Introduction
Fucoidan is a polysaccharide from the cell wall of
brown seaweed containing a substantial percentage of L-fucose and
sulfate ester groups (1). It has
numerous pharmacological properties as an anti-coagulant,
anti-tumor, anti-inflammatory and anti-oxidant agent (2–5). In
particular, its anti-tumor activity has recently attracted
considerable attention (6,7) as a potential therapeutic agent for
cancer. However, the anti-senescence effects and detailed mechanism
of action remains poorly understood in normal hepatic cells.
Treatment options for liver cancer focus primarily
on chemotherapy (8). While these
chemotherapeutic regimens are well established, the major drawback
remains their limited specificity for the tumor site.
Chemotherapeutic drugs, such as cisplatin, induce senescence by
enhancing the activity of the tumor suppressor p16INK4a
in cancer and normal cells, which results in increased toxicity to
normal cells, which require balanced expression of
p16INK4a for growth and differentiation to maintain cell
homeostasis. Thus, cancer cell-specific expression of
p16INK4a would be a valuable therapeutic strategy for
cancer treatment (9).
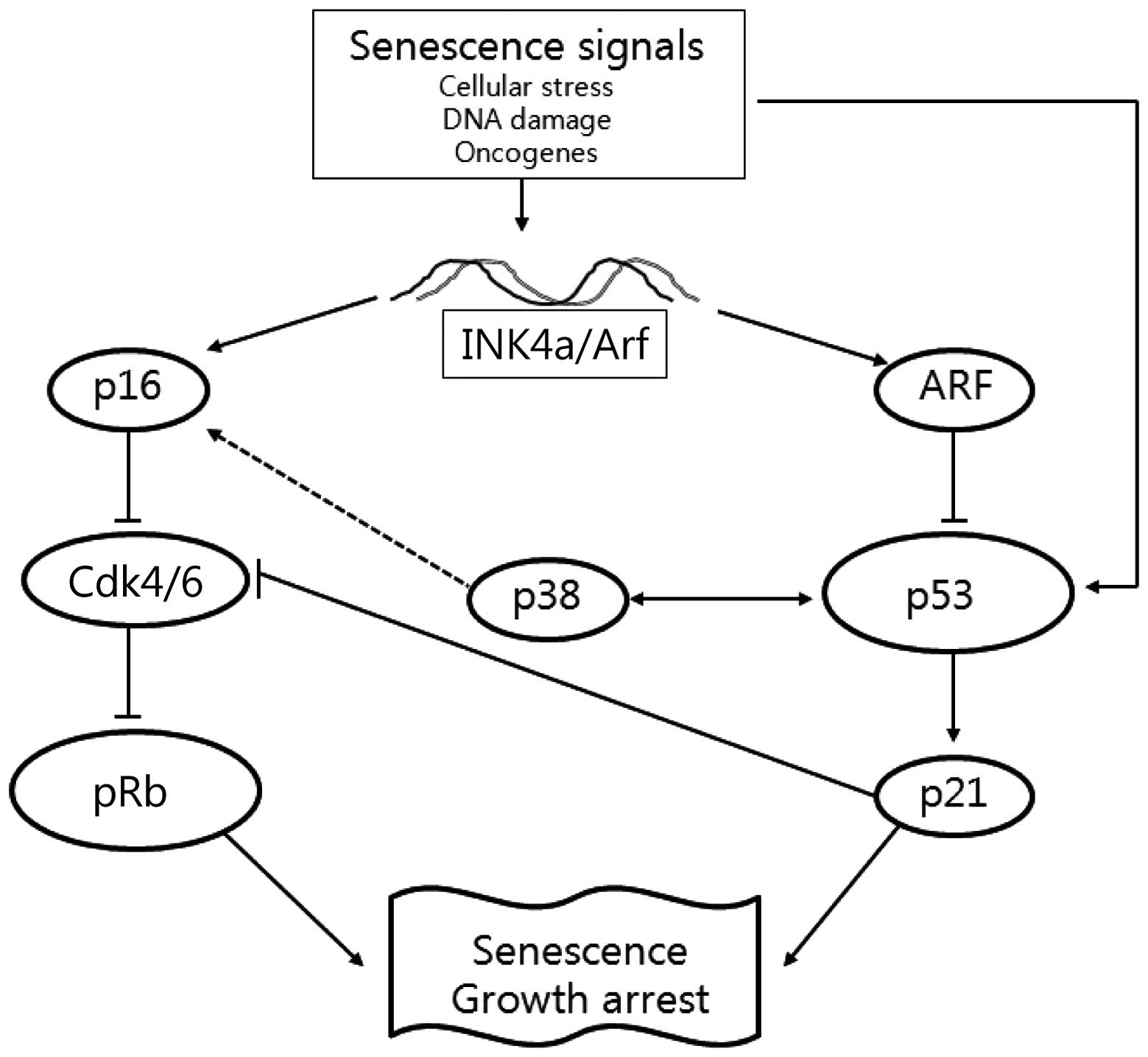
Cellular senescence, leading to cell death through
the prevention of regular cell renewal, is associated with the
upregulation of p16INK4a in most mammalian tissues
(10). This process requires
activation of several signaling pathways, including phosphorylated
retinoblastoma (pRb) and p14Arf-p53 (11). Phosphorylation of the Rb protein
results in increased p16INK4a expression to inhibit
cyclin-dependent kinase (Cdk) 4/6. This leads to increased levels
of hypophosphorylated Rb that decrease p16INK4a
expression (12). Although there
is a feedback loop between p16INK4a and Rb,
p16INK4a expression does not change appreciably during
the cell cycle to correlate with the activation status of Rb
(13). However, increased
expression of p16INK4a leads to senescence and cancer
cells inactivate p16INK4a by homozygous deletion or
hypermethylation to overcome its effects. The tumor suppressor
p14Arf (p19Arf in mouse cells) has emerged as
an interesting candidate linking transformation and senescence
responses. Arf is the second protein, in addition to p16, expressed
from the INK4a/Arf locus, but bears no homology to
p16 or any other cyclin-dependent kinase inhibitors (CKIs)
(14). Arf neutralizes the ability
of MDM2 to promote p53 degradation, leading to the stabilization
and accumulation of p53 (15).
Increased expression of Arf also causes growth arrest, one of the
hallmarks of premature senescence (16). Thus, the tumor suppressor proteins
of the INK4a/Arf locus function in distinct
anticancer pathways as p16INK4a directly regulates pRb,
while p14Arf directly regulates p53 and indirectly
regulates pRb (Fig. 1).
Inactivation of the p53 and pRb pathways through a variety of
mechanisms occurs in the majority of, if not all, human cancers.
Although these pathways play important roles in differentiation,
development and DNA repair, the INK4a/Arf locus
responds largely to aberrant growth or oncogenic stress. Therefore,
the INK4a/Arf locus appears to function as a
dual-pronged brake to malignant growth, which engages two potent
anti-proliferative pathways represented by p16INK4a-pRb
and p14Arf-p53 signaling (17).
The p38 mitogen-activated protein kinase (MAPK)
pathway regulates cellular processes that directly contribute to
tumor suppression, including oncogene-induced senescence and
replicative senescence (18), as
well as proliferation and tumorigenesis (19). Senescence is also accompanied by
markers associated with replicative exhaustion of normal cells,
such as senescence-associated β-galactosidase (SA-β-gal) activity
and the induction of p21, p16INK4a and/or
p14Arf (16,20). A previous study suggested that
expression of α-2-macroglobulin (α2M) can be used as a
biomarker of aging in cultured human fibroblasts, can be measured
easily by reverse-transcriptase polymerase chain reaction (RT-PCR)
with a limited sample, and is a more suitable biomarker candidate
of aging then the well-known senescence-associated genes such as
p16INK4a (21). In this
study, we determined the expression of α2M as a
biomarker of cellular senescence to assess the anti-senescence
effects of fucoidan in normal human liver cells.
Materials and methods
Fucoidan
Commercially available fucoidan purified form F.
vesiculosus (F5631) was purchased from Sigma-Aldrich, Inc. (St.
Louis, MO, USA).
Cell culture
The human hepatocellular carcinoma cell line (HepG2;
HB-8065) and the human normal liver cell line (Chang-L; CCL-13)
were obtained from the American Type Culture Collection (ATCC, GA,
USA). Cells were cultured in MEM medium supplemented with 10% fetal
bovine serum, penicillin (100 U/ml) and streptomycin (100
μg/ml) at 37°C in a humidified 5% CO2
incubator.
Cell viability assay
Cell viability was determined by the
Cyto™ cell viability assay kit (LPS solution, Daejeon,
Korea). Cells were seeded at a density of 1×104
cells/well in a 96-well plate. After 24 h, the cells were treated
with the phosphate-buffered saline (PBS) vehicle or 100, 250 and
500 μg/ml fucoidan. The Cyto solution was added to each well
and incubated for 4 h. The formazan product was estimated by
measuring absorbance at 450 nm in a microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA). The viability of
fucoidan-treated cells was expressed as a percentage of
vehicle-treated control cells considered 100% viable.
Cell cycle analysis
Cells were seeded at a density of 1×104
cells/well and treated with various concentrations of fucoidan for
24 h. Control and treated cells were harvested, washed in cold PBS,
fixed in 70% ethanol and stored at 4°C. The resulting cells were
stained with 200 μl of Muse™ cell cycle reagent
at room temperature for 30 min in the dark before analysis. DNA
content was assessed by Muse™ cell analyzer (EMD
Millipore Co., CA, USA).
Apoptosis analysis
The Muse Annexin V and Dead cell kit (EMD Millipore
Co., MA, USA) was used for the apoptosis assay. HepG2 cells and
Chang-L cells plated at a density of 1×106 cells/well
were treated with varying concentrations of fucoidan for 24 h.
Cells were harvested by trypsinization, washed twice with PBS, and
re-suspended in Annexin V and 7-aminoactinomycin D (7-AAD) for 20
min at room temperature in the dark. The cells were evaluated
immediately by Muse cell analyzer. The percentage of apoptotic
cells was assessed using the Muse™ software.
Western blotting
Samples were analyzed by western blotting, as
described previously (20).
Whole-cell lysates were prepared by lysing cell pellets in a NETN
lysis buffer [0.5% Nonidet P-40, 1 mM EDTA, 50 mM Tris (pH 7.4), 12
mM NaCl, 1 mM DTT, 10 mM NaF, 2 mM Na3VO4, 1
mM PMSF]. Samples (50 μg) were resolved by SDS-PAGE and
transferred to Immobilion-P transfer membranes (Millipore Co., MA
USA). The primary antibodies used included monoclonal
anti-p16INK4a, polyclonal anti-Cdk4 and -Cdk6,
polyclonal anti-p21, polyclonal anti-p38, monoclonal anti-p-p38,
polyclonal anti-p53 and polyclonal anti-pRb (1:1,000, Santa Cruz
Biotechnology, Inc., TX, USA). Membranes were washed and incubated
with the corresponding HRP-conjugated secondary antibody at
1:10,000. Bound secondary antibody was detected using a
chemiluminescence substrate (Advansta, Menlo Park, CA, USA) and
visualized on GeneSys imaging system (SynGene Synoptics, Ltd.,
London, UK).
Real-time PCR
Cells were harvested 24 h after treatment with PBS,
100, 250 or 500 μg/ml fucoidan. Total-RNA was extracted from
HepG2 and Chang-L cells using the QIAzol lysis reagent (Qiagen
Sciences, Inc., Germantown, MD, USA). RNA quality was evaluated by
measuring absorbance at 260 and 280 nm to calculate the
concentration and to assess the purity of RNA, respectively.
Agarose electrophoresis was used to detect RNA purity and
integrity.
The GoScript™ Reverse Transcription
System (Promega Corp., Madison, WI, USA) was used to prepare cDNA
according to the manufacturer’s instructions; the samples were
stored at −20°C. The quality of cDNA was assessed by amplifying an
internal reference gene, glyceraldehyde 3-phosphate dehydrogenase
(GAPDH), by PCR and the results were confirmed by 2% agarose gel
electrophoresis. The products were examined by computerized gel
imaging system (Bio-Rad, Hercules, CA, USA).
Quantitative PCR was conducted in 20 μl
reactions containing QuantiMix SYBR kit (PhilKorea Technology,
Inc., Daejeon, Korea) using the Illumina Eco™ real-time
PCR system (Illumina, Inc., Hayward, CA, USA). The oligonucleotide
primers for p16INK4a, p14Arf, p21, p53, p38,
α2M and GAPDH are shown in Table I. Reaction mixtures were incubated
for an initial denaturation at 95°C for 10 min followed by 40
cycles of 95°C for 15 sec, 55°C for 15 sec and 72°C for 15 sec. For
each sample, the expression level of each mRNA was quantified as
the cycle threshold difference (ΔCt) to GAPDH mRNA. All
reactions were performed in triplicate and repeated with two
independent experiments.
 | Table I.Primers for real-time PCR. |
Table I.
Primers for real-time PCR.
| Forward | Reverse |
|---|
| GAPDH | TGC ACC ACC AAC TGC
TTA GC | GGC ATG GAC TGT GGT
CAT GAG |
|
p16INK4a | GCC CAA CGC CCC GAA
CTC TTT C | TGA AGC TAT GCC CGT
CGG TCT G |
|
p14Arf | GGA GGC GGC GAG AAC
AT | TGA ACC ACG AAA ACC
CTC ACT |
| p53 | GAG GGA TGT TTG GGA
GAT GTA A | CCC TGG TTA GTA CGG
TGA AGT G |
| p38 | GAG GAT GCC AAG CCA
TG | TCT TAT CTG AGT CCA
ATA CAA GCA TC |
| p21 | CCG CGA CTG TGA TGC
GCT AAT G | CTC GGT GAC AAA GTC
GAA GTT C |
| α2M | TTG TCA GTG ACG TTT
GCC TC | CAA ACT CAT CCG TCT
CGT AG |
Statistical analysis
SPSS software (Chicago, IL, USA) was used to perform
the statistical analysis. For comparisons for more than two groups,
data were analyzed by one-way analysis of variance (ANOVA),
followed by Duncan’s test for multiple comparisons. For all tests,
P<0.05 was considered to indicate significance.
Results
Effects of fucoidan on cell
viability
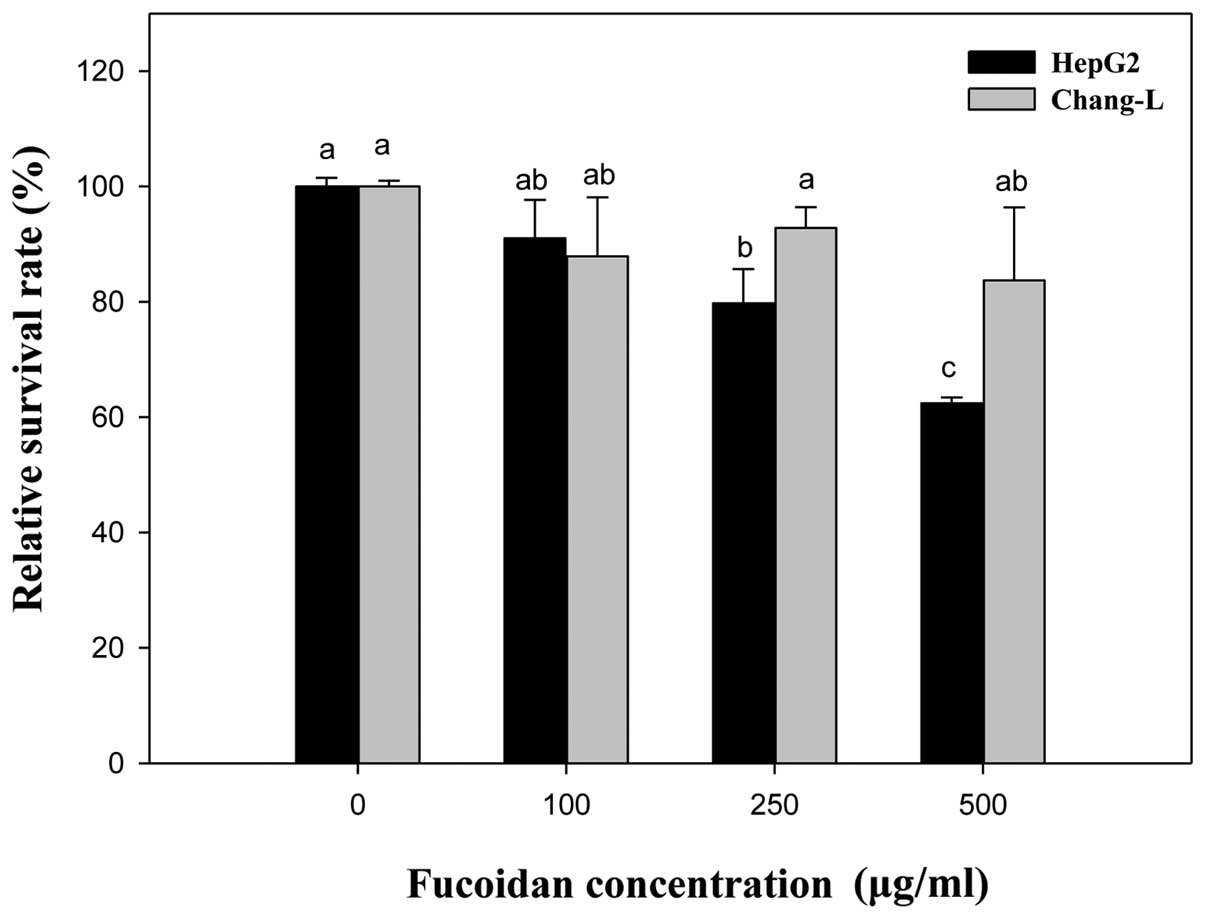
To verify the effects of fucoidan on cell viability,
cells were treated with fucoidan at the concentrations indicated
for 24 h (Fig. 2). Compared to the
untreated controls, Chang-L cells exhibited no cytotoxicity at
concentrations between 0 and 500 μg/ml. In contrast,
proliferation of HepG2 cells was dose-dependently inhibited by
fucoidan treatment (250 μg/ml, 79.75±9.94% inhibition; 500
μg/ml, 62.43±1.0% inhibition). Thus, the ability of fucoidan
to inhibit proliferation was significantly different between HepG2
and Chang-L cells.
Effects of fucoidan on apoptosis
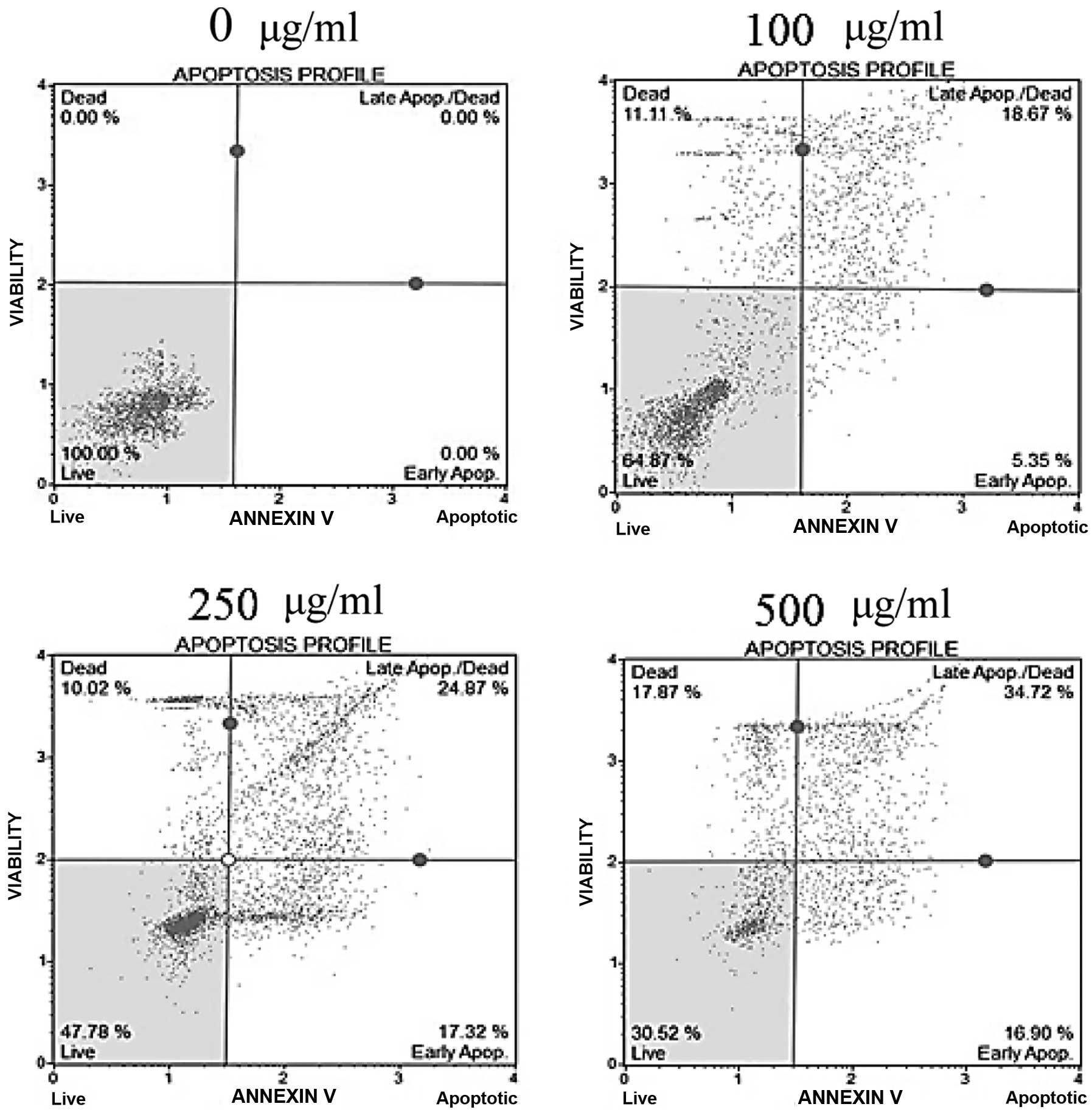
To determine whether the cytotoxicity of fucoidan
was caused by apoptosis, Annexin V/7-AAD double-staining was
performed. In fucoidan-treated HepG2 cells, the percentage of the
early apoptotic cells, as well as the total percentage of Annexin
V-positive cells indicating late apoptotic cells, was significantly
increased in a dose-dependent manner (Fig. 3). In Chang-L cells, the percentage
of apoptotic cells did not differ between fucoidan-treated groups
and controls (data not shown). These results indicate that fucoidan
had a strong antitumor effect on hepatocellular carcinoma cells and
is a potent apoptosis-inducing agent.
Effects of fucoidan on the cell
cycle
To determine whether fucoidan affected the cell
cycles of HepG2 and Chang-L cells, we performed cell analysis 24 h
after fucoidan treatment. Treatment with 500 μg/ml fucoidan
led to a significant decrease in the production of S and
G2/M phases and G0/G1 phase arrest
in HepG2 cells (43.12% in W/O, 77.78% in fucoidan-treated samples;
P<0.05). Treatment at concentrations between 0 and 500
μg/ml did not significantly change the cell cycles of
Chang-L cells (Table II). These
results suggest that the anti-proliferative effect of fucoidan on
HepG2 cells can be attributed to a blocking of the
G0/G1 phase of the cell cycle.
| Table II.Fractions of each cell cycle phase in
HepG2 and Chang-L cells cultured in the presence of fucoidan for 24
h. |
Table II.
Fractions of each cell cycle phase in
HepG2 and Chang-L cells cultured in the presence of fucoidan for 24
h.
| Fucoidan in
HepG2(μg/ml) |
G0/G1 (%) | S (%) | G2/M
(%) |
|
| 0 |
43.12±6.50a |
28.86±4.20a |
23.86±5.91a |
| 100 |
68.52±4.67b |
21.74±5.98a,b |
8.26±5.33b |
| 250 |
71.76±2.37b,c |
17.60±3.03b |
13.90±1.25b |
| 500 |
77.78±2.00c |
8.94±1.38c |
12.20±2.23b |
|
| Fucoidan in
Chang-L(μg/ml) |
G0/G1 (%) | S (%) | G2/M
(%) |
|
| 0 |
44.18±2.12a |
23.68±2.06a |
30.78±2.65a |
| 100 |
45.67±1.05a |
18.67±1.47b |
35.67±1.07b |
| 250 |
43.83±1.60a |
25.63±1.51a |
29.30±2.82a |
| 500 |
45.50±5.24a |
25.63±1.51a |
28.00±5.91a |
Expression of p16INK4a-pRb
pathway-related proteins in fucoidan-treated cells
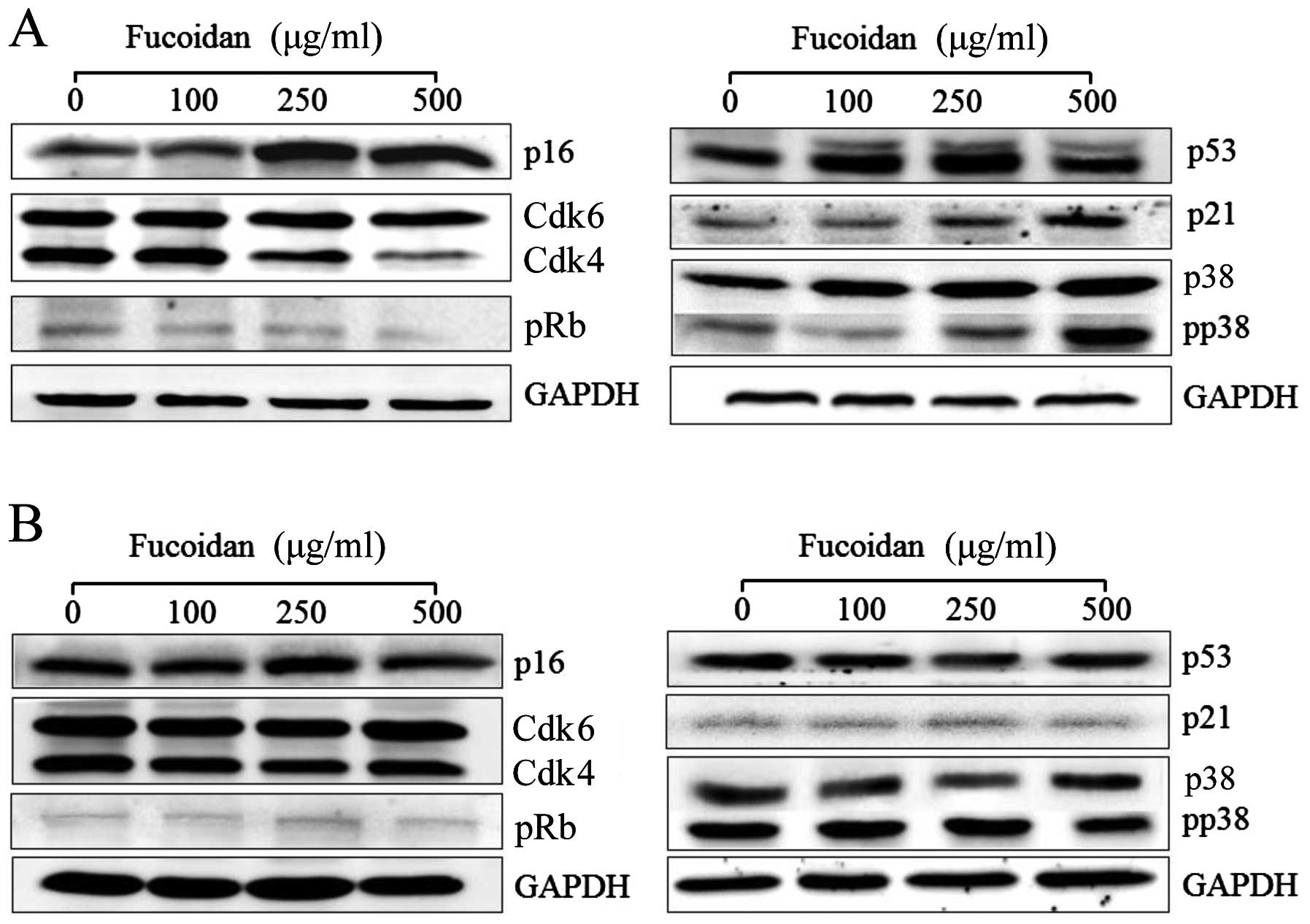
To evaluate the mechanism underlying the
tumor-suppressing activity of fucoidan, we examined the expressions
of p16INK4a, Cdk4/6 and pRb by western blotting in HepG2
cells after treatment with fucoidan for 24 h. We further explored
the mechanism of this antitumor action by evaluating mRNA
expression by real-time PCR.
The p16INK4a is a key component of the Rb
pathway that can inhibit the activity of Cdks, thereby preventing
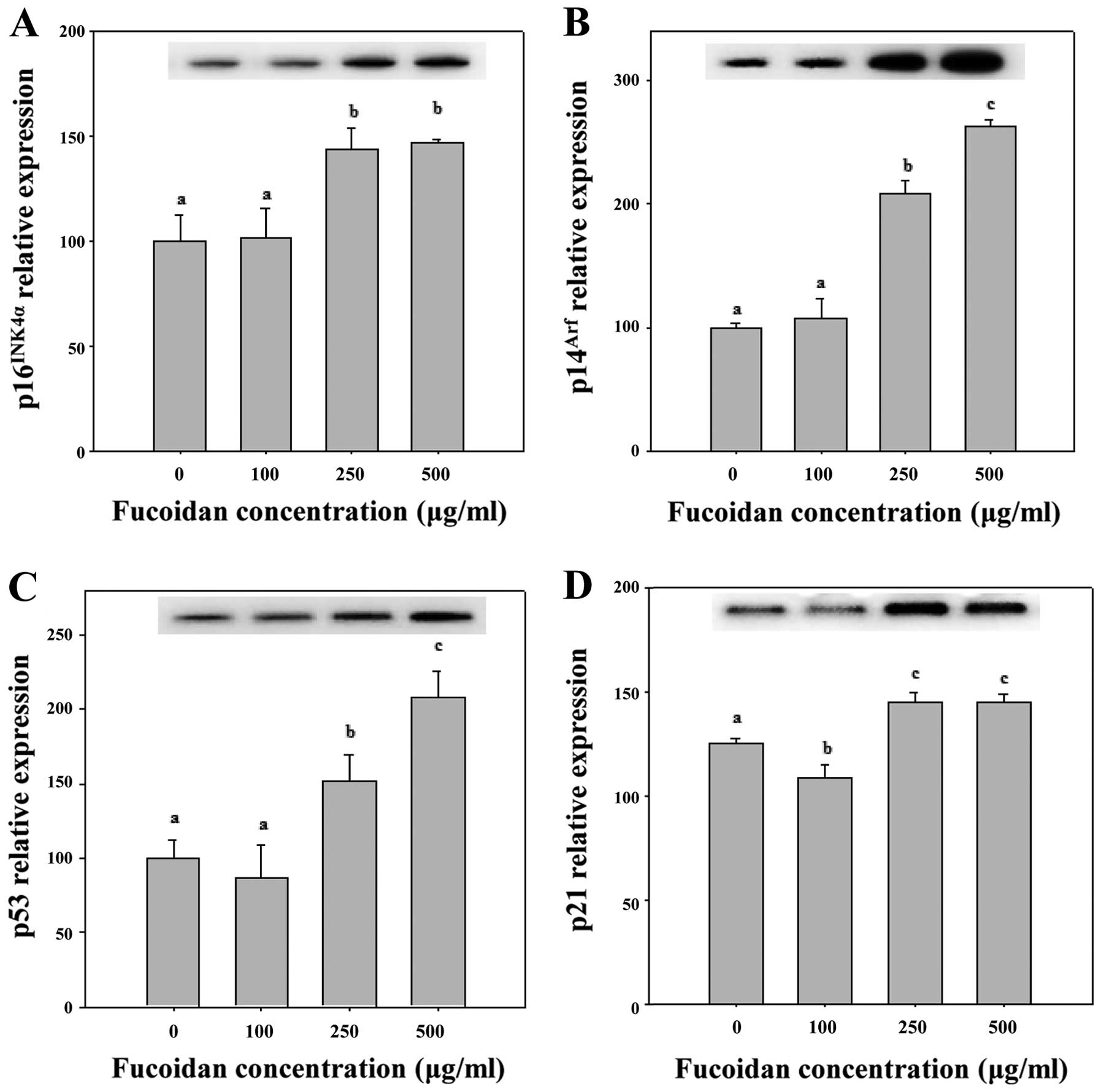
proliferating cells from entering the S phase (22). As shown in Figs. 4A and 5, 250 and 500 μg/ml fucoidan
significantly increased p16INK4a expression levels in
HepG2 cells (P<0.05). We determined that fucoidan likely
activates the Cdk4 and pRb pathway by triggering
p16INK4a overexpression and maintaining the
hypophosphorylated state of Rb (Figs.
4A and 5). Therefore, we
determined that p16INK4a arrests cells in the G1 phase
by inhibiting the activities of Cdk4 and pRb (Table II).
To identify the effects of fucoidan that regulate
cell growth arrest and thus promote cellular senescence in Chang-L
cells, we analyzed p16INK4a/Cdk4 and Cdk6/pRb, which are
primarily responsible for inhibiting cell growth and inducing
cellular senescence (23). The
activation of p16INK4a is a common step in the induction
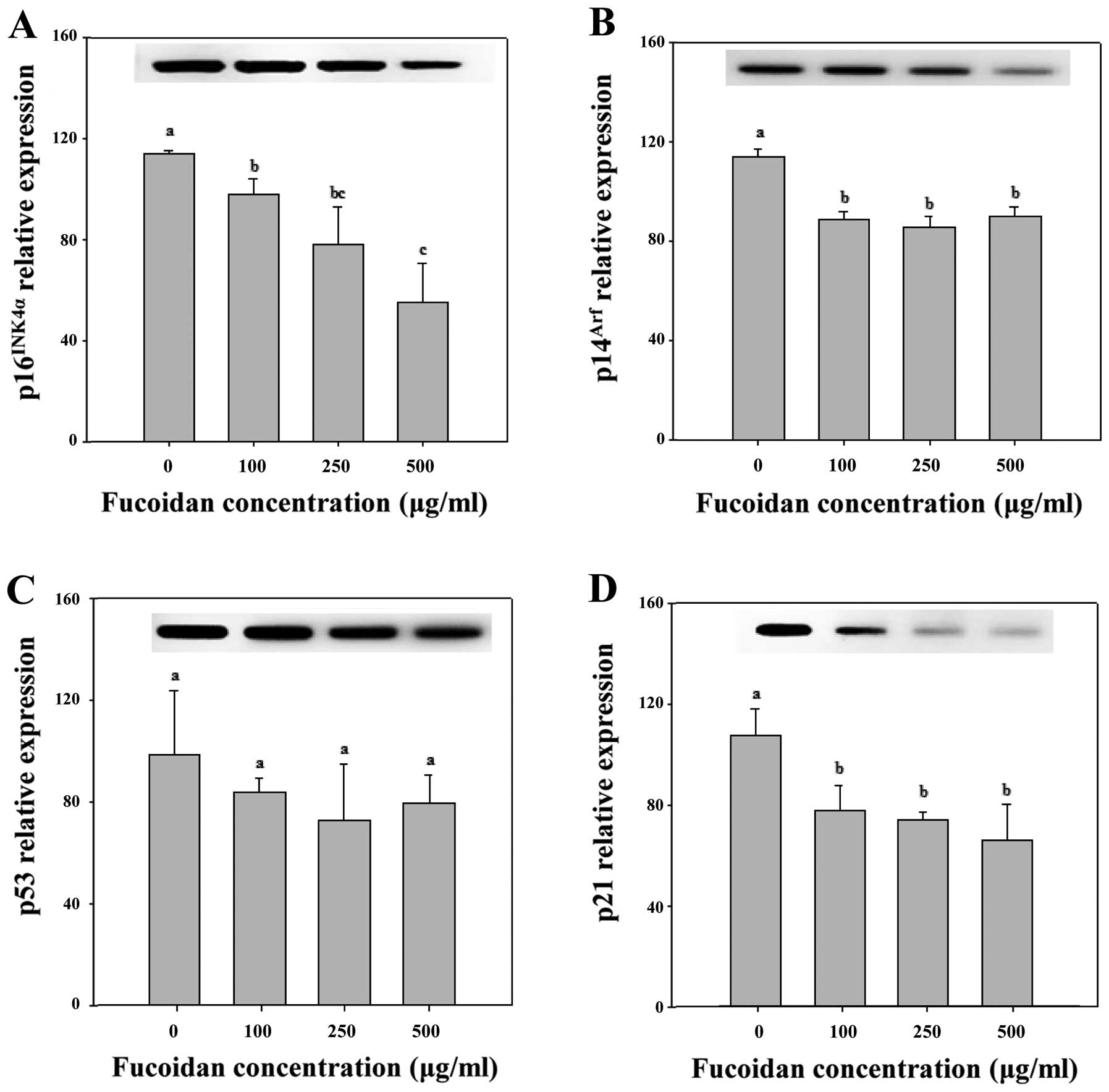
of senescence arrest. However, in Chang-L cells treated with
fucoidan, overexpression of p16INK4a protein was not
detected (Figs. 4B and 6). In addition, Cdk4-dependent activation
of pRb, which is important for cell cycle arrest, did not
significantly change (Fig. 4). The
p16INK4a also links several senescence-initiating
signals to p53 activation. However, fucoidan resulted in a
significant downregulation of p16INK4a compared to
non-treated Chang-L cells when analyzed by real-time PCR (Fig. 6).
Expression of p14Arf-p53
pathway-related proteins in fucoidan-treated cells
The p14Arf and p16INK4a are
key tumor suppressor genes that inactivate p53. To explore the
effects of the p14Arf-p53 pathway on maintaining
senescence arrest by fucoidan, we analyzed the expression of the
pathway in Chang-L cells by western blotting and real-time PCR. We
further investigated the tumor suppressor activity of fucoidan in
HepG2 cells through the p14Arf-p53 pathway.
Although p14Arf protein expression was
not detected by western blotting, the expression of
p14Arf mRNA was detected in both HepG2 and Chang-L
cells. Real-time PCR determined that fucoidan significantly
increased p14Arf mRNA expression in HepG2 cells at
concentrations of 250 and 500 μg/ml (Fig. 5) but significantly decreased
expression in Chang-L cells (Fig.
6). Hence, fucoidan suppresses p14Arf expression as
well as p16INK4a expression in Chang-L cells.
In parallel experiments, treatment of HepG2 cells
with 250 and 500 μg/ml fucoidan increased the protein
expression of p53 and p21, which are involved in the activation of
tumor suppressors (Fig. 4A), and
upregulated p53 mRNA. Furthermore, fucoidan-induced p53 mRNA
resulted in upregulation of p21 mRNA (Figs. 4A and 5).
In Chang-L cells, the mRNA levels of
p14Arf and p21, which are involved in cellular
senescence, were significantly lower at concentrations >100
μg/ml (Fig. 6). However,
fucoidan treatment did not significantly affect p53 mRNA compared
to controls (Fig. 6). Thus,
decreased expression of p14Arf and p21 induced
senescence arrest due to inactivation of the p53 pathway, and
significant changes in the cell cycle were observed after treatment
with fucoidan (Fig. 4B, Table II).
Expression of p38 MAPK in
fucoidan-treated cells
p38 MAPK can trigger premature senescence in primary
cells and permanent oncogene-induced proliferative arrest, which
has been proposed as an anti-tumorigenic defense mechanism that
induces p53 phosphorylation and upregulation of p16INK4a
(18). We defined whether
activation of p38 MAPK mediated tumor suppression and replicative
senescence, and examined the relationship between
fucoidan-stimulated activation of p16INK4a/p53 and p38
MAPK in HepG2 and Chang-L cells (Fig.
1). We further determined whether the expression of
phosphorylated p38 MAPK increased in both cell types. Fucoidan
dose-dependently elevated phospho-p38 MAPK (Fig. 4A) and p38 MAPK gene expression in
HepG2 cells (Fig. 7). In contrast,
it did not significantly affect p38 and phospho-p38 protein levels
in Chang-L cells (Fig. 4B).
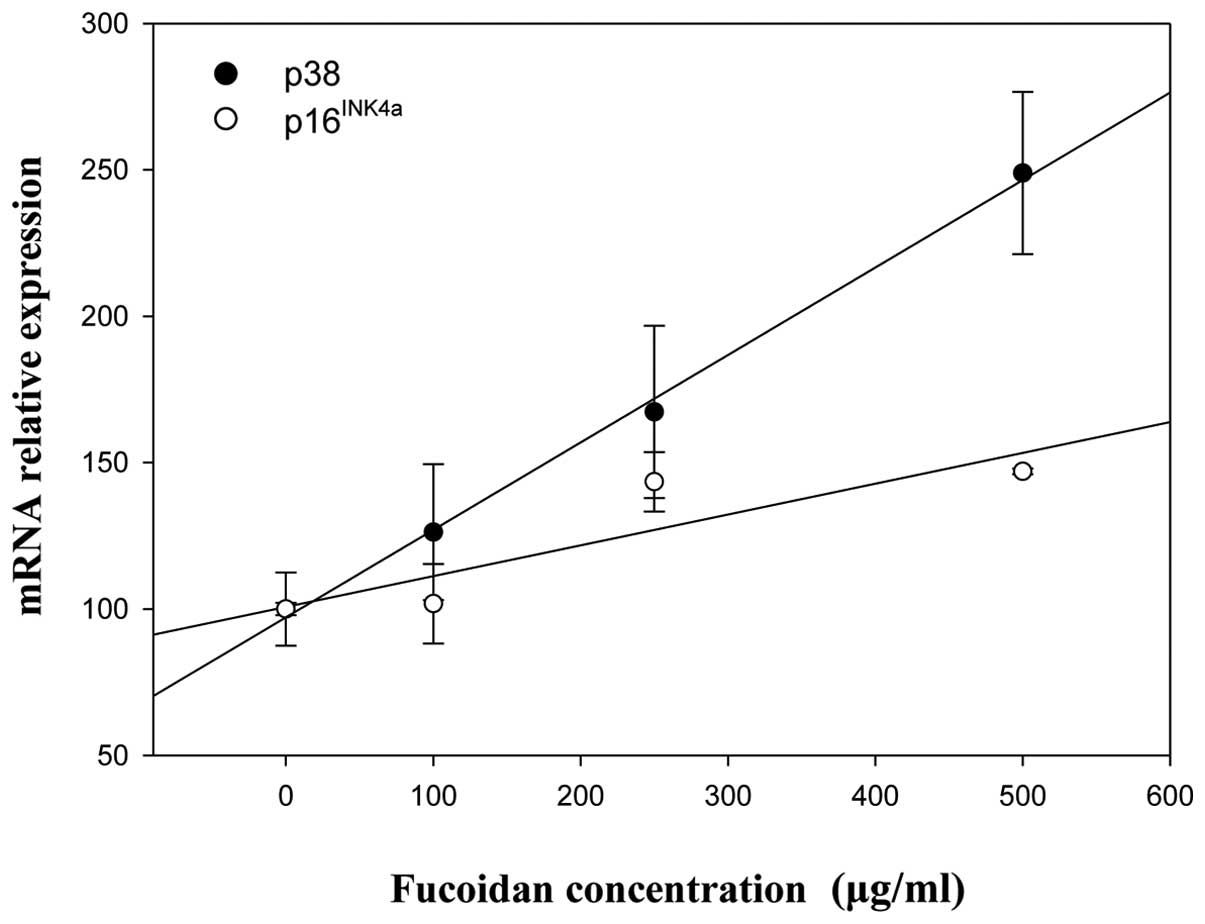
Correlation analysis was used to examine the
relevance of p38 MAPK, p16INK4a and p53 expression after
fucoidan treatment of HepG2 and Chang-L cells. Positive
correlations between p38 MAPK and p16INK4a/p53 were
found in HepG2 cells (Fig. 9),
whereas p38 MAPK was closely related with p16INK4a in
Chang-L cells (data not shown). However, the levels of p53 mRNA
were not associated with p38 MAPK (data not shown).
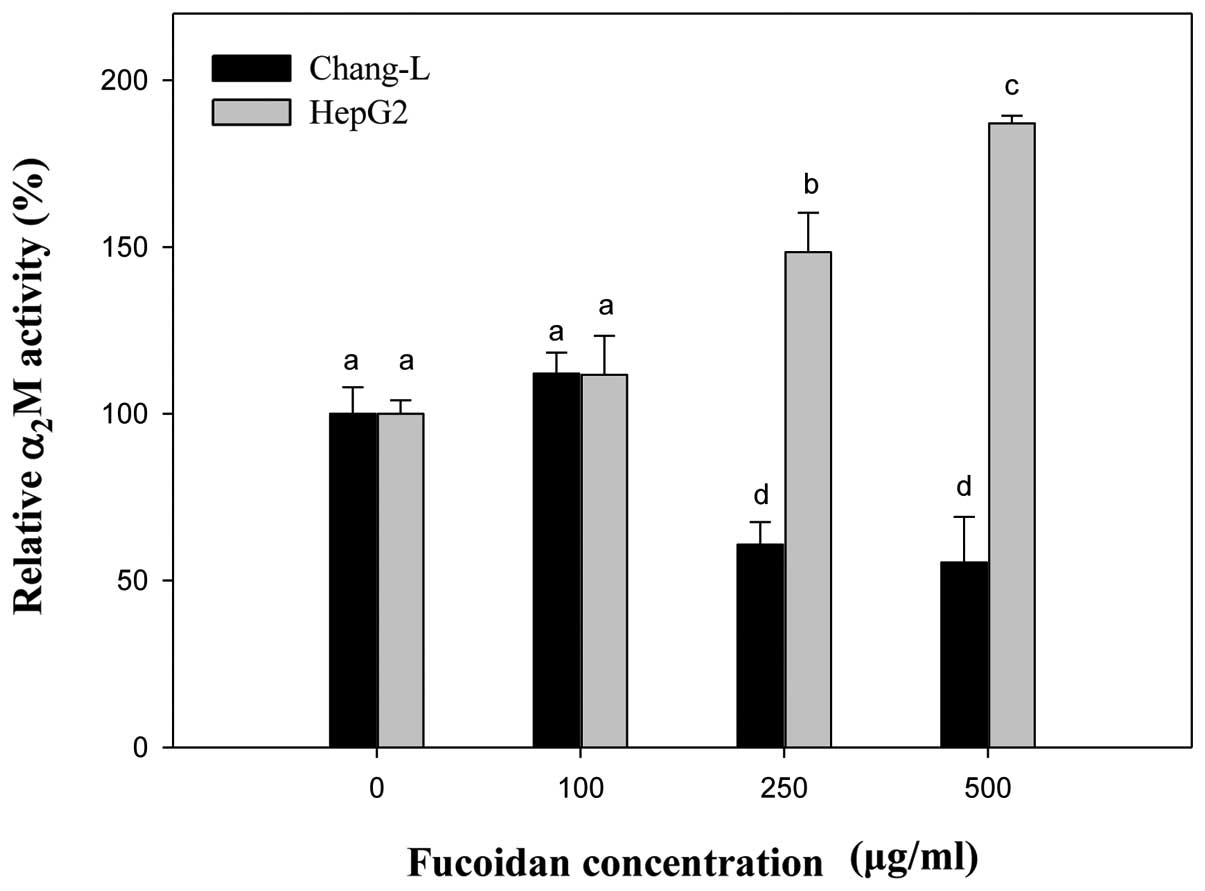
Gene expression of α2M as an
aging biomarker
The expression of α2M can easily be
measured by real-time PCR and it is a more suitable biomarker
candidate of aging then well-known senescence-associated genes such
as p16INK4a.
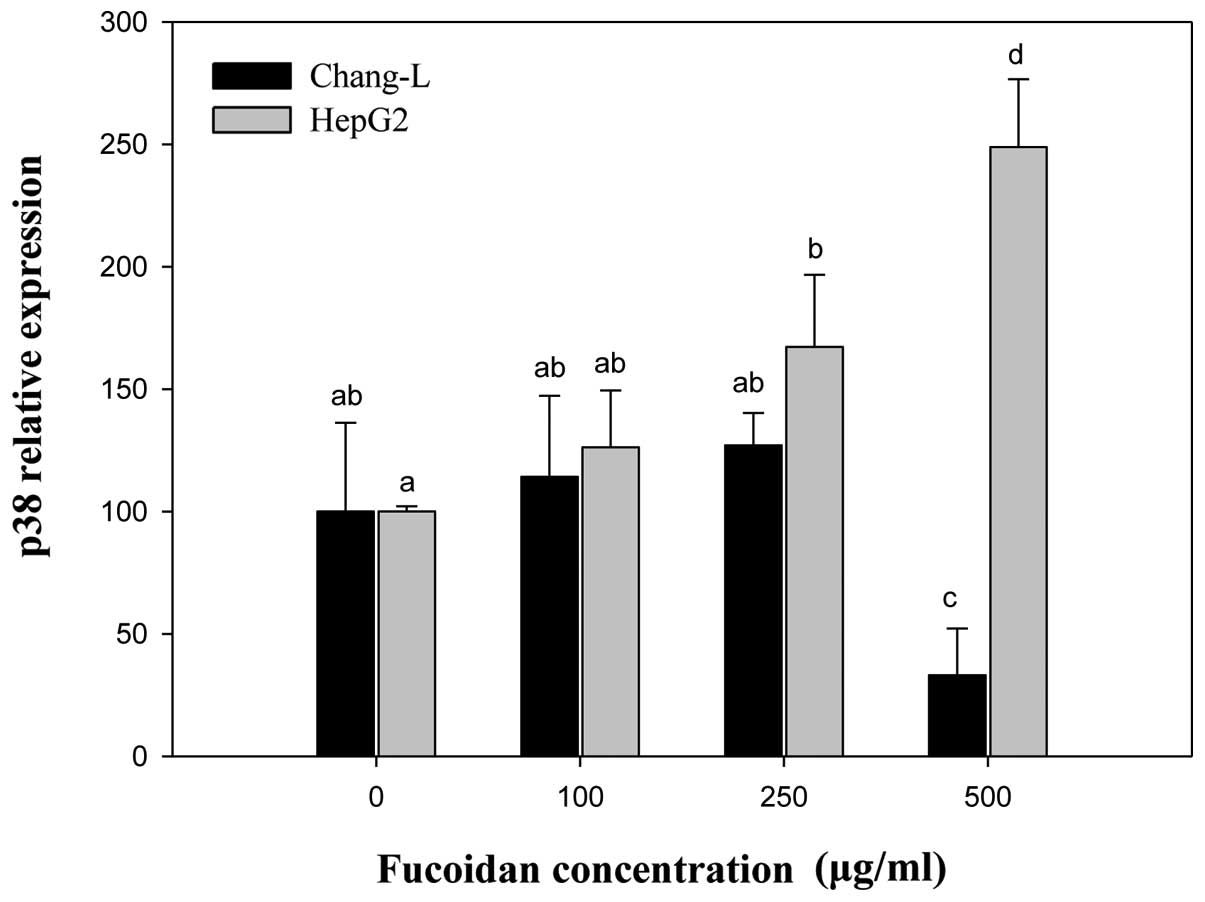
Compared to controls, the mRNA level of
α2M significantly increased in HepG2 cells treated with
fucoidan in a manner similar to p16INK4a expression
(Fig. 8). In Chang-L cells,
fucoidan treatment dose-dependently decreased α2M
expression, but not p16INK4a. When we compared HepG2 and
Chang-L cells, a significant difference in α2M mRNA
levels was noted after incubation with 250 and 500 μg/ml
fucoidan (Fig. 8).
Discussion
Natural products have played a pivotal role in the
quest to develop novel chemotherapeutic agents with enhanced
specificity and potency in liver cancer. Many marine compounds have
chemopreventive and chemotherapeutic effects through various
cell-signaling pathways involved in the transduction of mitogenic
signals and subsequent regulation of cell growth and proliferation
(24).
The diverse biological activities of fucoidan have
been studied intensively and include anti-oxidant,
immunomodulatory, anti-virus and anti-coagulant effects (3,4,25,26).
In particular, the anti-tumor activity has recently attracted
considerable attention and several studies have addressed its
anti-carcinogenic effects (7).
Fucoidan inhibits the growth of a wide variety of tumor cells
(3,26), which has become a focus of great
interest because it is expected to be a new candidate for
low-toxicity cancer therapy (7).
Cancer cells need to evade anti-proliferative
signals that negatively regulate growth and proliferation. Cancer
cells can avoid this control step by losing the physiological
function of the pRb, which controls all anti-proliferative signals
(24). Consequently, natural
product compounds that inhibit constitutive hyper-phosphorylation
of pRb contribute efficiently to the reestablishment of regulated
growth in cancer (24). In cancer
cells, the hyper-proliferation stress response tends to be
suppressed, allowing the continued proliferation of cells carrying
overactive mitogenic signals. Hyper-proliferation signals lead to
increased levels of p16INK4a and p14Arf
resulting in cell cycle arrest or cell death through the pRb and
p53 pathways. In many cell types, particularly in humans, the Cdk
inhibitor p16INK4a contributes to the cell cycle arrest
that occurs after hyper-proliferation stress. Thus, the loss of
p16INK4a, which occurs in many cancers, helps abolish
this response in some cell types. Some cancer-associated
chromosomal deletions disrupt both p16INK4a and
p14Arf genes, thereby knocking out regulators of both
the pRb and p53 pathways. Loss of p53 function is a remarkably
common event in tumor cells because it allows cell proliferation to
continue following different forms of stress and DNA damage
(27). In addition, the
INK4/Arf locus gene, p16INK4a and
p14Arf (or p19Arf in mouse), which is
upregulated during aging (28) has
been genetically linked to numerous aging-associated diseases in
humans (29). Accordingly, we
anticipated that marine compounds such as fucoidan could have
chemopreventive and chemotherapeutic effects by regulating the
expression of p16INK4a, p14Arf and p53 in
cancer cells.
Fedorov et al showed that the natural marine
chamigrane-type sesquiterpenoid dactylone inhibited cyclin D, Cdk4
expression and pRb phosphorylation (30). The inhibition of these cell cycle
components was followed by cell cycle arrest at the G1-S
transition, with subsequent p53-independent apoptosis in human
cancer cells. Park et al described the suppression of U937
human monocytic leukemia cell growth by dideoxypetrosynol A, a
polyacetylene from the sponge Petrosia sp., via induction of
the Cdk inhibitor p16INK4a and downregulation of pRb
phosphorylation (31).
Interestingly, the wild-type p53 gene is often
inactivated in HepG2 cells (32).
However, we determined that fucoidan significantly upregulated the
expression of p53 in HepG2 cells, while simultaneously inducing
apoptosis with inhibition of cellular proliferation (Figs. 2 and 3). Real-time PCR and western blotting
studies correlated increased mRNA and protein expression for
p16INK4a and p21 (Figs.
4A and 5). These results
suggest that the growth arrest in hepatocellular carcinoma cells
results from an increase in p53-mediated p21 expression as cells
enter senescence, followed by a sustained elevation of
p16INK4a (Fig. 5).
The p21 gene is a cell cycle inhibitor and tumor
suppressor downstream of p53. When cells are damaged, p53 and p21
act together to inactivate the cyclin-Cdk complex, which could
mediate G1 and G2/M arrest (33). In this study, induction of
G1 arrest by fucoidan was accompanied by a large
accumulation of p53 and p21. In particular, p21 sustains
hypophosphorylated Rb and arrests cells in the G1 phase.
Therefore, we can deduce that the anti-proliferative effect of
fucoidan regulates pRb- or p53-mediated cell cycle arrest in HepG2
cells (Table II). This supports
the idea that p16INK4a-pRb-mediated G1 arrest
by fucoidan is elicited by p53 and p21 upregulation.
Fujii et al suggested that the expression of
INK4a/Arf locus genes p16INK4a and
p14Arf could be related to cellular senescence and
apoptosis and reported that expression of this locus was increased
by valproic acid treatment and induced apoptosis in sphere cells
from rat sarcomas (34). We also
demonstrated that fucoidan caused induction of apoptosis and tumor
suppression by p16INK4a and p14Arf
overexpression in HepG2 cells but did not affect normal Chang-L
cells. Resistance to apoptosis by cancer cells can be acquired
through a variety of strategies, including p53 tumor suppressor
gene inactivation. The p53 can lead to induction of the apoptotic
cascade (24). However, numerous
studies have determined that p38 MAPK may be correlated with
apoptosis in various cancer cells (35). The p38 MAPK can regulate cell
proliferation and apoptosis through phosphorylation of p53,
increased c-myc expression and regulation of Fas/FasL-mediated
apoptosis (36). The impact of p38
MAPK on cell cycle regulators plays a crucial role in
oncogene-induced senescence involved in the suppression of
tumorigenesis and replicative senescence (18). Indeed, we found that fucoidan
induced the activation of p38 protein and mRNA in HepG2 cells. Our
results show that activation of p38 leads to increased expression
of p16INK4a, similarly to previous reports on
oncogene-induced senescence (19).
These studies indicate that p53 is a downstream effector of p38
MAPK, which has been proposed to function as an anti-tumorigenic
defense mechanism by inducing p53 and upregulating
p16INK4a.
We found that fucoidan treatment induced
phosphorylation of p38 MAPK and concurrently increased p38 MAPK in
HepG2 cells. Consistent with these results, a previous study
determined that the anti-tumor activity of fucoidan was mediated by
the induction of apoptosis through the activation of p38 MAPK in
human colon carcinoma cells (37).
Interestingly, in a previous study, honokiol, a novel antitumor
agent isolated from a plant, increased the phosphorylated p38 MAPK
without affecting p38 expression in HepG2 cells (35). Numerous studies have demonstrated
that increased levels of phosphorylated p38 are correlated with
malignancy in various cancers (38,39).
These data support the hypothesis that fucoidan may have
therapeutic potential for cancer treatment.
In summary, in HepG2 cells, it is apparent that
p16INK4a upregulation is a key event in anti-tumor
activity, and that fucoidan-induced overexpression of p38 MPAK is
associated with the p14Arf-p53 pathway during apoptosis.
This suggests that fucoidan treatment can induce growth-suppressive
signals from both p16INK4a-Rb and p14Arf-p53
pathways, a valuable therapeutic strategy for cancer treatment
(Fig. 4A). However, in Chang-L
cells, no increase in these pathway-related proteins was noted and
no evidence for apoptosis was observed after fucoidan treatment
(Fig. 4B). Among the marine
compounds, lactone spongistatin induces the degradation of XIAP
(40), an anti-apoptotic protein
that is overexpressed in chemoresistant cancer cells (41). This compound, similar to our
results, does not induce apoptosis in healthy peripheral blood
cells (40). Therefore, we suggest
that fucoidan could have selective chemotherapeutic effects.
Cellular senescence is an aging mechanism that
prevents cell renewal, leading to apoptosis and increased
expression of the tumor suppressor gene p16INK4a
(42). As proof of the stochastic
model, early fibroblasts have low levels of p16INK4a but
aging fibroblasts show significantly increased p16INK4a
expression (43). The loss or
inactivation of p16INK4a is correlated with cell
immortality (44). Given the
postulated importance of p16INK4a in cell senescence, it
is expected that inhibition of p16INK4a would extend the
proliferative life span of cultured cells (45). In the present study, we
investigated in detail the pathways induced by fucoidan in a normal
liver cell line. The expression of p16INK4a and
p14Arf mRNA significantly decreased with increasing
fucoidan concentration (Fig. 6).
Carnero et al used a strategy to express antisense
p16INK4a and p19Arf (p14Arf in
human) RNA in primary mouse embryonic fibroblasts (MEFs) (46). Consequently, the lifespan of MEFs
was extended, and a percentage of these cells eventually became
immortal. Their study suggested that cellular immortality derived
from p16INK4a acts through the Rb pathway, whereas
p19Arf acts through both the p53 and Rb pathways.
Furthermore, Jung et al determined that the
p14Arf-p53-p21 pathway, in addition to the
p16INK4a-Rb pathway, controls senescence (47). Phosphorylation of Rb results in
increased p16INK4a expression, which inhibits Cdk4/6
resulting in increased levels of hypophosphorylated Rb and
decreased p16INK4a expression (12,13).
The p14Arf-p53 pathway is important as part of the
normal life cycle of many cells; it regulates a cell’s entrance
into senescence (48). In the
present study, the expression of p14Arf and p21 mRNA was
significantly downregulated, regardless of protein expression in
Chang-L cells. However, the expression of p53 did not appear to
change significantly under the same conditions. In agreement,
several experimental studies have observed p14Arf in
senescent human fibroblasts, independent of p53 (16,48).
Moreover, the upregulation of p21 in aging and senescent human
fibroblasts is well documented (11,49).
It should be noted that INK4a/Arf expression
is both a biomarker and effector of aging. Our results suggest that
fucoidan prevented senescence in hepatoma cells by mediating a
process that included a decrease in p14Arf expression as
cells entered quiescence followed by a decline in the level of
p16INK4a. We must be clear that these results do not
negate an anti-aging effect of fucoidan in normal hepatic cells,
but only indicate that reduced levels of these proteins are not
enough to cause cellular longevity. Therefore, we examined other
clinical biomarkers of aging. Like oncogene-induced senescence,
replicative senescence is identified by senescence biomarkers such
as SA-β-gal and α2M. The α2M and SA-β-gal
expression is accompanied by increased expression of negative
growth regulators including p53, p21, p16INK4a and
p14Arf (20).
α2M is a major plasma protein that functions as a
panprotease inhibitor. Ma et al showed that expression of
p16INK4a at each passage was exponentially correlated
with cumulative population doubling level (PDL) (21), in agreement with previous reports
on p16INK4a (45), and
provided further evidence that mRNA expression of α2M
had a positive linear correlation with cumulative PDL, similar to
p16INK4a. These results, including the positive
relationship between α2M and p16INK4a,
suggest that α2M mRNA expression can be used as a
biomarker of cellular senescence (21). Another study found that the amount
of α2M fragment derived from culture medium increased as
the cells aged (50). We attempted
to identify changes that are essential for a cellular anti-aging
response by comparing the effects of fucoidan on α2M
mRNA expression in HepG2 and Chang-L cells. We found that
expression of α2M was upregulated in HepG2 cells, but
significantly downregulated in Chang-L cells after incubation with
fucoidan. These results suggest that fucoidan has the
anti-senescence effect in normal hepatic cell line. As we have
seen, there are several independent pathways that control
replicative senescence in human cells.
In conclusion, fucoidan arrests cells in the
G1 phase through p16INK4a and
p14Arf in HepG2 cells. It also increases the activity of
tumor suppressor proteins p53 and p38 MAPK, which play a critical
role in the regulation of apoptosis. Accordingly, in addition to
directly inhibiting the proliferation of tumor cells, fucoidan may
also restrain the development of tumor cells by inducing apoptosis.
Fucoidan also affected the senescence of Chang-L cells by
decreasing mRNA expression of p16INK4a,
p14Arf, p21 and the senescence biomarker α2M.
These findings suggest that fucoidan may offer substantial
therapeutic potential for cancer treatment without inducing
senescence in normal cells, and that it may be possible to use
fucoidan therapeutically.
Acknowledgements
This research was supported by Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science and
Technology (2012R1A6A1028677).
References
|
1.
|
Li Y, Nichols MA, Shay JW and Xiong Y:
Transcriptional repression of the D-type cyclin-dependent kinase
inhibitor p16 by the retinoblastoma susceptibility gene product
pRb. Cancer Res. 54:6078–6082. 1994.PubMed/NCBI
|
|
2.
|
Dürig J, Vruhn T, Zurborn KH, Gutensohn K,
Bruhn HD and Bèress L: Anticoagulant fucoidan fractions from
Fucus vesiculosus induce platelet activation in
vitro. Thromb Res. 85:479–491. 1997.PubMed/NCBI
|
|
3.
|
Maruyama H, Tamauchi H, Hasimoto M and
Nakano T: Antitumor activity and immune response of Mekabu fucoidan
extracted from Sporophyll of Undaria pinnatifida. In vivo.
17:245–249. 2003.PubMed/NCBI
|
|
4.
|
Wang J, Zhang Q, Zang Z and Li Z:
Antioxidant activity of sulfated polysaccharide fractions extracted
from Laminaria japonica. Int J Biol Macromol. 42:127–132.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hu T, Liu D, Chen Y, Wu J and Wang S:
Antioxidant activity of sulfated polysaccharide fractions extracted
from Undaria pinnitafida in vitro. Int J Biol Macromol.
46:193–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lee H, Kim JS and Kim E: Fucoidan form
seaweed Fucus vesiculosus inhibits migration and invasion of
human lung cancer cells via PI3K-Akt-mTOR pathways. PLoS One.
7:e506242012.PubMed/NCBI
|
|
7.
|
Xue M, Ge Y, Zhang J, et al: Anticancer
properties and mechanisms of fucoidan on mouse breast cancer in
vitro and in vivo. PLoS One. 7:e434832012. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kim NY, Sun JM, Kim YJ, et al:
Cisplatin-based combination chemotherapy for advanced
hepatocellular carcinoma: a single centre experience before the
sorafenib era. Cancer Res Treat. 42:203–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Rayess H, Wang MB and Srivatsan ES:
Cellular senescence and tumor suppressor gene p16. Int J Cancer.
130:1715–1725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Krishnamurthy J, Torrice C, Ramsey MR, et
al: Ink4a/Arf expression is a biomarker of aging. J Clin Invest.
114:1299–1307. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Stein GH, Drullinger LF, Soulard A and
Dulić V: Differential roles for cyclin-dependent kinase inhibitors
p21 and p16 in the mechanisms of senescence and differentiation in
human fibro-blasts. Mol Cell Biol. 19:2109–2117. 1999.PubMed/NCBI
|
|
12.
|
Li B, Lu F, Wei X and Zhao R: Fucoidan:
structure and bioactivity. Molecules. 13:1671–1695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Hara E, Smith R, Parry D, Tahara H, Stone
S and Peters G: Regulation of p16CDKN2 expression and its
implications for cell immortalization and senescence. Mol Cell
Biol. 16:859–867. 2008.PubMed/NCBI
|
|
14.
|
Haber DA: Splicing into senescence: the
curious case of p16 and p19Arf. Cell. 91:555–558. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Pomerantz J, Schreiber-Agus N, Liégeois
NJ, et al: Ink4a tumor suppressor gene product, p19Arf,
interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell.
92:713–723. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dimri GP, Itahana K, Acosta M and Campisi
J: Regulation of a senescence checkpoint response by the E2F1
transcription factor and p14Arftumor suppressor. Mol
Cell Biol. 20:273–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sharpless NE: INK4a/ARF: A multifunctional
tumor suppressor locus. Mutat Res. 576:22–38. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Han J and Sun P: 2007. The pathways to
tumor suppression via route p38. Trends Biochem Sci. 32:364–371.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bulavin DV and Fornace AJ Jr: p38 MAPK
kinase’s emerging role as a tumor suppressor. Adv Cancer Res.
92:95–118. 2004.
|
|
20.
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell.
88:593–602. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ma H, Li R, Zhang Z and Tong T: mRNA level
of alpha-2-macro-globulin as an aging biomarker of human
fibroblasts in culture. Exp Gerontol. 39:415–421. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Serrano M: The INK4a/ARF locus in murine
tumorigenesis. Carcinogenesis. 21:865–869. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Schmitt CA, Fridman JS, Yang M, et al: A
senescence program controlled by p53 and
p16INK4acontributes to the outcome of cancer therapy.
Cell. 109:335–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Schumacher M, Kelkel M, Dicato M and
Diederich M: Gold from the sea: marine compounds as inhibitors of
the hallmarks of cancer. Biotechnol Adv. 29:531–547. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ishii H, Iwatsuki M, Ieta K, et al: Cancer
stem cells and chemoradiation resistance. Cancer Sci. 99:1871–1877.
2008. View Article : Google Scholar
|
|
26.
|
Alekseyenko TV, Zhanayeva SY, Venediktova
AA, et al: Antitumor and antimetastatic activity of fucoidan, a
sulfated polysaccharide isolated from the Okhotsk Sea Fucus
evanescens brown alga. Bull Exp Biol Med. 143:730–732. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Meek DW: 2009. Tumour suppression by p53:
a role for the DNA damage response? . Nat Rev Cancer. 9:714–723.
2009.PubMed/NCBI
|
|
28.
|
Ressler S, Bartkova J, Niederegger H, et
al: p16INK4ais a robust in vivo biomarker of cellular
aging in human skin. Aging Cell. 5:379–389. 2006.
|
|
29.
|
Melzer D: Genetic polymorphisms and human
aging: association studies deliver. Rejuvenation Res. 11:523–526.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Fedorov SN, Shubina LK, Bode AM, Stonik VA
and Dong Z: Dactylone inhibits epidermal growth factor-induced
transformation and phenotype expression of human cancer cells and
induces G1-S arrest and apoptosis. Cancer Res. 67:5914–5920. 2007.
View Article : Google Scholar
|
|
31.
|
Park C, Kim GY, Kim GD, et al: Suppression
of U937 human monocytic leukemia cell growth by dideoxypetrosynol
A, a polyacetylene from the sponge Petrosia sp., via induction of
Cdk inhibitor p16 and down-regulation of pRB phosphorylation. Oncol
Rep. 16:171–176. 2006.PubMed/NCBI
|
|
32.
|
Müller M, Strand S, Hug H, et al:
Drug-induced apoptosis in hepatoma cells is mediated by the CD95
(APO-1/Fas) receptor/ligand system and involves activation of
wild-type p53. J Clin Invest. 99:403–413. 1997.PubMed/NCBI
|
|
33.
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Fujii H, Honoki K, Tsujiuchi T, et al:
Reduced expression of INK4a/ARF genes in stem-like sphere cells
from rat sarcomas. Biochem Biophysic Res Commun. 362:773–778. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Deng J, Qian Y, Geng L, et al: Involvement
of p38 mitogen-activated protein kinase pathway in honokiol-induced
apoptosis in a human hepatoma cell line (HepG2). Liver Int.
28:1458–1464. 2008. View Article : Google Scholar
|
|
36.
|
Wang YX, Xu XY, Su WL, et al: Activation
and clinical significance of p38 MAPK signaling pathway in patients
with severe trauma. J Surg Res. 161:119–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hyun JH, Kim SC, Kang JI, et al: Apoptosis
inducing activity of fucoidan in HCT-15 colon carcinoma cells. Biol
Pharm Bull. 32:1760–1764. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Iyoda K, Sasaki Y, Horimoto M, et al:
Involvement of the p38 mitogen-activated protein kinase cascade in
hepatocellular carcinoma. Cancer. 97:3017–3026. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Schyschka L, Rudy A, Jeremias I, Barth N,
Pettit GR and Vollmar AM: Spongistatin 1: a new chemosensitizing
marine compound that degrades XIAP. Leukemia. 22:1737–1745. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Igney FH and Krammer PH: Death and
anti-death: tumor resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Verzola D, Gandolfo MT, Gaetani G, et al:
Accelerated senescence in the kidneys of patients with type 2
diabetic nephropathy. Am J Physiol Renal Physiol. 295:F1563–F1573.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Tsygankow D, Liu Y, Sanoff HK, Sharpless
NE and Elston TC: A quantitative model for age-dependent expression
of the p16INK4atumor suppressor. Proc Natl Acad Sci USA.
106:16562–16567. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Huschtscha LI and Reddel RR:
p16INK4aand the control of cellular proliferative life
span. Carcinogenesis. 20:921–926. 1999.
|
|
45.
|
Duan J, Zhang Z and Tong T: Senescence
delay of human diploid fibroblast induced by anti-sense
p16INK4aexpression. J Biol Chem. 276:48325–48331.
2001.PubMed/NCBI
|
|
46.
|
Carnero A, Hudson JD, Price CM and Beach
DH: p16INK4Aand p19ARF act in overlapping pathways in
cellular immortalization. Nat Cell Biol. 2:148–155. 2000.
|
|
47.
|
Jung YS, Qian Y and Chen X: Examination of
the expanding pathway for the regulation of p21 expression and
activity. Cell Signal. 22:1003–1012. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Collado M, Blasco MA and Serrano M:
Cellular senescence in cancer and aging. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Tahara H, Sato E, Noda A and Ide T:
Increase in the expression level of
p21sdi1/cip1/waf1 with increasing division age
in both normal and SV40-transformed human fibroblasts. Oncogene.
10:835–840. 1995.
|
|
50.
|
Kondo T, Sakaguchi M and Namba M:
Two-dimensional gel electrophoresis studies on the cellular aging:
accumulation of alpha-2-macroglobulin in human fibroblasts with
aging. Exp Gerontol. 36:487–495. 2001. View Article : Google Scholar : PubMed/NCBI
|