Introduction
Two syndromes are associated with the major part of
inherited colorectal cancer, hereditary non-polyposis colorectal
cancer (HNPCC) and familial adenomatous polyposis (FAP). In Lynch
syndrome families harbour a deleterious mutation in one of the
mis-match repair (MMR) genes, MLH1, MSH2, MSH6
or PMS2, and these families constitute 15–25% of families
with HNPCC. In the major part of families with HNPCC no
disease-causing mutations in known CRC predisposing genes can be
found. Classical FAP is characterized by the development of
multiple colorectal adenomas and is caused by mutations in the
APC gene. It is today possible to detect nearly all
causative mutations responsible for this syndrome. However, for the
majority of patients who develop colorectal adenomas in the range
from a few to approximately 100, the genetic basis of disease is
yet to be unravelled. A limited number of these patients have
attenuated FAP (AFAP) with less polyps and higher age at onset. In
these cases the disease is caused by less severe mutations in the
APC gene. Attenuated polyposis can also be caused by
bi-allelic mutations in the MUTYH gene as seen in the
recessively inherited MAP syndrome (MUTYH-associated
polyposis).
Recently a new CRC syndrome, polymerase
proofreading-associated polyposis was described (1). This syndrome is characterized by a
dominantly inherited predisposition to the development of a
variable number of colorectal adenomas and carcinomas. The
proofreading (exonuclease) activity of the genes, POLE and
POLD1 (encoding the proofreading domain of polymerase ɛ and
δ, respectively), are impaired by mutations in patients with this
syndrome. The phenotypical expression of the disease varies;
carriers of mutations in the POLD1 gene developed
endometrial tumours besides development of colorectal adenoma and
carcinoma while the POLE mutation p.Leu424Val conferred a
highly-penetrant predisposition only to colorectal adenoma and
carcinoma in carriers.
We have identified a family with an amino-acid
substitution p.Asn363Lys in POLE, in the same exonuclease
domain of polymerase ɛ as the p.Leu424Val substitution. In this
family mutation carriers are affected by a broad spectrum of
tumours in addition to CRC. The substitution has a drastic affect
on the proofreading capacity of the polymerase and we therefore
suggest that a genotype to phenotype correlation might exist for
this gene.
Materials and methods
Patients
All patients have given their consent and the study
has been approved by the local ethics committee at the University
of Gothenburg, Sweden.
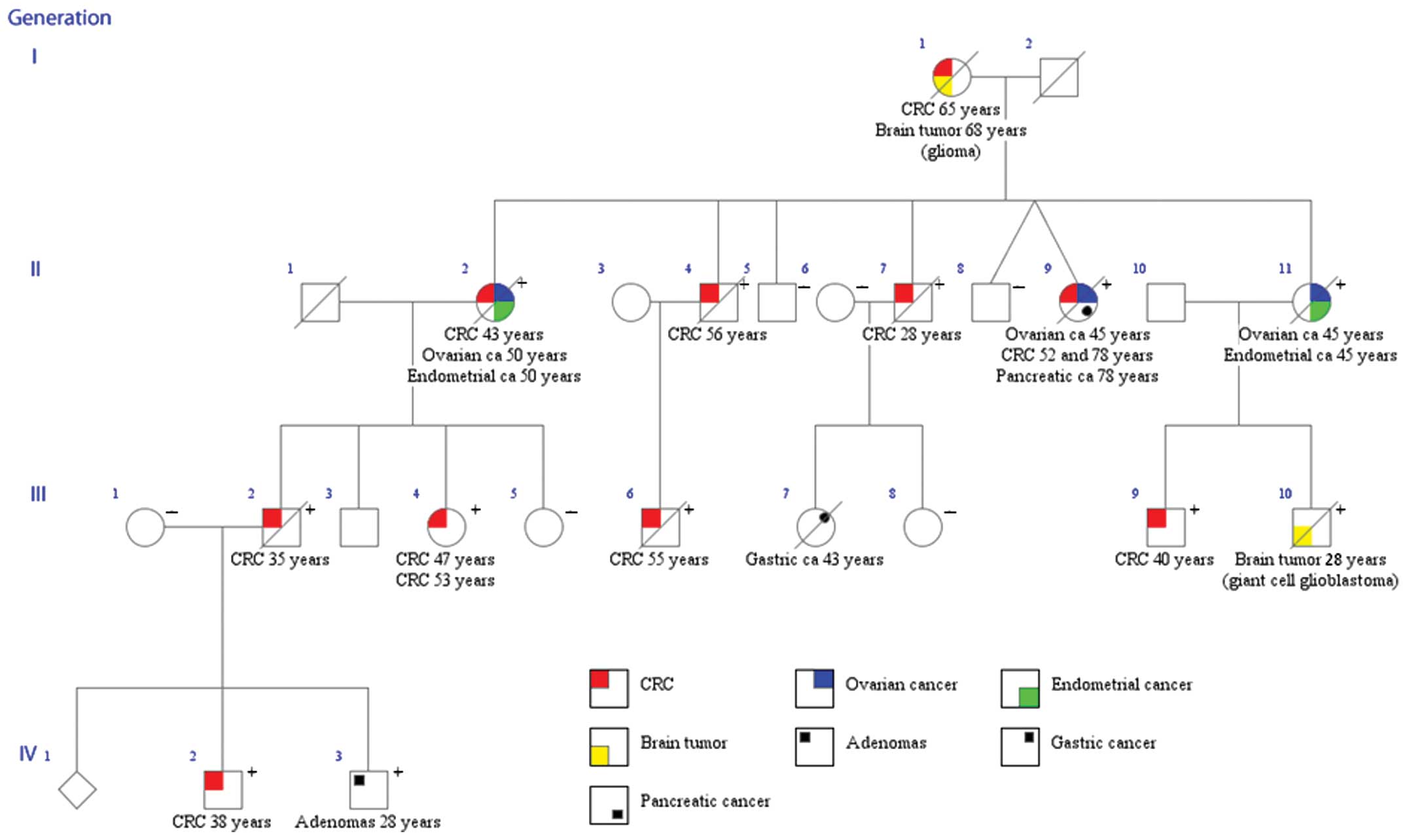
Family member III:4 (Fig. 1) had a prophylactic colectomy with
ileo-rectal anastomosis (IRA) at 47 years with an accidental
unexpected finding of CRC. She also developed a malign rectal
tumour at 53 years. Two affected individuals in the family, I:1 and
III:10 developed primary brain tumours, a glioma in one case and a
giant cell glioblastoma in the other. Six family members >50
years of age, without sign of disease did not carry the mutation.
Two of these individuals had normal barium enema at 59 (II:6) and
54 years (II:8) and they both reached an age of 84 years without
any sign of the disease. Family member III:8 had normal colonoscopy
at 46 years and member III:5 had prophylactic colectomy and IRA at
49 years and two normal rectoscopies after that at 54 and 58 years.
Unfortunately family member III:7 with gastric cancer could not be
tested due to no available material. Mutation testing was carried
out by Sanger sequencing and/or TaqMan analyses on blood samples in
most cases but for some individuals only paraffin-embedded tissue
were available.
DNA extraction and mutation
screening
Genomic DNA was extracted using the BioRobot EZ1
(Qiagen, Hilden, Germany) with the EZ1 DNA Blood 350 μl kit
(Qiagen). DNA from adenomas, tumours and paraffin embedded tissues
were extracted using QiAmp DNA microkit (Qiagen) following the
manufacturer’s recommendations. Family members III:4 and III:9
(Fig. 1) were screened for
mutations in the known CRC-predisposing genes, APC,
MUTYH, MLH1, MSH2, MSH6 and PMS2
using Sanger sequencing and MLPA (Multiplex Ligation-dependent
Probe Amplification, MRC-Holland, Amsterdam, The Netherlands) as
described in (2). Further details
on primer sequences and MLPA probe kits can be obtained on
request.
Affymetrix genome-wide human SNP array
6.0
The GeneChip 6.0 platform includes about 906,600
SNP- and about 900,000 copy-number probes, covering the whole
genome with an average spacing of approximately 1.3 kb. Samples
were prepared according to standard conditions (Affymetrix, Santa
Clara, CA). Purification of PCR products were performed using
Magnetic Beads (Agencourt Bioscience Corporation, Beverly, MA).
Hybridization, washing and staining of arrays were performed as
described by the supplier.
Exome capture and sequencing
DNA samples from family members III:1, III:4, IV:2
and IV:3 were quantified using the Qubit system (Life Technologies,
Carlsbad, CA). DNA, were fragmented using the Covaris S2
Ultrasonicator (Covaris, Woburn, MA), where after the samples were
analyzed on the Bioanalyzer (Agilent Technologies, Santa Clara, CA)
for correct fragment sizes. Libraries were constructed using the
Agilent SureSelectXT Human All Exon 50Mb library kit for Illumina
Paired End Sequencing (v3 protocol version 1.4.1, Agilent
Technologies) according to the manufacturer’s instruction. The
concentration of each library was determined using the Qubit and
the Bioanalyzer. Whole exome sequencing was performed on the
Illumina HiScanSQ with 2×76 bp paired end reads using TruSeq SBS
HS-v3 clustering and sequencing chemistry (Illumina, San Diego,
CA).
Analysis of sequencing data
Quality assessment of the sequence reads was
performed by generating QC statistics with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc).
Read alignment to the reference human genome (hg19,UCSC assembly,
February 2009) was done using BWA (3) with default parameters. After removal
of PCR duplicates (Picard tools, http://picard.sourceforge.net) and file conversion
(Samtools) (4), quality score
recalibration, indel realignment and variant calling were performed
with the GATK package (5).
Variants were annotated with Annovar (6) using a wide range of databases such as
dbSNP build 135 (7), dbNSFP
(8), KEGG (9), the Gene Ontology project (10), MITOMAP (11) and tracks from the UCSC (12). The targeted region of the exome
analysis included the complete coding region of the POLE
gene. Reference sequence used for POLE was NM_006231.2.
Data filtering
All common variants among the three affected
individuals were filtered against one unaffected individual from
the family. An in-house control data set (38 exomes of patients
with different disease phenotypes), provided by Genomics Core
facility (University of Gothenburg) was then applied for filtering
of platform specific technical artefacts and polymorphic
variants.
Analysis of the c.1089C>A, p.Asn363Lys
mutation
DNA from blood and/or paraffin-embedded tissue from
family members were analyzed for the presence of the mutation using
Sanger sequencing and TaqMan and the control material was analyzed
using TaqMan. The mutation was confirmed on a second PCR sample.
Custom TaqMan Assay Design Tool (Life Technologies) was used to
design the probe. The analysis was preformed according to the
manual instructions in 96-well plates on an Applied Biosystems
7900HT fast real-time PCR system (Applied Biosystems, Foster City,
CA). The analyses were carried out at the Genomics Core Facility at
the University of Gothenburg.
Results
The first attempts to identify the genetic cause of
the disease in the family were by screening known CRC-predisposing
genes (including APC, MUTYH, MLH1,
MSH2, MSH6 and PMS2) by conventional Sanger
sequencing and MLPA. These analyses all turned out negative and
further attempts to isolate a possibly new CRC predisposing gene
were done in 2007 when a linkage analysis was performed with
Affymetrix GeneChip Mapping 10K Array. However, these analyses did
not reveal any region of linkage and recently a re-analysis of the
10K array revealed a low SNP coverage and poor marker informativity
in the POLE region for family members. In fact POLE
maps distal to the most terminal chromosome 12p marker on this
array (position Chr12:133,200,348–133,263,945; Genome map build
GRCh37/hg19). Re-analyses of family members using an array with an
approximately 100 times denser marker set (Affymetrix Genome-Wide
Human SNP Array 6.0) revealed a shared haplotype between the
flanking markers rs1051219 at position 132,237,750 and rs2291256 at
position 133,393,323, and at least one crossover beyond the
coverage of the Affymetrix 10K array which explains why linkage
could not be established with the 10K array. A linkage analysis of
the detected mutation using Array 6.0 data gives a maximum LOD=3.7,
which is genome-wide significant. Using whole-exome sequencing
performed on four individuals in the family (Fig. 1) we identified a missense mutation
in the POLE gene, NM_006231.2:c.1089C>A, p.Asn363Lys, in
affected individuals (Fig. 1). A
local control material consisting of 642 healthy blood donors (i.e.
1284 alleles) from the western part of Sweden was also negative for
the mutation. The mutation is not present in the NHLBI GOExome
Sequencing Project (ESP) (https://esp.gs.washington.edu/drupal/) nor in dbSNP
(NCBI) (http://www.ncbi.nlm.nih.gov/SNP/). The human
POLE sequence contains a synonymous SNP (rs146639652 C/T) at
the same position with a minor allele frequency in Americans of
European decent of 0.0001 (based on ESP6500). The TCGA (The Cancer
Genome Atlas, http://cancergenome.nih.gov/) data set was examined
for possible somatic presence of the mutation in colon
adenocarcinoma, endometrial tumours, serous cystadenocarcinoma and
brain low grade glioma without any findings.
The substitution p.Asn363Lys has a profound effect
on the substrate binding at the active site of the proofreading
exonuclease domain. The structural homology of this domain is well
conserved between species. In comparing eukaryotes and prokaryotes,
the metal-coordinating active-site residues and the invariant
p.Asn363 can be superimposed with a root-mean-square deviation of
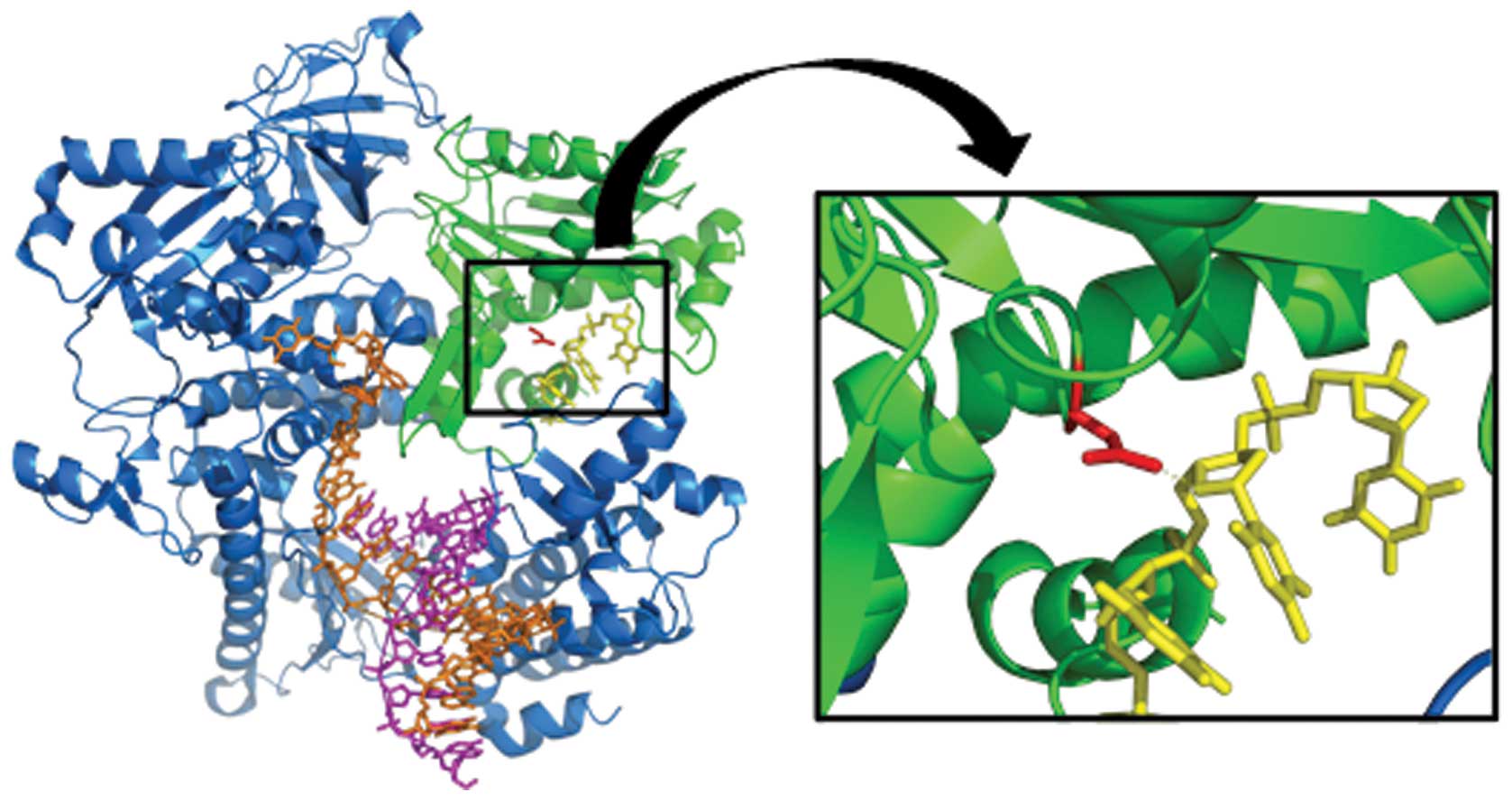
0.4 Å. A high-resolution structure of the human polymerase ɛ is
lacking, but the structure of the yeast Saccharomyces
cerevisiae polymerase δ (Pol δ also known as Pol3) (3IAY.pdb,
Protein Data Bank, www.rcsb.org) and the Escherichia
coli Klenow fragment (KF) with bound ssDNA substrate (1D8Y.pdb,
Fig. 2), provides, on the atomic
level, understanding of the pathogenic properties of the amino acid
substitution that we report. The side chain NH2-group of the Asn
can form a direct hydrogen bond with the 4’ oxygen of the substrate
backbone. This interaction is important for exonuclease activity
(13). In the KF, the rate for
excision of 3’ nucleotide residues drops up to 100-fold if the
corresponding Asn (Asn420 in the KF) is substituted for Ala
(14), and the exonuclease
activity drops a 1,000-fold when Asn is replaced with Asp in the
DNA polymerase of the thermophilic Thermococcus kodakarensis
(15). These mutations emphasize
the necessity of the precise conservation that exists in the wt
enzyme. The Ala side chain is small and does not impose steric
hinders. Similarly, the Asp side chain is of the same size as the
wt Asn, but, neither Asp nor Ala nor any of the other amino acids
(possibly with the exception of Ser) can readily form the required
stabilizing hydrogen bond to the substrate.
The very high degree of conservation of the
p.Asn363, strictly conserved in the exonuclease domain (Fig. 3), and the p.Leu424 (only replaced
with Met in the KF) in the B type polymerases demonstrates their
importance for enzyme functionality. The side chains of p.Tyr362
and p.Leu424 together form a platform for the single-stranded
primer DNA and in S. cerevisiae, the Leu479Ser
(corresponding to p.Leu424) mutant completely lacks exonuclease
activity (16). However, when the
Leu is substituted for Ala in the T4 DNA polymerase, approximately
25% exonuclease activity is retained (17), and the severity of the potentially
milder p.Leu424Val substitution is difficult to assess correctly.
The difference in phenotype of our family with the apparently more
severe p.Asn363Lys mutation compared to the phenotype of the family
with the p.Leu424Val mutation (only CRC in carriers) can probably
be correlated with to what extent the activity of the exonuclease
function is perturbed in the two mutant proteins.
Family members carrying the p.Asn363Lys mutation
demonstrate a highly penetrant predisposition not only to CRC but
also to extra-intestinal tumours such as ovarian, endometrial and
brain tumours. In one case a mutation carrier also developed
pancreatic cancer at age 78 (Fig.
1, family member II:9). The mutation was present in
heterozygous form in 12 family members. A limited number of
colorectal adenomas in the range resembling AFAP or
MUTYH-associated polyposis were found in mutation carriers
and several of them had undergone polypectomy prior to development
of CRC. To investigate the incidence of POLE mutations in
the population from the western part of Sweden (from where the
family originates), 87 patients with a history of familial CRC or
disease onset at young age were examined for mutations in the
exonuclease domain of POLE (exons 3–14, codons 69–491). The
patients had no colorectal adenoma or a number of adenomas in the
range between a few and approximately 100. They had all been shown
to be negative for mutations in either the mismatch-repair genes,
MSH2, MLH1, MSH6 and PMS2 and/or
APC and MUTYH. No mutation in POLE was found
in any of these patients, and it was concluded that mutations in
this gene only is responsible for a minor fraction of inherited CRC
in our region.
Two colorectal tumours, one from each of family
members III:9 and IV:2, and also a colorectal adenoma from member
IV:3, were examined for second hits in POLE using
whole-exome sequencing and loss of heterozygosity (LOH) of the wt
allele was examined with microarray analyses, but no aberrations
were identified. Second hits in POLE have been reported
(Cancer Genome Atlas Network) (1,18–21)
in some CRC tumours which could imply a tumour suppressor activity
of the gene but the question whether POLE act as a classical
tumour gene is not yet clear. Five colorectal tumours from three
affected individuals were all microsatellite stable (MSS) which is
in line with previous findings that tumours with mutations in
POLE are MSS and are hypermutated with mutations in other
genes involved in the classical tumorigenesis pathway (1,21).
Discussion
The family presented here has been a research
subject since 1980 due to the striking dominant inheritance of
tumours and a clinical description of the family was published 1984
(22). A first report relating to
the genetic cause of disease on chromosomal level was published in
1989 (23). During the years the
family has been screened for deleterious mutations in known
CRC-predisposing genes without any findings. Exome sequencing
finally made it possible to identify the genetic cause of disease
in the family. The mutation is likely to have a profound effect on
the proofreading activity of the exonuclease function of
POLE as is shown by mutagenesis studies performed in
microorganisms and further corroborated by the high degree of
conservation at this amino acid location.
It has been suggested that a possible genotype to
phenotype correlation might exist in the polymerase
proofreading-associated polyposis. Palles et al (1) reported that the mutation,
p.Ser478Asn, in POLD1, encoding the exonuclease domain of
polymerase δ predisposes carriers not only to colorectal tumours
but also to endometrial cancer and possibly also to brain tumours.
The apparent difference in tumour spectrum between carriers of the
POLE p.Leu424Val and the POLD1 p.Ser478Asn
substitutions was unexplained and the authors mention that similar
differences are found in Lynch syndrome where endometrial cancer
seems to be more frequent in carriers of MSH6 mutations.
However, the p.Asn363Lys mutation reported on here clearly
demonstrates that mutations in POLE can predispose carriers
to a broad spectrum of tumours. When comparing the effect of the
two amino acid substitutions, the p.Leu424Val reported (1) and the p.Asn363Lys presented here, it
is possible to more accurately suggest that a genotype to phenotype
correlation exist for this gene. The p.Asn363Lys substitution that
we now report on have a more drastic effect on the proofreading
activity which seemingly has the potential to initiate the
development of various tumours in carriers, while the p.Leu424Val
mutation seems to have a more limited effect which restricts
carriers to the development of colorectal tumours. We thus suggest
that a genotype to phenotype correlation needs to be considered in
the clinical follow-up of POLE gene mutation carriers.
Acknowledgements
We are grateful for the contribution
and support from the family members. This study was supported by
grants from The Swedish Cancer Society, The Swedish state under the
LUA/ALF agreement concerning research and education of doctors,
(no. 76310), The Health & Medical Care Committee of the Region
Västra Götaland, Project grant from the Laboratory division
Sahlgrenska University Hospital, The Nilsson-Ehle Foundation, The
Assar Gabrielsson Foundation, The Wilhelm and Martina Lundgren
Research Foundation and The Sahlgrenska University Hospital
Foundation. We acknowledge the Cancer Genome Atlas Research Network
for generating the TCGA datasets. The exome analyses were carried
out at the Genomics Core Facility at the University of
Gothenburg.
References
|
1.
|
Palles C, Cazier JB, Howarth KM, et al:
Germline mutations affecting the proofreading domains of POLE and
POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet.
45:136–144. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kanter-Smoler G, Bjork J, Fritzell K, et
al: Novel findings in Swedish patients with MYH-associated
polyposis: mutation detection and clinical characterization. Clin
Gastroenterol Hepatol. 4:499–506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Li H, Handsaker B, Wysoker A, et al: The
Sequence Alignment/Map format and SAMtools. Bioinformatics.
25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
McKenna A, Hanna M, Banks E, et al: The
Genome Analysis Toolkit: a MapReduce framework for analyzing
next-generation DNA sequencing data. Genome Res. 20:1297–1303.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wang K, Li M and Hakonarson H: ANNOVAR:
functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Sherry ST, Ward MH, Kholodov M, et al:
dbSNP: the NCBI database of genetic variation. Nucleic Acids Res.
29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Liu X, Jian X and Boerwinkle E: dbNSFP: a
lightweight database of human nonsynonymous SNPs and their
functional predictions. Hum Mutat. 32:894–899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kanehisa M, Goto S, Sato Y, Furumichi M
and Tanabe M: KEGG for integration and interpretation of
large-scale molecular data sets. Nucleic Acids Res. 40:102012.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ashburner M, Ball CA, Blake JA, et al:
Gene ontology: tool for the unification of biology. The Gene
Ontology Consortium Nat Genet. 25:25–29. 2000.PubMed/NCBI
|
|
11.
|
Ruiz-Pesini E, Lott MT, Procaccio V, et
al: An enhanced MITOMAP with a global mtDNA mutational phylogeny.
Nucleic Acids Res. 35:182007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Dreszer TR, Karolchik D, Zweig AS, et al:
The UCSC Genome Browser database: extensions and updates 2011.
Nucleic Acids Res (Database issue). 40:D918–D923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Beese LS and Steitz TA: Structural basis
for the 3’-5’ exonuclease activity of Escherichia coli DNA
polymerase I: a two metal ion mechanism. EMBO J. 10:25–33.
1991.
|
|
14.
|
Lin TC, Wang CX, Joyce CM and Konigsberg
WH: 3’-5’ Exonucleolytic activity of DNA polymerases: structural
features that allow kinetic discrimination between ribo- and
deoxyribonucleotide residues. Biochemistry. 40:8749–8755. 2001.
|
|
15.
|
Nishioka M, Mizuguchi H, Fujiwara S, et
al: Long and accurate PCR with a mixture of KOD DNA polymerase and
its exonuclease deficient mutant enzyme. J Biotechnol. 88:141–149.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Murphy K, Darmawan H, Schultz A, Fidalgo
da Silva E and Reha-Krantz LJ: A method to select for mutator DNA
polymerase deltas in Saccharomyces cerevisiae. Genome.
49:403–410. 2006. View
Article : Google Scholar
|
|
17.
|
Abdus Sattar AK, Lin TC, Jones C and
Konigsberg WH: Functional consequences and exonuclease kinetic
parameters of point mutations in bacteriophage T4 DNA polymerase.
Biochemistry. 35:16621–16629. 1996.PubMed/NCBI
|
|
18.
|
Flohr T, Dai JC, Buttner J, Popanda O,
Hagmuller E and Thielmann HW: Detection of mutations in the DNA
polymerase delta gene of human sporadic colorectal cancers and
colon cancer cell lines. Int J Cancer. 80:919–929. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Da Costa LT, Liu B, el-Deiry W, et al:
Polymerase delta variants in RER colorectal tumours. Nat Genet.
9:10–11. 1995.PubMed/NCBI
|
|
20.
|
Yoshida R, Miyashita K, Inoue M, et al:
Concurrent genetic alterations in DNA polymerase proofreading and
mismatch repair in human colorectal cancer. Eur J Hum Genet.
19:320–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hasselgren PO, Hulten L and Karlstrom L:
‘Cancer family syndrome’ - description of a family with a
hereditary incidence of malignant tumors of the colon and other
organs. Lakartidningen. 81:2451–2453. 1984.(In Swedish).
|
|
23.
|
Kopf I, Islam MQ, Friberg LG and Levan G:
Familial occurrence of cancer and heteromorphism of the
heterochromatic segment of chromosome 1. Hereditas. 110:79–83.
1989. View Article : Google Scholar : PubMed/NCBI
|