and autophagy have opposite roles on imatinib-induced
K562 leukemia cell senescence. Cell Death Dis. 3:e3732012.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Drullion C, Lagarde V, Gioia R, et al:
Mycophenolic acid overcomes imatinib and nilotinib resistance of
chronic myeloid leukemia cells by apoptosis or a senescent-like
cell cycle arrest. Leuk Res Treatment. 2012.8613012012.PubMed/NCBI
|
|
22.
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Han W, Sun J, Feng L, et al: Autophagy
inhibition enhances daunorubicin-induced apoptosis in K562 cells.
PLoS One. 6:e284912011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kuroda J, Shimura Y, Yamamoto-Sugitani M,
Sasaki N and Taniwaki M: Multifaceted mechanisms for cell survival
and drug targeting in chronic myelogenous leukemia. Curr Cancer
Drug Targets. 13:69–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Targeting survival cascades induced by activation of
Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for
effective leukemia therapy. Leukemia. 22:708–722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
McCubrey JA, Steelman LS, Bertrand FE, et
al: Multifaceted roles of GSK-3 and Wnt/beta-catenin in
hematopoiesis and leukemogenesis: opportunities for therapeutic
intervention. Leukemia. 28:15–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Chiara F and Rasola A: GSK-3 and
mitochondria in cancer cells. Front Oncol. 3:162013. View Article : Google Scholar
|
|
28.
|
Maurer U, Charvet C, Wagman AS, Dejardin E
and Green DR: Glycogen synthase kinase-3 regulates mitochondrial
outer membrane permeabilization and apoptosis by destabilization of
MCL-1. Mol Cell. 21:749–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Morel C, Carlson SM, White FM and Davis
RJ: Mcl-1 integrates the opposing actions of signaling pathways
that mediate survival and apoptosis. Mol Cell Biol. 29:3845–3852.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wang R, Xia L, Gabrilove J, Waxman S and
Jing Y: Down-regulation of Mcl-1 through GSK-3beta activation
contributes to arsenic trioxide-induced apoptosis in acute myeloid
leukemia cells. Leukemia. 27:315–324. 2013. View Article : Google Scholar : PubMed/NCBI
|