Introduction
The Src family kinases (SFKs) are non-receptor
tyrosine kinases that are membrane associated through a
myristoylation site near their N-terminus. There are nine members
in the family and five of which (Fyn, c-Src, Yes, Lyn and Lck) are
expressed in human gliomas (1–5).
Each member of the SFKs contains a unique N-terminal sequence,
followed by four SH (Src homology) domains, and a C-terminal
negative regulatory sequence. Structural study of c-Src has
revealed that intra-molecular interactions occur between the
phosphotyrosine 530 (pY530) in the C-terminus and the SH2 domain,
and between the kinase domain and the SH3 domain, that cause the
c-Src molecule to assume an inactive closed configuration (6). When pY530 is dephosphorylated, c-Src
molecules become open and active, with the potential for
autophosphorylation and phosphorylation of Src substrates. SFKs
interact with multiple cell surface receptors including integrin,
EGFR, PDGFR, VEGFR (2,3,7–9) and
are activated rapidly upon receptor engagement resulting in the
regulation of signaling events involving cell adhesion, migration,
invasion, proliferation, apoptosis and angiogenesis (10,11).
The role of SFKs in glioma development and
progression was demonstrated in transgenic mice of v-Src, a
constitutively active mutant of Src (11–13).
The v-Src transgenic mice, in which v-Src expression is under the
control of the GFAP promoter, developed glial tumors with
morphological and molecular characteristics that mimic human
glioblastoma multiforme (GBM) (14,15).
Although v-Src has not been reported in human glioblastoma, we now
know that members of SFKs are effector molecules of EGFR, PDGFR,
VEGFR and c-kit, many of which are overexpressed or constitutively
activated in GBM (15). In
addition, inhibition of SFKs by the tumor suppressor gene PTEN
(phosphatase and tensin homologue deleted on chromosome 10) is
abolished in gliomas due to mutation or loss of PTEN (16). Kinome profiling of clinical GBM
specimens revealed that SFKs were highly activated (17,18).
The SFKs specific inhibitor, PP2 or dasatinib, has been found to
suppress migration, proliferation, and induce autophagy and cell
death of glioma cells (15,17,19).
These findings collectively suggest that SFKs represent an
important target for glioma therapy.
Recent research has revealed that glioma stem cells
(GSCs) are resistant to chemotherapy and radiation and are
responsible for tumor recurrence (20,21).
Effective therapies which target GSCs are needed. Consequently, we
investigated the expression of SFKs in GSC and examined whether
inhibitors of SFK could effectively inhibit the growth and
migration of GSC. Since GSCs only account for a fraction of cells
in a glioma tumor mass, high levels of SFKs in glioma tumors may
not accurately reflect their levels in GSCs. In this study, we
obtained GSCs and primary glioma cells (PGCs) from the same human
GBM tumors xenografted in mice, and examined the expression and
function of several members of SFKs in these two cell populations.
We found that SFKs were highly expressed in GSCs and the expression
patterns were different from that of PGCs. Fyn, Yes and c-Src were
consistently expressed in both GSCs and PGCs while Lck was only
expressed in PGCs. SFKs inhibitor dasatinib significantly inhibited
migration of GSCs, but failed to inhibit their growth or
self-renewal. These results suggest that SFKs represent an
effective target for GSCs migration but not their growth.
Materials and methods
Culture of primary glioma cells and
glioma stem cells from human GBMs xenografted in mice
All glioma xenografts were established by direct
implantation of freshly resected human GBM tissue into the flanks
of immunocompromised athymic nude (nu/nu) mice and maintained by
serial transplantation as described previously (22). The University of Alabama at
Birmingham Institutional Animal Care and Use Committee approved the
use of all animal subjects. GSCs D456, JX6, JX10 and JX12 were
cultured as we have described previously (22). To establish glioma primary and stem
cell culture, xenograft tumors were harvested from the flank of
mice and washed five times with PBS to remove excess blood. Tumors
were separately minced finely with #11 scalpel blades and minced
tumor was disaggregated in an enzyme solution [5 mg collagenase-I
(Worthington Biochemical Corp., Lakewood, NJ, USA), 0.5%
trypsin/0.53 mM EDTA (Gibco, Carlsbad, CA, USA), and 2.5 mg DNase-I
(Worthington Biochemical Corp.)] in a sterile, vented, trypsinizing
flask (20 min, room temperature). At 20 min intervals,
approximately half of the cell suspension was removed and
transferred to a centrifuge tube containing 0.5 ml of FBS. Fresh
enzyme solution was added to the trypsinizing flask and the
harvests were repeated four to five times, pooling the cells at
each harvest. Cells were pelleted (200 × g, 8 min) and washed twice
with DMEM/F12 (MediaTech) on ice. Dead cells were removed by
density gradient centrifugation (1,500–1,800 × g, 20 min, 20°C) of
5×107 cells in 35 ml carefully layered onto 10 ml of
lymphocyte separation medium (Gibco). Cells collected at the
interface were washed twice with DMEM/F12 to remove LSM. A portion
of cells were added to glioma medium (DMEM/F12 50:50, 9% FBS, 1 mM
glutamine, 10/ml penicillin and 10 μg/ml streptomycin) for
culture of primary glioma cells. The rest of cells were added to
stem cell medium [NeuroBasal-A medium, supplemented with 1% B-27,
1X N-2 (Invitrogen, Carlsbad, CA, USA), bFGF and EGF (Peprotech,
Inc., Rocky Hill, NJ, USA) at 10 ng/ml each, 10 U/ml penicillin and
10 μg/ml streptomycine] and grow for 24–48 h before the
CD133+ GSCs were isolated by FACS (see below). The
isolated CD133+ GSCs were grown in stem cell medium
exchanged every 3–4 days, and passaged with Accutase (Invitrogen)
weekly.
FACS analyses and separation of
CD133+ glioma stem cells
CD133+ GSCs in glioma spheres were
analysed and isolated by FACS. Briefly, spheroids were harvested
from culture, pelleted by centrifugation (300 × g, 8 min),
resuspended in 1 ml of Accutase and incubated (37°C, 5–10 min) with
occasional shaking. The cell suspension was gently triturated
several times with a wide bore pipette to dissociate any remaining
glioma spheres. Cells were washed twice with DMEM/F12 50:50 and
counted. Viable cells (4×106) were resuspended in 80
μl of FACS buffer containing ice-cold PBS with 5% FBS and
0.1% of sodium azide. FCR blocking reagent (20 μl, Miltenyi
Biotech) and allophycocyanin (APC)-conjugated anti-CD133/2 antibody
(10 μl, Miltenyi Biotech) were added. Cells were incubated
(15 min, 4°C), washed and resuspended in 1 ml ice-cold PBS with
0.1% sodium azide. Cells were analyzed immediately by FACS and
CD133+ cells isolated at UAB Flow Cytometry Core
Facility. The side-scatter versus forward light scatter profiles of
a control sample was used to set gates. Data of FACS were analyzed
using Flo-Jo software and results were expressed as a percentage of
gated cells for each cell type identified by antibody binding.
GSC self-renewal and differentiation
assay
For GSC self-renewal assay, a single cell suspension
was obtained from a single sphere by repetitive pipeting up and
down. Cells were diluted in stem cell medium at a concentration of
10 cells/ml, and 100 μl was plated into each well of a
96-well plate and grown for 1 week when 100 μl of fresh stem
cell medium was added. Glioma spheres in each well were counted 2
weeks after plating. For differentiation assay, GSCs were dispersed
into single cell suspension and grown on cover slips in DMEM/F12
(50:50) supplemented with 2% FBS. Cells were grown for 7 days
before staining with antibodies against glial marker GFAP (Rabbit
polyclonal IgG; Dako, Carpinteria, CA, USA), neuronal marker
βIII-tubulin (mouse monoclonal IgG; Chemicon, Temecula, CA, USA)
and oligodendrocytic marker MBP (myelin basic protein, mouse
monoclonal IgG; Abcam, Cambridge, MA, USA). Glioma spheres were
immunostained with anti-Sox2 antibody (rabbit polyclonal IgG; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Alexa-488 goat
anti-rabbit IgG and Alexa 595 goat anti-mouse IgG (Invitrogen) were
used as secondary antibody for immuno labeling. Cells were
visualized under fluorescent microscopy.
Immunoblotting
Cultured cells were lysed in M-PER mammalian protein
extraction buffer (Thermo Scientific) in the presence of 1X Halt
proteinase and phosphatase inhibitor cocktail (Thermo Scientific)
(100 μg/ml aprotinin, 10 μg/ml leupeptin, 2 mg/ml
AEBSF hydrochloride, 50 μg/ml Bestatin, 200 μg/ml
E-64, 100 mg/ml EDTA, 10 μg/ml Pepstatin A). Protein
concentrations were determined by BCA protein assay kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). Equivalent amounts of
protein (20 μg) were resolved on an 8–16% gradient Precise
Protein Gel (disulfide-reduced SDS-PAGE gel; Thermo Scientific),
transferred to a nitrocellulose membrane (Millipore, Billerica, MA,
USA), blocked with 5% non-fat milk in TBST (1 h, 22°C), and reacted
with the primary antibody at 4°C overnight followed by a secondary
antibody conjugated to horseradish peroxidase (Sigma, St. Louis,
MO, USA). Membrane-bound antibodies were detected by enhanced
chemiluminescence (BD Biosciences, Franklin Lakes, NJ, USA).
Relative band intensities were determined by averaging the
intensity readings from three independent experiments using
QuantityOne software with a VersaDoc imaging system (Bio-Rad,
Hercules, CA, USA). Src family member-specific rabbit anti-Lyn,
anti-Fyn, anti-Yes and anti-Lck IgG were purchased from Santa Cruz
Biotechnology, Inc., and rabbit anti-c-Src IgG, mouse anti-Src
monoclonal antibody (clone GD11) and mouse anti-Src family (WTAPE)
antibody were purchased from Millipore. Rabbit anti-phosphorylated
(pY416)-Src polyclonal antibody was from Cell Signaling Technology,
Inc. (Danvers, MA, USA) and mouse anti-actin was from Sigma. Each
antibody was used from one lot and the optimal concentration was
determined. The following antibody concentrations were used for
blotting: rabbit anti-c-Src, anti-Lyn and anti-Fyn IgG (0.1
μg/ml), rabbit anti-Yes and anti-Lck IgG (0.2 μg/ml),
rabbit anti-phosphospecific Src IgG (pY416-Src, 0.01 μg/ml)
and mouse anti-actin (0.1 μg/ml).
Proliferation and cell viability
assay
PGCs in glioma medium with 2% FBS were seeded at a
density of 2.5×103 cell/well in a 96-well plate
overnight and treated with dasatinib (100 μM) the next
morning. Medium was changed every 2 days and viable cells were
determined at day 2, 4 and 6 after treatment. For GSCs, single
cells obtained from neurospheres were cultured in T-25 flasks in
the presence and absence of dasatinib, samples of 100 μl
were drawn with a large bore pipette tip and subjected to cell
viability assay on day 2, 4 and 6 after treatment. In this assay,
cellular ATP levels were assessed using the ViaLight Plus kit
(Lonza, Rockland, ME, USA). This assay measured ATP levels in the
lysates by a luciferase-catalyzed production of light from
luciferin. Emitted light, which is linearly related to the amount
of ATP present in the lysates, was measured in a Spectrafluor Plus
machine (Tecan, Männedorf, Switzerland). The relative cell amount
represents the averages of three experiments in triplicates. In
addition, viable cells of GSCs and PGCs under each growth condition
were counted using hemocytometer by trypan blue exclusion. PGCs
(100,000) were plated into a T-25 flask in glioma medium containing
2% FBS with or without dasatinib, and cells were counted with
trypan blue exclusion on day 2, 4 and 6. For GSCs, glioma spheres
were collected, digested with Accutase into single cells, and
counted on day 2, 4 and 6 of the dasatinib treatment.
Migration assay
Two-well Boyden-type chambers with 8-μm pore
non-coated polycarbonate filters (Costar) were coated on the lower
filter surface with laminin (10 μg/ml, Invitrogen) at 37°C
overnight, followed by blocking with 2% BSA in PBS (37°C, 1 h).
Migration assay buffer (DMEM/F12 50:50 with 0.1% BSA, 1 mM
L-glutamine) supplemented with EGF and FGF (20 ng/ml) as a
chemoattractant was added to the bottom chamber. PGCs (harvested
with Versene, Invitrogen) and GSCs (obtained by digesting the
glioma spheres with acccutase) were washed and resuspended in
migration assay buffer at 200,000 cells/ml. Cell suspension
aliquots (100 μl) were loaded onto the upper chamber and
incubated at 37°C (5% CO2). At 3 h, cells on the upper
filter surface were removed by wiping with Q-tips and cells on the
lower filter surface were fixed (4% buffered paraformaldehyde, 10
min), washed, stained (1% crystal violet, 15 min) and quantified by
counting cells in 5 random high power fields (1-mm2
fields in a 1-cm grid). Cell numbers were averaged from three
independent experiments. Some of the filters with migrated cells
fixed on the lower surface were used for immunofluorescent staining
(see below).
Immunofluorescent staining
The cells were allowed to migrate onto the laminin
coated filter surface as above. The filters were fixed by
paraformaldehyde, blocked in PBS containing 5% each of BSA and
horse serum (25°C, 60 min) and reacted with rabbit anti-CD133
polyclonal IgG (Abcam) at 4°C for overnight. Cells were washed,
incubated with AlexFluor-488 conjugated goat anti-rabbit IgG
(1:1000, 22°C, 60 min). After rinsing in PBS, cells were stained
with DAPI (50 ng/ml in PBS, 2 min) and visualized under an Olympus
fluorescence microscope equipped with a digital camera. Monochrome
images from each color channel were acquired separately and then
merged. Fluorescence images were processed using Adobe Photoshop
(Adobe Systems, San Jose, CA, USA).
siRNAs and transfection
Control siRNA or siRNAs (100 pmol) specific for
human c-Src, Yes and Fyn (SMARTpool; Dharmacon, Lafayette, CO, USA)
was transfected into 4×105 GSCs by using Lipofectamine
2000 (Invitrogen) according to protocol recommended by the
manufacture. Cells were plated and cultured for 72 h prior to
immunoblot analysis for target knock down and migration assay.
Statistical analysis
Two-tailed Student’s t-tests were used to determine
the statistical significance between two groups (Sigma Plot 2000;
SPSS Inc., Chicago, IL, USA), and were expressed as mean ± standard
errors (SE). All experiments were repeated at least three times. A
P-value of <0.05 was considered to be statistically
significant.
Results
Culture of glioma stem and primary glioma
cells
Four pairs of GSCs and PGCs were obtained from four
human GBM xenografted in mice (D456, JX6, JX10 and JX12) as
described previously (22). The
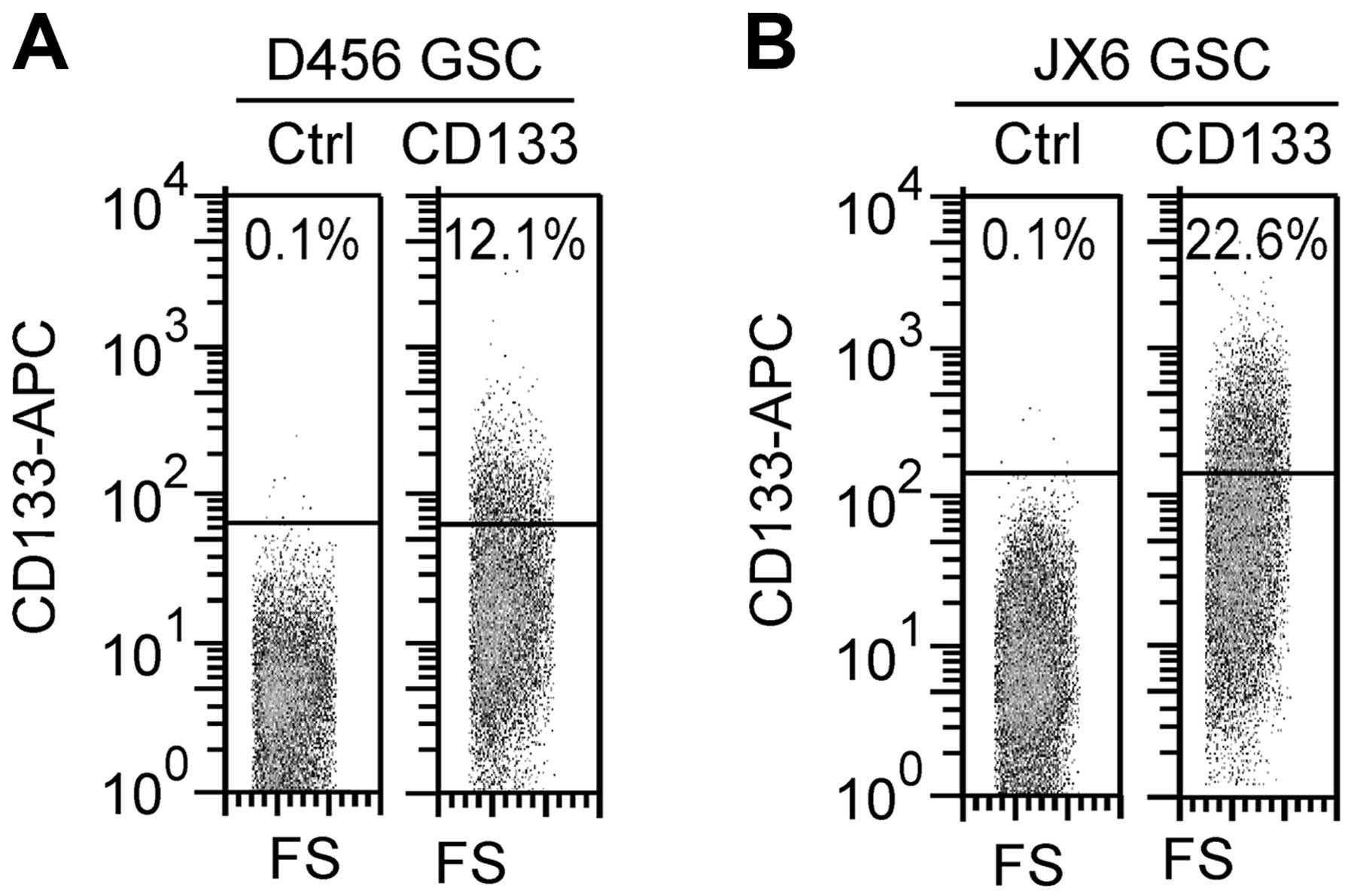
CD133+ population in GSC D456 and JX6 reached a steady
level of 12.1±1.04% and 22.6±1.51%, respectively, after 4 passages
and remained stable for at least up to 8 passages (Fig. 1). The CD133+ cells in
GSC JX12 and JX10 were determined to be 36.2 and 9.1%,
respectively, at passage 4, but the two cell lines decreased their
CD133+ populations with each passage and became growth
arrested after 8 passages. The GSC JX10 and JX12 were excluded from
further analysis due to instability of CD133+
population. The results of CD133+ populations in these
GBM xenografts are consistent with the previous data (22). For culture of PGCs, cells obtained
from the GBM xenografts were grown in regular medium containing 9%
FBS. PGCs grew as a monolayer and the CD133+ populations
were <2% (see data below).
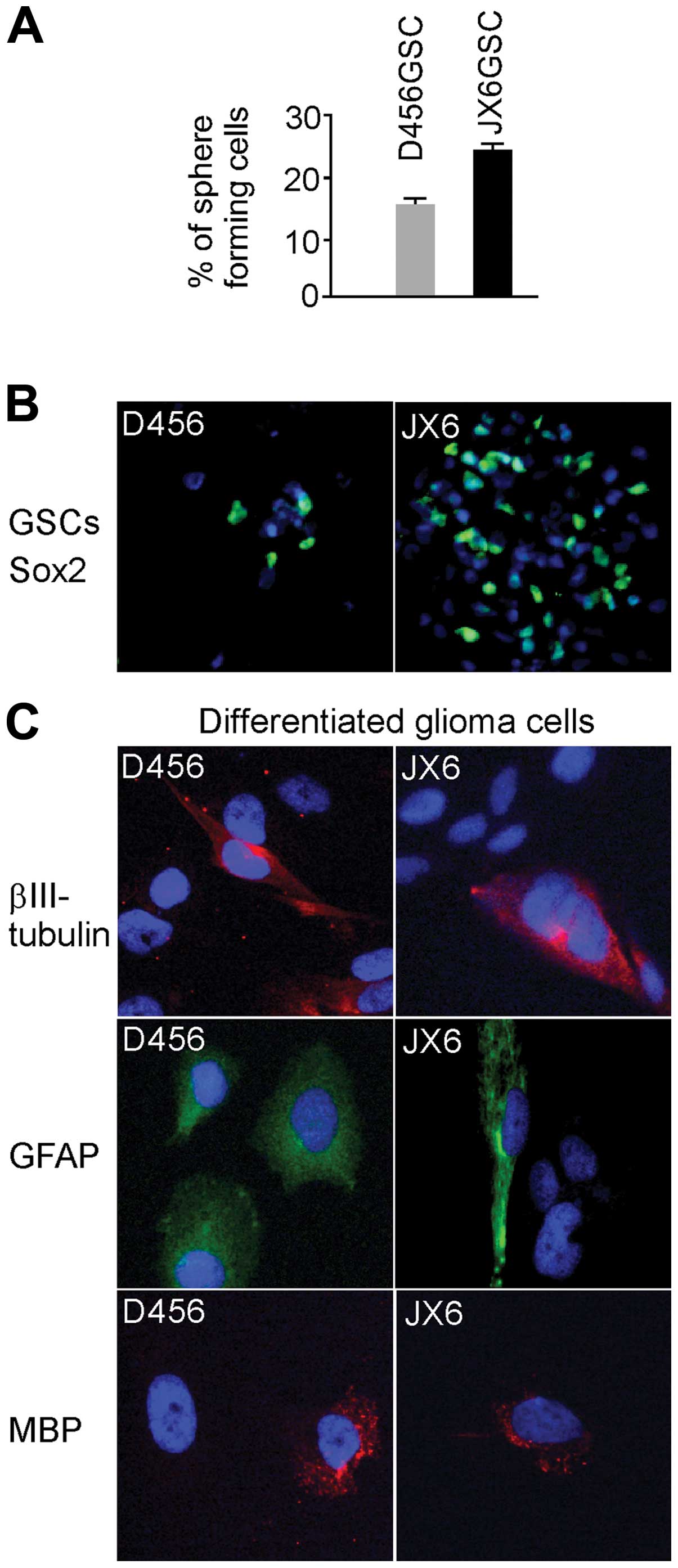
The self-renewal capacity of the GSCs was determined
by the self-renewal assay. We found that 16±0.84% of single cells
from GSC D456 and 24±1.2% of cells from GSC JX6 formed spheres
(Fig. 2A). The cells in daughter
spheres of both GSC D456 and JX6 had the same potential to form
spheres as their parent. Next, we determined the differentiation
capacity of these GSCs. Cells in glioma spheres were immunostained
with anti-Sox2 (a glioma stem cell marker) and the results are
shown (Fig. 2B). Cells from glioma
spheres were grown in serum-containing medium to induce
differentiation before they were stained for specific markers for
astrocytes (GFAP), oligodendrocytes (MBP) and neurons
(βIII-tubulin) (Fig. 3C). All
three lineages of neuroepithelial cell including astrocytes,
oligodendrocytes and neurons were present. The glioma spheres were
tumorigenic as these GBM xenografts were maintained by sequential
re-inoculation of the cells from glioma spheres obtained from the
immediate prior generation of xenografts. These results
demonstrated that cells grown in stem cell medium that had stable
CD133 expression contained the ability to self-renew and
differentiate, both features of GSCs.
SFKs are expressed and active in
GSCs
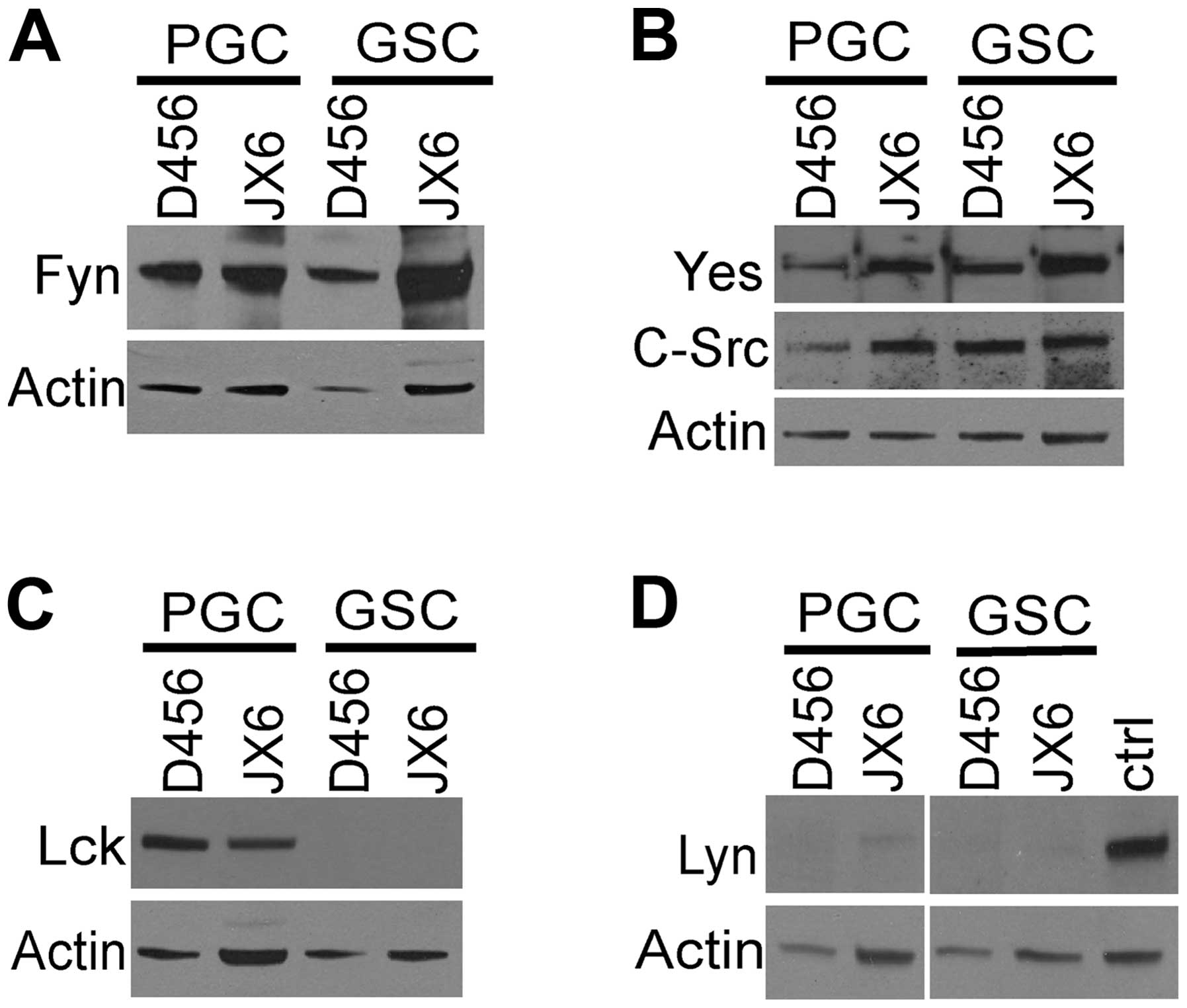
The expression of SFKs in GSCs was determined in
comparison to that of the paired PGCs which served as references.
We harvested the glioma spheres and the PGCs and determined Fyn,
Yes, c-Src, Lyn and Lck (Fig. 3).
Fyn, Yes and c-Src were detected in both GSCs and PGCs (Fog. 3A and
B). Expression of Lck showed a differential pattern, it was absent
in both GSCs whereas it was present in the PGCs (Fig. 3C). Expression of Lyn was very low
in the GSCs and PGCs (Fig. 3D).
These results demonstrate that Fyn, Yes and c-Src were the three
major SFKs present in D456 and JX6 GSCs.
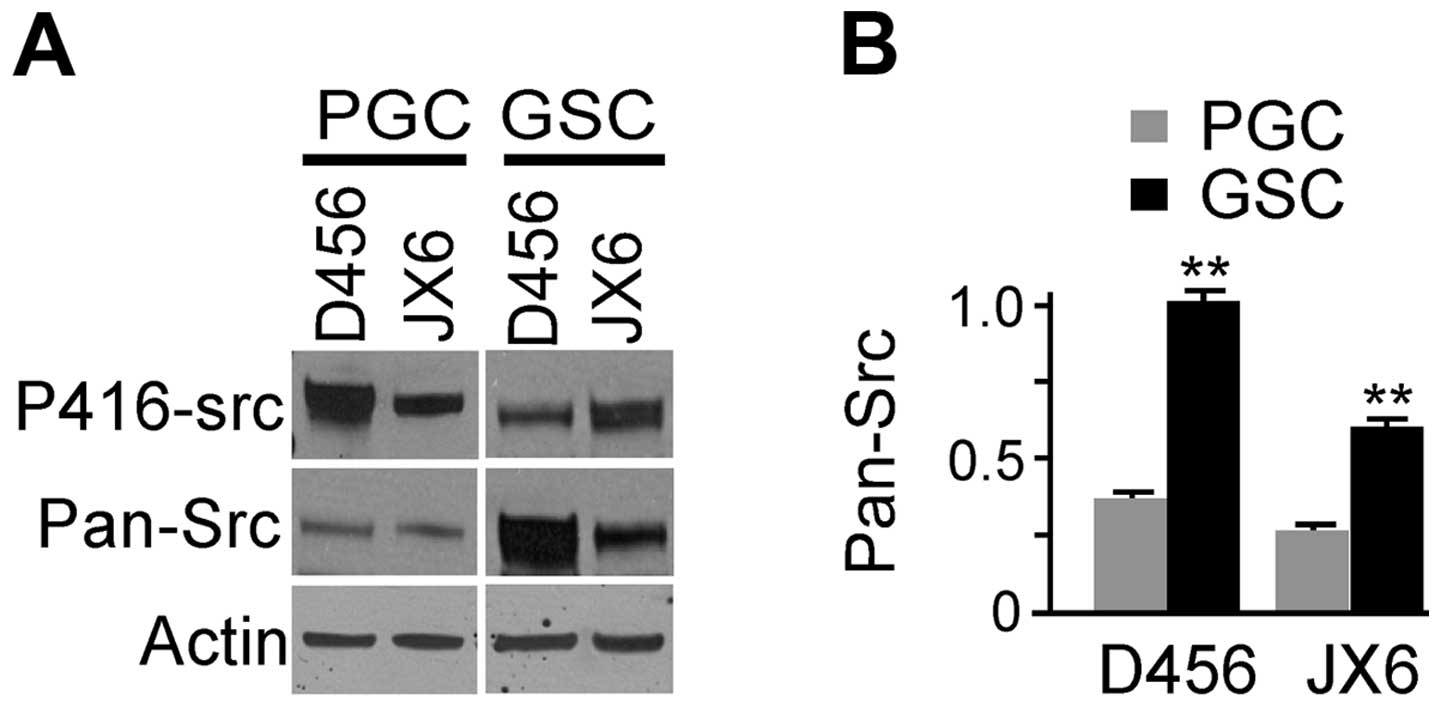
The active SFKs in GSC were evaluated by detecting
the amount of pY416 residue with anti-pY416-Src (Fig. 4A, upper panel). Anti-pY416-Src
reacts with multiple members of SFKs (Fyn, Yes, c-Src, Lyn and Lck)
when activated and phosphorylated at equivalent sites. p-416-Src
was detected in all cells and it appeared higher in PGCs than that
of corresponding GSCs. Total SFKs were assessed in GSCs by
immunoblotting using mouse anti-Src family (WTAPE) antibody, which
react with multiple members of SFKs including Src, Yes, Fyn and
others (Fig. 4A, middle panel).
Anti-Src family antibody detected bands around ∼60 kDa which
represented members of SFKs. The expression levels of SFKs
(pan-src) were normalized by β-actin and the relative levels of
SFKs in each GSCs and PGCs are shown (Fig. 4B). The levels of pan-Src in GSCs
were significantly higher than that of their paired PGCs. Taken
together, the results indicate that the total SFKs are expressed at
higher levels in GSCs compare with PGCs.
Dasatinib effectively penetrates into
glioma spheres and inhibits SFKs in GSCs
It is well established that dasatinib inhibits SFKs
in many cell lines that grow as monolayer or single cell
suspension, including glioma cells (8,15,17,19).
However, it is unclear whether dasatinib can effectively inhibit
the SFKs in GSCs because these cells grow as neurospheres that may
not be well penetrable by dasatinib. In addition, GSCs may employ
mechanisms such as the ABC transporter to eliminate intracellular
small molecules as seen in normal neural stem cells (23). To address the impact of dasatinib
on the growth of GSCs, we first examined whether dasatinib could
inhibit the activation of SFKs in GSCs. GSCs were grown in stem
cell medium without or with dasatinib at 100 nM, a concentration
similar to the clinical plasma trough concentration at therapeutic
dose (24). At 4 and 72 h of the
treatments, the cell lysates were prepared for analyzing pY416-Src,
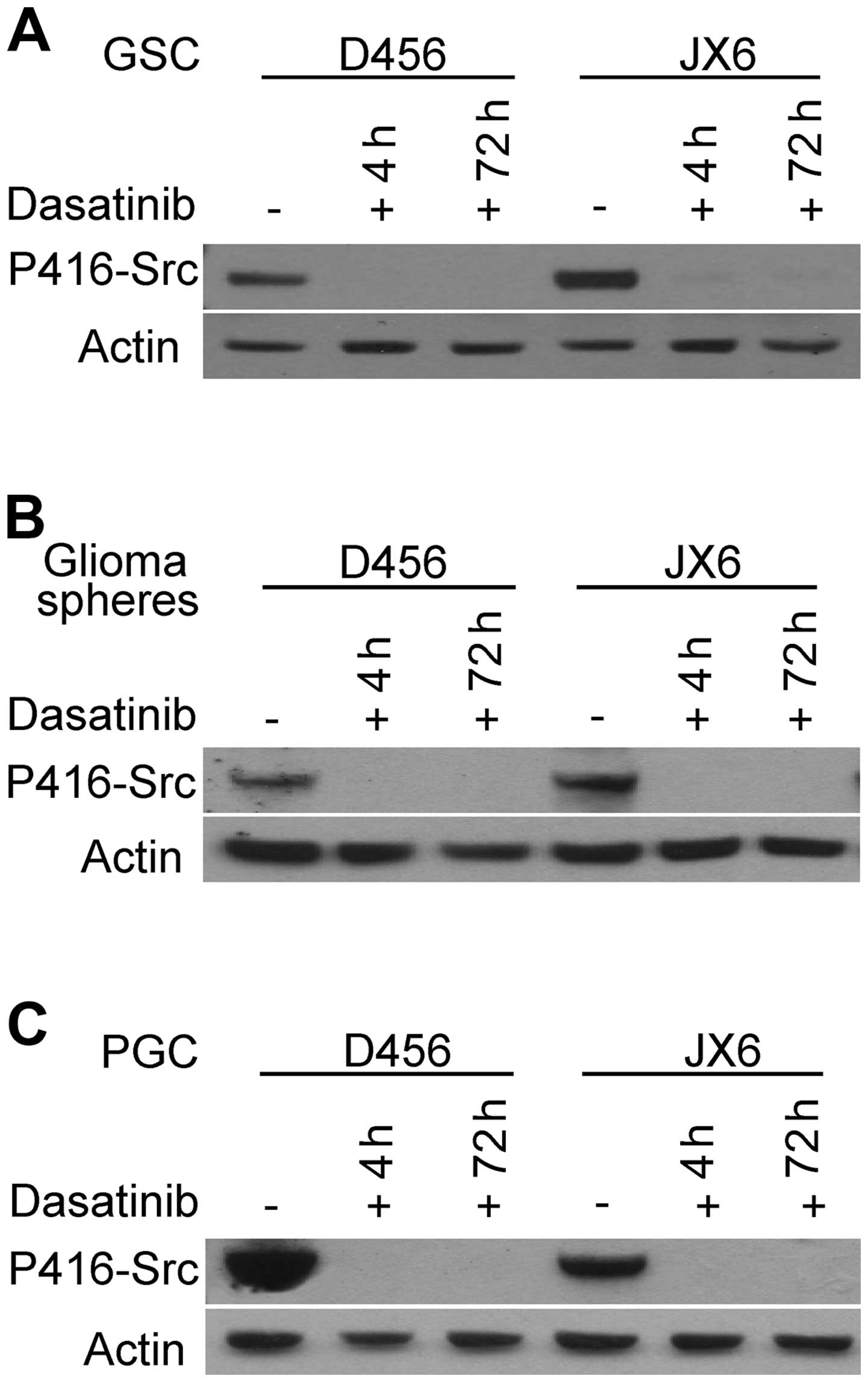
the activated form of SFKs. As shown in Fig. 5A, pY416-Src was undetectable 4 h
after addition of dasatinib, and the inhibitory effect persisted 72
h afterward in the GSCs. The results demonstrate that dasatinib
effectively inhibited SFKs in GSCs grown as single cells or small
neurospheres. We then examined whether dasatinib would be able to
penetrate into large neurospheres. To do this, single stem cells
suspension were allowed to grow for 14 days without splitting to
form large glioma neurospheres before dasatinib treatment. As shown
in Fig. 5B, dasatinib effectively
abolished the pY416-Src in all the glioma neurospheres. As a
control, we examined the effect of dasatinib on p416-Src in PGCs
and found that it was completely inhibited as expected (Fig. 5C). These results demonstrated that
dasatinib, at a clinical therapeutic serum concentration, is able
to effectively penetrate into GSCs and inhibit SFKs.
Inhibition of SFKs by dasatinib fails to
suppress the growth or self-renewal of GSCs
It has been known that dasatinib inhibits the growth
of PGCs, glioma cell lines and gliomas in animal models (17,18,25),
however, the effects of dasatinib on the growth and self-renewal of
GSCs have not been reported. The effect of SFKs inhibition by
dasatinib on the growth of GSCs was assessed in GSC D456 and JX6 in
comparison to the paired PGCs. We plated 1500 cells into each well
of a 96-well plate in culture medium containing 2% FBS with or
without dasatinib (100 nM). Viable cells were determined on day 2,
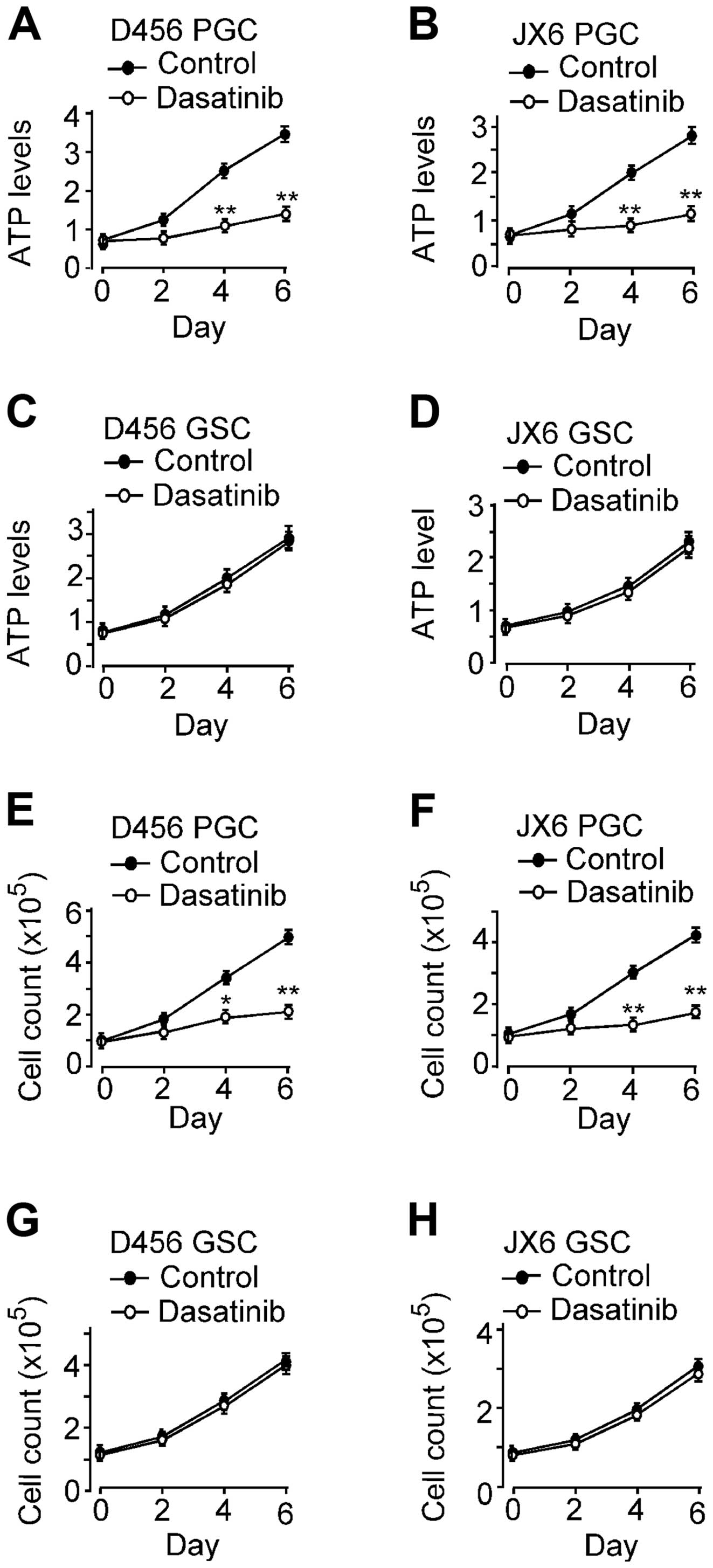
4 and 6 of the treatment (Fig. 6A and
B). As expected, the growth of PGC D456 and JX6 were
significantly inhibited by dasatinib. By day 6, PGCs D456 and JX6
grew by 380±18% and 322±16%, respectively, in control, whereas
cells in the dasatinib containing medium grew by 105±4.8% and
74.1±3.8%, respectively. These results are consistent with the
previous reports that dasatinib inhibits the growth of glioma cells
(15,17,19).
The effect of dasatinib on the growth of GSC D456 and JX6 were
determined by cell viability assay (Fig. 6C and D). The growth of GSC D456 and
JX6 cell lines was not affected by dasatinib. By day 6, GSC D456
and JX6 grew by approximately 260±11% and 210±9%, respectively in
the controls. The growth rate was not significantly affected by
treatment of dasatinib. The effect of dasatinib on cell
proliferation was further confirmed by cell counting. GSCs and PGCs
grown under the same conditions as above were counted with trypan
blue exclusion on day 2, 4 and 6 (Fig.
6E–H). By day 6, the PGC D456 and JX6 in control medium grew by
about 370±16% and 320±13%, respectively, but the cells in dasatinib
containing medium grew only by about 120±4.9% and 70±2.8%,
respectively. However, the growth of GSC D456 and JX6 GSC was
unaffected by treatment of dasatinib. These results were consistent
with those obtained by cell viability assay. Taken together, the
data demonstrated that the growth of GSCs was not affected by SFKs
inhibition as opposed to that of PGCs, suggesting that SFKs are not
critical in the growth of GSCs.
Inhibition of SFKs by dasatinib did not
change the proportion of CD133+ GSCs in glioma
spheres
To assess whether the proportion of
CD133+ GSCs in glioma spheres were affected by exposure
to dasatinib, we examined the population of CD133+ GSCs
by FACS. The CD133+ cell population in GSC D456 and JX6
were 12.1 and 22.6%, respectively (Table I). The proportions were essentially
the same after treatment with dasatinib for 14 days in both GSCs.
The results support that the growth of GSCs, in contrary to PGCs,
are intrinsically insensitive to SFKs inhibition by dasatinib. For
comparison, we treated PGCs with 100 nM dasatinib for 14 days and
determined the CD133+ cell population prior and after
treatment (Table I). We found that
the CD133+ population expanded in D456 and JX6 PGCs,
likely as a result of preferential inhibition of differentiated
PGCs.
 | Table I.Dasatinib treatment did not change
the proportion of CD133+ populations in GSCs. |
Table I.
Dasatinib treatment did not change
the proportion of CD133+ populations in GSCs.
| Dasatinib | CD133+
GSCs (%)
|
|---|
| − | + |
|---|
| GSC D456 | 12.1±1.04 | 12.9±1.14 |
| GSC JX6 | 22.6±1.51 | 23.2±1.90 |
| PGC D456 | 0.72±0.09 | 3.76±0.24a |
| PGC JX6 | 0.52±0.08 | 1.62±0.12a |
Inhibition of SFKs by dasatinib
significantly suppresses the migration of GSCs
One of the most important functions of SFKs is its
role in cancer cell migration and invasion. It is technically
difficult to examine the migration of free floating GSCs. Recent
research demonstrates that GSCs grow as a monolayer on laminin or
other extracellular matrix coated flask without changing the stem
cell properties (26,27). When the two GSC cell lines (D456
and JX6) were plated in flasks pre-coated with laminin (10 mg/ml,
37°C overnight), the GSCs grew as monolayer and were readily
reversible to spheres in the non-coated flask. To evaluate whether
SFKs affected GSC migration, cell migration assay was performed on
laminin coated transwells and the cells that migrated in the
control and dasatinib treated samples were visualized and counted
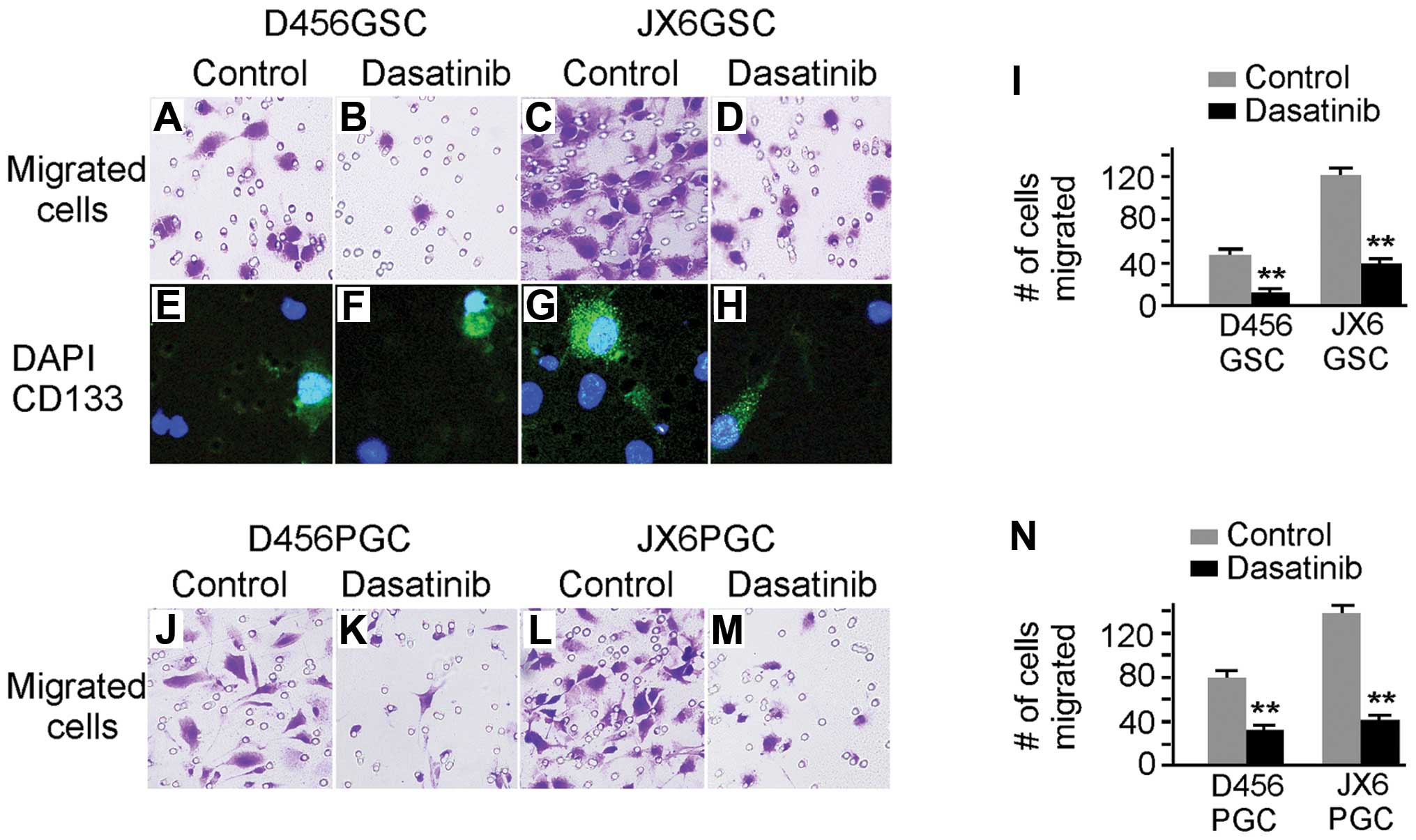
(Fig. 7). The migration of D456
and JX6 GSCs were significantly inhibited by dasatinib treatment
(Fig. 7A–D). To confirm that the
migrated cells contain CD133+ GSCs, we immunostained the
migrated cells with rabbit anti-CD133 antibody followed by
Alexor-488-conjugated goat anti-rabbit IgG (Fig. 7E–H). The CD133+ GSCs
were present in the migrated cell population regardless of
dasatinib. The inhibitory effects of dasatinib on migration were
quantified and shown in Fig. 7I.
As a control, we examined the effect of dasatinib on migration of
PGCs, and observed reduced migration (Fig. 7I and N).
c-Src and Yes are involved in the
migration of GSCs
To dissect the role of c-Src, Yes and Fyn in the
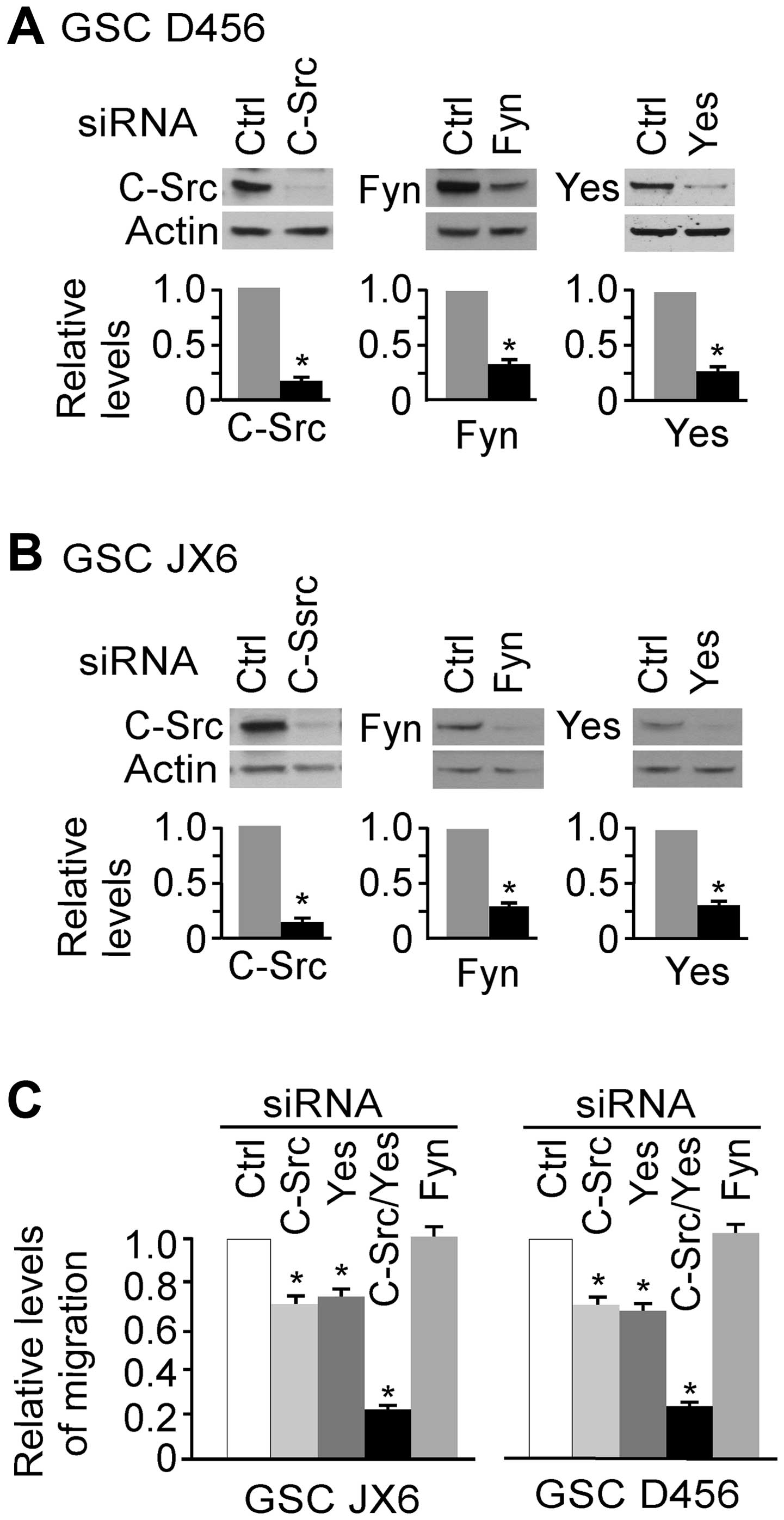
migration of GSCs, we knocked down each of these three targets by
siRNA and the results are shown in Fig. 8A and B. c-Src, Fyn and Yes were
knocked down by 86, 72 and 75%, respectively in D456 GSCs, and by
82, 71 and 70%, respectively in JX6 GSCs. Cells with target
knockdown were used for transwell migration assays and the results
are shown in Fig. 8C. Knocking
down of either c-Src or Yes significantly inhibited the migration
of GSCs in each of the GSCs. However, knockdown of Fyn did not
alter the migration capacity of these cells. Furthermore,
simultaneous knockdown of Yes and c-Src inhibited cell migration by
approximately 80% in both GSCs. These results indicate that c-Src
and Yes are directly involving in migration of these cells on a
laminin coated surface.
Discussion
SFKs are important kinases involved in glioma cell
migration and invasion, and SFKs inhibitors have been in
investigation for anti-glioma therapy. Recent data indicate that
GSCs are important for glioma initiation, treatment resistance and
recurrence. In this study, we demonstrate that several members of
SFKs (Fyn, c-Src and Yes) are expressed and active in GSCs.
Inhibition of SFKs by dasatinib has no effects on the growth or
self-renewal of GSCs, but significantly inhibited the migration of
GSCs. Knockdown of c-Src and Yes significantly inhibited migration
of GSCs indicating that these SFKs are important for GSCs
migration.
Five members of SFKs, Fyn, c-Src, Yes, Lyn and Lck,
were reported to be expressed in glioma cells (1–5), and
our results in PGCs are consistent with these reports. Previous
studies demonstrated that c-Src and Fyn interact with and
phosphorylate the cytoplasmic tail of CD133 in medulloblastoma stem
cells (28), and it is possible
that Fyn, c-Src and Yes directly interact with CD133 in GSCs;
however inhibiting SFKs by dasatinib did not alter CD133 expression
patterns in GSC lines. Interestingly, Lck appears to be expressed
in PGCs but not in the paired GSCs. A recent study showed that Lck
is activated in the glioma cell lines U87 and U373 after exposure
to radiation and the activation is important for expansion of GSCs
after radiation treatment (29).
Lck may play a specific role in radiation induced expansion of
GSCs, however, our results suggest that Lck activity is not
necessary for the maintenance of GSCs.
Dasatinib is an ATP-competitive, dual SFKs/ABL
inhibitor, and it inhibits all members of SFKs including c-Src,
Lck, Fyn and Yes (IC50<1.1 nM) (30–32).
At higher concentrations (3–28 nM), dasatinib also inhibits Abl,
PDGFR, c-kit and EphA2 (31).
Previous studies demonstrated that PDGFRs are frequently expressed
in malignant glioma (33,34) as well as EphA2 (35,36).
However, expression of c-kit and c-abl is rarely identified in
glioblastoma (34). The
concentration we used is based on clinical serum trough level
(24) and is a concentration that
will inhibit all the above targets if present. It should be noted
that the concentration of dasatinib in cerebrospinal fluid (CSF) at
a clinically therapeutic dose is undefined. However, dasatinib
likely reaches a therapeutic concentration in CSF because it has
demonstrated remarkable activity in patients with CNS metastases
from Ph+ leukemia (37).
Inhibition of SFKs by dasatinib significantly
suppressed proliferation of PGCs but had no significant inhibitory
effect on the growth of GSCs. The mechanism for GSC insensitivity
to dasatinib-mediated inhibition of SFKs and possible other
receptor tyrosine kinases is unclear. Previous studies demonstrated
that GSCs are intrinsically more resistant to chemotherapeutic
agent, temozolomide, as well as radiation treatment (20,21).
Insensitivity to SFK inhibition is likely yet another intrinsic
property of at least some GSCs. GSCs are maintained and regulated
in niches close to blood microvessels (20,21),
and various factors such as cell-to-cell communication, various
secreted factors and signals, and oxygen tension all influence
whether GSCs self-renew, proliferate or migrate (38). These niches are rich in laminin
which strongly interacts with α6, an integrin receptor highly
expressed in normal neural stem cells as well as GSCs (39,40).
The laminin/α6 integrin mediated signaling could be inhibited by
dasatinib leading to inhibition of migration of GSCs. The
inhibition of anchorage/migration has no direct effect on the
ability of GSC to self-renew or proliferate. Dasatinib
significantly inhibited the growth of PGCs and the growth
inhibition was time sensitive. Freshly isolated primary cell lines
from xenografts and the first few passages appeared more sensitive
to dasatinib treatment than did later passages (data not shown).
While the mechanism is unknown, it is clear that cells isolated
from tumors change dramatically after a few passages in serum
containing medium when compared to cells grown in defined
serum-free medium designed to support GSCs (41). It is likely that the cells are
initially under stress after disaggregation and an additional
stress superimposed by treatment of dasatinib resulted in a greater
effect on cell growth and survival. Dasatinib significantly
inhibited the migration of both GSC lines in transwell migration
assays. The effect is mediated at least in part by c-Src and Yes
because the knockdown either molecule decreased the migration
capacity of GSCs. It would be useful to know whether α6β1
preferentially interacts with c-Src or Yes in laminin mediated
anchorage and migration of GSCs.
In conclusion, our results demonstrate that SFKs are
expressed and active in GSCs which are important for migration of
GSCs. Inhibition of SFKs by dasatinib inhibited the growth of PGCs
but was ineffective at inhibiting the growth of GSCs. The results
suggest that SFK inhibitors such as dasatinib remain valuable
agents for targeting some glioma cells; however, SFK inhibitor
alone is likely to have a limited effect on growth and self-renewal
of GCCs.
Acknowledgements
NIH NCI Grants P30CA013148 (UAB
Comprehensive Cancer Center Core Support Grant); P50CA097247,
P20CA151129 (G.Y.G.); St. Baldrick’s Foundation and the Rally
Foundation (G.K.F.); VA Merit Review (P.H.K.).
References
|
1.
|
Irby RB and Yeatman TJ: Role of Src
expression and activation in human cancer. Oncogene. 19:5636–5642.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Stettner MR, Wang W, Nabors LB, et al: Lyn
kinase activity is the predominant cellular SRC kinase activity in
glioblastoma tumor cells. Cancer Res. 65:5535–5543. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ding Q, Stewart J Jr, Prince CW, et al:
Promotion of malignant astrocytoma cell migration by osteopontin
expressed in the normal brain: differences in integrin signaling
during cell adhesion to osteopontin versus vitronectin. Cancer Res.
62:5336–5343. 2002.
|
|
4.
|
Summy JM and Gallick GE: Src family
kinases in tumor progression and metastasis. Cancer Metastasis Rev.
22:337–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Frame MC: Src in cancer: deregulation and
consequences for cell behaviour. Biochim Biophys Acta.
1602:114–130. 2003.PubMed/NCBI
|
|
6.
|
Yamaguchi H and Hendrickson WA: Structural
basis for activation of human lymphocyte kinase Lck upon tyrosine
phosphorylation. Nature. 384:484–489. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Guarino M: Src signaling in cancer
invasion. J Cell Physiol. 223:14–26. 2010.
|
|
8.
|
Lu KV, Zhu S, Cvrljevic A, et al: Fyn and
SRC are effectors of oncogenic epidermal growth factor receptor
signaling in glioblastoma patients. Cancer Res. 69:6889–6898. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Lund CV, Nguyen MT, Owens GC, Pakchoian
AJ, Shaterian A, Kruse CA and Eliceiri BP: Reduced glioma
infiltration in Src-deficient mice. J Neurooncol. 78:19–29. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Summy JM and Gallick GE: Treatment for
advanced tumors: SRC reclaims center stage. Clin Cancer Res.
12:1398–1401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Yeatman TJ: A renaissance for SRC. Nat Rev
Cancer. 4:470–480. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Takeya T and Hanafusa H: DNA sequence of
the viral and cellular src gene of chickens. II. Comparison of the
src genes of two strains of avian sarcoma virus and of the cellular
homolog. J Virol. 44:12–18. 1982.PubMed/NCBI
|
|
13.
|
Jove R and Hanafusa H: Cell transformation
by the viral src oncogene. Annu Rev Cell Biol. 3:31–56. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Weissenberger J, Steinbach JP, Malin G,
Spada S, Rulicke T and Aguzzi A: Development and malignant
progression of astrocytomas in GFAP-v-src transgenic mice.
Oncogene. 14:2005–2013. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
de Groot J and Milano V: Improving the
prognosis for patients with glioblastoma: the rationale for
targeting Src. J Neurooncol. 95:151–163. 2009.PubMed/NCBI
|
|
16.
|
Dey N, Crosswell HE, De P, Parsons R, Peng
Q, Su JD and Durden DL: The protein phosphatase activity of PTEN
regulates SRC family kinases and controls glioma migration. Cancer
Res. 68:1862–1871. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sikkema AH, Diks SH, den Dunnen WF, et al:
Kinome profiling in pediatric brain tumors as a new approach for
target discovery. Cancer Res. 69:5987–5995. 2009. View Article : Google Scholar
|
|
18.
|
Du J, Bernasconi P, Clauser KR, et al:
Bead-based profiling of tyrosine kinase phosphorylation identifies
SRC as a potential target for glioblastoma therapy. Nat Biotechnol.
27:77–83. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nomura N, Nomura M, Sugiyama K and Hamada
J: Src regulates phorbol 12-myristate 13-acetate-activated
PKC-induced migration via Cas/Crk/Rac1 signaling pathway in
glioblastoma cells. Int J Mol Med. 20:511–519. 2007.
|
|
20.
|
Bao S1, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Liu G, Yuan X, Zeng Z, et al: Analysis of
gene expression and chemoresistance of CD133+cancer stem
cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Friedman GK, Langford CP, Coleman JM,
Cassady KA, Parker JN, Markert JM and Yancey Gillespie G:
Engineered herpes simplex viruses efficiently infect and kill
CD133+human glioma xenograft cells that express CD111. J
Neurooncol. 95:199–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lin T, Islam O and Heese K: ABC
transporters, neural stem cells and neurogenesis - a different
perspective. Cell Res. 16:857–871. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bradeen HA, Eide CA, O’Hare T, et al:
Comparison of imatinib mesylate, dasatinib (BMS-354825), and
nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based
mutagenesis screen: high efficacy of drug combinations. Blood.
108:2332–2338. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Milano V, Piao Y, LaFortune T and de Groot
J: Dasatinib-induced autophagy is enhanced in combination with
temozolomide in glioma. Mol Cancer Ther. 8:394–406. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Fael Al-Mayhani TM, Ball SL, Zhao JW,
Fawcett J, Ichimura K, Collins PV and Watts C: An efficient method
for derivation and propagation of glioblastoma cell lines that
conserves the molecular profile of their original tumours. J
Neurosci Methods. 176:192–199. 2009.PubMed/NCBI
|
|
27.
|
Pollard SM, Yoshikawa K, Clarke ID, et al:
Glioma stem cell lines expanded in adherent culture have
tumor-specific phenotypes and are suitable for chemical and genetic
screens. Cell Stem Cell. 4:568–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Boivin D, Labbé D, Fontaine N, Lamy S,
Beaulieu E, Gingras D and Béliveau R: The stem cell marker CD133
(prominin-1) is phosphorylated on cytoplasmic tyrosine-828 and
tyrosine-852 by Src and Fyn tyrosine kinases. Biochemistry.
48:3998–4007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kim RK, Yoon CH, Hyun KH, et al: Role of
lymphocyte-specific protein tyrosine kinase (LCK) in the expansion
of glioma-initiating cells by fractionated radiation. Biochem
Biophys Res Commun. 402:631–636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Burgess MR, Skaggs BJ, Shah NP, Lee FY and
Sawyers CL: Comparative analysis of two clinically active BCR-ABL
kinase inhibitors reveals the role of conformation-specific binding
in resistance. Proc Natl Acad Sci USA. 102:3395–3400. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Lombardo LJ, Lee FY, Chen P, et al:
Discovery of
N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide
(BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor
activity in preclinical assays. J Med Chem. 47:6658–6661. 2004.
View Article : Google Scholar
|
|
32.
|
Shah NP, Tran C, Lee FY, Chen P, Norris D
and Sawyers CL: Overriding imatinib resistance with a novel ABL
kinase inhibitor. Science. 305:399–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hermanson M, Funa K, Hartman M,
Claesson-Welsh L, Heldin CH, Westermark B and Nistér M:
Platelet-derived growth factor and its receptors in human glioma
tissue: expression of messenger RNA and protein suggests the
presence of autocrine and paracrine loops. Cancer Res.
52:3213–3219. 1992.
|
|
34.
|
Haberler C, Gelpi E, Marosi C, Rössler K,
Birner P, Budka H and Hainfellner JA: Immunohistochemical analysis
of platelet-derived growth factor receptor-alpha, -beta, c-kit,
c-abl, and arg proteins in glioblastoma: possible implications for
patient selection for imatinib mesylate therapy. J Neurooncol.
76:105–109. 2006. View Article : Google Scholar
|
|
35.
|
Wang LF, Fokas E, Bieker M, et al:
Increased expression of EphA2 correlates with adverse outcome in
primary and recurrent glioblastoma multiforme patients. Oncol Rep.
19:151–156. 2008.PubMed/NCBI
|
|
36.
|
Wykosky J, Gibo DM, Stanton C and Debinski
W: EphA2 as a novel molecular marker and target in glioblastoma
multiforme. Mol Cancer Res. 3:541–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Porkka K, Koskenvesa P, Lundán T, et al:
Dasatinib crosses the blood-brain barrier and is an efficient
therapy for central nervous system Philadelphia chromosome-positive
leukemia. Blood. 112:1005–1012. 2008. View Article : Google Scholar
|
|
38.
|
Gilbertson RJ and Rich JN: Making a
tumour’s bed: glioblastoma stem cells and the vascular niche. Nat
Rev Cancer. 7:733–736. 2007.
|
|
39.
|
Shen Q, Wang Y, Kokovay E, et al: Adult
SVZ stem cells lie in a vascular niche: a quantitative analysis of
niche cell-cell interactions. Cell Stem Cell. 3:289–300. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Lathia JD, Gallagher J, Heddleston JM, et
al: Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem
Cell. 6:421–432. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Lee J, Kotliarova S, Kotliarov Y, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006. View Article : Google Scholar : PubMed/NCBI
|