Introduction
Topoisomerases are essential enzymes in all
organisms that are involved in the topological homeostasis of DNA
molecules during DNA replication, transcription and chromosomal
segregation. Topoisomerases are classified into two classes,
topoisomerase I and II, which create single and double-strand
breaks, respectively. Because topoisomerases are crucial enzymes in
DNA replication, they have served as primary targets for the
development of anticancer agents (1,2).
Topoisomerase II inhibitors such as etoposide, teniposide and
doxorubicin have been extensively used in clinical cancer
treatment. However, these agents also have undesirable side
effects, including immunosuppression, myelosuppression,
gastrointestinal toxicity and the development of secondary leukemia
(3). Thus, the discovery of new
topoisomerase inhibitors with fewer side effects from natural
products has received a great deal of attention (4,5).
Daurinol is a natural arylnaphthalene lignan
isolated from the traditional medicinal plant Haplophyllum
dauricum (6). According to an
ethnopharmacological study, this plant has been used to treat
tumors in Russia (7). The chemical
structure of daurinol is similar to that of the clinical anticancer
agent VP-16 (also known as etoposide phosphate). Recently, we
suggested that daurinol could be a promising antitumor agent with
minimal side effects, compared to etoposide, based on in
vitro and in vivo results. Daurinol suppressed the
growth of human colorectal cancer cells through the inhibition of
human topoisomerase IIα in vitro and dramatically inhibited
the growth of HCT116 tumors in a nude mouse xenograft model.
Moreover, daurinol did not show severe side effects such as loss of
body weight and hematological toxicity, i.e., loss of white blood
cells and red blood cells, and decreased hemoglobin content
(5).
However, this previous study revealed some
limitations of daurinol that must be resolved to develop this agent
as a novel alternative to etoposide in clinical trials. First, the
availability of natural daurinol from the plant H. dauricum
is limited because its content is quite low (0.013% of the plant
dry weight) (6). Second, daurinol
inhibits human topoisomerase IIα activity in the millimolar range
(5). We neither elucidated the
inhibitory activity against other topoisomerases, including
topoisomerase I, nor identified the detailed biochemical mechanism
underlying the daurinol-induced inhibition of topoisomerase IIα.
Last but most important, we did not examine its effect on other
cancer cell types besides colorectal cancer, even though etoposide
is also frequently used to treat other cancers such as ovarian,
small-cell lung and testicular cancer (3,8).
To address these limitations, in the present study,
we used synthetic daurinol prepared by a regioselective chemical
synthesis. In addition, we have proposed a biochemical mechanism
underlying the inhibitory action of daurinol against human
topoisomerase IIα based on a computational molecular docking study
and biochemical experiments. Finally, we tested the anticancer
activity of synthetic daurinol against human ovarian, small-cell
lung and testicular cancer cells and investigated the effects of
daurinol on cell cycle distribution and cell morphology in the
selected cancer cells in comparison to etoposide.
Materials and methods
Reagents
Daurinol used in this study was chemically
synthesized according to recently reported methods (9). Natural daurinol was isolated from
Haplophyllum dauricum as previously described (6). Dimethyl sulfoxide (DMSO), etoposide,
novobiocin, propidium iodide (PI) and ATP disodium salt were
purchased from Sigma (St. Louis, MO, USA). Camptothecin was
purchased from TopoGEN, Inc. (Port Orange, FL, USA). Daurinol,
etoposide and camptothecin were dissolved in DMSO for cellular
treatment. Antibodies against E2F-1 (3742), cyclin A (4656) and
Cdk4 (2906) were purchased from Cell Signaling Technology (Danvers,
MA, USA). Antibodies against Cdk2 (sc-163), cyclin E (sc-247),
cyclin D1 (sc-753) and β-actin (sc-47778) were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Secondary anti-rabbit and
anti-mouse antibodies were purchased from Santa Cruz
Biotechnology.
Molecular docking analysis
The crystal structure of human topoisomerase IIα
(ATP2ATPase) complexed with AMPPNP (a non-hydrolyzable ATP analog)
from the Protein Data Bank (PDB code 1ZXM) was used for the docking
simulation. Daurinol was built using the Maestro build panel and
minimized using the impact module of Maestro in the Schrödinger
suite program. The starting coordinates of the ATP2ATPASE were
further modified for the prediction of daurinol binding. The
protein structure was minimized using the Protein Preparation
Wizard by applying an OPLS force field. For grid generation, the
binding site was defined as the centroid of the AMPPNP. Ligand
docking into the active site of ATP2ATPASE was performed using the
Schrödinger docking program Glide (Schrödinger, Inc., USA).
Energy-minimized daurinol was docked into the prepared receptor
grid. The best-docked poses were selected based on the lowest Glide
scores. The molecular graphics of the inhibitor-binding pocket and
refined docking model for daurinol were generated using the PyMol
package (http://www.pymol.org).
Measurement of human topoisomerase IIα
catalytic activity
The inhibitory activity of daurinol against human
topoisomerase IIα was measured using the Topoisomerase II Drug
Screening kit (TopoGEN, Inc.). The standard reaction mixture (20
μl) contained 250 ng of supercoiled DNA (pHOT1), 0.7 μl of
topoisomerase IIα, and different concentrations of ATP (0.5, 1 or 2
mM) and the tested compounds dissolved in DMSO. The final
concentration of DMSO was 1%, and the topoisomerase II poison
etoposide was used as a positive control. Reactions were initiated
by the addition of the supercoiled DNA substrate. The reaction
mixture was incubated at 37°C for 30 min. Other procedures were
performed as described previously (5).
Measurement of human topoisomerase I
catalytic activity
The inhibitory activity of daurinol against human
topoisomerase I was measured using the Topoisomerase I Assay kit
(TopoGEN, Inc.). The standard reaction mixture (20 μl) contained 10
mM Tris-HCl (pH 7.9), 1 mM EDTA, 150 mM NaCl, 0.1% bovine serum
albumin (BSA), 0.1 mM spermidine, 5% glycerol, 250 ng of
supercoiled DNA (pHOT1), 0.7 μl of topoisomerase I, and the test
compound dissolved in DMSO. The final concentration of DMSO was 1%,
and the topoiso-merase I poison camptothecin was used as a positive
control. Reactions were initiated by the addition of the
supercoiled DNA substrate. The reaction mixture was incubated at
37°C for 30 min, and 2 μl of 10% sodium dodecyl sulfate was added
to stop the reaction. Additional procedures were performed using
procedures similar to the topoisomerase II assay, as described
previously (5).
ATPase activity assay
The DNA-dependent ATP hydrolysis activity of human
topoisomerase IIα was determined by quantifying hydrolyzed
inorganic phosphate using the malachite green assay with slight
modifications (10–12). Briefly, the standard reaction
mixture (20 μl) contained 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10
mM MgCl2, 0.5 mM dithiothreitol, 30 μg/ml BSA, 200 ng of
supercoiled DNA (pHOT-1), 2.0 μl of human topoisomerase IIα
(TopoGEN, Inc.); and 100 μM daurinol or 400 μM novobiocin dissolved
in DMSO. The final concentration of DMSO was 1%, and novobiocin was
used as a positive control (12,13).
The reaction was initiated by adding ATP at a final concentration
of 2 mM (Sigma) and was incubated at 37°C for 30 min. The reaction
was stopped by the addition of the Lanzetta reagent (80 μl, freshly
prepared mixture of 0.035% malachite green-HCl and 4.2% ammonium
molybdate in 4 N HCl at a ratio of 3:1 and 0.2% CHAPS), and the
color development was read immediately using a Synergy HT
Multi-Mode Microplate Reader (BioTek Instruments, Winooski, VT,
USA) at 620 nm. The amount of inorganic phosphate was calculated
using a phosphate (potassium dihydrogen phosphate,
KH2PO3) standard curve. Enzyme activity was
expressed as μM phosphate produced per min reaction.
Cell culture
SK-OV-3 and NIH:OVCAR-3 human ovarian cancer; and
NTERA-2 cl.D1 (NT2-D1), NCCIT human testicular cancer cell lines
were obtained from American Type Culture Collection (ATCC;
Rockville, MD, USA). The SNU-840 human ovarian cancer; and NCI-H69,
NCI-H146, NCI-H187 and NCI-H417 human small-cell lung cancer cell
lines were obtained from the Korean Cell Line Bank (Seoul, Korea).
These cells were cultured in RPMI-1640 supplemented with 25 mM
HEPES, 10% (v/v) heat-inactivated fetal bovine serum (FBS), 100
U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained
at sub-confluence in a 95% air and 5% CO2 humidified
atmosphere at 37°C.
Measurement of anti-proliferative
activity
Cancer cells (5×103 cells/well) were
plated in 96-well plates, incubated at 37°C for 24 h, and treated
with daurinol for 48 h. Cell viability was determined using the
EZ-Cytox cell viability assay kit (Daeil Lab Service, Ltd., Seoul),
as previously described (14).
Cell cycle analysis
NIH:OVCAR-3 (5×105 cells/well), SNU-840
(3×105 cells/well) and NCCIT (3×105
cells/well) cells were plated in 6-well plates, incubated at 37°C
for 24 h, and treated with daurinol or etoposide for 24 and 48 h.
The cells were stained with PI, and their DNA contents were
analyzed using a FACSCalibur flow cytometer (Becton-Dickinson, San
Jose, CA, USA) and Modfit LT V3.0 software (Verity Software House,
Topsham, ME, USA) as previously described (5).
Western blot analysis
SNU-840 cells (5×105) were seeded on
60-mm dishes, incubated for 24 h, and then treated with daurinol
for 24 and 48 h. Additional procedures were performed as previously
described (15). The relative
protein expression was measured by densitometry.
Evaluation of cell and nuclear size
To evaluate the effect of daurinol and etoposide on
cell and nuclear size, we performed fluorescence pulse signal
analysis of PI-stained SNU-840 cells as previously described
(15,16). The treatment and flow cytometric
DNA content analysis were performed as described above in the
section of cell cycle analysis. FSC-H and FL2-W values, which
correlate with cell and nuclear size, respectively, were measured
using flow cytometry. The mean values of FSC-H and FL2-W were
calculated using the histogram statistics tool from CellQuest Pro
software (Becton-Dickinson). To compare the FSC-H and FL2-W
distributions of the control and chemical-treated cells, we used
the Kolmogorov-Smirnov statistics tools from CellQuest Pro
software. We also observed SNU-840 cell morphology after treatment
with daurinol or etoposide using an Olympus CK40 phase contrast
microscope (Tokyo, Japan).
Statistical analysis
The values represent the mean ± standard deviation
(SD). Statistical analyses were performed using GraphPad Prism 5
software (La Jolla, CA, USA). A paired two-tailed Student’s t-test
was used to compare the IC50 values of daurinol and
etoposide in each cell line. One-way analysis of variance (ANOVA)
followed by Dunnett’s or Tukey’s multiple comparison test was used
to analyze all other data. P<0.05 was considered statistically
significant.
Results
Synthetic daurinol and natural daurinol
have equivalent anticancer activity in HCT116 cells
Because only a minute amount of natural daurinol is
present in the plant H. dauricum, large-scale production of
daurinol is one of the key hurdles to its development as a clinical
anticancer drug. Thus, we prepared synthetic daurinol using
recently described regioselective chemical synthesis methods
(9) and tested its anticancer
activity in various cancer cells, including HCT116 cells. The
anti-proliferative activity (IC50 value) of synthetic
daurinol was equivalent to that of natural daurinol in HCT116
cells. In addition, synthetic daurinol also clearly induced cell
cycle arrest at S phase in HCT116 cells (data not shown). These
results were in accordance with our previous report (5). Based on these data, we used synthetic
daurinol as a substitute for natural daurinol in the present
study.
Molecular docking predicts that daurinol
binds to the ATP-binding site of human topoisomerase IIα
We used molecular docking simulations to evaluate
the biochemical mechanism underlying the inhibitory activity of
daurinol against human topoisomerase IIα. The results indicated the
formation of hydrogen-bond interactions between daurinol and the
polar side chains in the catalytic site of topoisomerase IIα. As
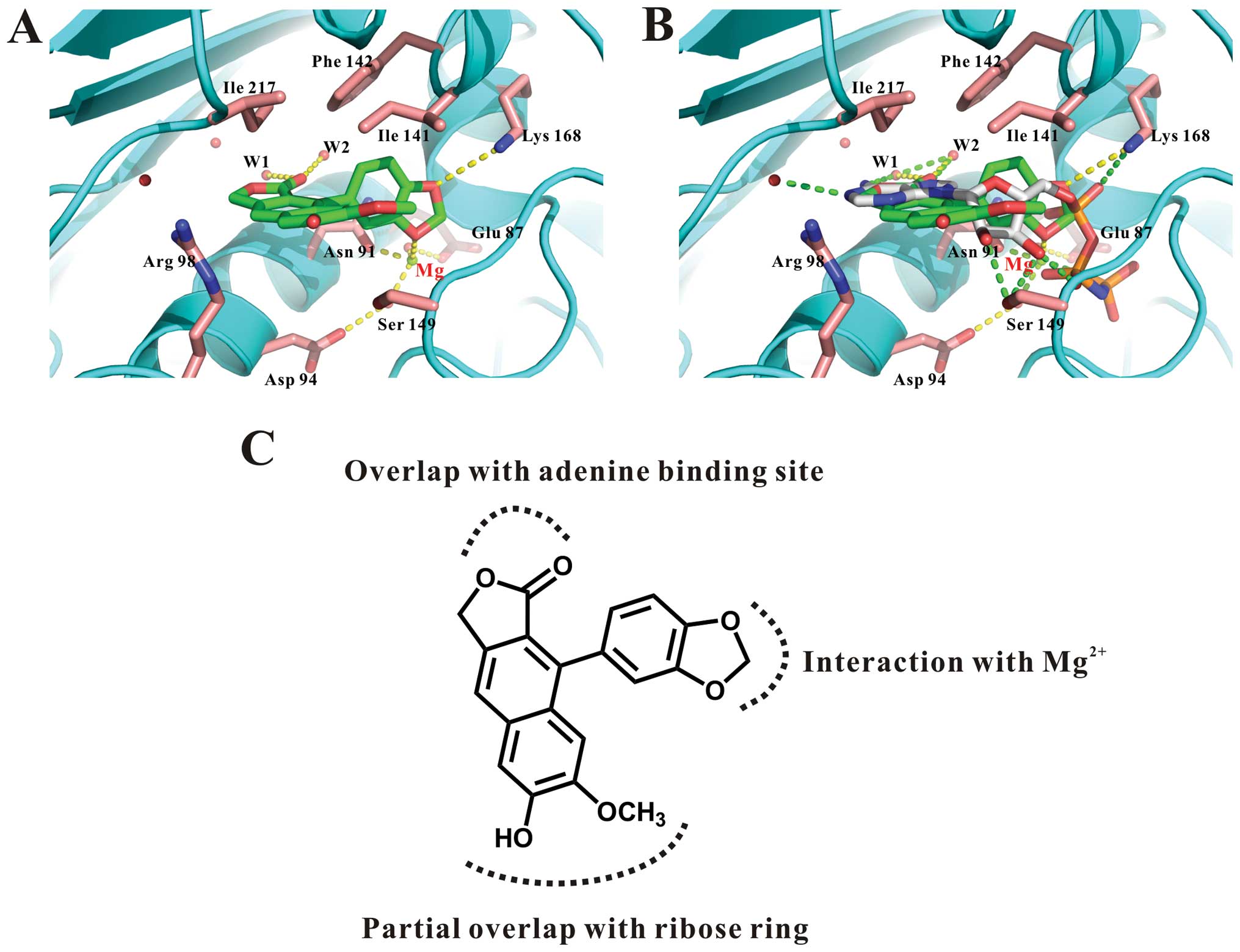
shown in Fig. 1A, the model of
daurinol bound to topoisomerase IIα indicated the following
important interactions: the hydrogen bond between the benzodioxol
functional group of daurinol and the Lys168 side chain of the
enzyme and the interactions between the naphthofuranone ring of
daurinol and water in topoisomerase IIα (Fig. 1A and C). Most importantly, the
binding site of daurinol is identical to the binding site of
AMP-PNP, a non-hydrolyzable ATP analog (Fig. 1B). Therefore, we speculated that
daurinol inhibits human topoisomerase IIα by targeting its ATPase
domain.
Daurinol inhibits human topoisomerase IIα
catalytic activity in an ATP concentration-dependent manner
To verify the hypothesis from the molecular docking
study, we measured human topoisomerase IIα activity in the presence
of different concentrations of ATP. We previously reported the
inhibitory activity of daurinol against topoisomerase IIα to be in
the millimolar range (5). To more
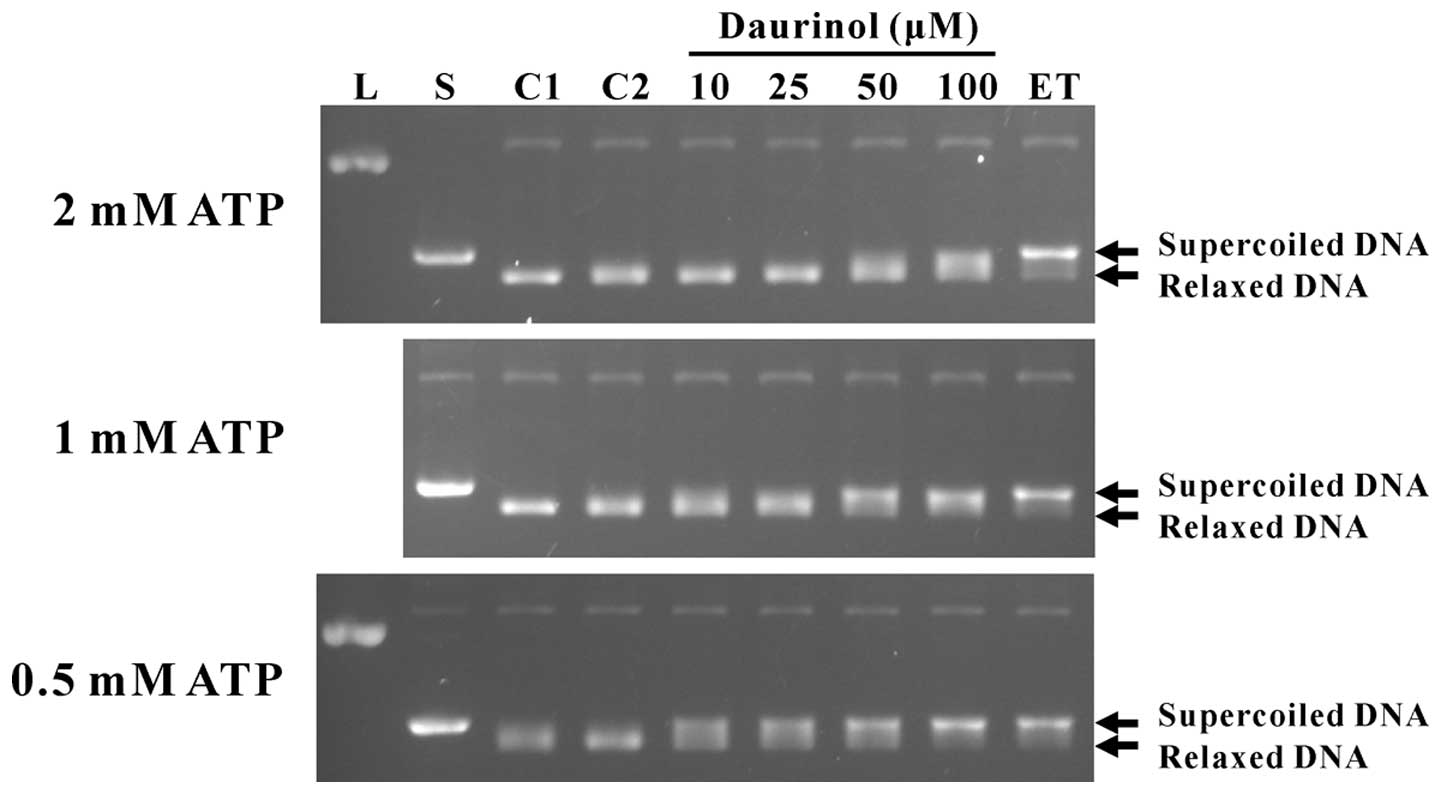
accurately evaluate topoisomerase IIα inhibition, we tested
micromolar concentrations of daurinol in this study. As shown in
Fig. 2, micromolar concentrations
of daurinol potently inhibited the catalytic activity of human
topoisomerase IIα and prevented the relaxation of the supercoiled
DNA substrate. Notably, increasing the ATP concentration resulted
in decreased inhibitory activity. When the enzyme reaction was
performed in the presence of 0.5 mM ATP, daurinol inhibited the
enzyme activity at concentrations of 10–100 μM. However, in the
presence of 2 mM ATP, only 100 μM daurinol clearly inhibited the
catalytic activity of topoisomerase IIα (Fig. 2). These data are in agreement with
the molecular docking predictions.
Daurinol does not inhibit human
topoisomerase I catalytic activity
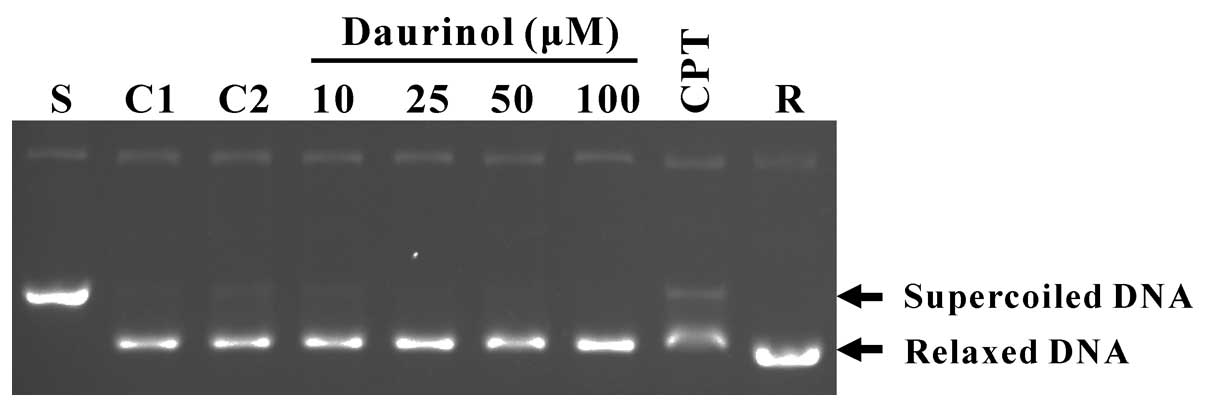
To further confirm that daurinol inhibits human
topoisomerase IIα by targeting the ATP-binding pocket, we measured
the inhibitory activity of daurinol against human topoisomerase I
using an in vitro biochemical assay. Because topoisomerase I
does not contain an ATPase domain and its catalytic activity is
ATP-independent (17), we
anticipated that daurinol, which targets ATP binding, would not
inhibit human topoisomerase I activity. As expected, daurinol did
not severely inhibit the catalytic activity of topoisomerase I at
the concentrations (up to 100 μM) tested, whereas camptothecin (a
topoisomerase I inhibitor) clearly inhibited the catalytic activity
of topoisomerase I (Fig. 3).
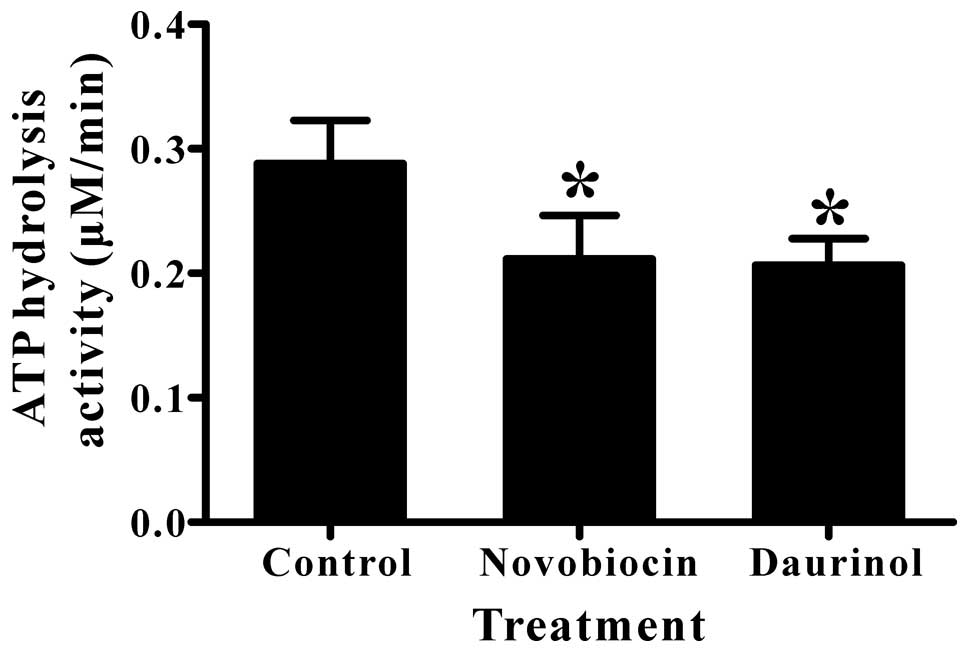
Daurinol inhibits the ATP hydrolysis
activity of human topoisomerase IIα
We also measured the ATP hydrolysis activity of
human topoisomerase IIα in the presence or absence of daurinol
using the malachite green method to confirm that daurinol inhibits
topoisomerase IIα by targeting its ATPase domain. Both daurinol and
novobiocin (a topoisomerase II inhibitor that blocks ATP binding)
(12,18) significantly inhibited the ATP
hydrolysis activity of human topoisomerase IIα (Fig. 4). Taken together, these results
demonstrate that micromolar daurinol selectively inhibits human
topoisomerase IIα by targeting the ATPase domain.
Daurinol inhibits SNU-840 human ovarian
cancer cells in vitro
Next, we measured the anti-proliferative activity of
daurinol in various human cancer lines. We previously reported the
anticancer activity of daurinol in colorectal cancer cells compared
to etoposide. However, etoposide is usually used to treat various
cancers such as ovarian, small cell lung, and testicular cancer
rather than colorectal cancer. Therefore, to evaluate the
overwhelming anticancer effects of daurinol as a novel alternative
to etoposide, we measured the anti-proliferative activity in other
cancer cell types. We tested a total of 9 different cell lines (3
ovarian cancers, 4 small cell lung cancers and 2 testicular
cancers). Daurinol potently inhibited ovarian cancer cell lines,
particularly NIH:OVCAR-3 and SNU-840 cells, compared to etoposide.
Daurinol also inhibited small-cell lung cancer cells, but the
potency of daurinol was similar to or weaker than that of etoposide
(Table I). Because the strongest
anti-proliferative activity of daurinol was observed in SNU-840
cells compared to etoposide, we selected SNU-840 for further
investigation.
 | Table IAntiproliferative activity of daurinol
and etoposide in human ovary, small-cell lung and testicular cancer
cell lines. |
Table I
Antiproliferative activity of daurinol
and etoposide in human ovary, small-cell lung and testicular cancer
cell lines.
| | IC50
(μM) | |
|---|
| |
| |
|---|
| Cancer type | Cell line | Daurinol | Etoposide | Statistical
difference |
|---|
| Ovarian | SK-OV-3 | 15.3±2.7 | 10.2±0.9 | * |
| NIH:OVCAR-3 | 4.6±0.4 | 47.7±3.2 | ** |
| SNU-840 | 0.6±0.2 | 37.5±3.3 | ** |
| Small cell lung
cancer | NCI-H69 | 3.3±1.0 | 8.8±2.4 | |
| NCI-H146 | 4.0±1.3 | 2.7±1.4 | |
| NCI-H187 | 4.6±0.1 | 0.3±0.0 | *** |
| NCI-H417 | 7.2±1.1 | 7.8±1.9 | |
| Testicular | NT2-D1 | 3.3±0.3 | 0.1±0.0 | ** |
| NCCIT | 0.7±0.3 | 18.1±3.0 | ** |
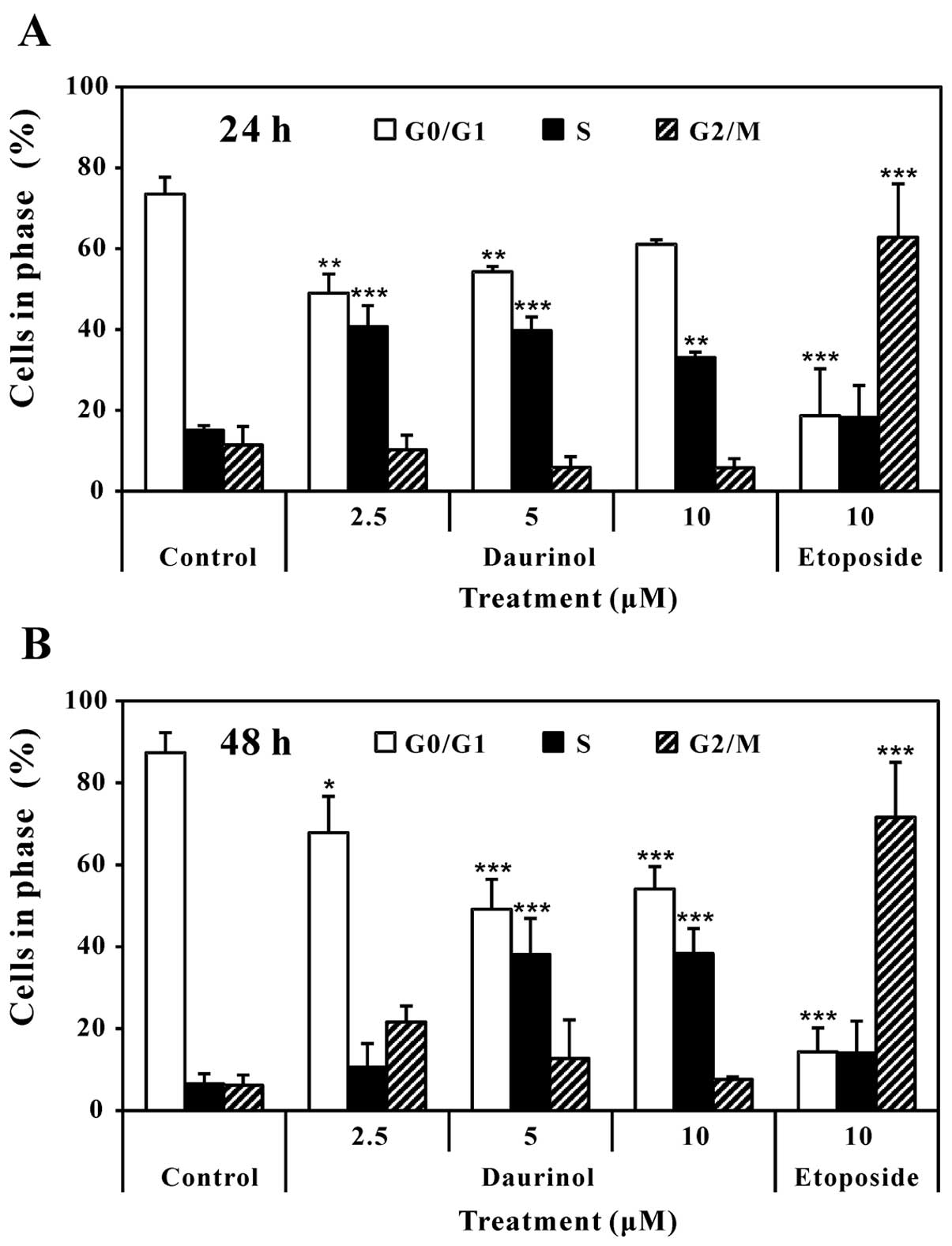
Daurinol induces cell cycle arrest in S
phase in SNU-840 cells
We evaluated the effects of daurinol on cell cycle
distribution in human ovarian cancer cells using flow cytometric
DNA content analysis. Treatment with daurinol for 24 or 48 h
apparenly induced cell cycle arrest in S phase. By contrast,
etoposide significantly induced cell cycle arrest in the G2/M phase
(Fig. 5). Daurinol also induced an
accumulation of the cell population in S phase in NCCIT human
testicular cancer cells (data not shown).
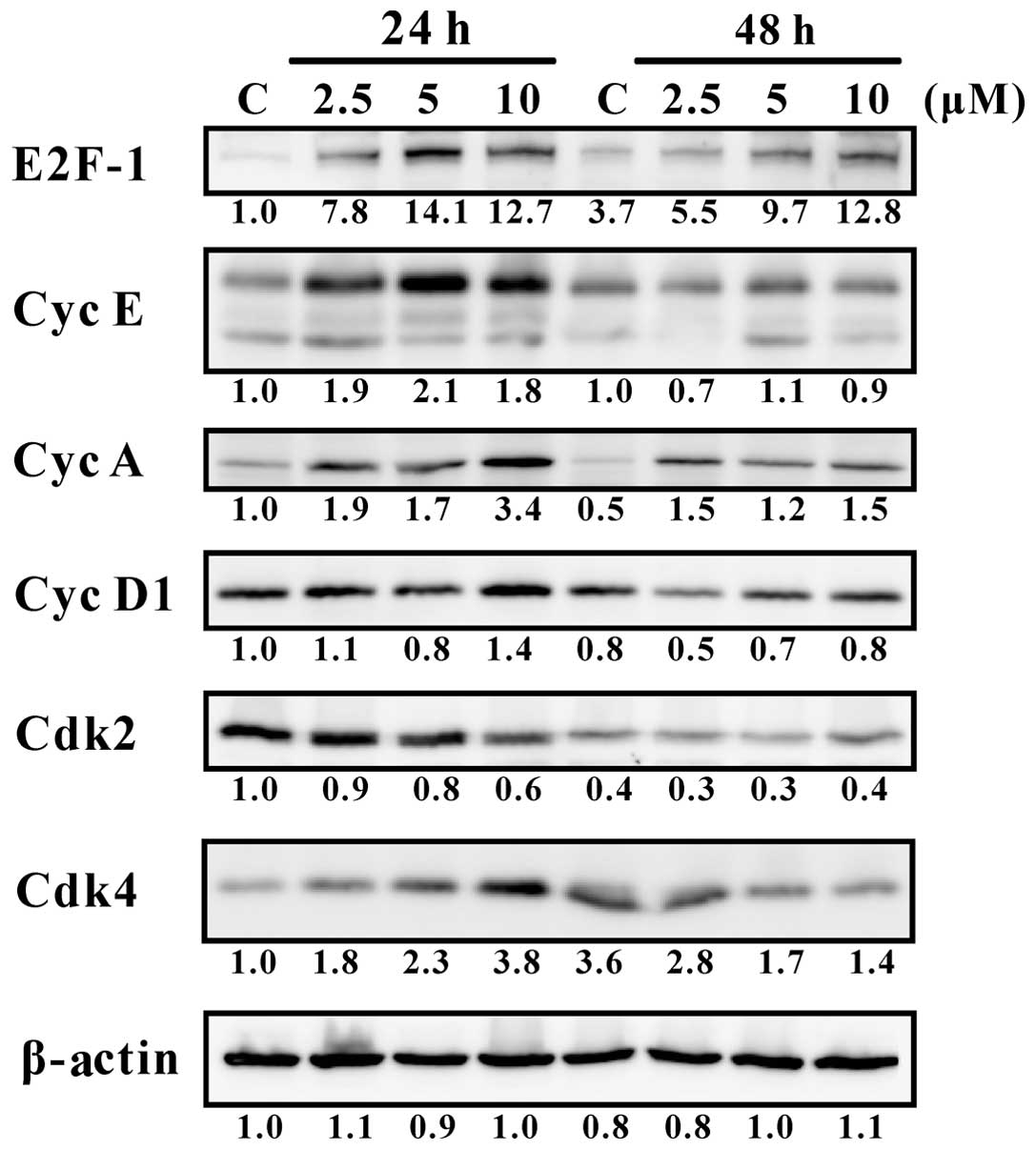
Daurinol induces increased expression of
cyclin E, cyclin A and E2F-1 in SNU-840 cells
We also investigated the expression of cell cycle
regulatory proteins such as cyclins and cyclin-dependent kinases to
elucidate the molecular mechanism underlying the S phase arrest
induced by daurinol in SNU-840 cells. Daurinol increased the
expression of cyclins E and A (Fig.
6), which associate with Cdk2. The resulting complexes cyclin
E/Cdk2 and cyclin A/Cdk2 regulate the initiation and progression of
S phase, respectively (19,20).
The expression of cyclin E was increased after 24 h of treatment;
thus, we speculated that daurinol accelerated the initiation of S
phase at 24 h. The expression of cyclin A was increased at both 24
and 48 h, suggesting that daurinol persisted in the enhanced S
phase progression. In addition, daurinol increased E2F-1 expression
at 24 and 48 h (Fig. 6). E2F-1 is
a transcription factor that promotes the transcription of cyclin E
and A (21). In summary, the
enhanced expression of cyclin E, cyclin A and E2F-1 appear to be
primarily responsible for the S phase arrest induced by
daurinol.
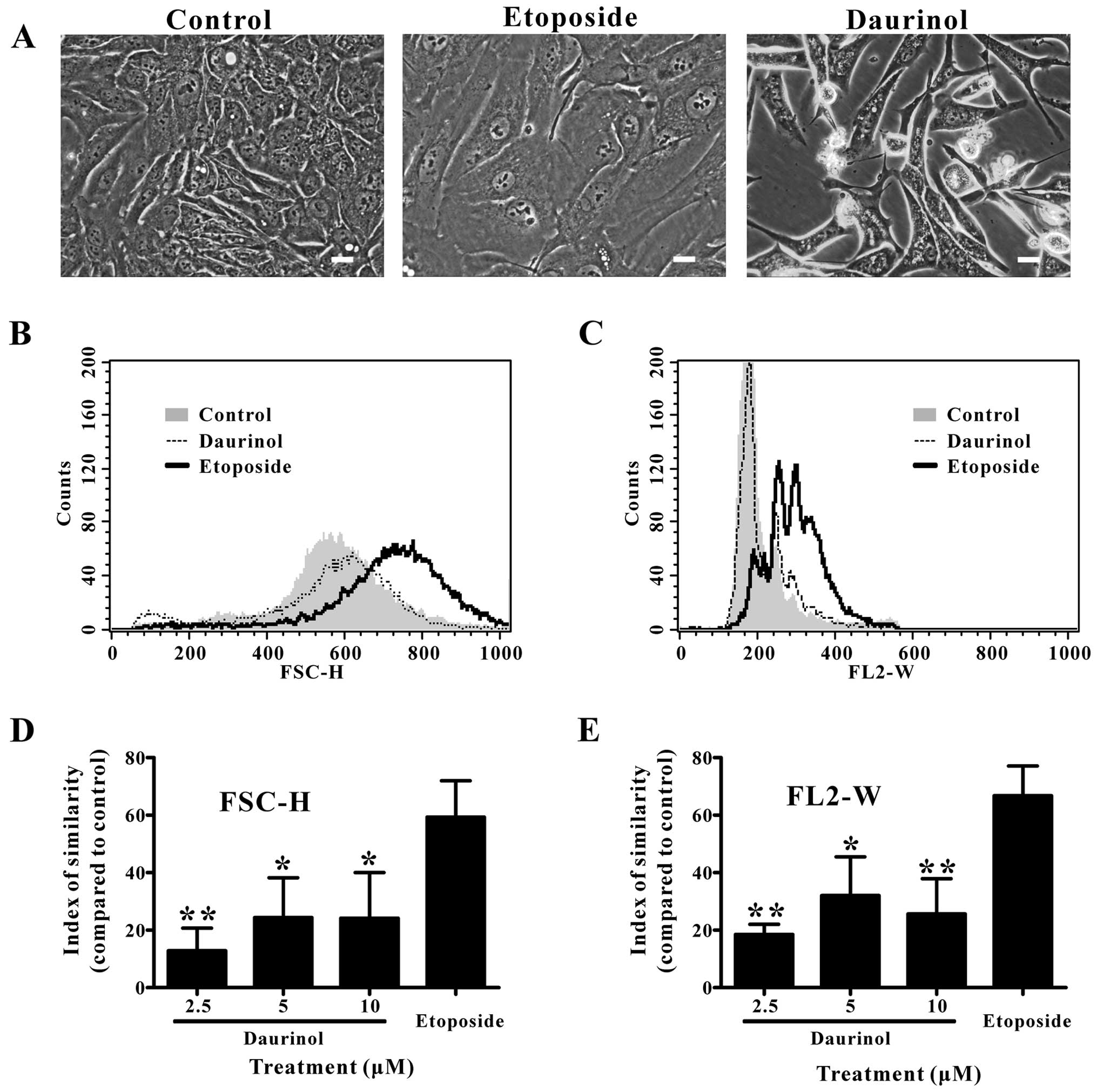
Daurinol does not induce cell or nuclear
enlargement in SNU-840 cells compared to etoposide
Finally, we investigated the effect of daurinol and
etoposide on the cell and nuclear size of SNU-840 cells using
microscopy and flow cytometry (15). Abnormal cell and nuclear
enlargement was previously correlated with the side effects of
anticancer agents (5). Microscopic
observation showed that etoposide induced cell and nuclear
enlargement in SNU-840 cells, while daurinol did not (Fig. 7A). To more accurately evaluate this
phenomenon, we performed flow cytometric analysis of the
fluorescence pulse signal in cells stained with DNA-binding
fluorescent dye. The FSC-H values, which indicate the cell size, of
etoposide-treated cells were higher than those of control or
daurinol-treated cells (Fig. 7B);
the mean values of FSC-H were 554.8±10.5, 587.3±19.6, 578.2±28.0,
604.6±44.2 and 743.3±33.9 for the control, 2.5, 5 and 10 μM
daurinol and 10 μM etoposide-treated cells, respectively.
Similarly, the FL2-W values (pulse width of PI fluorescence), which
indicate the nuclear size (16),
of etoposide-treated cells were higher than those of the control-
or daurinol-treated cells (Fig.
7C); the mean FL2-H values were 197.4±4.5, 218.9±7.9,
237.0±14.4, 224.9±10.3 and 313.0±21.9 for the control, 2.5, 5 and
10 μM daurinol- and 10 μM etoposide-treated cells, respectively.
Kolmogorov-Smirnov statistical analysis demonstrated that the FSC-H
and FL2-W distributions of etoposide-treated cells were
significantly different from those of the control or
daurinol-treated cells. There was no significant difference between
the control and daurinol-treated cells (Fig. 7D and E). Based on these data, we
concluded that daurinol did not induce abnormal the cell or nuclear
enlargement of SNU-840 cells, in contrast to etoposide.
Discussion
In the present study, we suggest the biochemical
mechanism underlying the inhibitory action of micromolar daurinol
against human topoisomerase IIα based on molecular docking studies
and in vitro biochemical experiments. First, computational
simulations predicted that daurinol might bind to the ATP-binding
pocket of the enzyme. Second, a biochemical topoisomerase IIα assay
revealed that increasing the concentration of ATP resulted in
decreased inhibitory activity of daurinol against topoisomerase
IIα. Third, determination of the amount of inorganic phosphate in
the enzyme reaction mixture using the malachite green assay
demonstrated that daurinol inhibited the ATP hydrolysis activity of
topoisomerase IIα. Fourth, daurinol did not inhibit the activity of
human topoisomerase I, which does not contain an ATP-binding
domain. These properties of daurinol are similar to those reported
for D11, a novel glycosylated diphyllin derivative (22). Taken together, these data
demonstrate that daurinol inhibits human topoisomerase IIα by
targeting its ATP-binding domain.
The molecular docking study also predicted that the
benzodioxol functional group of daurinol interacts with a magnesium
ion and the Lys168 residue of the enzyme; the naphthofuranone ring
of daurinol overlaps with the adenine-binding site (Fig. 1C). Based on these data, we are
synthesizing daurinol derivatives to test the binding model and to
optimize inhibitory activity of topoisomerase and anticancer
effects ultimately.
When optimizing lead compounds, several key factors
must be considered. These factors include anticancer potency in
appropriate cancer types; side effects such as hematological
toxicity and drug-induced secondary leukemia; pharmacokinetic
parameters; and physicochemical properties (solubility and
stability). Especially, a rapid method for evaluating side effects
is required prior to preclinical and clinical toxicological
assessment. In a previous study, we suggested that abnormal nuclear
enlargement was correlated with the side effects of the
topoisomerase inhibitors daurinol and etoposide in a human
colorectal cancer model (5). In
the present study, we confirmed that daurinol did not induce
nuclear enlargement in SNU-840 human ovarian cancer cell line, in
contrast to etoposide, which dramatically induced nuclear
enlargement of SNU-840 cells. Therefore, we speculate that daurinol
may be a potential candidate maintenance drug to treat ovarian
cancer with minimal side effects. However, this method of assessing
toxicity is limited because it uses cancer cells rather than normal
tissue cells. To overcome this limitation, we suggest performing
cell and nuclear size analysis in normal cells originating from
human tissue or in a model animal such as Caenorhabditis
elegans (23,24). C. elegans is frequently used
for toxicity assessments of pharmaceutical compounds (24) and is compatible with flow
cytometric DNA content analysis (25).
Etoposide-related secondary leukemia was recently
reviewed. Although etoposide has been used successfully and
extensively for the treatment of various cancers, secondary
leukemia is a critical problem in clinical cancer treatment using
topoisomerase inhibitors (3). The
rearrangement of the mixed lineage leukemia (MLL) gene on
chromosome 11q23 is associated with secondary leukemia. Similar to
the author of that review, we also have hypothesized that prolonged
maintenance of the abnormal enlarged nuclear status induced by
etoposide may cause MLL gene rearrangement and the development of
secondary leukemia. Because the previous (5) and present study confirmed that
daurinol did not induce abnormal nuclear enlargement in either
HCT116 or SNU-8 cells, daurinol may serve as a novel alternative to
etoposide for the treatment of various cancers.
To evaluate daurinol as a novel alternative to the
clinical anticancer drug etoposide, we investigated the
anti-proliferative activity of ovarian, small-cell lung, and
testicular cancer cells in addition to colorectal cancer, as
etoposide is usually used to treat cancers other than colorectal
cancer (5). Among the tested
cancer cell lines, daurinol exerted more significant
anti-proliferative activity than etoposide in human ovarian SNU-840
cells. Daurinol induced cell cycle arrest in S phase in SNU-840
cells, while etoposide induced G2/M phase arrest. These results are
similar to previous reports in HCT116 human colorectal cancer cells
(5).
In summary, we demonstrated that daurinol inhibited
human topoisomerase IIα by interfering with ATP binding during the
catalytic cycle of the enzyme. We also verified the potent
anti-proliferative activity of daurinol in SNU-840 human ovarian
cancer cells. Daurinol induced cell cycle arrest in S phase through
the enhanced expression of cyclin E, cyclin A and E2F-1 in SNU-840
cells. However, daurinol did not induce abnormal nuclear
enlargement compared to etoposide. Based on these data, we suggest
that daurinol could be a novel alternative to the clinical
anticancer agent etoposide. We hope that the results of this study
can be exploited to develop daurinol and its derivatives as
anticancer drugs to avoid the side effects of other topoisomerase
II inhibitors, including hematological toxicity and secondary
leukemia.
Acknowledgements
We gratefully acknowledge Dr Dulamjav Batsuren and
Dr Jigjidsuren Tunsag of the Institute of Chemistry and Chemical
Technology, Mongolia, for kindly providing natural daurinol. The
present study was supported by an intramural grant from KIST
(2Z04220), a grant from the Center Project for Korea-Mongolia
Science and Technology Cooperation (Ministry of ICT, Science and
Future Planning, Korea; 2U04650) and a grant from Gangneung Asan
Hospital (Biomedical Research Center Promotion Fund).
References
|
1
|
Bailly C: Contemporary challenges in the
design of topoisomerase II inhibitors for cancer chemotherapy. Chem
Rev. 112:3611–3640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pogorelcnik B, Perdih A and Solmajer T:
Recent advances in the development of catalytic inhibitors of human
DNA topo-isomerase IIα as novel anticancer agents. Curr Med Chem.
20:694–709. 2013.PubMed/NCBI
|
|
3
|
Ezoe S: Secondary leukemia associated with
the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int
J Environ Res Public Health. 9:2444–2453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen W, Qiu J and Shen YM: Topoisomerase
IIα, rather than IIβ, is a promising target in development of
anti-cancer drugs. Drug Discov Ther. 6:230–237. 2012.
|
|
5
|
Kang K, Oh SH, Yun JH, et al: A novel
topoisomerase inhibitor, daurinol, suppresses growth of HCT116
cells with low hematological toxicity compared to etoposide.
Neoplasia. 13:1043–1057. 2011.PubMed/NCBI
|
|
6
|
Batsuren D, Batirov EK, Malikov VM,
Zemlyanskii VN and Yagudaev MR: Arylnaphthalene lignans of
Haplophyllum dauricum. The structure of daurinol. Chem Nat
Compd. 17:223–225. 1981.
|
|
7
|
Graham JG, Quinn ML, Fabricant DS and
Farnsworth NR: Plants used against cancer - an extension of the
work of Jonathan Hartwell. J Ethnopharmacol. 73:347–377. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hande KR: Etoposide: four decades of
development of a topoisomerase II inhibitor. Eur J Cancer.
34:1514–1521. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park JE, Lee J, Seo SY and Shin D:
Regioselective route for arylnaphthalene lactones: convenient
synthesis of taiwanin C, justicidin E, and daurinol. Tetrahedron
Lett. 55:818–820. 2014. View Article : Google Scholar
|
|
10
|
Lanzetta PA, Alvarez LJ, Reinach PS and
Candia OA: An improved assay for nanomole amounts of inorganic
phosphate. Anal Biochem. 100:95–97. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang K, Jin YM, Jeon H, et al: The three
proline residues (P25, P242, and P434) of Agrobacterium CP4
5-enolpyruvylshikimate-3-phosphate synthase are crucial for the
enzyme activity. Plant Biotechnol Rep. 4:329–334. 2010. View Article : Google Scholar
|
|
12
|
Jun KY, Lee EY, Jung MJ, et al: Synthesis,
biological evaluation, and molecular docking study of
3-(3′-heteroatom substituted-2′-hydroxy-1′-propyloxy) xanthone
analogues as novel topoisomerase IIα catalytic inhibitor. Eur J Med
Chem. 46:1964–1971. 2011.
|
|
13
|
Robinson MJ, Corbett AH and Osheroff N:
Effects of topoisomerase II-targeted drugs on enzyme-mediated DNA
cleavage and ATP hydrolysis: evidence for distinct drug interaction
domains on topoisomerase II. Biochemistry. 32:3638–3643. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang K, Lee HJ, Kim CY, et al: The
chemopreventive effects of Saussurea salicifolia through
induction of apoptosis and phase II detoxification enzyme. Biol
Pharm Bull. 30:2352–2359. 2007.
|
|
15
|
Kang K, Lee HJ, Yoo JH, et al: Cell and
nuclear enlargement of SW480 cells induced by a plant lignan,
arctigenin: evaluation of cellular DNA content using fluorescence
microscopy and flow cytometry. DNA Cell Biol. 30:623–629. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kang K, Lee SB, Yoo JH and Nho CW: Flow
cytometric fluorescence pulse width analysis of etoposide-induced
nuclear enlargement in HCT116 cells. Biotechnol Lett. 32:1045–1052.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roca J: The mechanisms of DNA
topoisomerases. Trends Biochem Sci. 20:156–160. 1995. View Article : Google Scholar
|
|
18
|
Larsen AK, Escargueil AE and Skladanowski
A: Catalytic topoisomerase II inhibitors in cancer therapy.
Pharmacol Ther. 99:167–181. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eastman A: Cell cycle checkpoints and
their impact on anticancer therapeutic strategies. J Cell Biochem.
91:223–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masai H, Matsumoto S, You Z,
Yoshizawa-Sugata N and Oda M: Eukaryotic chromosome DNA
replication: where, when, and how? Annu Rev Biochem. 79:89–130.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
DeGregori J, Kowalik T and Nevins JR:
Cellular targets for activation by the E2F1 transcription factor
include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol.
15:4215–4224. 1995.PubMed/NCBI
|
|
22
|
Gui M, Shi DK, Huang M, et al: D11, a
novel glycosylated diphyllin derivative, exhibits potent anticancer
activity by targeting topoisomerase IIα. Invest New Drugs.
29:800–810. 2011.PubMed/NCBI
|
|
23
|
Schmeisser S, Schmeisser K, Weimer S, et
al: Mitochondrial hormesis links low-dose arsenite exposure to
lifespan extension. Aging cell. 12:508–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dengg M and van Meel JC: Caenorhabditis
elegans as model system for rapid toxicity assessment of
pharmaceutical compounds. J Pharmacol Toxicol Methods. 50:209–214.
2004. View Article : Google Scholar
|
|
25
|
van den Heuvel S and Kipreos ET: C.
elegans cell cycle analysis. Methods Cell Biol. 107:265–294.
2012.
|