1. Introduction
Cancer is classically viewed as a genetic disease
where the biology, pathophysiology and clinical features of
neoplasia are almost entirely defined by events occurring within
the genome of the cancer cell. This paradigm has been challenged in
recent years, and today cancer is considered as an
ecological disease, involving a dynamic interplay between
malignant and non-malignant cells. Although changes in the genome
and epigenome of the cancer cell continues to be considered as
essential for the onset and evolution of the disease, there is a
growing interest in the heterogeneous array of cells existing
within the tumor mass. Pathologists have long recognized that
tumors may be densely infiltrated by cells of both the innate and
adaptive arms of the immune system, and nowadays it is clear that
virtually every neoplastic lesion contains variable quantities of
infiltrating immune cells. Whereas in the past the immune
infiltrating cells have been thought to reflect an attempt by the
immune system to eradicate the growing tumor, in more recent years
it is emerging that the immune infiltrate can actually sustain
cancer growth, by directly providing growth and mitogenic factors
to the tumor mass and by indirectly contributing to the genetic and
epigenetic alterations that occur during tumor progression
(1).
It is important to remark that this model represents
one among many views concerning the liaison between chronic
inflammation and cancer. There is clear clinical and
epidemiological evidence of the impact of chronic inflammation on
cancer. The observation that cancer often arises at sites of
chronic inflammation and the indirect evidence of a protective
effect of the chronic use of non-steroidal anti-inflammatory drugs
(NSAIDs) against both colorectal and prostate cancers are important
examples supporting a model whereby inflammatory cells play a
causative role in cellular and molecular oncogenesis (2). Moreover, two major pathways leading
to inflammation in the cancer microenvironment have been described
by Allavena et al (3) and
Colotta et al (4).
In the intrinsic pathway, the genetic events
in cancer cells may trigger inflammation-related programs that
promote the assembly of an inflammatory milieu; conversely, in the
extrinsic pathway, inflammation may speed up the early
neoplastic transformation of normal tissue cells, thus facilitating
the oncogenic process in a given tissue. In other words, the
trigger of inflammation leading to neoplasia may originate in the
tumor stroma (in the extrinsic pathway) or inside the cancer cell
itself (in the intrinsic pathway). Finally, chronic inflammation
may affect cancer at the different stages of initiation, promotion
and progression (5,6).
Our article addresses the role of chronic
inflammation in the initiation step; intestinal cancer was chosen
as a model, since some of the most convincing examples of
carcinogenesis induced by chronic inflammation are seen in the
gastrointestinal tract (7).
The anatomy of the intestinal epithelial lining is
uniquely suited to study adult stem cells in their niche. The bowel
crypt, in particular the Lieberkühn crypt of the small intestine,
is indeed an ideal developmental biology system, as proliferation,
differentiation and cell migration are distributed linearly along
the long axis of the crypt (8).
Crypts are lined with younger transit-amplifying
epithelial cells that proliferate and migrate towards the surface
of the mucosa and the villi, progressively differentiating and
acquiring diverse secretory, enteroendocrine and absorptive
functions. Mucosal enterocytes, Paneth and goblet cells derive from
stem cells located at the bottom of the crypts, as a result of a
finely regulated process involving proliferation, migration and
ultimate differentiation of these cells. Interestingly, crypt stem
cells are regarded today as the most likely targets of neoplastic
transformation in bowel cancer (9,10).
2. Epidemiology, histology, pathogenesis and
prognosis of colitis-associated colorectal cancer in humans
The general term inflammatory bowel disease (IBD)
includes two distinct severe chronic inflammatory conditions of the
intestine, Chron’s disease (CD) and ulcerative colitis (UC).
Similar to individuals affected by familial forms of
colorectal cancer (CRC) - i.e., adenomatous polyposis and
hereditary non-polyposis-associated CRC- IBD patients are at
increased risk for developing colitis-associated colorectal cancer
(CAC), when compared to sporadic colorectal cancer (CRC) occurring
in the general population.
Epidemiology
As a matter of fact, the exact extent of the risk of
developing CRC associated with IBD has not been quantified yet, due
to the variable design of the epidemiological studies conducted so
far. Nevertheless, there is substantial agreement that the risk of
developing CRC in IBD patients exceeds that of subjects without IBD
by a factor of approximately 3–5-fold, and that the duration of
IBD, its extent, the severity of inflammation, the patient’s
gender, the family history of sporadic CRC and the presence of
concomitant sclerosing cholangitis are major risk factors,
concurring to varying extent to the onset of CRC in patients
suffering from IBD (11).
A widely cited meta-analysis of 116 studies
performed on a total of 54,478 patients, reports that the estimated
cumulative incidence of CRC in UC is 2% after 10 years, 8% after 20
years, and 18% after 30 years of disease (results are from a
sub-analysis of 19 studies) (12).
Although risk assessment studies have mostly focused
on ulcerative colitis patients, it is emerging that the magnitude
of the risk for developing CRC in Crohn’s disease patients is
similar to that for UC (13).
Since more recent analyses have claimed lower
incidence data (14–17), it has been postulated that the
absolute risk of developing CRC is declining, mainly due to
increasing worldwide diffusion of endoscopic surveillance
protocols.
Histology
Carcinogenesis in IBD proceeds through increasing
grades of dysplasia, generally defined as unequivocally neoplastic
but non-invasive epithelium. Remarkably, the sequence of
histo-pathological events described in CAC is in sharp contrast
with the development of non-IBD-associated familial or sporadic
colorectal carcinomas, arising in most cases from single, monofocal
adenomatous lesions (18–20).
A widely accepted model of colitis-associated
oncogenesis is based on the progressive acquisition of neoplastic
features through transition from low-grade dysplasia (LGD) to
high-grade dysplasia (HGD) and, ultimately, to multifocal
adenocarcinoma. Multifocal CAC lesions emerge from dysplastic
mucosa as the result of a field-effect, with a frequency of
multiple sychronous lesions ranging between 10 and 30%, compared to
3–5% in the general non-CAC population (18,19).
Dysplastic lesions may be broadly classified as flat
or raised, the latter being endoscopically visible. Raised
macroscopic lesions may present as resectable adenomas or polyps,
or as endoscopically unresectable dysplasia-associated
lesion-or-mass (DALM) (19).
Microscopic diagnosis of IBD-associated dysplasia is
often difficult, and is characterized by a high degree of
inter-observer variability. The histological features of dysplasia
include distorted crypts showing cellular atypia and sometimes
containing increased numbers of dystrophic goblet cells. In LGD,
moderate enlargement, hyperchromasia and mild stratification of the
nuclei of crypt epithelial cells are observed. Moreover,
polarization is maintained, and nuclei are seen in the basal
one-third of epithelial cells. In HGD, nuclear enlargement,
hyperchromasia, increased nuclear/cytoplasmic ratio and
stratification are more pronounced, there is increasing loss of
cellular polarity, and nuclei and mitotic figures are frequently
visible in the upper half of epithelial cells (19,21).
At the microscopic level, CAC is characterized by a high frequency
of mucinous or signet-ring cell phenotypes (22).
Pathogenesis
Clinically detectable IBD always precedes, sometimes
by decades, tumor initiation in CAC. Whereas CAC risk directly
correlates with the severity and extent of active inflammatory
disease, both familial and sporadic CRC do not arise in the context
of preceding inflammation. In other words, whereas in CAC chronic
inflammation likely drives the onset of neoplasia from its early
stages, in sporadic or familial CRC chronic inflammation may be
evoked by the cancer cell itself, in a later phase of the
neoplastic progression (5,23,24).
Current models of bowel oncogenesis are based on a
well established multistep sequence of alterations in oncogenes or
tumor suppressor genes occurring within crypt stem cells.
Chromosomal and/or microsatellite instability may further
contribute to the full neoplastic transformation and clonal
expansion of tumor cells (9,25,26).
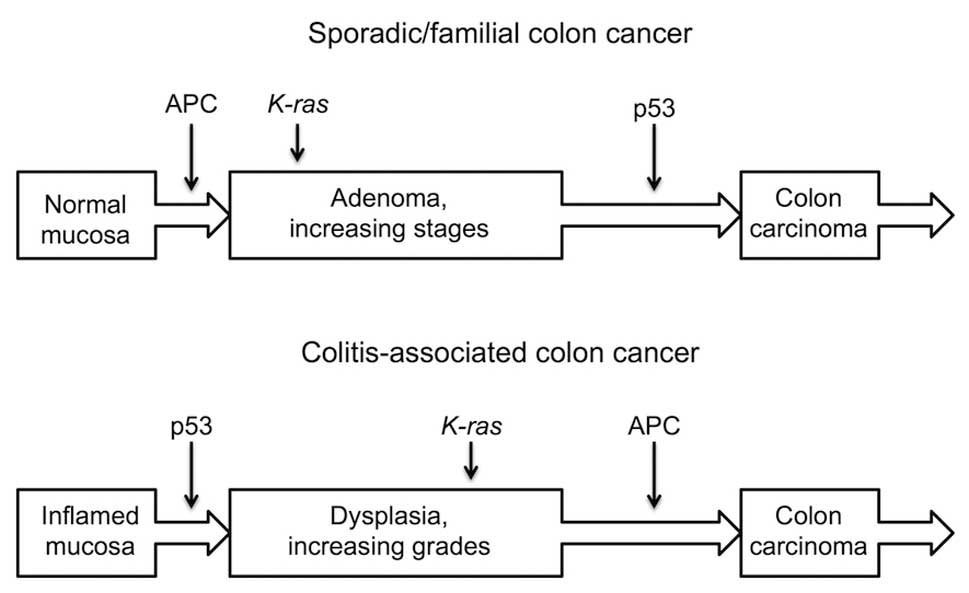
Interestingly, whereas genes like β-catenin, APC,
p53, K-ras and c-src are known to play a pivotal role in tumor cell
initiation, promotion and progression, it has become evident that
the pattern of sequential activation of these genes in CAC
significantly differs from that of familial and sporadic CRC
(Fig. 1). The Wnt/β-catenin
signaling pathway has been extensively investigated, due to its key
role in the renewal of intestinal epithelium and in the fine
regulation of normal and malignant cell proliferation. In familial
or sporadic CRC, the mutational inactivation of APC, a negative
regulator of the Wnt/β-catenin signaling pathway, appears to be an
immediate-early event, occurring in over 90% of cancers and
triggering the formation of focal early adenomas. Besides APC,
GSK3β, a kinase that controls APC, and β-catenin were also found to
be mutated, albeit with a lower frequency.
Despite their pivotal role in bowel tumor
development, genetic alterations of the Wnt/β-catenin signaling
machinery were shown to occur at a late stage of colitis-associated
oncogenesis. In particular, APC loss of function, occurring through
gene/protein truncation or allelic loss, was shown to take place
during the transition from high-grade dysplasia to frank carcinoma
(Fig. 1) (18,26).
Remarkably, several lines of evidence suggest that
different inflammatory pathways can enhance β-catenin signaling
during the early phases of colitis-associated carcinogenesis in the
absence of activating mutations.
At variance with the Wnt/β-catenin signaling
pathway, which is altered in the early steps of carcinogenesis,
p53 alterations are considered as later events in familiar
or sporadic colon oncogenesis, occurring during the final
adenoma-carcinoma transition (Fig.
1). Conversely, p53 alterations are apparently taking place in
the early stages of the genesis of CAC. It should be stressed that
these models are in no way univocal, since some authors describe
p53 mutational inactivation as an early step in both sporadic and
colitis-associated oncogenesis (27).
Interestingly, both models of colitis-associated and
familial/sporadic carcinogenesis involve mutational activation of
K-ras and c-src activation as intermediate steps, causing the
transition towards increasingly transformed dysplastic or
adenomatous lesions, ultimately resulting in the onset of frank
carcinoma (26,28).
Prognosis and management of colon
cancers
The prognosis for sporadic CRC and CAC is similar,
with a 5-year survival of approximately 50% (11). The diagnosis and grading of colonic
dysplasia in endoscopic surveillance biopsies play a key role in
the management of patients with IBD, as patients with dysplasia are
3–30-fold as likely to have cancer anywhere in the colorectum,
compared to patients not showing dysplastic lesions (29,30).
Moreover, it was demonstrated that a diagnosis of CRC is made in
20–50% of IBD patients previously diagnosed with colorectal
dysplasia (31,32), and that dysplasia is found together
with carcinoma in over 90% of resected surgical specimens (26).
In the presence of HGD, prophylactic proctocolectomy
is usually recommended, whereas no consensus has been reached
regarding a surgical indication upon detection of LGD. This issue
is further complicated by the very high rate of inter-observer
variability (50–60%) in the diagnosis of LGD (26,33,34).
In any case, a DALM with HGD or LGD, or pancolonic disease, or
active disease for over 10 years are considered as indications for
proctocolectomy (35).
3. Does chronic inflammation play a role in
the early stages of colon carcinogenesis? An overview of
established and putative mechanisms
The current literature highlights a potential role
for chronic inflammation in virtually all steps of carcinogenesis,
including tumor initiation and promotion, as well as progression
(4,5,23,24).
The classical definition of initiation is a set of events that
introduce changes into the genome and/or epigenome of an otherwise
normal cell, driving its neoplastic transformation. Promotion
instead is the action of a substance or stimulus supporting the
clonal growth or survival of a previously initiated cell by
increasing proliferation or by inhibiting apoptosis; this process
is also facilitated by the onset of angiogenesis within the
incipient tumor. Finally, progression is the gain of novel
molecular and functional features by the in situ cancer,
that further direct neoplastic growth and tumor mobilization,
eventually leading to invasion and metastasis.
Notably, chronic inflammation may extend its
influence far beyond the invasion and metastasis steps. In fact,
inflammation also drives systemic metabolic alterations such as
cachexia (36). Inflammation is
traditionally understood to play a role in the tumor promotion step
of carcinogenesis. Hence, the preventive effect of NSAIDs on cancer
development is likely due to the dampening of chronic inflammation
at the stage of tumor promotion (37). In addition, a pivotal role of
chronic inflammation in tumor progression is now quite well
established (38), for example
during the epithelial mesenchymal transition (EMT), which is a key
trigger of subsequent invasion and metastasis (39). Yet, an impact of chronic
inflammation during the initiation step of carcinogenesis remains
experimentally elusive and hence more controversial.
The research group led by Michael Karin has
thoroughly investigated the relationship between chronic
inflammation and intestinal cancer (5,23,24,27).
Their experimental approach has been to alter, by tissue-specific
transgenesis, the expression of pivotal players of the inflammatory
machinery (e.g., ablation of the NF-κB pathway by conditional
knockout of IKKβ) in intestinal epithelial cells (IECs) or in cells
of the myeloid lineage. After exposure to a CAC-inducing regimen
(see below), significant variations in the rate of cancer onset
have been observed in transgenic mice, when compared to parental
animals. If transgenesis were found to alter the median number of
tumor foci per mouse rather than the tumor median size, it could be
implied that inflammation plays a major role in tumor initiation
(inflammatory ‘field-effect’) (40); conversely, if a variation in the
tumor median size were observed, it might be supposed that
inflammation acts mainly as a tumor promoter.
Whether an inflammatory cue that stimulates and
sustains proliferation in a cell that is not initiated, but is at
risk of neoplastic transformation because of its stemness, should
be considered as an initiator or a promoter is a matter of debate.
As a matter of fact, experimental approaches quite often do not
produce clear-cut results and, accordingly, the distinction between
initiation and promotion is not always straightforward.
The mechanisms whereby a state of chronic
inflammation could initiate the neoplastic process in the intestine
(24) may be approximately
classified into four groups. Experimental evidence is available
supporting the first two mechanisms, but the last two are rather
hypothetical and at present ill-defined.
Inflammation may act as an initiator of oncogenesis
by directly inducing DNA damage and mutagenesis through production
of reactive oxygen species (ROS) and reactive nitrogen
intermediates (RNI). This issue is well established, since chronic
colitis produces local and systemic genotoxic effects, whereby
macrophages and neutrophils recruited by inflammation act as the
principal sources of ROS and RNI. Within this framework, mutations
in the p53 gene have been identified not only in areas of
dysplasia or carcinoma, but also in inflamed, but otherwise normal
intestinal mucosa. Moreover, it has been unambiguously demonstrated
that prolonged chronic inflammation itself can induce detectable
DNA damage and intestinal tumors in murine models, in the absence
of exposure to mutagens (24 and references therein).
Inflammatory signaling may stimulate
hyper-proliferation in non-initiated IECs, thus increasing the risk
of neoplastic transformation. This is an issue of major concern for
our discussion. Notably, inflammatory mediators, such as cytokines
IL-1β and TNFα (41–43), or soluble mediators such as
prostaglandin E2 (44), may act in
some circumstances as growth or survival stimuli. The principal
pathways targeted by these agents are Wnt/β-catenin, Akt, NF-κB and
STAT3. Interestingly, in most cases the final aftermath of these
signal transduction pathways is β-catenin nuclear accumulation even
in the absence of APC mutations. A prominent exception is the STAT3
pathway, triggered by IL-6 as well as by other cytokines, where the
main effect on IECs is inhibition of apoptosis, which in turn
enhances cell survival rates even without involving the
Wnt/β-catenin pathway (45,46).
It remains to be ascertained whether
hyper-proliferation of IECs, occurring mainly through a
dysregulated Wnt pathway, needs to be continuously stimulated by
the pro-inflammatory microenvironment or is rather self-sustained
as a consequence of an epigenetic switch. The normal gut microbiota
could play an important role in intestinal epigenetic homeostasis
by producing high amounts of butyrate, a short chain fatty acid
acting mainly through the inhibition of histone deacetylases. In
this scenario, a dysbiotic state could cause inadequate butyrate
production by the microbiota, thereby facilitating the
establishment of pro-cancerous epigenetic tags in IECs (47).
Chronic intestinal inflammation may start a
breakdown of protective intestinal barriers, which causes increased
accessibility of IECs to food-borne mutagens. Accordingly,
inactivation of Muc2, a major component of the mucus layer in the
colon, causes spontaneous intestinal inflammation (48), that progresses to CAC even without
further exposure to external carcinogens or mutagens (49). A large body of evidence supports
the view that alterations in the gut mucus layer, in the integrity
of IEC tight junctions and/or in the gut microbiota may lead to
increased susceptibility to intestinal inflammation and cancer
(24,50,51).
Inflammation may cause inactivation of genes
encoding DNA proofreading proteins, or enzymes involved in repair
mechanisms, such as mismatch repair (MMR). Likewise, inflammation
may increase synthesis and activity of activation induced cytidine
deaminase AID, which is thought to cause mutations and genetic
instability (24).
Although experimental evidence indicating a role for
the mechanisms summarized in points i-iv at the very onset of
neoplasia has been reported, obtaining a definitive proof of these
concepts will be challenging, for reasons that mostly concern the
epigenetics of cancer. Evidence accumulated during the past few
years indicates that epigenetic changes are strongly associated
with cancer development (52).
Epigenetic alterations are now recognized to be a driving force of
the processes at all steps of carcinogenesis (53). Whether these mechanisms might be
involved in very early alterations in cancer, thus anticipating
genetic mutations, or whether, most probably, the two types of
hereditary alterations concur to cancer development, is still
debated. However, it is well known that demethylation of DNA causes
mutations and chromosome instability, and aberrant DNA methylation
has been observed in many human diseases (54).
Three main epigenetic processes are now recognized
to remodel genetic expression programs during development and
differentiation: DNA methylation, histone modifications and the
activation of the miRNA pathway. Chronic inflammation has been
shown to induce epigenetic alterations, and these alterations are
also observed during inflammation-induced tumorigenesis,
contributing to the processes whereby a normal cell becomes
neoplastic (4).
Hypermethylation of the cytosine (5′meC) in CpG
islands (CGI) of the promoter regions or in the gene body of tumor
suppressors, leading to gene repression, has been observed in
several diseases that can degenerate into cancers, including: i)
Helicobacter pylori-associated gastritis [hypermethylated
genes being CDNK2A (p16), FLNC, HAND1, HRASLS, LOX, THBD]; ii)
ulcerative colitis (CDNK2A and MyoD1); iii) virus-induced hepatitis
(HBV and HCV; CDNK2A, RUNX3); and iv) reflux esophagitis (Barret’s
esophagus) (CDNK2A) (for detailed reviews see 55,56).
Interestingly, in Helicobacter pylori-associated gastritis,
the frequency of cells harboring mutations is lower than the
frequency of cells with aberrant DNA methylation, suggesting that
in gastric mucosae DNA methylation alterations might be important
inducers of cell transformation (57). By using glutathione peroxidase
double knockout (Gpx1/2-KO) mice as a model of inflammatory bowel
disease that predisposes to cancer, Hahn et al have shown
that 60% of the Polycomb (PcG) target genes that are found to be
methylated in tumors were already methylated in the inflamed normal
tissue. Moreover, hypermethylation of CGIs in the ileum of
Gpx1/2-KO mice is often associated with loss of trimethylation of
histone H3 lysine 27 (H3K27) at the same loci. These data suggest
that PcG proteins might direct an aberrant inflammatory DNA
methylation and histone signature that is observed later in the
transformed tissue (57).
A number of studies suggest that cross-signaling
between epithelial and stromal cells leads to autocrine and
paracrine networks of diffusible factors, resulting in intense
cross-talk that contributes to tumor progression (58). Using an animal model in which TGF-β
signaling had been deleted in stromal fibroblasts to induce
inflammation and DNA damage in the neighbouring epithelia of the
forestomach, Achyut et al observed loss of transcription of
cell cycle-dependent kinase inhibitors p15 and p16 and p21waf1/cip1
hypermethylation (59). Most
interestingly, these events were preventable by treating the mice
with the selective COX2 inhibitor celecoxib, suggesting a direct
role of inflammation in the silencing process.
Although overexpression of IL-1β has been found to
be associated with an increased risk of human gastric cancer
(60–62), and although upregulation of
cytokines have been found in gerbil gastric mucosae infected with
H. pylori (Cxcl2, IL-1β, Nos2 and TNF) and in human
hepatitis and ulcerative colitis (TGF-β, IL-10, TNFα, IL-1β, Nos2)
(56,63,64),
the mechanism by which cytokine signaling is able to produce
epigenetic changes is not yet fully understood.
Global DNA hypomethylation is also observed during
tumor induction following inflammation. Repetitive sequences of the
genome, such as LINE1, Alu and Satα, amounting to over 40% of the
human genome and often used as surrogate markers of genome-wide DNA
methylation, are induced to lose 5′meC in gastric mucosae of
individuals affected by H. pylori infection (65).
Finally, another point of major concern is the role
played by hypoxia and hypoxia-induced transcription factors (HIFs)
in both models of colon carcinogenesis, i.e., CRC and CAC. Chronic
hypoxia regularly occurs in solid tumors, including CRC and CAC; in
addition, increases in HIF-dependent signaling can also ensue from
activation of oncogenic pathways, including the Wnt/β-catenin,
Ras/Raf/MAPK and PI3K/Akt/mTOR pathways, as well as from tumor
suppressor gene silencing (66,67).
On the other hand, a chronic increase in HIF signaling in colon
epithelial cells has been reported to trigger an inflammatory
response through direct activation of genes encoding
pro-inflammatory cytokines (68,69)
and through extensive cross-talk with inflammatory transcription
factors, such as STAT3 and NFκB (70). Conversely, an inflammatory
microenvironment can sustain high levels of HIF signaling through
both oxygen-dependent and -independent mechanisms (71–74).
Hypoxic adaptation has long been acknowledged as a major driving
force for tumor progression (75),
but its role in tumor initiation is still debated. While HIF
activation has been reported to lead to acquisition of stem
cell-like properties by tumor cells (76), hard evidence for a similar role in
non-initiated cells is elusive. However, hypoxia-induced epigenetic
silencing (via colonic expression of the transcriptional repressor
DEC-1) leading to MMR deficiency and genomic instability has been
demonstrated in a mouse IBD model (77), suggesting that HIF-dependent gene
expression might also impact on the initiation phase.
In conclusion, chronic inflammation can produce
profound alterations in the epigenome in normal tissues that might
be causative of neoplastic transformation.
4. Intestinal stem cells and their possible
involvement in the early onset of bowel cancer
The different regions of the epithelial monolayer
lining the gut mucosa display correspondingly different
anatomo-functional features and cell composition. Whereas the
epithelium of the small intestine is organized into large numbers
of self-renewing crypt-villus units, the surface of the large
intestine still retains the crypts, yet it is devoid of villi. In
both the small and the large intestine crypts are lined with
transit-amplifying TA cells which proliferate and migrate towards
the lumen surface of the mucosa, progressively differentiating into
four main types of post-mitotic cells. Mucosal enterocytes,
enteroendocrine cells, goblet and Paneth cells derive from stem
cells located at the bottom of the crypts, as a result of a finely
regulated process involving proliferation, migration and ultimate
differentiation into post-mitotic cells. Of the utmost relevance to
this context, Paneth cells are normally found exclusively in the
small intestine, and are absent from all other parts of the
gastrointestinal tract (78).
The intestinal epithelium represents the most
vigorously renewing tissue in adult mammals. Since the unique
anatomy of the intestinal crypt epithelium makes it one of the most
accessible models for the study of adult stem cell biology, the
research on this subject started almost four decades ago (8). However, the stem cells that fuel the
gut self-renewal process have been identified only recently. In
2007 Barker et al identified Lgr5 as putative marker of
crypt stem cells. Lgr5 is an orphan receptor of unknown function,
and is targeted by the Wnt signaling pathway (79).
Through an elegant series of lineage tracing
experiments, using a knocked-in, tamoxifen-inducible Cre
recombinase, it was demonstrated that Lgr5 positive cells lie at
the bottom of the crypt, intermingled with terminally
differentiated Paneth cells. Most importantly, it was shown that
such cells could differentiate into all four main cell types found
within the intestinal epithelium meeting the definition of a stem
cell. Counter-intuitively, Lgr5 cells are not nearly as quiescent,
as stem cells would be expected to be, but divide every day. Lgr5
daughter cells constitute the transit-amplifying (TA) crypt
compartment. TA cells divide every 12–16 h, generating some 300
cells per crypt every day; they reside within the crypts for
approximately 48 h, undergoing up to five rounds of cell division
while migrating upwards. When committed TA cells reach the
crypt/villus junction, they rapidly differentiate into absorptive
enterocytes, enteroendocrine cells or mucosecreting goblet cells,
while continuing their upward migration. In contrast, Paneth cells
escape this flow and reside for 3–6 weeks at the base of the
crypt.
Despite the enthusiasm raised by the discovery of
Lgr5 as the definitive marker of crypt stem cells, further research
reignited the long lasting controversy over the exact location of
intestinal stem cells. In fact, the exact identity of intestinal
stem cells has proven controversial over the last 30 years, with
two opposing models dominating the literature (80).
In the ‘+4 position’ model it was assumed that the
base of the crypt is exclusively populated by terminally
differentiated Paneth cells, and that stem cells should therefore
be located just above Paneth cells at the +4 position. Instead, the
more recent ‘stem cell zone’ model states that small,
undifferentiated, cycling cells, intermingled with Paneth cells,
are likely to be the true intestinal stem cells. These cells were
termed crypt base columnar (CBC) cells, and are currently
identified by the Lgr5 surface marker.
Moreover, in 2008, using an approach similar to the
one implemented by Clevers et al, Sangiorgi and Capecchi
identified Bmi1 as a further marker for adult intestinal stem cells
(10). Remarkably, Bmi1-positive
cells appear to occupy the previously described +4 position, and
therefore are distinct from Lgr5 positive CBC cells. Moreover,
whereas Lgr5 positive (Lgr5+) cells are known to be
rapidly dividing, in most cases Bmi1 positive cells are quiescent
or slowly replicating.
These findings have stimulated rethinking of the
biology of the stem cell niche: distinct quiescent and active stem
cell compartments may coexist within rapidly self-renewing tissues
like the gut, skin and bone marrow, in separate yet adjoining
locations. Actively dividing stem cell types (Lgr5+
cells in the intestine) could serve to maintain the regenerative
capacities of these tissues under homeostatic conditions, whereas
quiescent cells, less affected by environmental stresses
(Bmi1+ in the intestine), could be held in reserve,
playing the role of ‘backup’ cells (81).
There has been considerable discussion concerning
whether cancer arises from adult stem cells. This issue has been
thoroughly investigated in murine models of carcinogenesis in the
small intestine. The contribution of adult intestinal stem cells to
colorectal cancer initiation has been studied by transgenic
induction of the conditional deletion of APC exclusively in the
Lgr5+ intestinal stem cell compartment. This resulted in
rapid transformation of Lgr5+ stem cells, which
persisted and fuelled the growth of large adenomas mainly in the
small intestine, but also in the colon. In contrast, and
remarkably, APC deletion in non-stem TA cell populations failed to
sustain significant adenoma growth (9).
The issue of the cell of origin of intestinal cancer
was further investigated in quiescent intestinal stem cells marked
as Bm1-positive (Bmi1+), where transgenic ectopic
expression of an oncogenic variant of β-catenin was also found to
promote intestinal neoplasia in mice (10). Overall, these observations support
the hypothesis that stem cells are the predominant cell-of-origin
of colorectal cancer.
Two models for the histogenesis of colorectal cancer
in humans have been suggested: the ‘top-down’ model, proposing that
dysplastic cells located in the intracryptal region eventually
spread laterally and downwards to form new crypts, and the
‘bottom-up’ model, which posits that the inceptive cancer derives
from the base of the crypt spreading upwards (82,83).
A number of findings in murine models of bowel
tumorigenesis lend support to the bottom-up hypothesis for the
generation of adenomas.
Whereas the outstanding, clear-cut results described
above indicate that the main cell of origin for cancer in the small
intestine of mice is the adult stem cell, the current, dynamic view
of the cancer stem cell hypothesis, that somehow also applies to
bowel cancer models, maintains that intestinal stem cells are not
the only cells from which cancer may originate. It is now clear
that cancer-propagating cells, viz. the cancer stem cells,
may regenerate during neoplastic progression from TA to cancer
cells, most notably during the epithelial-mesenchymal transition
(EMT). In other words, the present view is that almost any somatic
cell, normal or cancerous, has the potential to trigger or
propagate the neoplasia in the presence of appropriate genetic or
epigenetic conditions (84,85).
In continuously renewing ‘labile’ tissues, such as
the epidermis, the gut and the bone marrow, it is now accepted that
cancer arises mostly from adult, normal stem cells. However, the
case of stable tissues (a most investigated example is the liver)
is different, in that a wider range of cell types can be the
target, and then the origin, of tumorigenesis (86).
5. The impact of pro-inflammatory signaling
on bowel adult stem cells. Unanswered questions and possible
experimental approaches
In order to draw a more conclusive picture of the
events linking inflammation to the early steps of oncogenesis in
intestinal stem cells, the tumorigenic potential of ectopic,
chronic release of pro-inflammatory cytokines in the adult
intestinal stem cell niche should be investigated in detail.
Specific in vivo models should be designed, to convey
chronic inflammation in the close proximity of adult intestinal
stem cells with the prospect of initiating neoplastic
transformation. Targeted ectopic expression of pro-inflammatory
cytokines in Paneth cells may offer the opportunity to redirect
adult intestinal stem cells toward oncogenesis by acting directly
on the stem cell niche. To illustrate this hypothesis a standard
cre-lox tamoxifen-inducible strategy of transgenic expression in a
cell of interest may be designed, as described in Fig. 2 (87).
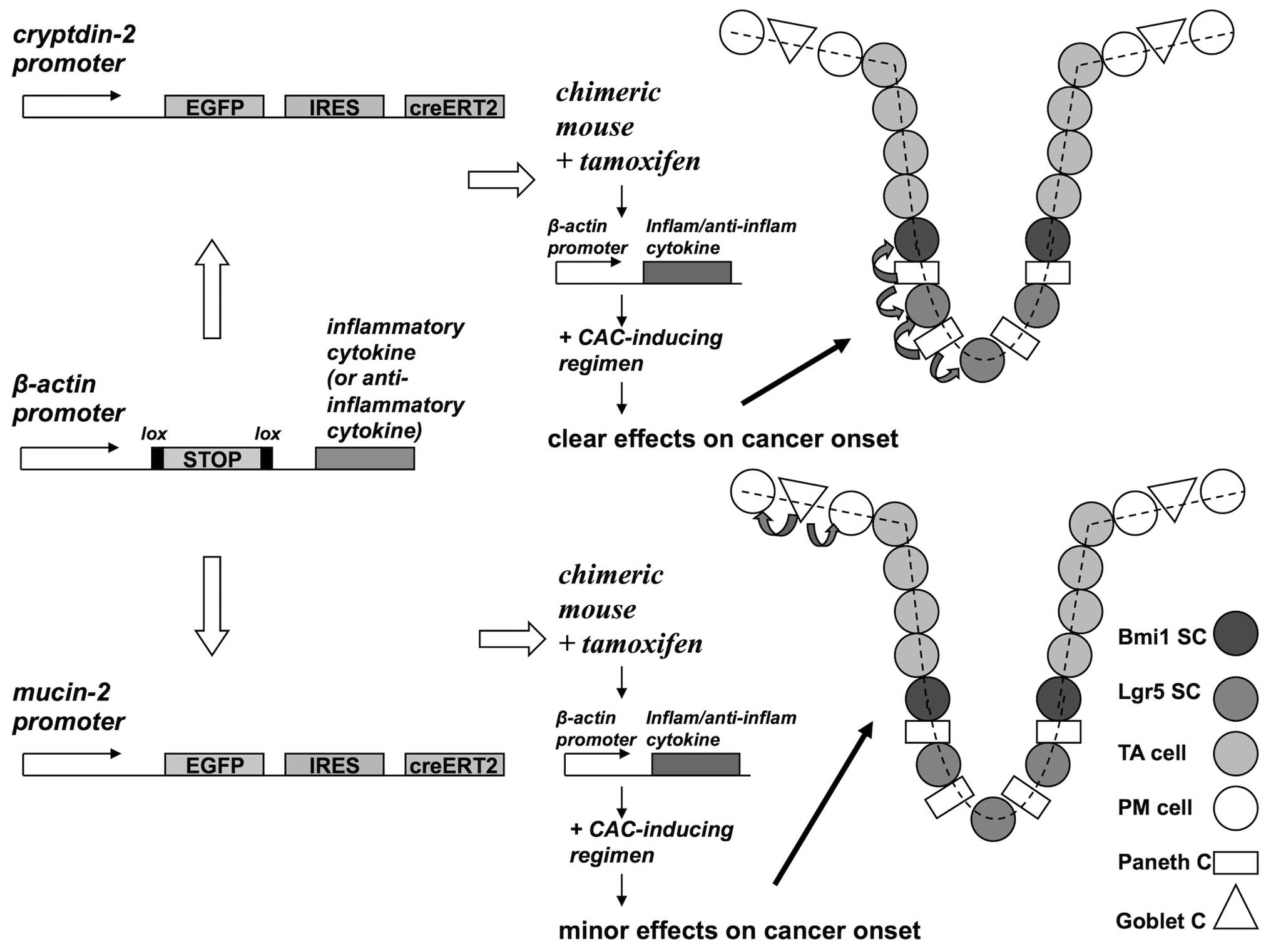 | Figure 2Investigation of intestine
carcinogenesis by ectopic expression of pro/anti-inflammatory
cytokine transgenes in Paneth cells. To obtain the spatial and
temporal control of transgene expression, a creERT2
construct encoding the cre recombinase fused to a
tamoxifen-activated ERT2 mutant estrogen ligand binding domain may
be used (87). A cell-specific
promoter region [cryptdin-2 for Paneth cells (102) or mucin-2 for goblet cell
(103)] drives the transcription
of the fusion recombinase whereas the gene of interest (coding for
a pro-inflammatory or an anti-inflammatory cytokine) is cloned
downstream a housekeeping promoter (β-actin), but it is not
expressed due to a loxP-flanked (floxed) STOP cassette. In chimeric
mice that bear both trasgenes, exposure to tamoxifen may drive the
cre-mediated deletion of the floxed STOP cassette, and hence the
conditional expression of the cytokine in the cell of interest
(i.e., Paneth or goblet cells). In this figure the direct targets
of the ectopic cytokine are adjacent epithelial cells;
alternatively, the action of the cytokine may be indirectly
mediated through a stromal cell, most likely of myeloid lineage, as
reported by Tu et al (95).
Candidate pro-inflammatory or anti-inflammatory cytokines for
transgenic expression may be TNFα or IL-10, respectively. In order
to produce a substantial effect on cancer onset, this transgenic
model should be coupled with a treatment predisposing to cancer in
the intestine (e.g., a CAC regimen). In the figure stem and
trans-amplifying cells are shown in different gray shades, whereas
fully differentiated post-mitotic cells are shown in white. |
We believe that an in-depth examination of the
rationale and feasibility of such an experimental approach could
raise a number of interesting issues regarding tumor biology in the
intestine and, more broadly, the interplay between chronic
inflammation and cancer stem cells.
In this regard, the following points seem
particularly worthy of discussion:
It is well known that in humans gut cancer is most
frequently found in the colorectum. On the other hand, our intended
target of transgenesis, i.e., the Paneth cell, is restricted to the
crypts of the small intestine in both humans and mice.
Consequently, in this case neoplasia is expected to be confined to
the small intestine of mice. Actually, this circumstance does not
constitute a difficulty for the experimental approach under
scrutiny, as most murine models of bowel cancer show neoplastic
growth chiefly in the small intestine (49,88–90).
The pathophysiology of the gut is different in
humans and mice, as demonstrated, for example, by the fact that
when aged C57BL/6 mice were checked for spontaneous bowel cancer,
the majority of the neoplastic lesions were located in the small
intestine (91). Nevertheless,
rodent models recapitulate quite faithfully the adenoma-carcinoma
sequence that is found in human familial or sporadic CRC. Hence,
murine models of intestine cancer have been always regarded as
informative for human colon cancer (92).
Paneth cells serve as multifunctional ‘guardians’ of
crypt stem cells, both by secreting bactericidal products and by
providing essential niche signals. This second feature of Paneth
cells has been demonstrated recently: co-culturing of sorted
intestinal stem cells with Paneth cells markedly improved ex
vivo crypt formation, suggesting that the latter contributes to
the construction of the stem cell niche (93,94).
Experimental strategies should be aimed at
‘hijacking’ the crypt stem cell niche biology by transforming the
function of the Paneth cell from homeostasis keeper to
local inducer of chronic inflammation. To this end, the
choice of the paracrine ectopic inflammatory stimulus is a relevant
aspect. An obvious option could be an inflammatory cytokine though,
to our knowledge, only a paper by Tu et al has reported the
chronic, ectopic expression of an inflammatory cytokine in a
cancer-prone epithelial layer of the gastrointestinal tract
(95).
The results of Tu et al suggest that the
overexpression of a single cytokine, IL-1β, in the gastric mucosa
may be sufficient to induce cancer in that site. Moreover, in that
experiment tumorigenesis was triggered by a myeloid population
recruited by IL-1β, suggesting that the inflammatory infiltrate
contributed not only to cancer progression but also to the earlier
stages of carcinogenesis. The important conclusion that can be
drawn from this murine transgenic model is that in the bowel
inflammation alone suffices for tumor development.
For the choice of the inflammatory cytokine to be
ectopically expressed in Paneth cells, IL-1β or TNFα could be
interesting candidates, as in vitro experiments showed that
these cytokines may over-stimulate in a paracrine fashion the
Wnt/β-catenin pathway in colon carcinoma cell lines (41–43).
Conversely, Paneth cell-targeted overexpression of an
anti-inflammatory cytokine, such as IL-10, could create a ‘shield’,
protecting the stem cell niche and attenuating the oncogenic
potential of a CAC-inducing regimen on the whole mucosa. The fact
that IL-10-deficient mice may spontaneously develop colitis, and
subsequently colorectal tumors supports the use of this cytokine in
this context (96).
Another point worthy of investigation could be the
most likely cell source of oncogenic inflammatory stimuli in
vivo. While it is generally agreed that myeloid cells may play
a role in this respect, it would be important to investigate
whether IECs, or more specifically Paneth cells, may secrete
significant amounts of inflammatory cytokines in healthy or
diseased gut mucosal tissues. This is in fact the case of the
preferential secretion by Paneth cells among IECs of TNFα leading
to IBD, particularly Chron’s disease, and possibly to cancer, even
if this last aspect has not been addressed (97).
At present the issue of cytokine production and/or
response by the epithelia lining the mucosal surface is rather
ill-investigated. Some key questions for future investigation
should be: what is known about secretion of cytokines by mucosal
epithelial cells? Is this secretion directional? What is the
identity of the cellular target of cytokines secreted by IECs,
bystander cells or the same epithelial cells? What is the function,
in healthy or diseased tissues, of cytokine secretion into mucosal
fluids, for example, saliva?
Another important aspect is that in most cases the
published transgenic mouse models of gut cancer show an intrinsic
low rate of cancer. Where this happens, in order to observe a
significant increase of tumor incidence over the parental controls,
it is mandatory to challenge the animals with a CAC-inducing
regimen or to produce double transgenic mice. To this aim, a most
widely used carcinogenic regimen consists of azoxymethane AOM, a
colonotropic mutagen, followed by dextran sulfate sodium DSS, an
intestine irritant (90).
Finally, models of transgenic mice harboring
targeted modification of goblet cells overexpressing inflammatory
cytokines would be expected to display a low rate of gut cancer in
animals exposed to a CAC-inducing regimen, since goblet cells in
the epithelial monolayer lining the gut mucosa of the small
intestine reside far outside the crypt niche (98).
6. Concluding remarks
At present, little is known about the roles of crypt
stem cells in colitis-induced colon cancer, even in mice. The
principal concern of this article is to hypothesize the likely
outcome of a bowel ectopic stem cell niche redirected toward a
pro-inflammatory state. By answering the questions that arise from
the targeted trasgenesis of Paneth cells, a gain in comprehension
could be achieved on the origin and pathogenesis of bowel cancer,
even in humans.
It is hotly debated to what extent stemness is a
cell-autonomous, intrinsic property of cells or, rather, the result
of inputs coming from the intestinal niche (99). In this respect, the small intestine
crypt represents an outstanding model, due to the unique role of
Paneth cells in the fine-tuning of stem cell niche homeostasis.
These cells secrete substantial quantities of anti-microbial
peptides that protect the niche environment from the invasion by
the gut microbiota. In addition and intriguingly, Paneth cells
secrete factors that sustain and fuel the crypt stem cell
compartment (93).
The idea of a targeted expression of pro- or
anti-inflammatory cytokines in Paneth cells should be evaluated in
the context of the evidence reviewed in the present article. In
this regard, the construction of a ‘precancer niche’ has been
hypothesized as a necessary early step required for initiated cells
to survive and evolve toward cancer (100).
From the early reports by the Clevers’s lab, the
widespread significance of Lgr5 as a stemness marker has been
further endorsed. In mice, Lgr5 is a stem cell marker also in the
crypt of the large intestine. Moreover, in humans, Lgr5 is a
selective marker for human colorectal cancer stem cells (101).
It should be stressed that the results obtained in
mice indicating Lgr5+ and/or Bmi1+ crypt stem
cells as the cells-of-origin of bowel cancer most likely pertain to
the familiar or sporadic CRC.
In conclusion, genetic studies with specific stem
cell markers or the targeted trasgenesis of Paneth cells herein
discussed should provide more information in the near future on the
cell-of-origin of colitis-associated cancer in mice, but even in
humans.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thun MJ, Henley SJ and Gansler T:
Inflammation and cancer: an epidemiological perspective. Novartis
Found Symp. 256:6–21. 2004. View Article : Google Scholar
|
|
3
|
Allavena P, Garlanda C, Borrello MG, Sica
A and Mantovani A: Pathways connecting inflammation and cancer.
Curr Opin Genet Dev. 18:3–10. 2008. View Article : Google Scholar
|
|
4
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar
|
|
5
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar
|
|
6
|
Trinchieri G: Cancer and inflammation: an
old intuition with rapidly evolving new concepts. Annu Rev Immunol.
30:677–706. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boland C, Luciani M, Gasche C and Goel A:
Infection, inflammation, and gastrointestinal cancer. Gut.
54:1321–1331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Potten CS, Gandara R, Mahida YR, Loeffler
M and Wright NA: The stem cells of small intestinal crypts: Where
are they? Cell Prolif. 42:731–750. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barker N, Ridgway RA, van Es JH, et al:
Crypt stem cells as the cells-of-origin of intestinal cancer.
Nature. 457:608–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sangiorgi E and Capecchi MR: Bmi1 is
expressed in vivo in intestinal stem cells. Nat Genet. 40:915–920.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dyson JK and Rutter MD: Colorectal cancer
in inflammatory bowel disease: what is the real magnitude of the
risk? World J Gastroenterol. 18:3839–3848. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eaden JA, Abrams KR and Mayberry JF: The
risk of colorectal cancer in ulcerative colitis: a meta-analysis.
Gut. 48:526–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Canavan C, Abrams KR and Mayberry J:
Meta-analysis: colorectal and small bowel cancer risk in patients
with Crohn’s disease. Aliment Pharmacol Ther. 23:1097–1104.
2006.
|
|
14
|
Rubio CA, Befrits R, Ljung T, Jaramillo E
and Slezak P: Colorectal carcinoma in ulcerative colitis is
decreasing in Scandinavian countries. Anticancer Res. 21:2921–2924.
2001.
|
|
15
|
Jess T, Loftus EV Jr, Velayos FS, et al:
Risk of intestinal cancer in inflammatory bowel disease: a
population-based study from Olmsted county, Minnesota.
Gastroenterology. 130:1039–1046. 2006. View Article : Google Scholar
|
|
16
|
Winther KV, Jess T, Langholz E, Munkholm P
and Binder V: Long-term risk of cancer in ulcerative colitis: a
population-based cohort study from Copenhagen County. Clin
Gastroenterol Hepatol. 2:1088–1095. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loftus EV Jr: Epidemiology and risk
factors for colorectal dysplasia and cancer in ulcerative colitis.
Gastroenterol Clin North Am. 35:517–531. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ullman TA and Itzkowitz SH: Intestinal
inflammation and cancer. Gastroenterology. 140:1807–1816. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harpaz N and Polydorides AD: Colorectal
dysplasia in chronic inflammatory bowel disease: pathology,
clinical implications, and pathogenesis. Arch Pathol Lab Med.
134:876–895. 2010.
|
|
20
|
Riddell RH, Goldman H, Ransohoff DF, et
al: Dysplasia in inflammatory bowel disease: standardized
classification with provisional clinical applications. Hum Pathol.
14:931–968. 1983. View Article : Google Scholar
|
|
21
|
Friedlich MS, Guindi M and Stern HS: The
management of dysplasia associated with ulcerative colitis:
colectomy versus continued surveillance. Can J Surg. 4:212–214.
2004.PubMed/NCBI
|
|
22
|
Kulaylat MN and Dayton MT: Ulcerative
colitis and cancer. J Surg Oncol. 101:706–712. 2010. View Article : Google Scholar
|
|
23
|
Grivennikov SI and Karin M: Inflammation
and oncogenesis: a vicious connection. Curr Opin Genet Dev.
20:65–71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grivennikov S: Inflammation and colorectal
cancer: colitisassociated neoplasia. Semin Immunopathol.
35:229–244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie J and Itzkowitz SH: Cancer in
inflammatory bowel disease. World J Gastroenterol. 14:378–389.
2008. View Article : Google Scholar
|
|
27
|
Terzić J, Grivennikov S, Karin E and Karin
M: Inflammation and colon cancer. Gastroenterology. 138:2101–2114.
2010.
|
|
28
|
Itzkowitz SH and Harpaz N: Diagnosis and
management of dysplasia in patients with inflammatory bowel
diseases. Gastroenterology. 126:1634–1648. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taylor BA, Pemberton JH, Carpenter HA, et
al: Dysplasia in chronic ulcerative colitis: implications for
colonoscopic surveillance. Dis Colon Rectum. 35:950–956. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gorfine SR, Bauer JJ, Harris MT and Kreel
I: Dysplasia complicating chronic ulcerative colitis: is immediate
colectomy warranted? Dis Colon Rectum. 43:1575–1581. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bernstein CN, Shanahan F and Weinstein WM:
Are we telling patients the truth about surveillance colonoscopy in
ulcerative colitis? Lancet. 343:71–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rutter MD, Saunders BP, Wilkinson KH, et
al: Thirty-year analysis of a colonoscopic surveillance program for
neoplasia in ulcerative colitis. Gastroenterology. 130:1030–1038.
2006.PubMed/NCBI
|
|
33
|
Connell WR, Lennard-Jones JE, Williams CB,
Talbot IC, Price AB and Wilkinson KH: Factors affecting the outcome
of endoscopic surveillance for cancer in ulcerative colitis.
Gastroenterology. 107:934–944. 1994.PubMed/NCBI
|
|
34
|
Melville DM, Jass JR, Morson BC, et al:
Observer study of the grading of dysplasia in ulcerative colitis:
comparison with clinical outcome. Hum Pathol. 20:1008–1014. 1989.
View Article : Google Scholar
|
|
35
|
Sjöqvist U: Dysplasia in ulcerative
colitis - clinical consequences? Langenbecks Arch Surg.
389:354–360. 2004.PubMed/NCBI
|
|
36
|
Deans C and Wigmore SJ: Systemic
inflammation, cachexia and prognosis in patients with cancer. Curr
Opin Clin Nutr Metab Care. 8:265–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Philip M, Rowley DA and Schreiber H:
Inflammation as a tumor promoter in cancer induction. Semin Cancer
Biol. 14:433–439. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
DeNardo DG, Johansson M and Coussens LM:
Immune cells as mediators of solid tumor metastasis. Cancer
Metastasis Rev. 27:11–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kalluri R: EMT: when epithelial cells
decide to become mesenchymal-like cells. J Clin Invest.
119:1417–1419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luo Y, Yu M and Grady WM: Field
cancerization in the colon: a role for aberrant DNA methylation?
Gastroenterol Rep. 2:16–20. 2014.(Epub ahead of print).
|
|
41
|
Kaler P, Godasi BN, Augenlicht L and
Klampfer L: The NFkappaB/AKT-dependent induction of Wnt signaling
in colon cancer cells by macrophages and IL-1beta. Cancer
Microenviron. 2:69–80. 2009. View Article : Google Scholar
|
|
42
|
Lee G, Goretsky T, Managlia E, et al:
Phosphoinositide 3-kinase signaling mediates beta-catenin
activation in intestinal epithelial stem and progenitor cells in
colitis. Gastroenterology. 139:869–881. 2010. View Article : Google Scholar
|
|
43
|
Oguma K, Oshima H, Aoki M, et al:
Activated macrophages promote Wnt signalling through tumor necrosis
factor-alpha in gastric tumor cells. EMBO J. 27:1671–1681. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a Gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Corvinus FM, Orth C, Moriggl R, et al:
Persistent STAT3 activation in colon cancer is associated with
enhanced cell proliferation and tumor growth. Neoplasia. 7:545–555.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: role of STAT3 in the
tumor microenvironment. Nature Rev Immunol. 7:41–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Van Immerseel F, Ducatelle R, De Vos M, et
al: Butyric acid-producing anaerobic bacteria as a novel probiotic
treatment approach for inflammatory bowel disease. J Med Microbiol.
59:141–143. 2010.
|
|
48
|
Van der Sluis M, De Koning BA, De Bruijn
AC, et al: Muc2-deficient mice spontaneously develop colitis,
indicating that MUC2 is critical for colonic protection.
Gastroenterology. 131:117–129. 2006.
|
|
49
|
Velcich A, Yang W, Heyer J, et al:
Colorectal cancer in mice genetically deficient in the mucin Muc2.
Science. 295:1726–1729. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Petersson J, Schreiber O, Hansson GC, et
al: Importance and regulation of the colonic mucus barrier in a
mouse model of colitis. Am J Physiol Gastrointest Liver Physiol.
300:G327–G333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Arthur JC, Perez-Chanona E, Mühlbauer M,
et al: Intestinal inflammation targets cancer-inducing activity of
the microbiota. Science. 338:120–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
53
|
Suvà ML, Riggi N and Bernstein BE:
Epigenetic reprogramming in cancer. Science. 339:1567–1570.
2013.
|
|
54
|
Chiba T, Marusawa H and Ushijima T:
Inflammation-associated cancer development in digestive organs:
mechanisms and roles for genetic and epigenetic modulation.
Gastroenterology. 143:550–563. 2012. View Article : Google Scholar
|
|
55
|
Niwa T and Ushijima T: Induction of
epigenetic alterations by chronic inflammation and its significance
on carcinogenesis. Adv Genet. 71:41–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hahn MA, Hahn T, Lee DH, et al:
Methylation of polycomb target genes in intestinal cancer is
mediated by inflammation. Cancer Res. 68:10280–10289. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cirri P and Chiarugi P:
Cancer-associated-fibroblasts and tumor cells: a diabolic liaison
driving cancer progression. Cancer Metastasis Rev. 31:195–208.
2012. View Article : Google Scholar
|
|
59
|
Achyut BR, Bader DA, Robles AI, et al:
Inflammation-mediated genetic and epigenetic alterations drive
cancer development in the neighboring epithelium upon stromal
abrogation of TGF-β signaling. PLoS Genet.
9:e10032512013.PubMed/NCBI
|
|
60
|
El-Omar EM, Carrington M, Chow WH, et al:
Interleukin-1 polymorphisms associated with increased risk of
gastric cancer. Nature. 404:398–402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
El-Omar EM, Carrington M, Chow WH, et al:
The role of interleukin-1 polymorphisms in the pathogenesis of
gastric cancer. Nature. 412:992001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chan AO, Chu KM, Huang C, et al:
Association between Helicobacter pylori infection and
interleukin 1beta polymorphism predispose to CpG island methylation
in gastric cancer. Gut. 56:595–597. 2007.
|
|
63
|
Niwa T, Tsukamoto T, Toyoda T, et al:
Inflammatory processes triggered by Helicobacter pylori
infection cause aberrant DNA methylation in gastric epithelial
cells. Cancer Res. 70:1430–1440. 2010.
|
|
64
|
McLaughlan JM, Seth R, Vautier G, et al:
Interleukin-8 and inducible nitric oxide synthase mRNA levels in
inflammatory bowel disease at first presentation. J Pathol.
181:87–92. 1997. View Article : Google Scholar
|
|
65
|
Yoshida T, Yamashita S, Takamura-Enya T,
et al: Alu and Satα hypomethylation in Helicobacter
pylori-infected gastric mucosae. Int J Cancer. 128:33–39.
2011.
|
|
66
|
Semenza G: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar
|
|
67
|
Giles RH, Lolkema MP, Snijckers CM, et al:
Interplay between VHL/HIF1alpha and Wnt/beta-catenin pathways
during colorectal tumorigenesis. Oncogene. 25:3065–3070. 2006.
View Article : Google Scholar
|
|
68
|
Shah YM, Ito S, Morimura K, Chen C, et al:
Hypoxia-inducible factor augments experimental colitis through an
MIF-dependent inflammatory signaling cascade. Gastroenterology.
134:2036–2048. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Imtiyaz HZ, Williams EP, Hickey MM, Patel
SA, et al: Hypoxia-inducible factor 2alpha regulates macrophage
function in mouse models of acute and tumor inflammation. J Clin
Invest. 120:2699–2714. 2010. View Article : Google Scholar
|
|
70
|
Chaturvedi MM, Sung B, Yadav VR, Kannappan
R and Aggarwal BB: NF-κB addiction and its role in cancer: ‘one
size does not fit all’. Oncogene. 30:1615–1630. 2011.
|
|
71
|
Colgan SP, Curtis VF and Campbell EL: The
inflammatory tissue microenvironment in IBD. Inflamm Bowel Dis.
19:2238–2244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Colgan SP and Taylor CT: Hypoxia: an alarm
signal during intestinal inflammation. Nat Rev Gastroenterol
Hepatol. 7:281–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Clambey ET, McNamee EN, Westrich JA, et
al: Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3
drives regulatory T-cell abundance and function during inflammatory
hypoxia of the mucosa. Proc Natl Acad Sci USA. 109:E2784–E2793.
2012. View Article : Google Scholar
|
|
74
|
Olaru AV, Selaru FM, Mori Y, et al:
Dynamic changes in the expression of MicroRNA-31 during
inflammatory bowel disease-associated neoplastic transformation.
Inflamm Bowel Dis. 17:221–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gillies RJ and Gatenby RA: Hypoxia and
adaptive landscapes in the evolution of carcinogenesis. Cancer
Metastasis Rev. 26:311–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Covello KL, Kehler J, Yu H, et al:
HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell
function, embryonic development, and tumor growth. Genes Dev.
20:557–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Edwards RA, Witherspoon M, Wang K, et al:
Epigenetic repression of DNA mismatch repair by inflammation and
hypoxia in inflammatory bowel disease-associated colorectal cancer.
Cancer Res. 69:6423–6429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Clevers H and Batlle E: SnapShot: the
intestinal crypt. Cell. 152:1198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Barker N, van Es JH, Kuipers J, et al:
Identification of stem cells in small intestine and colon by marker
gene Lgr5. Nature. 449:1003–1007. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Barker N, van de Wetering M and Clevers H:
The intestinal stem cell. Genes Dev. 22:1856–1864. 2008. View Article : Google Scholar
|
|
81
|
Li L and Clevers H: Coexistence of
quiescent and active adult stem cells in mammals. Science.
327:542–545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shih IM, Wang TL, Traverso G, et al:
Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci
USA. 98:2640–2645. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Preston SL, Wong WM, Chan AO, et al:
Bottom-up histogenesis of colorectal adenomas: origin in the
monocryptal adenoma and initial expansion by crypt fission. Cancer
Res. 63:3819–3825. 2003.
|
|
84
|
Ward RJ and Dirks PB: Cancer stem cells:
at the headwaters of tumor development. Annu Rev Pathol. 2:175–189.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Scheel C and Weinberg RA: Phenotypic
plasticity and epithelial-mesenchymal transitions in cancer and
normal stem cells? Int J Cancer. 129:2310–2314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Roskams T: Liver stem cells and their
implication in hepatocellular and cholangiocarcinoma. Oncogene.
25:3818–3822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
El Marjou F, Janssen KP, Chang BH, et al:
Tissue-specific and inducible Cre-mediated recombination in the gut
epithelium. Genesis. 39:186–193. 2004.PubMed/NCBI
|
|
88
|
Haigis KM, Hoff PD, White A, Shoemaker AR,
Halberg RB and Dove WF: Tumor regionality in the mouse intestine
reflects the mechanism of loss of Apc function. Proc Natl Acad Sci
USA. 101:9769–9773. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Shaked H, Hofseth LJ, Chumanevich A, et
al: Chronic epithelial NF-kappaB activation accelerates APC loss
and intestinal tumor initiation through iNOS up-regulation. Proc
Natl Acad Sci USA. 109:14007–14012. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kanneganti M, Mino-Kenudson M and
Mizoguchi E: Animal models of colitis-associated carcinogenesis. J
Biomed Biotechnol. 2011:3426372011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Rowlatt C, Franks LM, Sheriff MU and
Chesterman FC: Naturally occurring tumors and other lesions of the
digestive tract in untreated C57BL mice. J Natl Cancer Inst.
43:1353–1364. 1969.PubMed/NCBI
|
|
92
|
Preston SL, Leedham SJ, Oukrif D, et al:
The development of duodenal microadenomas in FAP patients: the
human correlate of the Min mouse. J Pathol. 214:294–301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sato T, van Es JH, Snippert HJ, et al:
Paneth cells constitute the niche for Lgr5 stem cells in intestinal
crypts. Nature. 469:415–418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Clevers HC and Bevins CL: Paneth cells:
maestros of the small intestinal crypts. Annu Rev Physiol.
75:289–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tu S, Bhagat G, Cui G, et al:
Overexpression of interleukin 1β induces gastric inflammation and
cancer and mobilizes myeloid-derived suppressor cells in mice.
Cancer Cell. 14:408–419. 2008.
|
|
96
|
Sturlan S, Oberhuber G, Beinhauer BG, et
al: Interleukin-10-deficient mice and inflammatory bowel disease
associated cancer development. Carcinogenesis. 22:665–671. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lala S, Ogura Y, Osborne C, et al: Crohn’s
disease and the NOD2 gene: a role for Paneth cells.
Gastroenterology. 125:47–57. 2003.
|
|
98
|
Medema JP and Vermeulen L:
Microenvironmental regulation of stem cells in intestinal
homeostasis and cancer. Nature. 474:318–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Scadden DT: The stem-cell niche as an
entity of action. Nature. 441:1075–1079. 2006. View Article : Google Scholar
|
|
100
|
Barcellos-Hoff MH, Lyden D and Wang TC:
The evolution of the cancer niche during multistage carcinogenesis.
Nat Rev Cancer. 13:511–518. 2013. View Article : Google Scholar
|
|
101
|
Kemper K, Prasetyanti PR, De Lau W,
Rodermond H, Clevers H and Medema JP: Monoclonal antibodies against
Lgr5 identify human colorectal cancer stem cells. Stem Cells.
30:2378–2386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Vaishnava S, Behrendt CL, Ismail AS,
Eckmann L and Hooper LV: Paneth cells directly sense gut commensals
and maintain homeostasis at the intestinal host-microbial
interface. Proc Natl Acad Sci USA. 105:20858–20863. 2008.
View Article : Google Scholar
|
|
103
|
Gum JR Jr, Hicks JW, Gillespie AM, et al:
Goblet cell-specific expression mediated by the MUC2 mucin gene
promoter in the intestine of transgenic mice. Am J Physiol.
276:G666–G676. 1999.PubMed/NCBI
|