Introduction
Epithelial-mesenchymal transition (EMT) is a
crucially important phenotypic conversion in which epithelial cells
dissociate from each other, largely lose cell-cell adhesion and
acquire a migratory, fibroblast-like phenotype (1). EMT occurs at various stages of
embryonal development, including such early events as the
generation of mesoderm and the formation of the neural crest.
Typically, EMT entails the disassembly of epithelial cell-cell
junctions such as adherens junctions, tight junctions and
desmosomes, as well as a radical reorganisation of the
cytoskeleton. Cells lose basolateral-apical polarity and instead
acquire a leading edge-trailing edge polarity. At the level of gene
expression, genes coding for components of cell-cell junctions
become inactive, as well as those of certain intermediate filament
proteins, which are replaced by others. Typical markers for EMT are
loss of E-cadherin, the cell-cell adhesion molecule of epithelial
adherens junctions, and gain of expression of the intermediate
filament protein vimentin. A number of gene regulatory proteins
orchestrating these events have been identified, including Slug,
Snail and Twist, which are known to repress E-cadherin expression
(2). These in turn can be
activated by intracellular signalling pathways, notably the
TGFβ-induced Smad pathway and the MAP kinase cascade (3).
In recent years, a rapidly growing number of studies
have implicated EMT or EMT-like events in the development of
carcinomas, although this matter is still the subject of
considerable controversy (3).
Proponents of EMT as a mechanism for carcinogenesis point to the
loss of E-cadherin and gain of vimentin expression seen in cells at
the invasive front of epithelial tumours (4) and the frequent acquisition of
malignant properties in cells where EMT has been experimentally
induced (5,6). Moreover, recent findings indicate
that cells generated by EMT share characteristics with cancer stem
cells (7).
E-cadherin has long been regarded as a crucial
target of EMT-induced changes in gene expression. In epithelial
cells, E-cadherin is the transmembrane protein responsible for
homophilic binding between structures of adherens junctions of
neighbouring cells. On its cytoplasmic side, E-cadherin is
associated with the actin cytoskeleton by interaction with β- or
γ-catenin. It is generally believed that these adaptor molecules
bind to actin filaments by interaction with α-catenin (8), although the precise role for this
protein has been vigorously debated in recent years (9). E-cadherin has also been identified as
a tumour suppressor protein, based on the observations that its
expression often is lost during carcinogenesis and forced
re-expression has been seen to reverse malignant properties of
carcinoma cells (10). Loss of
E-cadherin expression is also generally viewed as a decisive event
in EMT where it is widely assumed to be the direct cause of
cell-cell detachment (1).
We have previously (11,12)
described the induction of EMT in a cellular model for mammary
carcinogenesis caused by c-erbB2 (HER2), an oncogenic receptor
tyrosine kinase (RTK) over-expressed in a subset of mammary
cancers. This overexpression is thought to cause activation of
c-erbB2 by ligand-independent homodimerisation (contrasting with
its normal function in ligand-induced heterodimerisation with other
EGF receptor family members) (13). In our model, high c-erbB2
signalling is induced in an immortalised human luminal mammary
epithelial cell line [HB2, originally developed in the lab of Dr
Joyce Taylor-Papadimitriou (14)]
by means of a hybrid RTK, ‘trk-neu’. This hybrid consists of the
transmembrane and cytoplasmic domains of c-erbB2 fused to the
extracellular domain of the trkA nerve growth factor (NGF) receptor
(15). Expression of this
construct allows signalling from a homodimer of c-erbB2 to be
induced by treatment of the cells with NGF. In earlier studies we
observed that prolonged induction of c-erbB2 signalling in HB2
cells stably expressing the trk-neu hybrid receptor caused
irreversible EMT (5). Furthermore,
and as observed by other researchers (11,16,17),
the progression of EMT was significantly delayed when cells were
grown at high density, a finding which is in good accordance with
clinical studies preferentially detecting EMT markers at the
invasive front of a tumour, where cell density is at a minimum
(18,19). The phenomenon of density-dependent
inhibition of EMT certainly merits further investigation as it
offers insight into a mechanism whereby cancer cell invasiveness
might be counteracted. It was also observed that cells which had
recently scattered as a consequence of c-erbB2 signalling (i.e.,
commenced the EMT process) still expressed surface-bound E-cadherin
(11), calling into question the
key causal role ascribed to E-cadherin loss in EMT. Here we have
further analysed these phenomena by following the progression of
EMT while ectopically expressing wild-type or dominant-negative
E-cadherin in trk-neu expressing HB2 cells. EMT was not impeded,
nor were fibroblastic cells isolated after EMT affected in their
morphology by the presence of ectopically expressed wild-type
E-cadherin, suggesting that downregulation of E-cadherin expression
is indeed not required for the progression of EMT. In an attempt to
elucidate whether density-dependent inhibition of EMT was mediated
by intercellular E-cadherin engagement, a dominant-negative
E-cadherin mutant deficient in adhesiveness was expressed. While
cell-cell adhesion was weakened, the mutant failed to promote
c-erbB2-induced EMT at high cell density, suggesting that
E-cadherin signalling does not account for density-dependent
inhibition of EMT.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal
bovine serum (FBS) zeocin, blasticdin S, Geneticin, and the plasmid
vectors pcDNA3.1- and pcDNA5/TO were from Invitrogen (Carlsbad, CA,
USA). Insulin, hydrocortisone, doxycycline and Triton X-100 were
from Sigma (St. Louis, MO, USA). The monoclonal antibody (mAb)
HECD-1 against E-cadherin was from Takara Bio (Shiga, Japan). The
mAb Vim3B4 against vimentin was from Dakopatts AB (Älvsjö, Sweden)
and R-phycoerythrin-labeled goat anti-mouse antibody was from
Electra-Box Diagnostica (Tyresö, Sweden). Antibodies against
β-catenin, Slug and ZO-1 were from Cell Signalling Technology
(Danvers, MA, USA). The γ-catenin antibody (Transduction
Laboratories, Lexington, KY, USA) was a kind gift from Professor
Mikael Nilsson, University of Gothenburg. Antibodies against
α-tubulin and human fibronectin were from Sigma. Restriction
endonucleases were from Fermentas (Pittsburgh, PA, USA). The 2.5S
nerve growth factor (NGF) from mouse submaxillary gland was
purchased from Promega (Madison, WI, USA). The pIRES2-GFP plasmid
was from Clontech (Mountain View, CA, USA) and the pcDNA3.1- and
pcDNA5/TO plasmids were from Invitrogen. Full-length human
E-cadherin cDNA in pBluescript had previously been obtained as a
gift from the late Professor Margaret Wheelock.
Cell culture
MDA-MB-468 cells (no. HTB-132, ATCC, Manassas, VA,
USA) were grown in DMEM containing 10% FBS. Tr-ep cells were grown
in DMEM with 10% FBS 5 μg/ml hydrocortisone, 10 μg/ml
insulin, 5 μg/ml zeocin and 10 μg/ml blasticidin S.
Transfectants carrying IRES-GFP constructs also had 0.5 mg/ml
Geneticin added to the medium. Cells were grown at 37°C and 5%
CO2.
cDNA constructs
Expression constructs were based on
pcDNA3.1neo/TO-IRES-GFP, a vector constructed in the lab by first
combining the commercially available vectors pcDNA3.1- (neo
resistance) and pcDNA5/TO (zeocin resistance) using cleavage with
MluI and XmaI in both plasmids and combining the
tetracycline operator-containing fragment from pcDNA5/TO and the
neo resistance-containing fragment from pcDNA3.1-. The resulting
plasmid, pcDNA3.1neo/TO, was then further modified by inserting the
IRES-GFP sequence from pIRES2-GFP as an XhoI-XbaI
fragment, and then deleting duplicated polylinker restriction sites
by site-directed muta-genesis. Human E-cadherin cDNA was then
introduced into this vector to yield
pcDNA3.1neo/TO/E-cadherin-IRES-GFP. Further site-directed
mutagenesis was used to obtain the WV156-157AA (WV) E-cadherin
mutant. Transformants were screened for the introduction of a
NaeI site and selected clones were sequenced through the
coding region.
Transfections
Expression constructs were introduced by calcium
phosphate co-precipitation using standard techniques (20). Desired clones were identified by
fluorescence microscopy following brief (24 h) doxycycline (dox)
treatment and then recovered by detachment in cloning cylinders.
Further characterisation was performed by measuring GFP
fluorescence and binding of E-cadherin mAb HECD-1 in flow
cytometry.
Flow cytometry
Flow cytometric measurements were performed as
described (11) with the following
modifications: cells were detached with trypsin-EDTA, and where
permeabilisation was required, cells were treated with 0.5% Triton
X-100 (ice, 10 min). It should be noted that unless otherwise
indicated, non-permeabilised cells were used for analysis of
E-cadherin expression in order to exclusively detect plasma
membrane-bound E-cadherin. mAb HECD-1 (10 μg/ml) was used
for detection of E-cadherin and the mAb Vim3B4 (1:100 dilution) was
used to probe for vimentin. R-phycoerythrin-labeled goat anti-mouse
antibody was used as a secondary reagent. Dox-treated (i.e.,
GFP-expressing) cells incubated with secondary antibody only were
used as controls for compensation of leakage of GFP fluorescence
into the FL2 channel used to detect R-phycoerythrin fluorescence.
In some experiments, allophycocyanin-labeled goat anti-mouse
antibody was used as a secondary reagent; here, compensation was
not required.
EMT assays
Cells (7×104) were seeded into T75 cell
culture flasks and grown under the following conditions: no
treatment, NGF (50 ng/ml), or NGF + dox (1 μg/ml). In order
to ensure an even level of dox-induced expression, additional dox
corresponding to 0.5 μg/ml was supplied every second day.
When cells reached 50% confluence (typically after 3–4 days), they
were trypsinised and a new passage of cells was seeded as described
above. The remainder of the cells were analysed for E-cadherin and
vimentin expression in flow cytometry. This procedure was repeated
for at least four passages.
The significance of the changes in E-cadherin
expression was analysed as follows: a linear model was used to
analyse the data. Logarithmic fluorescence values were used as the
dependent variable and passage, treatment and replicate were
regarded as factors. An interaction term between passage and
treatment was used. Tests of difference for each time between the
respective treatments and control cells were performed. When
evaluating the percentage of vimentin-positive cells, the cutoff
fluorescence level for defining cells as positive was set to the
level where 99.5% of untreated cells were excluded.
Isolation of fibroblastic clones
From TrE-ep1 cells passaged at low density in the
presence of NGF and dox as described above for EMT assays,
fibroblastic clones were prepared by plating at 300–1,000 cells per
9-cm dish, growing in the continued presence of NGF and dox, and
isolating individual colonies with fibroblastoid cell morphology
using cloning cylinders.
Microscopy
Cells grown on glass coverslips in 24-well plates
were fixed in 4% paraformaldehyde in PBS for 20 min at room
temperature (RT), and where permeabilisation was required, cells
were treated with 0,5% Triton X-100 for 5 min on ice (in order to
restrict detection of E-cadherin to surface-bound molecules, only
non-permeabilised cells were used in these analyses). Cells were
further blocked in 20% FBS in PBS for 20 min in RT, incubated with
primary antibody (diluted in 5% FBS in PBS) for 1 h at RT, washed
2×5 min in PBS, incubated with secondary TRITC-conjugated antibody
diluted in 5% FBS in PBS, washed 2×5 min in PBS and mounted with
ProLong Antifade Gold (Molecular Probes, Eugene, OR, USA).
Fluoromicrographs were taken in a Zeiss Axioplan 2 microscope at
×40 magnification (Carl Zeiss, Oberkochen, Germany). At least three
different micrographs were evaluated for each condition and
representative details were processed with respect to brightness
and contrast (consistently between samples) using GraphicConverter
(Lemke Software, Peine, Germany).
Western blot analysis
For whole-cell extract preparation, cells were
treated with lysis buffer 150 mM NaCl, 50 mM Tris-HCl (pH 8), 1%
Triton X-100 and 1X complete protease inhibitors (Roche,
Indianapolis, IN, USA) for 30 min at 4°C. Protein concentrations
were determined by using Bio-Rad Protein Assay. A total of 40
μg protein/well was loaded and electrophoresed through a
NuPAGE 4 to 12% Bis-Tris sodium dodecyl sulfate-polyacrylamide gel
(Invitrogen) and subsequently electroblotted onto a Hybond-P filter
(GE Healthcare, Little Chalfont, UK).
Detergent extraction assay
Cells were trypsinised, dispersed and strained as
described for flow cytometry, and then incubated in 0.1 ml of FACS
diluent (DMEM, 10% FCS, 0.02% sodium azide) containing 1 μg
E-cadherin mAb HECD-1 for 1 h. After three washes with FACS
diluent, cells were extracted for 10 min on ice in 500 μl of
PBS containing 0.5% Triton X-100. Cells were again washed,
incubated with R-phycoerythrin-labeled goat anti-mouse antibody and
fluorescence was measured in flow cytometry as described above. The
E-cadherin levels were compared with parallel samples stained
without detergent extraction.
Dissociation assays
Cells were grown to confluence in 6-well plates with
no treatment, with dox (1 μg/ml) and/or NGF (50 ng/ml) for
2–3 days. Parallel samples were either washed with Puck’s saline
containing 0.02% EDTA and detached with trypsin (0.05%), or washed
with thermolysin buffer (10 mM HEPES, 142 mM NaCl, 6.7 mM KCl, 0.43
mM NaOH and 1 mM CaCl2) and detached with thermolysin
(0.5 mg/ml in thermolysin buffer). After mechanical dissociation by
passage 4 times through a 23G needle, the total number of cells was
counted in the trypsin-treated sample (where E-cadherin is
degraded) and the number of ‘particles’, i.e., single cells or cell
aggregates, was counted in the thermolysin-treated sample (where
E-cadherin is preserved) using a haemocytometer. Changes in the
degree of dissociation were expressed as a dissociation index,
calculated as DI =
(Pt/Ct)/(Pu/Cu) where P
and C represent thermolysin-detached particles and trypsin-detached
cells, respectively, and the indices u and t represent untreated
and treated cells, respectively.
Results
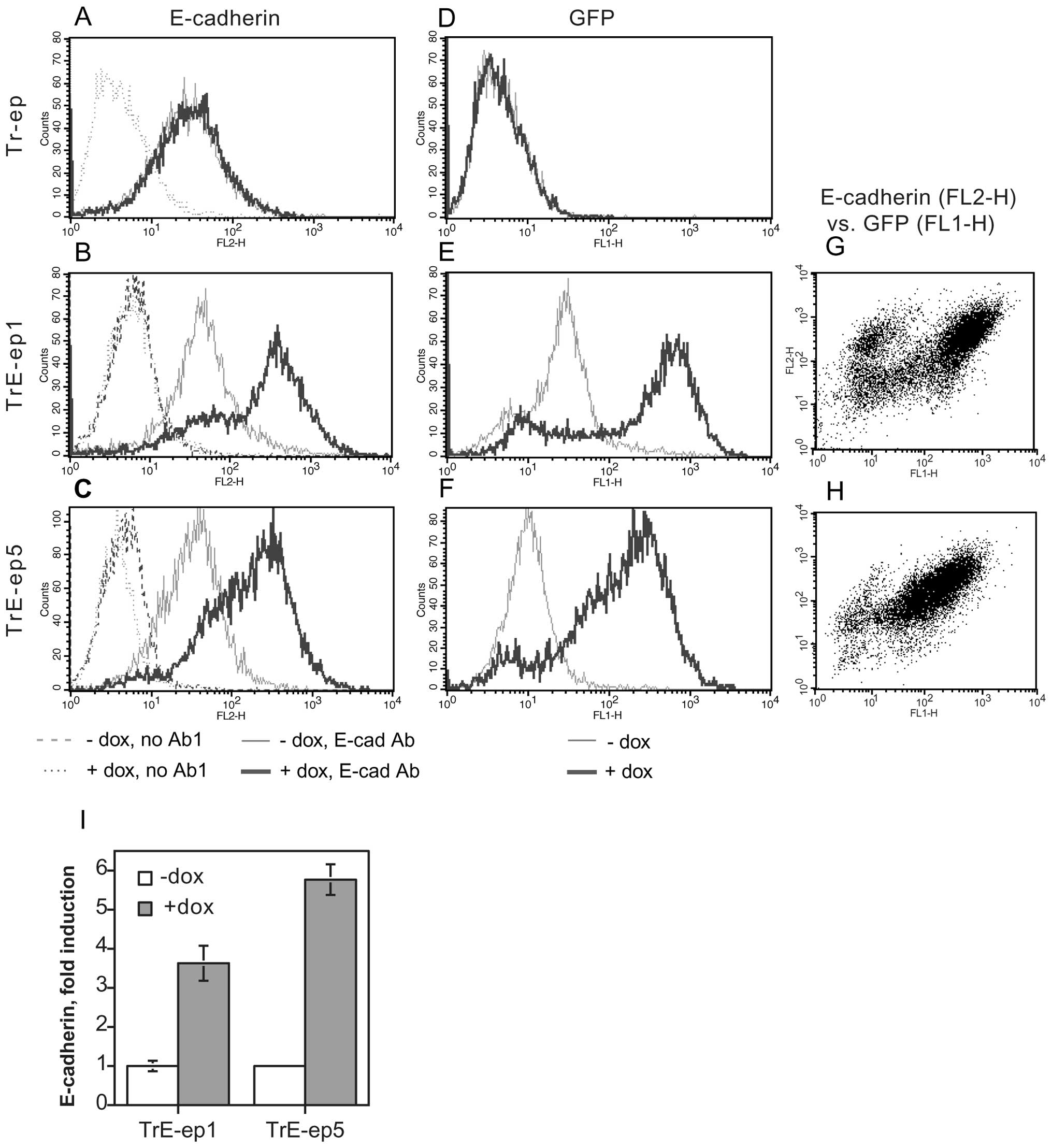
Generation of cells with
tetracycline-inducible expression of E-cadherin and GFP
The HB2/tnz34 cell line (5) is derived from the human luminal
mammary epithelial cell line HB2 (14) which has been transfected with the
hybrid ‘trk-neu’ receptor construct (15), allowing induction of c-erbB2
signalling by means of NGF treatment (see Table I for naming of transfectants and
clones). In order to elucidate the role of modulation of E-cadherin
expression in c-erbB2 induced EMT, a transfectant clone of
HB2/tnz34 stably expressing the tetracycline repressor (Tr-ep) was
further transfected with the pcDNA3.1neo/TO/E-cadherin-IRES-GFP
plasmid. Using this construct, bicistronic expression of wild-type
E-cadherin and GFP can be induced by addition of tetracycline or
its analogue, doxycycline (dox). Stable clones from this
transfection were selected and analysed for dox-induced E-cadherin
and GFP expression. Two clones, designated TrE-ep1 and TrE-ep5,
were chosen for further analysis (Fig.
1). The majority of cells in these clones overexpressed
E-cadherin upon dox treatment with concomitant GFP expression.
There was a good correlation between GFP and E-cadherin expression
levels (Fig. 1G–H). In a minority
of cells, E-cadherin overexpression was seen without GFP
fluorescence, possibly suggesting a longer half-life for E-cadherin
in the induced cells compared to GFP. The reverse expression
pattern, i.e., GFP expression without E-cadherin overexpression,
was not seen. Hence, practically all cells expressing GFP could be
assumed to overexpress E-cadherin.
 | Table I.Names and descriptions of cell lines
and subclones used in the study. |
Table I.
Names and descriptions of cell lines
and subclones used in the study.
| Cell line | Description |
|---|
| HB2 | Cell line
established from human luminal mammary epithelial cells (14) |
| HB2/tnz34 | HB2 cells stably
transfected with the trk-neu hybrid receptor (5) |
| Tr-ep | HB2/tnz34 cells
stably transfected with the tetracycline repressor; epithelial
morphology |
| Tr-fib | Tr-ep cells having
undergone EMT as a result of c-erbB2 signalling (NGF treatment);
fibroblastoid morphology |
| TrE-ep1
TrE-ep5 | Tr-ep cells stably
expressing wild-type E-cadherin and GFP under the tetracycline
operator; epithelial morphology |
| TrE-fib | Clone of TrE-ep1
cells having undergone EMT as a result of c-erbB2 signalling (NGF
treatment) with concomitant ectopic E-cadherin expression (dox
treatment); fibroblastoid morphology |
|
TrEWV-ep | Tr-ep cells stably
expressing the E-cadherin WV156-157AA mutant and GFP under the
tetracycline operator; epithelial morphology |
| TrGFP-ep | Tr-ep cells stably
expressing the ‘empty’ IRES-GFP construct under the tetracycline
operator; epithelial morphology |
Ectopic expression of E-cadherin does not
affect c-erbB2-induced EMT
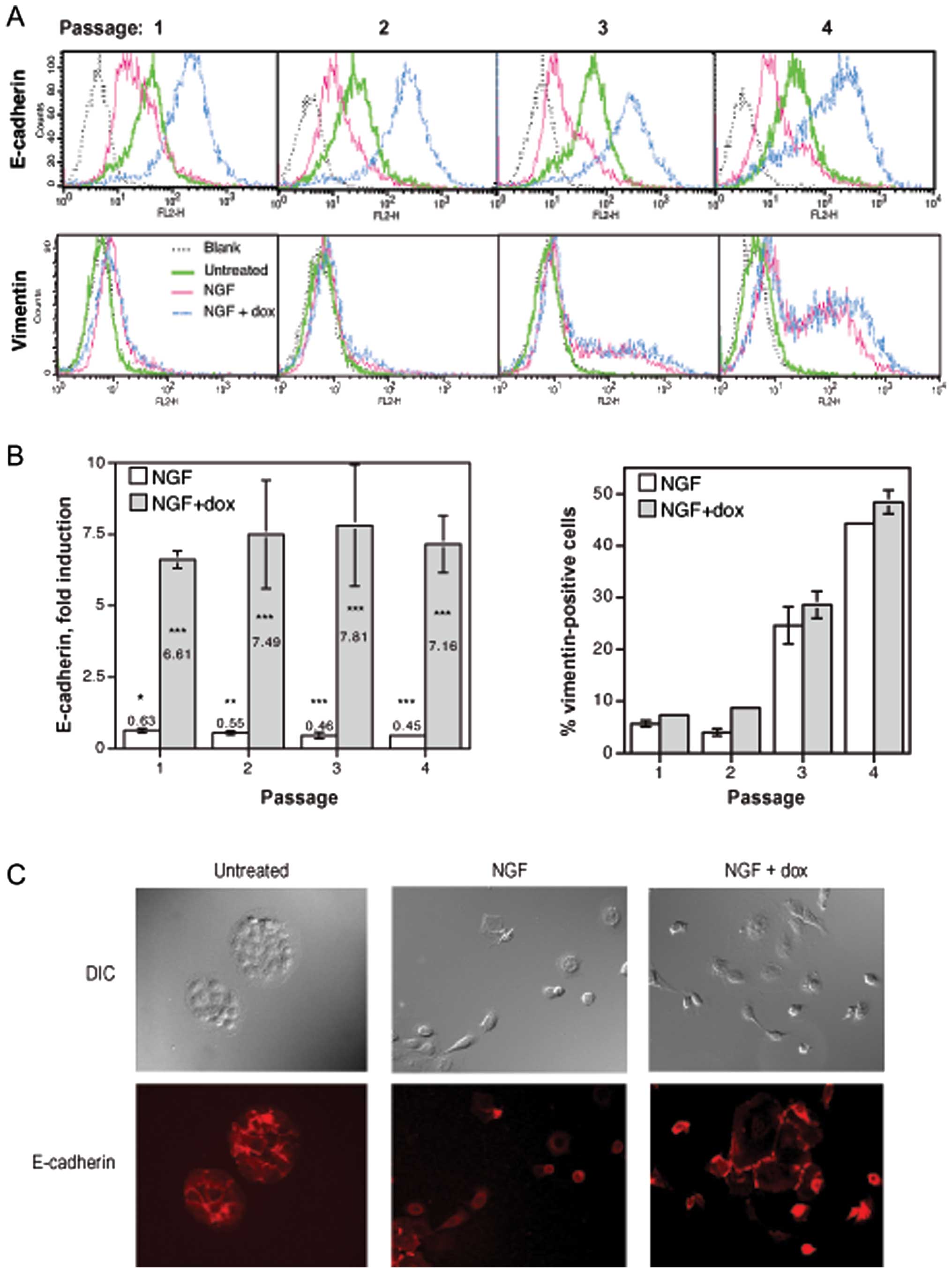
We next used the transfectant clone TrE-ep5 in order
to study the possible influence of ectopic E-cadherin expression
during the process of c-erbB2-induced EMT. Cells were grown at low
density for four passages with c-erbB2 signalling (NGF treatment),
with c-erbB2 signalling and ectopic E-cadherin expression (NGF and
dox treatment) or untreated. For each passage, EMT was monitored by
analysis of E-cadherin and vimentin expression as outlined under
EMT assays in Materials and methods. As expected, c-erbB2
signalling alone caused a reduction in E-cadherin expression and a
gradual increase in the appearance of vimentin-positive cells
(Fig. 2A and B, NGF). Combining
c-erbB2 signalling with dox treatment caused a substantial increase
in E-cadherin expression (also as expected), but the high level of
E-cadherin did not affect the increase in vimentin-positive cells
(Fig. 2A and B, NGF + dox) which
was highly similar to that seen with c-erbB2 signalling alone.
Ectopic E-cadherin expression thus did not appear to prevent the
process of c-erbB2-induced EMT.
We also used immunofluorescence microscopy to
analyse TrE-ep5 cells for E-cadherin surface expression (Fig. 2C). Treatments with NGF or NGF + dox
had been performed for 5 days in this experiment, which is the
earliest time point at which significant scattering can be
observed. Here, NGF-treated and NGF + dox-treated as well as
untreated cells all expressed detectable levels of E-cadherin.
Importantly, expression was evident also in scattering cells,
confirming that cell-cell dissociation was not prevented by
E-cadherin. At later passages, scattered cells treated only with
NGF largely lost E-cadherin expression indicating a transition to
the fully mesenchymal-like phenotype (data not shown).
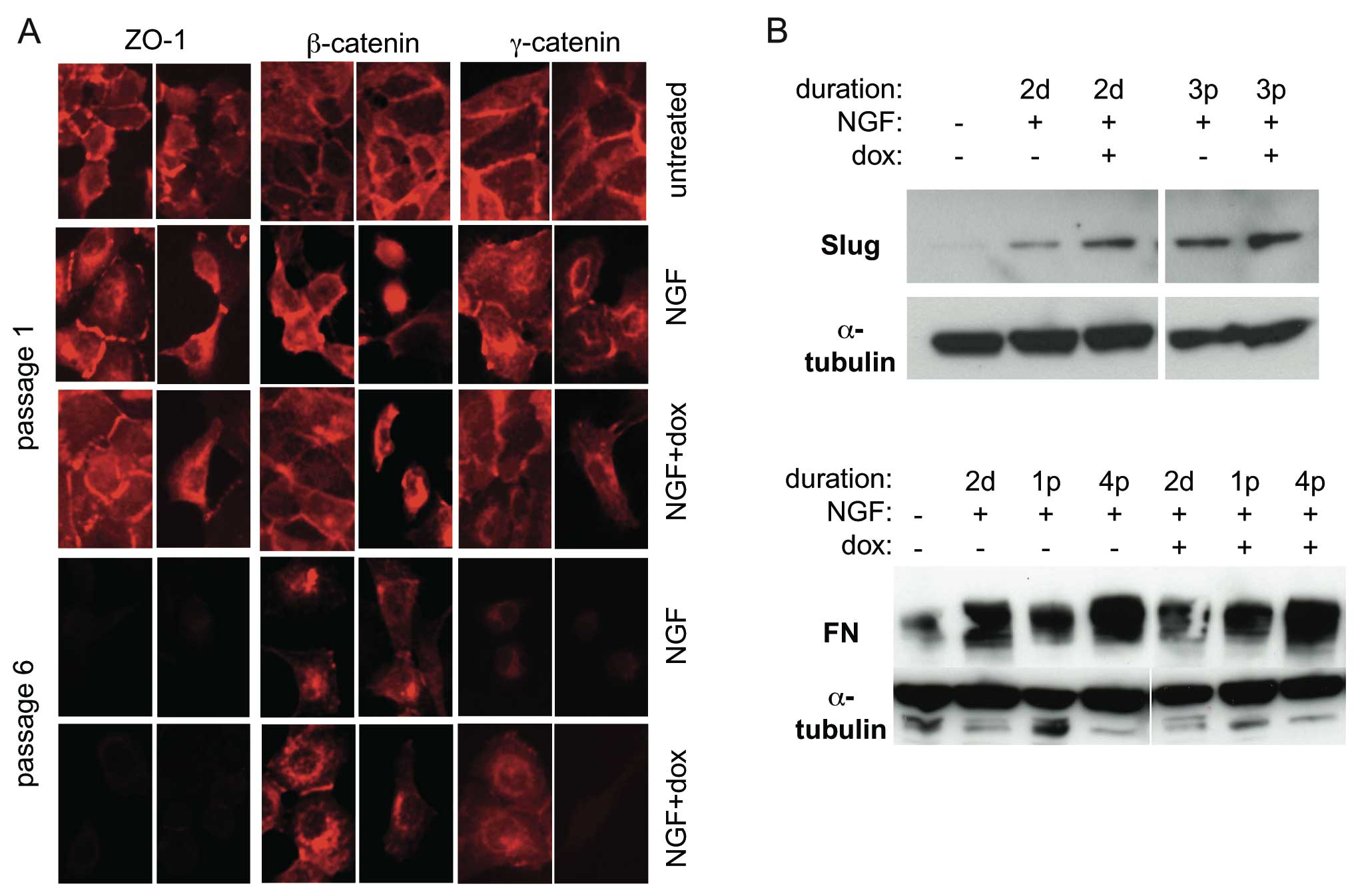
In order to further study the possible influence of
E-cadherin overexpression on EMT in this system, the expression of
EMT markers was analysed by immunofluorescence microscopy and
western blot analysis. Fig. 3A
shows expression and subcellular localisation of the tight
junction-associated protein ZO-1 as well as the
E-cadherin-associated cytoskeletal linker proteins β- and
γ-catenin. ZO-1 expression was maintained in recently scattered
cells but subsequently lost. β-catenin showed the redistribution
from plasma membrane-associated to cytoplasmic and
nuclear/perinuclear localisation typical of EMT, both when treated
with NGF and NGF + dox. γ-catenin was also relocalised but with a
concomitant sharp decrease in expression level at later passages.
These data suggest that ZO-1 and γ-catenin, like E-cadherin, are
not lost until after the initial cell-cell detachment phase of EMT.
In no case did dox-induced E-cadherin expression influence these
changes.
Western blot analysis showed an early (2 days after
induction of c-erbB2 signalling) upregulation of the EMT markers
Slug and fibronectin (Fig. 3B)
which persisted in subsequent passages. Again, E-cadherin
expression did not prevent these EMT-associated changes. Together,
these data indicate that ectopic expression of E-cadherin is not a
significant obstacle to EMT.
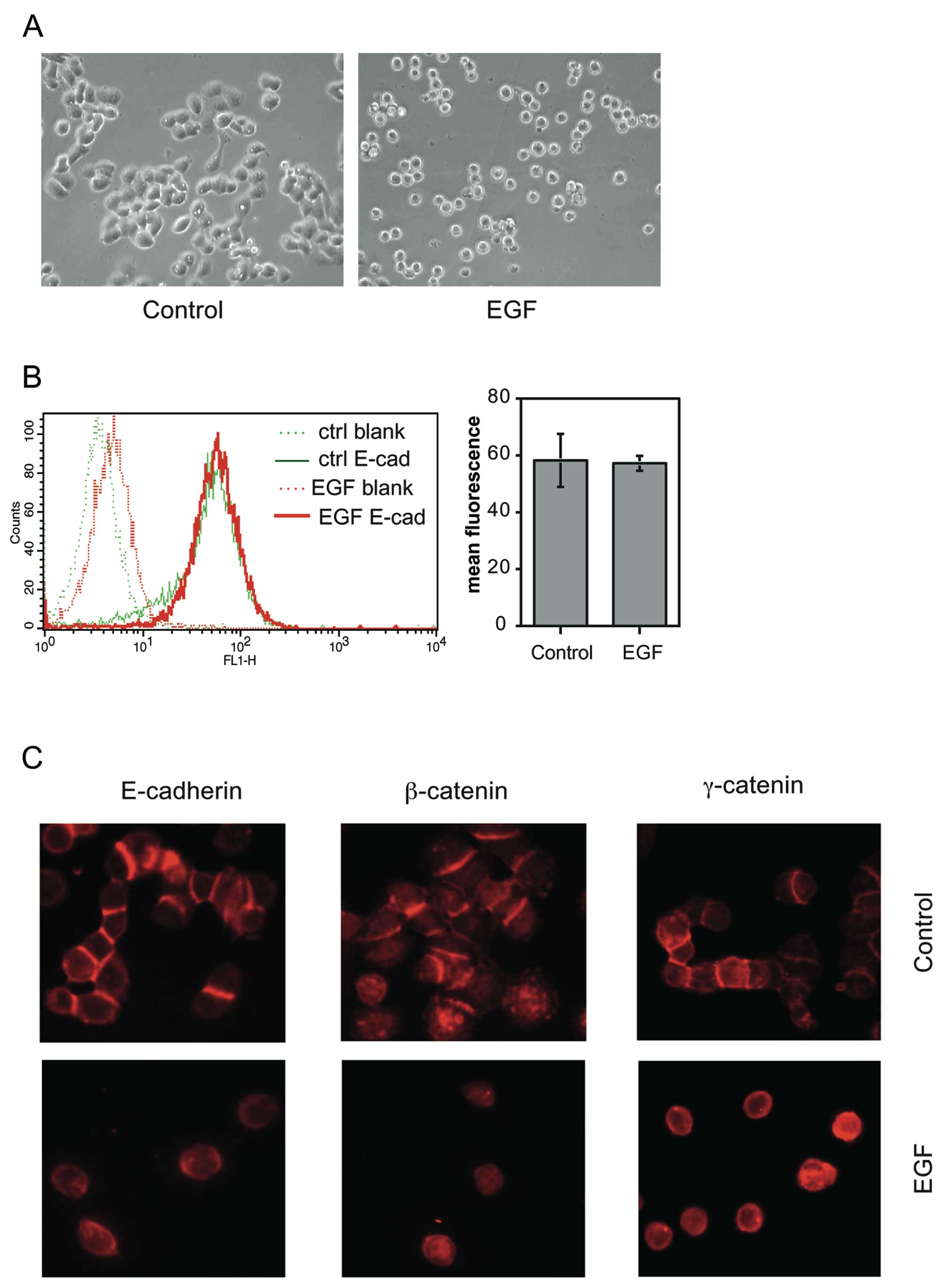
In order to verify that EMT-associated cell
scattering without reduction in cell surface E-cadherin expression
can occur also in other cell systems, we examined the early stages
of EMT in the mammary carcinoma cell line MDA-MB-468. This cell
line has previously been shown to undergo EMT upon EGF treatment
with only partial reduction in E-cadherin expression (21,22).
We found that overnight treatment of MDA-MB-468 cells with EGF
caused extensive cell-cell dissociation (Fig. 4A). Strikingly, cell surface
expression of E-cadherin (as measured by flow cytometry of
unpermeabilised cells) was virtually unchanged by EGF at this stage
(Fig. 4B). This finding was
confirmed by immunofluorescence microscopy, which furthermore
showed that β-and γ-catenin expression was also retained (Fig. 4C). These data suggest that cell
scattering without the prerequisite for downregulation of surface
E-cadherin expression may be a common feature in the early steps of
EMT caused by EGFR family receptor activation.
Properties of fibroblastic cells isolated
after c-erbB2-induced EMT in E-cadherin-overexpressing cells
In order to further study the properties of cells
having undergone EMT in the presence of E-cadherin, fibroblastic
clones were isolated from a population of TrE-ep1 cells that had
been treated with dox and NGF for several passages. One clone,
named TrE-fib, was selected for further study, as its induced
E-cadherin expression was comparable to that of parental epithelial
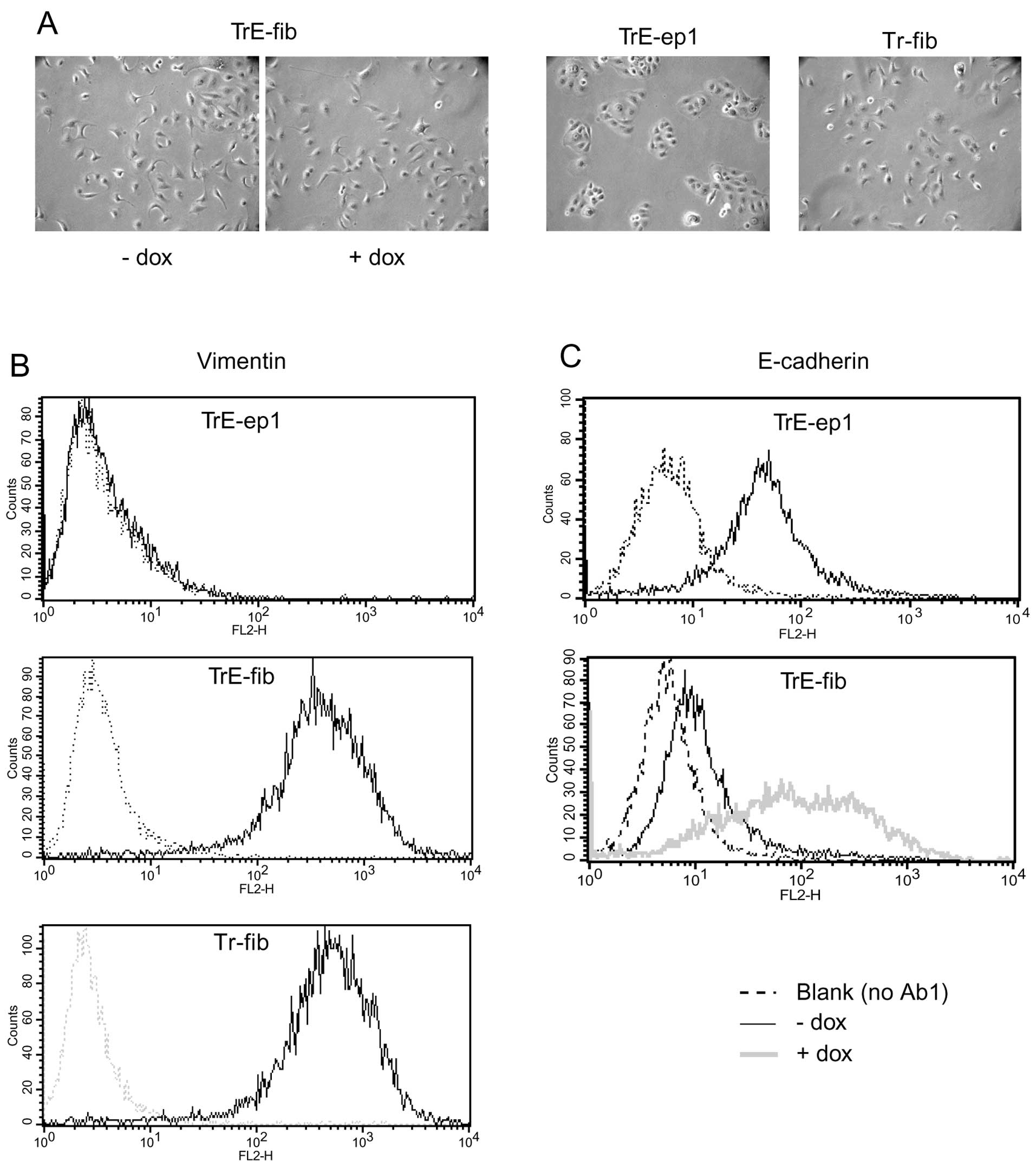
cells (see below). TrE-fib cells showed a fibroblastoid morphology
and strong vimentin expression (Fig.
5A and B) typical of HB2 cells after EMT (compare with Tr-fib
cells, fibroblastic cells obtained from Tr-ep cells by
c-erbB2-induced EMT but lacking the E-cadherin-IRES-GFP construct,
in Fig. 5A and B). E-cadherin
expression was negligible in dox-untreated cells, indicating that
the endogenous CDH1 gene had been silenced (Fig. 5C). These properties did not change
following prolonged culture without NGF or dox (data not shown),
suggesting an irreversible phenotypic conversion, in line with
previous results on EMT in HB2 cells (11). Upon dox treatment, E-cadherin
expression was readily induced (Fig.
5C). However, no changes in cell morphology were seen following
E-cadherin induction in this clone (Fig. 5A).
E-cadherin ectopically expressed in
fibroblastic cells after EMT is poorly attached to the
cytoskeleton
The apparent lack of effect of forced E-cadherin
expression on the phenotype of the fibroblastic cells emerging
after EMT raised the question whether E-cadherin was functional as
a cell adhesion molecule under these circumstances. We therefore
performed dissociation assays on cells from confluent layers of
TrE-ep5 and TrE-fib cells in the presence or absence of dox. In
striking contrast to the restoring effect on cell-cell adhesion
seen in dox-treated epithelial cells, dox-induced E-cadherin
expression in confluent fibroblastic TrE-fib cells failed to
influence intercellular adhesion (Fig.
6A). This result strengthened the notion that the function of
E-cadherin was impaired in the fibroblastic cells. We therefore
sought to elucidate the cause of this impairment.
Immunofluorescence microscopy of non-permeablilised, dox-treated
TrE-fib cells showed that E-cadherin was predominantly present at
cell-cell contacts in a manner roughly similar to that seen in
parental epithelial cells, although diffuse staining distributed
over the cell surface was also observed (Fig. 6B). This suggests that gross
abnormalities in the localisation of E-cadherin were not a cause of
malfunction.
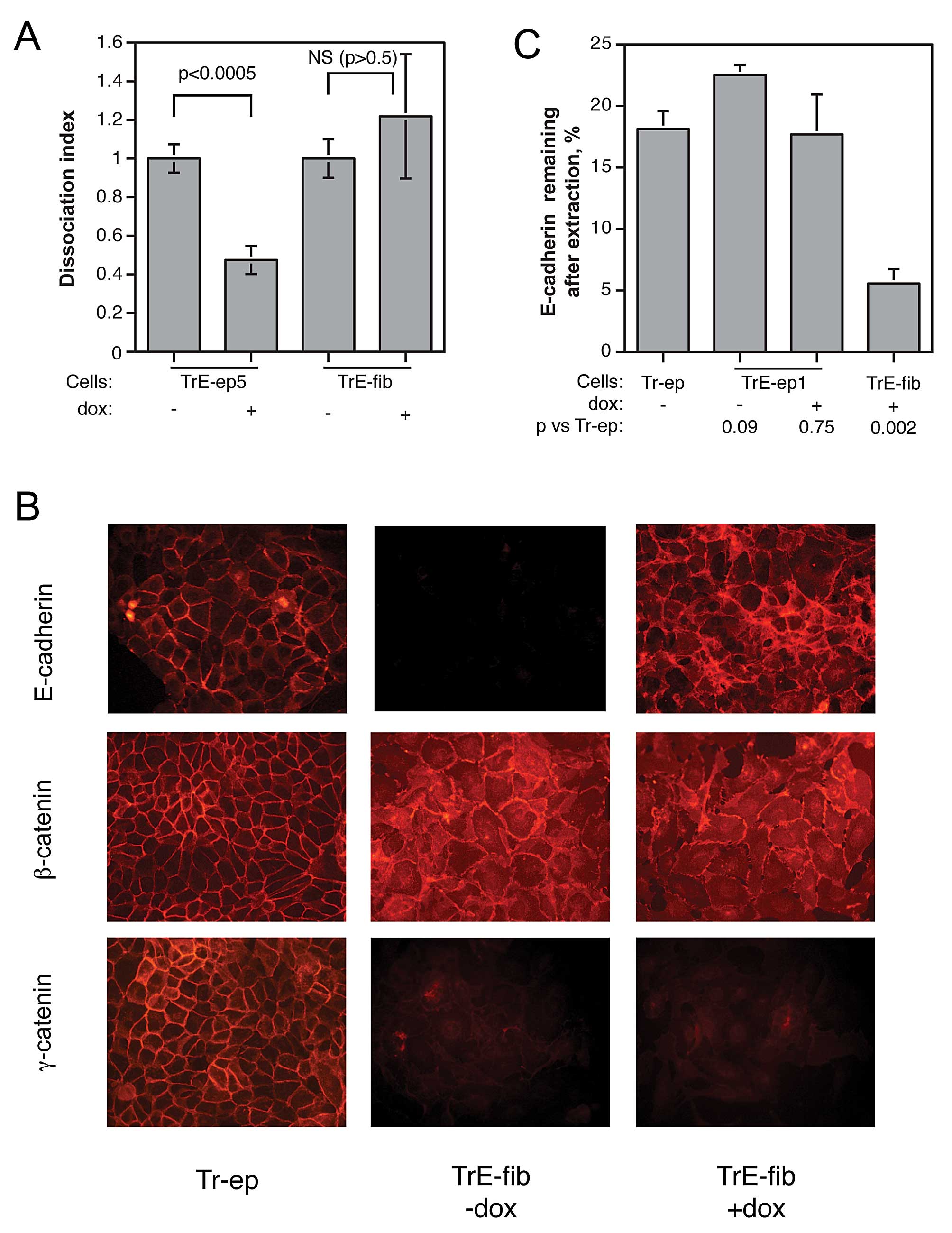 | Figure 6.Characterisation of fibroblastic
cells with respect to cell-cell adhesion and localisation and
cytoskeletal attachment of E-cadherin. (A) Influence of forced
E-cadherin expression on cell-cell adhesion, as measured by
dissociation assay, in epithelial TrE-ep5 and fibroblastic TrE-fib
cells (p-values obtained by Student’s t-test; NS, not significant).
(B) Immunofluorescence micrographs showing localisation of
E-cadherin, β-catenin and γ-catenin in Tr-ep cells and in TrE-fib
cells with and without dox treatment. For analysis of E-cadherin,
cells were not permeabilised before staining; thus, only cell
surface-bound E-cadherin is visualised. (C) Percentage of
E-cadherin detected after extraction of membrane lipids with Triton
X-100 in Tr-ep, TrE-ep1 and TrE-fib cells with and without dox
treatment as indicated. n, number of independent experiments; p,
p-value in Student’s t-test for comparison with results for Tr-ep
cells. Error bars, SEM. |
Another mechanism by which E-cadherin function could
be disrupted is loss of cytoskeletal attachment. The cytoskeletal
linker proteins β-catenin and γ-catenin were assayed in
immunofluorescence microscopy (Fig.
6B). β-catenin, as expected, showed increased cytoplasmic and
nuclear staining in the TrE-fib cells compared to control Tr-ep
cells, but also significant amounts close to the plasma membrane.
In contrast, γ-catenin expression was strongly decreased with
complete relocalisation to the cytoplasm and nucleus. These
EMT-induced changes in β- and γ-catenin expression and localisation
were not affected by ectopic E-cadherin expression (i.e., dox
treatment). We further examined the role of E-cadherin cytoskeletal
anchorage by measuring the percentage of surface-bound E-cadherin
still remaining after extraction of membrane lipids by Triton X-100
treatment. This procedure should remove cell surface proteins
attached only via interactions between the transmembrane domains
and the lipid bilayer, whereas proteins bound to the cytoskeleton
should be preferentially retained. As shown in Fig. 6C, the E-cadherin ectopically
expressed in fibroblastic cells isolated after EMT was much easier
to extract than E-cadherin in parental epithelial cells. These
results suggest that following EMT, ectopically expressed
E-cadherin has a poor cytoskeletal anchorage, offering a possible
explanation for its lack of effect on cell phenotype.
Expression of dominant-negative
E-cadherin fails to expedite EMT at high cell density
The density-dependent inhibition of EMT which has
previously been observed by us (11) and others (16,17)
indicates that cell-cell contact elicits intracellular signalling
events which impede the EMT process. The autoregulatory properties
of E-cadherin expression described by Conacci-Sorrell et al
(23) suggest that E-cadherin
itself may be a sensor for the degree of cell-contact experienced
by a cell. If this were indeed the case, interfering with the
proper function of E-cadherin as an adhesion molecule would
abrogate density-dependent inhibition of EMT and thus allow EMT to
occur at high cell density. In order to test this hypothesis, we
used the extracellular domain mutant WV156–157AA (WV), a
well-characterised mutant in which the adhesiveness of E-cadherin
is abolished (24). This WV mutant
was also expressed as a tetracycline-regulated bicistronic IRES-GFP
construct in Tr-ep cells. Stable transfectants harbouring this
construct were generated and the clone TrEWV-ep was
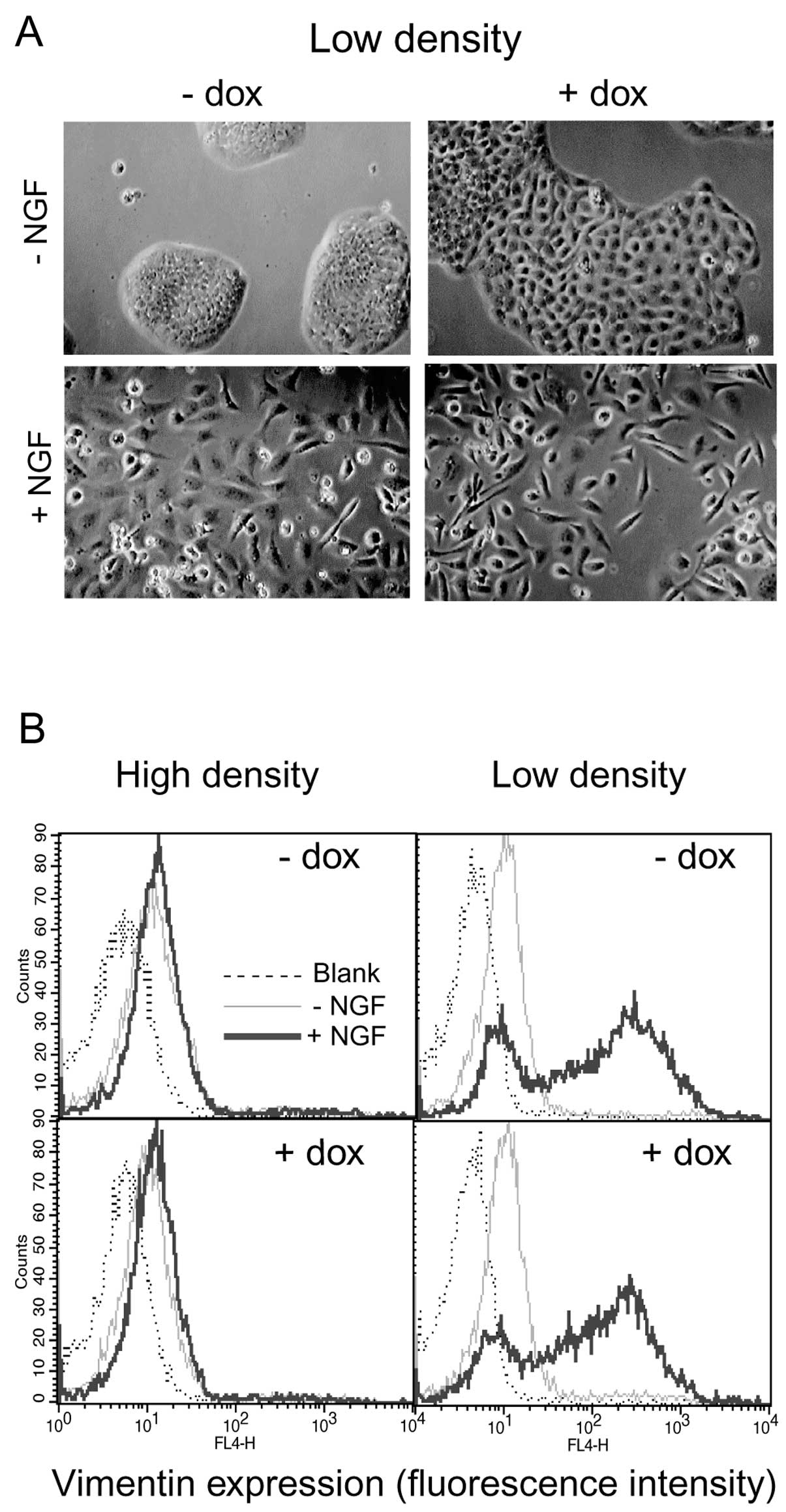
selected for further study. As shown in Fig. 7A, robust expression of the mutant
construct was induced upon dox treatment. A significant decrease in
cell-cell adhesion, as measured by cell dissociation assay
(Fig. 7B) was observed upon
induction of mutant expression.
When TrEWV-ep cells were grown at low
density, dox-induced mutant expression without concomitant c-erbB2
signalling caused some morphological changes in the cells: although
still largely epithelial in appearance, the dox-treated cells
occupied a much larger surface area per cell and the cell-cell
borders were more clearly visible (Fig. 8A, top row). At this low density,
NGF-induced c-erbB2 signalling caused the appearance of
fibroblast-like cells both with and without mutant expression
(Fig. 8A, lower panel). However,
c-erbB2-induced EMT at high cell density did not appear to be
derepressed upon expression of the non-adhesive E-cadherin.
Vimentin expression was virtually absent in cells grown at high
density irrespective of treatment with NGF, dox or both, whereas
NGF treatment caused the appearance of vimentin-positive cells at
low cell density, equally well with or without dox-induced mutant
expression (Fig. 8B). These data
do not support a role for E-cadherin as a sensor for cell-cell
contact in density-dependent inhibition of EMT.
Discussion
The possible role of EMT as a mechanism for
carcinogenesis, particularly in the generation of invasive,
metastatic and treatment-resistant cells, has been the subject of
intense study for the last few years and remarkable advances in our
understanding of these phenomena have been made (1). However, comparatively few recent
studies have been devoted to the interplay between cell-cell
contact/adhesion and EMT. Loss of E-cadherin expression is widely
seen as one of the most crucial and defining events in the progress
of EMT (25), and since EMT is
characterised by loss of cell-cell adhesion, it is intuitively easy
to assume that downregulation of E-cadherin expression is required
for EMT to occur. This notion has been reinforced by numerous
studies where E-cadherin has been re-expressed in carcinoma cells
yielding cells with variable degrees of restoration of epithelial
phenotype and non-malignant behaviour (26). In contrast, the necessity of loss
of E-cadherin expression for the actual progress of EMT has not
been extensively studied in a rigorous manner. Even less attention
has been paid to the mechanisms underlying density-dependent
inhibition of EMT, a highly interesting phenomenon noted in several
studies but rarely elaborated upon.
In an earlier publication (11), we noted that cells apparently
undergoing the first scattering phase in c-erbB2-induced EMT still
expressed E-cadherin, thus raising the question of whether loss of
E-cadherin is a cause or a consequence of EMT. In the present
study, we have addressed this question by creating cells which are
capable both of NGF-inducible c-erbB2 homodimer signalling and
dox-inducible E-cadherin expression. The E-cadherin ectopically
expressed in these cells consistently failed to prevent
c-erbB2-induced EMT from occurring (Fig. 2), strengthening the hypothesis that
the presence of E-cadherin is not an impediment to the progression
of EMT.
Our observation of extensive EGF-induced cell
scattering with unaltered E-cadherin surface expression in the
MDA-MB-468 cell line (Fig. 4)
strengthens the notion that surface-bound E-cadherin can be
rendered incompetent at maintaining cell-cell adhesion and that
EMT-associated cell scattering without requirement for prior
downregulation of E-cadherin is not a cell line-specific event.
Further investigation into this phenomenon and its possible
occurrence in other instances of EMT is clearly merited.
The finding that cell-cell dissociation occurs in
spite of the robust expression of a major cell-cell adhesion
molecule suggests that the adhesive function of E-cadherin becomes
compromised during EMT, before expression is lost. Several
mechanisms could account for such an inactivation, including
weakening of the cytoskeletal attachment or increased turnover or
endocytosis of E-cadherin. Influence from cytoskeletal
rearrangements on E-cadherin function is suggested by our results
with fibroblastic cells: here, E-cadherin expression had no effect
on cell-cell adhesion and the expressed E-cadherin was much more
readily extracted from the plasma membrane than in the epithelial
cells (Fig. 6). It is possible
that some component of the linkage between E-cadherin and the
cytoskeleton was inactivated in the fibroblastic cells, e.g. by
phosphorylation or ubiquitinylation of E-cadherin or β-catenin.
However, we have failed to detect changes in β-catenin
phosphorylation using site-specific mAbs (data not shown) although
the interpretation of such data is complicated by the
non-synchronous nature of c-erbB2-induced EMT in the system
studied.
Another cytoskeletal mechanism, more supported by
our data, could be envisioned where the combined influences of
c-erbB2 and low cell density trigger extensive cytoskeletal
rearrangements which make the cells unable to support strong
cell-cell adhesion, even in the presence of E-cadherin expression.
This suggestion is supported by previous data showing a dramatic
destabilisation of the cortical cytoskeleton following c-erbB2
activation in HB2 cells (27).
It is interesting to note that onset of cell
scattering prior to E-cadherin downregulation in EMT has been
observed in another system (28);
there, E-cadherin internalisation was offered as an explanation
(29). In our case, net removal of
E-cadherin from the plasma membrane is highly unlikely to be the
cause of impaired cell-cell adhesion as the measurements of
E-cadherin expression were made by flow cytometry of
non-permeabilised cells, which only detects surface-bound antigens,
although adherens junction destabilisation by increased E-cadherin
recycling cannot be excluded. For instance, the combination of Par3
downregulation and c-erbB2 signalling caused invasive and
metastatic behaviour in mammary epithelial cells which was
attributed to increased F-actin and E-cadherin turnover (30). Other studies have also reported the
persistence of E-cadherin expression during EMT (22,31,32),
but without direct reference to cell scattering. It has even been
suggested that surface expression of E-cadherin promotes
EMT-inducing β-catenin signalling (33). Very recently, Hollestelle et
al (34) have showed a lack of
consistent correlation between E-cadherin loss and expression of
EMT markers in a survey of 38 breast cancer cell lines as well as
in clinical tumour samples. In the same study, restoring E-cadherin
expression failed to influence the mesenchymal-like phenotype of
E-cadherin-negative cell lines. While not addressing the role of
E-cadherin expression during the actual EMT process, those data
strongly support the conclusions from the present study.
The other hypothesis tested here concerns the
density-dependent inhibition of EMT observed earlier by us and
several other researchers. This is a highly interesting phenomenon,
especially in the light of the proposed significance of EMT in
cancer progression: if invasion and metastasis indeed are dependent
on EMT-like phenomena, then a mechanism which inhibits EMT would be
of great potential value in combating cancer cell dissemination. A
key question in elucidating the signalling pathway responsible for
density-dependent EMT inhibition regards the nature of the sensor
for cell cell-contact. E-cadherin itself has recently been
identified as a mechanosensor (35) and the findings by Conacci-Sorrell
et al (23) suggest that
E-cadherin engagement is important for the maintenance of its own
expression. We therefore tested the influence of expression of a
dominant-negative E-cadherin mutant on our system. According to our
working hypothesis, that E-cadherin engagement suppresses EMT at
high cell density, the expression of non-adhesive E-cadherin mutant
would relieve this suppression. Although we found a significant
reduction in cell-cell adhesion upon expression of this mutant,
c-erbB2-induced EMT was still inhibited at high cell density as far
as could be measured in our assays. The residual cell-cell adhesion
seen upon expression of the E-cadherin mutant could be attributed
to cell-cell adhesion molecules other than E-cadherin, but our data
cannot rule out an incomplete inactivation of E-cadherin. This
should also be kept in mind when interpreting the results. An
alternative mechanism for density-dependent inhibition of EMT was
found to operate in mouse mammary epithelial cells, which underwent
EMT upon overexpression of matrix metalloproteinase 3 (MMP-3); in
these cells, the cytoskeletal effects of cell crowding seemed to
serve as an anti-EMT signal, as limiting the cell area by growing
single cells on a micropatterned substrate was sufficient to
prevent MMP-3- (but not TGF-β-) induced EMT (36). It is possible that cytoskeletal
effects are more important for regulation of EMT in our system as
well. In summary, our results indicate that E-cadherin does not
appear to have a key role in preventing c-erbB2-induced EMT, either
as a physical obstacle to cell-cell separation at low density or as
an mediator of density-dependent inhibition of EMT in confluent
cells.
Acknowledgements
The authors thank Edvin Atic and Oskar
Erlandsson for their contributions with plasmid construction and
establishment of cell lines. This study was supported by the
Swedish Cancer Fund, the Swedish Research Council, Magnus Bergvalls
Foundation, IngaBritt and Arne Lundberg’s Foundation, Carl
Trygger’s Foundation and Assar Gabrielsson’s Fund.
References
|
1.
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Hugo HJ, Kokkinos MI, Blick T, Ackland ML,
Thompson EW and Newgreen DF: Defining the E-cadherin repressor
interactome in epithelial-mesenchymal transition: The PMC42 model
as a case study. Cells Tissues Organs. 193:23–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Klymkowsky MW and Savagner P:
Epithelial-mesenchymal transition. A cancer researcher’s conceptual
friend and foe. Am J Pathol. 174:1588–1593. 2009.PubMed/NCBI
|
|
4.
|
Spaderna S, Schmalhofer O, Hlubek F, et
al: A transient, EMT-linked loss of basement membranes indicates
metastasis and poor survival in colorectal cancer.
Gastroenterology. 131:830–840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Baeckström D, Lu PJ and
Taylor-Papadimitriou J: Activation of the a2b1 integrin prevents
c-erbB2-induced scattering and apoptosis of human mammary
epithelial cells in collagen. Oncogene. 19:4592–4603.
2000.PubMed/NCBI
|
|
6.
|
Lee S-A, Lee S-Y, Cho I-H, et al:
Tetraspanin TM4SF5 mediates loss of contact inhibition through
epithelial-mesenchymal transition in human hepatocarcinoma. J Clin
Invest. 118:1354–1366. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mani SA, Guo W, Liao M-J, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Maiden SL and Hardin J: The secret life of
α-catenin: Moonlighting in morphogenesis. J Cell Biol. 195:543–552.
2011.
|
|
9.
|
Nelson WJ: Regulation of cell-cell
adhesion by the cadherincatenin complex. Biochem Soc T.
036:149–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Semb H: The tumor-suppressor function of
E-cadherin. Am J Hum Genet. 63:1588–1593. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Jenndahl LE, Isakson P and Baeckström D:
c-erbB2-induced epithelial-mesenchymal transition in mammary
epithelial cells is suppressed by cell-cell contact and initiated
prior to E-cadherin downregulation. Int J Oncol. 27:439–448.
2005.
|
|
12.
|
Jenndahl LE, Taylor-Papadimitriou J and
Baeckström D: Characterization of integrin and anchorage dependence
in mammary epithelial cells following c-erbB2-induced
epithelial-mesenchymal transition. Tumour Biol. 27:50–58. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Spears M, Taylor K, Munro A, et al: In
situ detection of HER2:HER2 and HER2:HER3 protein-protein
interactions demonstrates prognostic significance in early breast
cancer. Breast Cancer Res Treat. 132:463–470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Berdichevsky F, Alford D, D’Souza B and
Taylor-Papadimitriou J: Branching morphogenesis of human mammary
epithelial cells in collagen gels. J Cell Sci. 107:3557–3568.
1994.PubMed/NCBI
|
|
15.
|
Sachs M, Weidner KM, Brinkmann V, et al:
Motogenic and morphogenic activity of epithelial receptor tyrosine
kinases. J Cell Biol. 133:1095–1107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Paumelle R, Tulasne D, Leroy C, Coll J,
Vandenbunder B and Fafeur V: Sequential activation of ERK and
repression of JNK by scatter factor/hepatocyte growth factor in
Madin-Darby canine kidney epithelial cells. Mol Biol Cell.
11:3751–3763. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Valles AM, Tucker GC, Thiery JP and Boyer
B: Alternative patterns of mitogenesis and cell scattering induced
by acidic FGF as a function of cell density in a rat bladder
carcinoma cell line. Cell Regul. 1:975–988. 1990.PubMed/NCBI
|
|
18.
|
De Wever O, Pauwels P, De Craene B, et al:
Molecular and pathological signatures of epithelial-mesenchymal
transitions at the cancer invasion front. Histochem Cell Biol.
130:481–494. 2008.
|
|
19.
|
Rees JRE, Onwuegbusi BA, Save VE, Alderson
D and Fitzgerald RC: In vivo and in vitro evidence for transforming
growth factor-β1-mediated epithelial to mesenchymal transition in
esophageal adenocarcinoma. Cancer Res. 66:9583–9590. 2006.
|
|
20.
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: A Laboratory Manual. Cold Spring Harbor
Laboratory Press; Cold Spring Harbor, NY: 1989
|
|
21.
|
Lo H-W, Hsu S-C, Xia W, et al: Epidermal
growth factor receptor cooperates with signal transducer and
activator of transcription 3 to induce epithelial-mesenchymal
transition in cancer cells via up-regulation of TWIST gene
expression. Cancer Res. 67:9066–9076. 2007. View Article : Google Scholar
|
|
22.
|
Davis FM, Kenny PA, Soo ETL, et al:
Remodeling of purinergic receptor-mediated Ca2+signaling
as a consequence of EGF-induced epithelial-mesenchymal transition
in breast cancer cells. PLoS One. 6:e234642011.PubMed/NCBI
|
|
23.
|
Conacci-Sorrell M, Simcha I, Ben-Yedidia
T, Blechman J, Savagner P and Ben-Ze’ev A: Autoregulation of
E-cadherin expression by cadherin-cadherin interactions: the roles
of beta-catenin signaling, Slug, and MAPK. J Cell Biol.
163:847–857. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Chitaev N and Troyanovsky S: Adhesive but
not lateral E-cadherin complexes require calcium and catenins for
their formation. J Cell Biol. 142:837–846. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Baranwal S and Alahari SK: Molecular
mechanisms controlling E-cadherin expression in breast cancer.
Biochem Biophys Res Commun. 384:6–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Frixen UH, Behrens J, Sachs M, et al:
E-cadherin-mediated cell-cell adhesion prevents invasiveness of
human carcinoma cells. J Cell Biol. 113:173–185. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hedjazifar S, Jenndahl LE, Shimokawa H and
Baeckström D: PKB mediates c-erbB2-induced epithelial
β1integrin conformational inactivation through
Rho-independent F-actin rearrangements. Exp Cell Res. 307:259–275.
2005.PubMed/NCBI
|
|
28.
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor β-1 induces snail transcription factor
in epithelial cell lines. J Biol Chem. 278:21113–21123. 2003.
|
|
29.
|
Khew-Goodall Y and Wadham C: A perspective
on regulation of cell-cell adhesion and epithelial-mesenchymal
transition: Known and novel. Cells Tissues Organs. 179:81–86. 2005.
View Article : Google Scholar
|
|
30.
|
Xue B, Krishnamurthy K, Allred DC and
Muthuswamy SK: Loss of Par3 promotes breast cancer metastasis by
compromising cell-cell cohesion. Nat Cell Biol. 15:189–200. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Cao C, Chen Y, Massod R, Sinha UK and
Kobielak A: α-catulin marks the invasion front of squamous cell
carcinoma and is important for tumor cell metastasis. Mol Cancer
Res. 10:892–903. 2012.
|
|
33.
|
Howard S, Deroo T, Fujita Y and Itasaki N:
A positive role of cadherin in Wnt/β-catenin signalling during
epithelial-mesenchymal transition. PLoS One. 6:e238992011.
|
|
34.
|
Hollestelle A, Peeters J, Smid M, et al:
Loss of E-cadherin is not a necessity for epithelial to mesenchymal
transition in human breast cancer. Breast Cancer Res Treat.
138:47–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Le Duc Q, Shi Q, Blonk I, et al: Vinculin
potentiates E-cadherin mechanosensing and is recruited to
actin-anchored sites within adherens junctions in a myosin
II-dependent manner. J Cell Biol. 189:1107–1115. 2010.PubMed/NCBI
|
|
36.
|
Nelson CM, Khauv D, Bissell MJ and Radisky
DC: Change in cell shape is required for matrix
metalloproteinase-induced epithelial-mesenchymal transition of
mammary epithelial cells. J Cell Biochem. 105:25–33. 2008.
View Article : Google Scholar
|