Introduction
Inflammatory breast cancer (IBC) is an angioinvasive
form of breast cancer in which cancer cells block the lymphatic
vessels in the skin covering the breast and it is associated with a
high incidence of early nodal and systemic metastases (1). This type of breast cancer is called
‘inflammatory’ because the breast often looks swollen and red,
resembling an inflammatory condition (2). Although IBC accounts for only 2–5% of
all breast cancers, IBC tumors show a poor prognosis with 40%
five-years survival rate versus 87% for all breast cancers
combined, making IBC a priority area for the development of new
therapeutic strategies (3). IBC
tumors display a triple negative breast cancer (TNBC) phenotype
characterized by high chromosomal instability (CIN) and low levels
of expression of estrogen receptor α (ERα), progesterone receptor
(PR) and HER-2 tyrosine kinase receptor (4). Since the TNBC cells lack these
receptors necessary to promote tumor growth, common treatments such
as endocrine therapy and molecular targeting of HER-2 receptor are
ineffective for this subtype of breast cancer (5). Employing conventional chemotherapy to
treat TNBC tumors is still an effective option because TNBC tumors
may respond even better to chemotherapeutic agents in the earlier
stages than many other forms of breast cancer (6). Unfortunately, TNBC tumors will likely
develop chemoresistance leading to tumor progression, metastatic
spreading to distant organs and poor outcome (7).
Germline mutations in the BRCA1 and BRCA2 breast
cancer susceptibility genes have been associated with up to 15% of
TNBC, and TNBC accounts for 70% of breast tumors arising in BRCA1
mutation carriers and 16–23% of breast tumors in BRCA2 carriers
(8). Because BRCA1 and BRCA2 tumor
suppressor genes play a key role in the control of genomic
stability through regulation of DNA repair and centrosome
duplication (9–11), these findings explain the causal
role of BRCA1 and BRCA2 mutations in the development of high CIN
commonly observed in TNBC tumors. Moreover, TNBC tumors frequently
overexpress cyclin E, a late G1 cyclin that binds and activates
cyclin-dependent kinase 2 (Cdk2) leading to phosphorylation and
inactivation of retinoblastoma (Rb) essential for the G1-S phase
transition of the cell cycle (12,13).
Importantly, aberrant activation of cyclin E/Cdk2 complex plays a
key role in the development of centrosome amplification, CIN and
breast cancer progression and may represent an attractive
‘druggable oncogenic signaling’ for the treatment of TNBC tumors
lacking molecular targeted therapy (14–16).
Recent studies have demonstrated that breast carcinomas are
composed of a singular subpopulation of cells harboring stem-like
properties termed cancer stem cells (CSCs) (17). CSCs show activation of epithelial
to mesenchymal transition (EMT) programming and acquire a
basal-like CD44+/CD24low/− phenotype with
increased capacity for self-renewal, invasion, drug resistance and
tumor progression (18–20). The discovery of
CD44+/CD24low/− CSCs has generated excitement
because this subpopulation of cancer cells may represent a source
of therapeutic failure to anticancer drugs. Significantly, it has
been demonstrated that treatment of breast tumors with conventional
chemotherapeutic agents enriched the
CD44+/CD24low/− subpopulation conferring
resistance to the initial treatment and leading to the development
of distant metastases (21). In
agreement with these findings, TNBC tumors generally display a
basal-like CD44+/CD24low/− phenotype that
clarifies their resistance to conventional chemotherapy responsible
for high risk of recurrence and poor prognosis (22).
In this study we employed the luminal ER+
MCF-7 and the IBC SUM149PT breast cancer cells to establish the
extent to which high grade of CIN and chemoresistance was
mechanistically linked to the enrichment of
CD44+/CD24low/− CSCs. We demonstrate that
overexpression of cyclin E is restricted to the stem-like
CD44+/CD24low/− subpopulation and is
functionally linked to phosphorylation of retinoblastoma (Rb),
centrosome amplification and genomic instability. Significantly,
CD44+/CD24low/− CSCs displayed higher
sensitivity to a specific Cdk2 inhibitor than the bulk SUM149PT
cells indicating that aberrant activation of cyclin E/Cdk2
oncogenic signaling is essential for the maintenance and expansion
of CD44+/CD24low/− CSCs subpopulation in IBC.
In conclusion our findings highlight a novel therapeutic approach
based on the combination of conventional chemotherapy with small
molecule inhibitors of the Cdk2 cell cycle kinase to treat
chemoresistant IBC tumors.
Materials and methods
Human breast cancer cell lines
The human breast cancer cell line MCF-7 was obtained
from ATCC (Manassas, VA, USA), normal human mammary epithelial
cells HMEC and IBC SUM149PT cancer cells were kindly provided by Dr
Lingle and Dr Couch’s laboratories, respectively (Mayo Clinic,
Rochester, MN). All cell lines were maintained in EMEM medium
containing 5 mM glutamine, 1% penicillin/streptomycin, 20 microgram
insulin/ml and 10% FBS at 37°C in 5% CO2 atmosphere.
Cytogenetic and SKY analysis
Cell harvest and metaphase slide preparation for
routine cytogenetic and spectral karyotyping (SKY) analysis were
performed as previously described (23–25).
Hybridization, wash and detection of the human SKYPaint®
probe (Applied Spectral Imaging; Vista, CA) were performed as
recommended by the manufacturer. Image acquisition and spectral
analysis of metaphase cells were achieved by using the SD200
SpectraCube™ Spectral Imaging system (Applied Spectral Imaging)
mounted on a Zeiss Axioplan2 microscope (Carl Zeiss MicroImaging,
Inc., Thornwood, NY). Images were analyzed using HiSKY analysis
software (Applied Spectral Imaging).
FACS analysis
CD44 and CD24 antibodies (Abcam, Cambridge, MA, USA)
were employed to identify and isolate
CD44+/CD24− breast cancer initiating cells by
FACS sorting analysis as previously described (20).
Immunoblot and immunofluorescence
assays
Immunoblot and immunofluorescence studies were
performed as previously described (27). Antibodies employed for the
immunoblot and immunofluorescence assays were: pericentrin (Abcam),
cyclin E (Santa Cruz Biotechnology, Santa Cruz, CA), P~Rb and
β-actin (Sigma, St. Louis, MO). Results are derived from three
independent experiments.
Chemoresistance studies
For chemoresistance studies, 5×104 cancer
cells were plated in 6-well costar plates and cultured in complete
EMEM medium for 48 h. Following 48-h incubation, cancer cells were
treated with 1 μM methotrexate, 0.5 μM paclitaxel alone or in
combination with 1 μM SU9516 (Sigma) for additional 72 h.
Cytotoxicity of conventional chemotherapy alone and in combination
with SU9516 was tested by immunofluorescence employing a
cleaved-PARP antibody (Cell Signaling Technology, Danvers, MA) as a
marker of apoptosis. Results are derived from three independent
experiments.
Results
In view of the fact that IBC tumors commonly display
higher grade of CIN compared to luminal ERα+ breast
tumors, we employed luminal ERα+ MCF-7 and IBC SUM149PT
cancer cell lines to investigate their level of chromosomal
abnormalities; normal mammary epithelial HMEC cells were used as
control. The SUM149PT cancer cell line was isolated from an IBC
tumor and carries the 2288delT mutation that is linked to loss of
BRCA1 function and represents an excellent preclinical model
to study the molecular mechanisms responsible for the development
of chemoresistance and tumor progression in IBC tumors (23). We performed an integrative
karyotypic analysis of HMEC, MCF-7 and SUM149PT cells employing the
spectral karyotyping (SKY) technology and routine cytogenetic
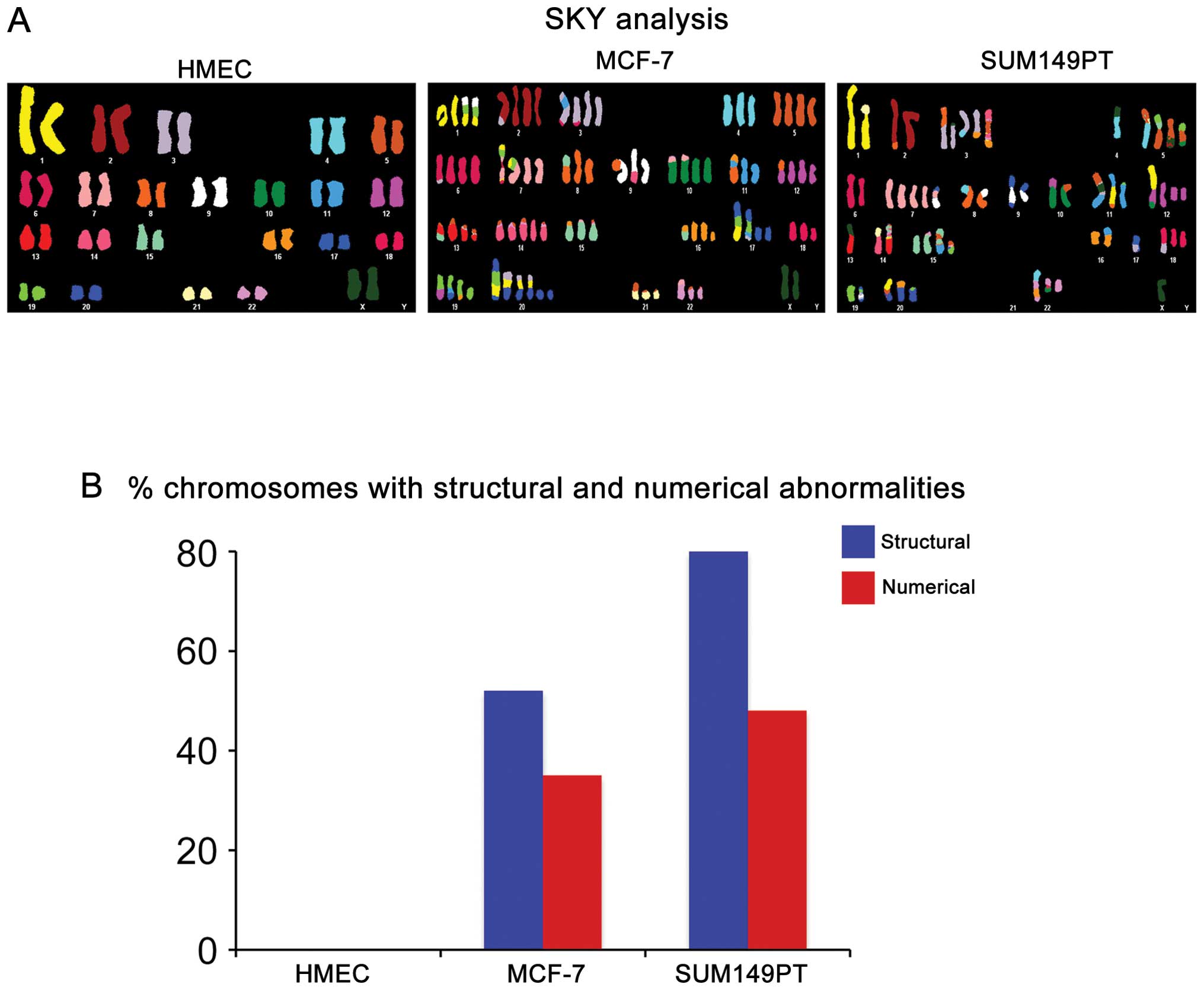
analysis (Fig. 1A). Breast cancer
cell lines were harvested and metaphase spreads for cytogenetic and
SKY analyses were prepared as previously described (24). Comparison of the three cell lines
showed that while MCF-7 and SUM149PT cancer cells displayed a
variety of chromosomal abnormalities, HMEC cells exhibited a normal
diploid karyotype. Significantly, SUM149PT cells displayed a higher
rate of CIN characterized by higher percentage of structural and
numerical chromosomal abnormalities compared to MCF-7 cells
(Fig. 1B).
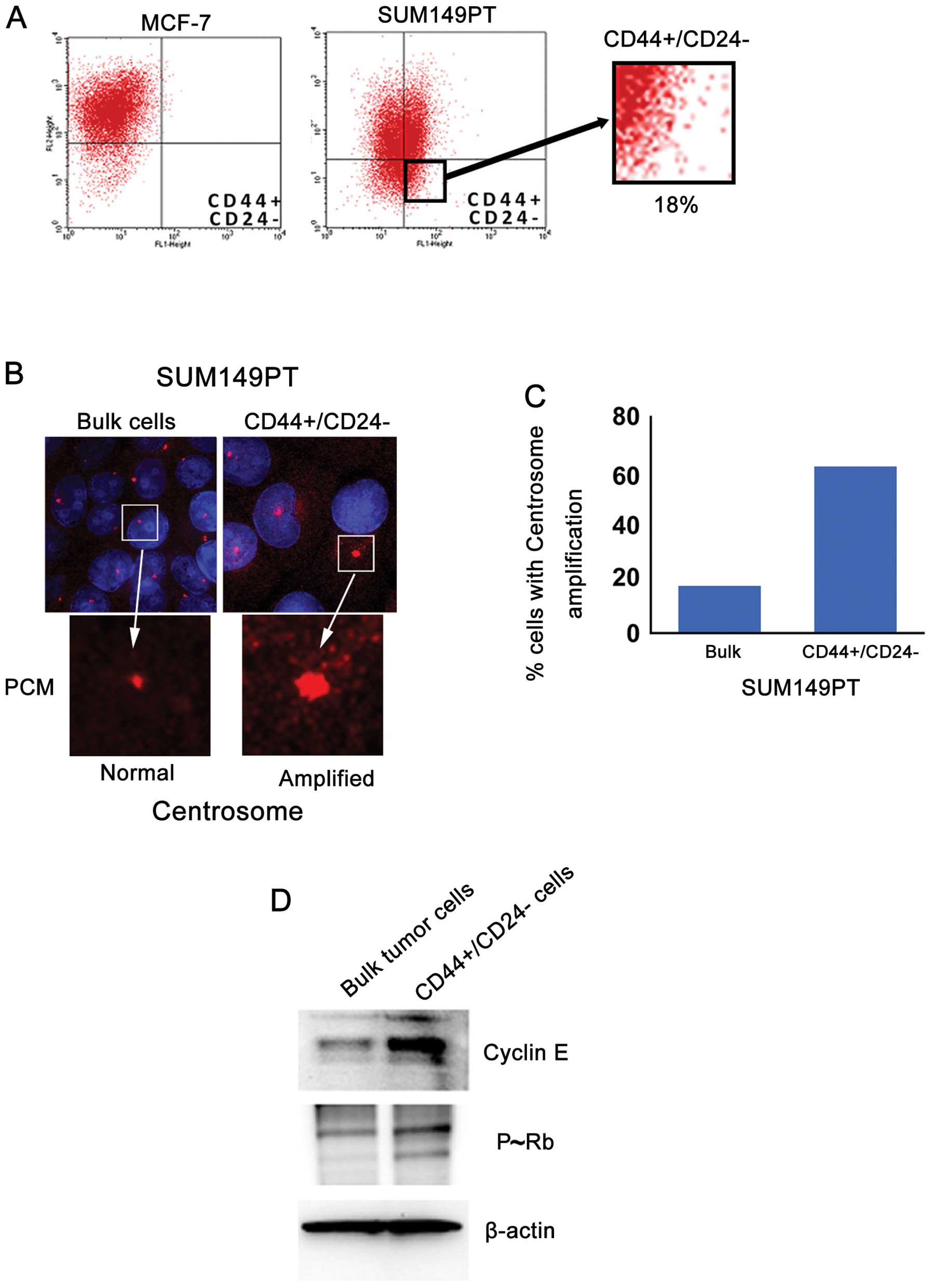
To establish the extent to which the higher level of
CIN observed in the SUM149PT cancer cells was functionally linked
to the presence of a stem-like
CD44+/CD24−/Low subpopulation, FACS analysis
was performed on MCF-7 and SUM149PT cancer cells to analyze the
percentage of cells displaying a
CD44+/CD24−/Low phenotype. While MCF-7 cells
showed mainly a luminal CD44−/CD24+
phenotype, 18% of SUM149PT cells exhibited a stem-like
CD44+/CD24−/Low phenotype (Fig. 2A). Because CIN in breast cancer is
mechanistically linked to development of centrosome abnormalities,
we analyzed the grade of centrosome amplification in bulk SUM149PT
cancer cells versus the stem-like
CD44+/CD24−/Low subpopulation isolated by
FACS sorting assay. Centrosome amplification in cancer cells was
examined by labeling the centrosome size with pericentrin, an
oncoprotein that is localized in the pericentriolar material
(25). Significantly, the
CD44+/CD24−/Low subpopulation revealed higher
centrosome amplification compared to bulk SUM149PT cancer cells
(Fig. 2B and C), suggesting that
the higher degree of CIN observed in SUM149PT cancer cells was
functionally linked to the genesis of
CD44+/CD24−/Low CSCs harboring amplified
centrosomes.
Next we investigated the molecular mechanisms
responsible for the development of centrosome abnormalities in
CD44+/CD24−/Low CSCs. Because the oncoprotein
cyclin E plays a key role in the development of centrosome
abnormalities and is frequently overexpressed in TNBC tumors, we
investigated the expression and activity of cyclin E in SUM149PT
cells versus the CD44+/CD24−/Low
subpopulation isolated by FACS sorting assay. Importantly,
immunoblot analysis showed that cyclin E was overexpressed in the
CD44+/CD24−/Low subpopulation compared to
bulk SUM149PT cells (Fig. 2D). To
investigate whether cyclin E overexpression was functionally linked
to aberrant Cdk2 kinase activity, we analyzed the phosphorylation
status of Rb tumor suppressor that is a downstream target of cyclin
E/Cdk2 oncogenic signaling and its inactivation leads to centrosome
aberrations (26). Higher
phosphorylation and consequent inactivation of Rb was observed in
the CD44+/CD24−/Low subpopulation compared to
bulk SUM149PT cancer cells (Fig.
2D). Taken together these findings reveal that centrosome
amplification was mechanistically linked to aberrant cyclin E/Cdk2
activity and consequent loss of Rb function in
CD44+/CD24−/Low CSCs.
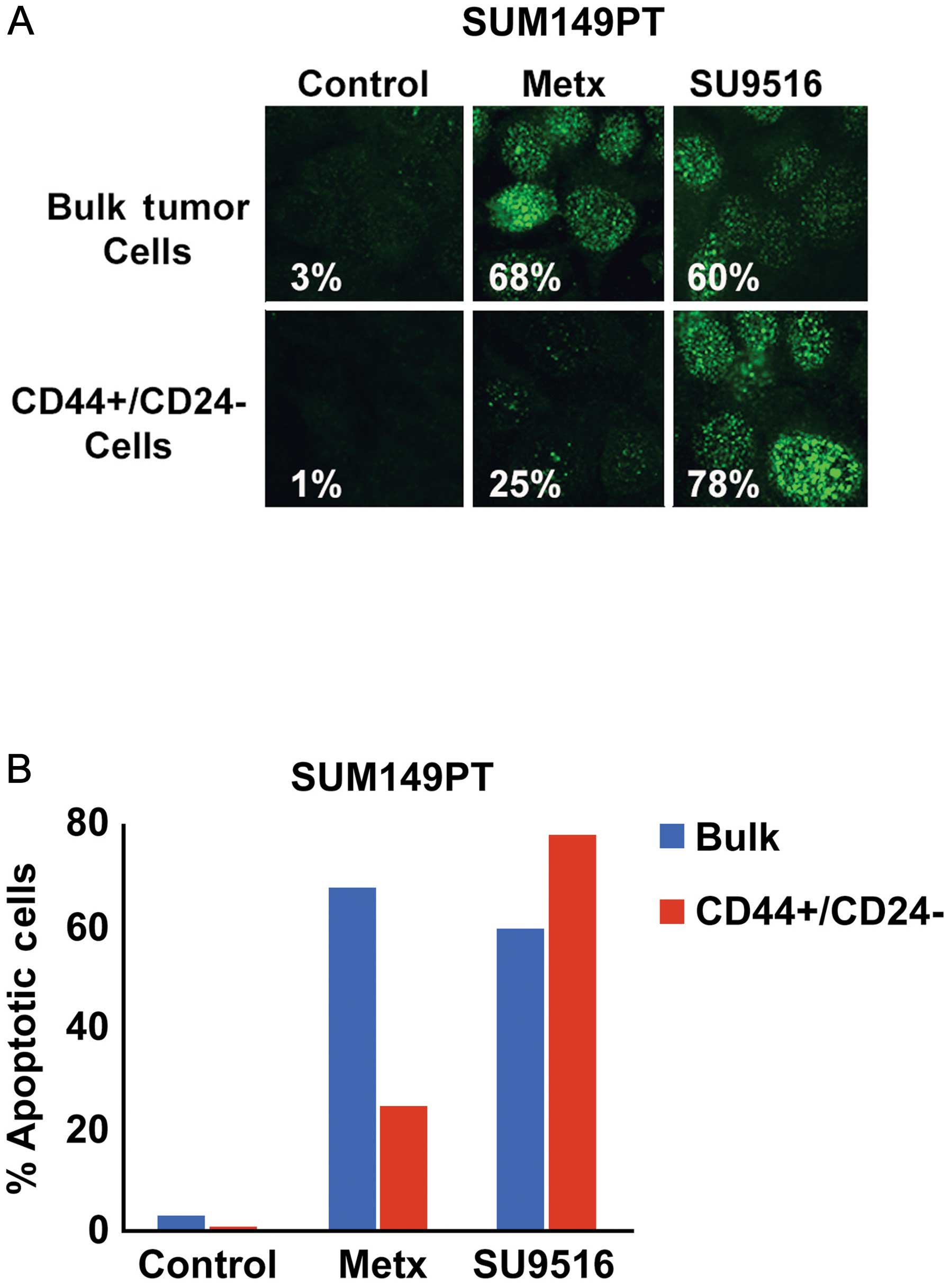
Because TNBC tumors display a poor prognosis that is
associated to higher chemoresistance and stemness properties
compared to luminal breast tumors, we investigated the extent to
which inhibition of Cdk2 kinase activity selectively targeted the
stem-like CD44+/CD24−/Low subpopulation and
restored chemosensitivity of SUM149PT cancer cells. Bulk SUM149PT
and CD44+/CD24−/Low cancer cells were treated
with the antimetabolite methotrexate (a conventional
chemotherapeutic drug) or SU9516 (a small molecule inhibitor of
Cdk2 activity) and cytotoxicity was quantified by analyzing the
percentage of cells harboring cleaved PARP as a marker of
apoptosis. Significantly, CD44+/CD24−/Low
CSCs displayed a higher resistance to methotrexate while they where
highly sensitive to the Cdk2 inhibitor SU9516 compared to bulk
SUM149PT cancer cells (Fig. 3).
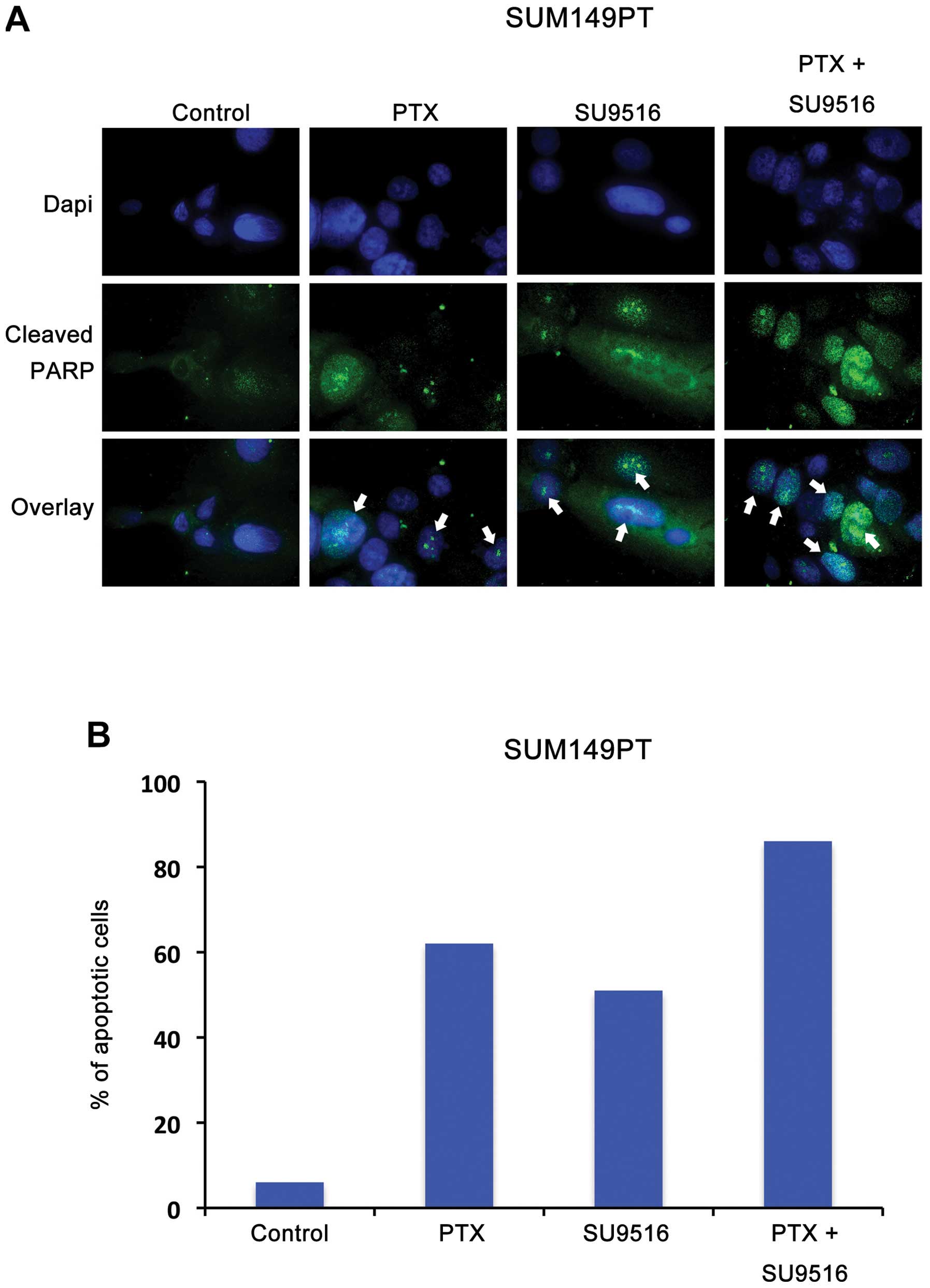
Finally, we tested in vitro the translational therapeutic
relevance of combining conventional anticancer agents with small
molecule inhibitors of Cdk2 activity to restore chemosensitivity of
TNBC tumors. SUM149PT cells were treated with paclitaxel, a taxane
commonly employed in the treatment of TNBC tumors, alone or in
combination with SU9516. Notably, our study revealed that
combination of paclitaxel with SU9516 induced a stronger cytotoxic
activity characterized by the majority of SUM149PT cells undergoing
apoptosis compared to treatment with paclitaxel or SU9516 alone
(Fig. 4).
Discussion
IBC tumors represent a rare and very aggressive
subtype of breast carcinomas that display a TNBC phenotype defined
as the absence of staining for ERα, PR and HER-2 receptors
(1–3). TNBC tumors are insensitive to some of
the most effective targeted therapies available for breast cancer
treatment including endocrine therapies such as tamoxifen,
aromatase inhibitors or fulvestrant and HER-2-directed therapy such
as trastuzumab (27). To date, not
a single targeted therapy has been approved for early-stage TNBC
tumors and combination of conventional cytotoxic chemotherapeutic
agents administered in a dose-dense or metronomic schedule remains
the standard therapy (28).
Significantly, a prospective analysis of 1,118 patients who
received neoadjuvant chemotherapy at a single institution, of whom
255 (23%) had TNBC, found that patients with TNBC tumors had higher
pathologic complete response (pCR) rates compared with non-TNBC
patients (29). Because pCR is
functionally linked to improved long-term outcomes, it is
imperative to develop innovative targeted therapeutic strategies
aimed to increase pCR rates with consequent benefits on the
disease-free and overall survival of TNBC patients. Moreover, the
use of anthracyclines (such as doxorubicin and epirubicin) and
taxanes (such as paclitaxel and docetaxel) as chemotherapeutic
agents for IBC tumors have been shown to improve outcomes (30). Although IBC tumors respond well
initially to conventional chemotherapy, they often develop
chemoresistance leading to tumor progression and poor outcome
(31).
Development of EMT and consequent activation of
stemness programming is responsible for invasion, tumor
self-renewal and drug resistance leading to breast cancer
progression, distant metastases and poor prognosis (32). For this reason the cancer stem
cell-like phenotype commonly observed in IBC tumors may contribute
to their aggressive nature but also may offer itself novel
therapeutic strategies to the selective targeting of
CD44+/CD24− CSCs from bulk tumors. Abrogation
of BRCA1 and BRCA2 genes function can in part explain the high
grade of CIN observed in inflammatory and non-inflammatory TNBC
tumors resulting from centrosome amplification and impairment of
the DNA repair machinery (10).
Breast tumors carrying BRCA1 mutations are considered to be
sensitive to inhibitors of poly (ADP-ribose) polymerases (PARPs)
that are nuclear enzymes implicated in cellular responses to DNA
injury provoked by genotoxic stress (33). PARP-1, the best characterized
member of the PARP family, is essential to the repair of DNA
single-strand breaks via the base excision repair pathway (34). Inhibitors of PARP-1 have been shown
to enhance the cytotoxic effects of ionizing radiation and
DNA-damaging chemotherapy agents, such as the methylating agents
and topoisomerase II inhibitors (35). Moreover, recent studies suggests
that PARP inhibitors could be used not only as chemo/radiotherapy
sensitizers, but also as single agents to selectively kill cancers
defective in DNA repair, specifically cancers with mutations in the
breast cancer-associated genes BRCA1 and BRCA2 (36). Although clinical trials indicate
that PARP-inhibitors have emerged as a promising new class of
antineoplastic agents, significant numbers of TNBC tumors still
recur (37). TNBC tumors
recurrence following treatment with PARP-inhibitors can be
explained by recent studies demonstrating that PARP-inhibitors
eliminate the bulk of tumor cells, but they have limited ability to
eliminate CD44+/CD24− CSCs (38). To date no preclinical study has
demonstrated the efficacy of PARP-inhibitors in selective targeting
of CD44+/CD24− CSCs in IBC tumors. For this
reason the discovery of oncogenic signalings responsible for the
maintenance and expansion of CD44+/CD24− CSCs
is crucial for the development of innovative-targeted therapies
aimed to drastically reduce the recurrence and poor outcome of IBC
tumors.
The findings presented in this study demonstrate
that IBC SUM149PT cancer cells exhibit higher CIN based on
structural and numerical chromosome abnormalities than luminal
ERα+ MCF-7 cancer cells. Moreover, we established that
only SUM149PT cancer cells contain a
CD44+/CD24−/Low CSCs subpopulation displaying
centrosome amplification that was functionally linked to cyclin E
overexpression and Rb phosphorylation. Several studies have shown
that elevated levels of cyclin E induce aberrant activation of Cdk2
kinase leading to Rb phosphorylation and inactivation with
deleterious consequences on the control of centrosome duplication
and CIN (14–16). Significantly, cyclin E is
overexpressed in aggressive breast tumors and it has been
associated with CIN, development of distant metastases and poor
outcome (39). Concurring with our
findings, it was previously demonstrated that SUM149PT cancer cells
display very high levels of cyclin E expression for the duration of
the cell cycle which is in contrast to cyclin E degradation
observed in the mid to late S phase of normal cells. In addition,
comparative genomic hybridization indicated that SUM149PT cells
exhibit many chromosome copy number alterations, which may reflect
prior or ongoing chromosome instability driven by cyclin E activity
(40). Because we have
demonstrated that cyclin E overexpression is restricted to the
subpopulation of CD44+/CD24− CSCs isolated
from SUM149PT cells, we treated bulk SUM149PT cells and
CD44+/CD24− CSCs with a conventional
anticancer drug (methotrexate) and a specific Cdk2 inhibitor
(SU9516) to abrogate the cyclin E/Cdk2 oncogenic signaling. CSCs
showed higher resistance to methotrexate than bulk SUM149PT cells,
nonetheless CSCs were more sensitive to the cytotoxic effects of
SU9516 indicating that molecular inhibition of Cdk2 kinase activity
selectively targeted CSCs in IBC. Finally, we showed that
combination of paclitaxel with SU9516 in vitro induces a
stronger cytotoxic effect characterized by activation of apoptosis
in SUM149PT cells. Importantly, the preclinical relevance of our
findings is justified by recent studies demonstrating that
administration of cyclin E siRNA in vivo inhibited breast
tumor growth in nude mice. Moreover, the authors demonstrate that
cyclin E siRNA synergistically enhanced the cell killing effects of
chemotherapeutic agents in vitro and this combination
greatly suppressed the tumor growth in nude mice. In conclusion our
study demonstrate for the first time that aberrant activation of
cyclin E/Cdk2 oncogenic signaling is restricted in CSCs derived
from IBC cells and combination of conventional chemotherapy with
small molecule inhibitors of Cdk2 kinase activity may represent a
novel targeted therapeutic approach to treat aggressive IBC tumors
with consequent benefits on the disease-free and overall survival
of patients.
Acknowledgements
This study was supported by USAMRMC BC022276 and
Intramural RECDA Award to A.B.D., NCI CA72836 to J.L.S., the Mayo
Clinic Breast Cancer Specialized Program of Research Excellence NIH
CA116201 to J.I., the Atwater Foundation to E.G. and the Mayo
Clinic School of Medicine. We also wish to acknowledge the
Cytogenetic Shared Resource and PCR core facilities of the Mayo
Clinic Comprehensive Cancer Center, for performing SKY, routine
cytogenetic and assisting us with the interpretation of the
results.
References
|
1
|
Fernandez SV, Robertson FM, Pei J,
Aburto-Chumpitaz L, Mu Z, Chu K, Alpaugh RK, Huang Y, Cao Y, Ye Z,
Cai KQ, Boley KM, Klein-Szanto AJ, Devarajan K, Addya S and
Cristofanilli M: Inflammatory breast cancer (IBC): clues for
targeted therapies. Breast Cancer Res Treat. 140:23–33. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yasumura K, Ogawa K, Ishikawa H, Takeshita
T, Nakagawa Y and Osamura RY: Inflammatory carcinoma of the breast:
characteristic findings of MR imaging. Breast Cancer. 4:161–169.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamauchi H, Woodward WA, Valero V, Alvarez
RH, Lucci A, Buchholz TA, Iwamoto T, Krishnamurthy S, Yang W,
Reuben JM, Hortobágyi GN and Ueno NT: Inflammatory breast cancer:
what we know and what we need to learn. Oncologist. 17:891–899.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liedtke C, Bernemann C, Kiesel L and Rody
A: Genomic profiling in triple-negative breast cancer. Breast Care
(Basel). 8:408–413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qiu M, Peng Q, Jiang I, Carroll C, Han G,
Rymer I, Lippincott J, Zachwieja J, Gajiwala K, Kraynov E, Thibault
S, Stone D, Gao Y, Sofia S, Gallo J, Li G, Yang J, Li K and Wei P:
Specific inhibition of Notch1 signaling enhances the antitumor
efficacy of chemotherapy in triple negative breast cancer through
reduction of cancer stem cells. Cancer Lett. 328:261–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cortazar P, Zhang L, Untch M, Mehta K,
Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L,
Valagussa P, Swain SM, Prowell T, Loibl S, Wickerham DL, Bogaerts
J, Baselga J, Perou C, Blumenthal G, Blohmer J, Mamounas EP, Bergh
J, Semiglazov V, Justice R, Eidtmann H, Paik S, Piccart M, Sridhara
R, Fasching PA, Slaets L, Tang S, Gerber B, Geyer CE Jr, Pazdur R,
Ditsch N, Rastogi P, Eiermann W and von Minckwitz G: Pathological
complete response and long-term clinical benefit in breast cancer:
the CTNeoBC pooled analysis. Lancet. Feb 14–2014.(Epub ahead of
print).
|
|
7
|
Crown J, O’Shaughnessy J and Gullo G:
Emerging targeted therapies in triple-negative breast cancer. Ann
Oncol. 23(Suppl 6): vi56–vi65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greenup R, Buchanan A, Lorizio W, Rhoads
K, Chan S, Leedom T, King R, McLennan J, Crawford B, Kelly Marcom P
and Shelley Hwang E: Prevalence of BRCA mutations among women with
triple-negative breast cancer (TNBC) in a genetic counseling
cohort. Ann Surg Oncol. 20:3254–3258. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Venkitaraman AR: Cancer susceptibility and
the functions of BRCA1 and BRCA2. Cell. 108:171–182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Farrugia DJ, Agarwal MK, Pankratz VS,
Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J,
Tavtigian SV, Goldgar DE and Couch FJ: Functional assays for
classification of BRCA2 variants of uncertain significance. Cancer
Res. 68:3523–3531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kais Z, Chiba N, Ishioka C and Parvin JD:
Functional differences among BRCA1 missense mutations in the
control of centrosome duplication. Oncogene. 31:799–804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kolupaeva V and Basilico C: Overexpression
of cyclin E/CDK2 complexes overcomes FGF-induced cell cycle arrest
in the presence of hypophosphorylated Rb proteins. Cell Cycle.
11:2557–2566. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reed SI: Control of the G1/S transition.
Cancer Surv. 29:7–23. 1997.
|
|
14
|
D’Assoro AB, Lingle WL and Salisbury JL:
Centrosome amplification and the development of cancer. Oncogene.
21:6146–6153. 2002.
|
|
15
|
D’Assoro AB, Busby R, Suino K, Delva E,
Almodovar- Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V,
Stivala F and Salisbury JL: Genotoxic stress leads to centrosome
amplification in breast cancer cell lines that have an inactive
G1/S cell cycle checkpoint. Oncogene. 23:4068–4075. 2004.PubMed/NCBI
|
|
16
|
Hanashiro K, Kanai M, Geng Y, Sicinski P
and Fukasawa K: Roles of cyclins A and E in induction of centrosome
amplification in p53-compromised cells. Oncogene. 27:5288–5302.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell
LL, Polyak K, Brisken C, Yang J and Weinberg RA: The epithelial
mesenchymal transition generates cells with properties of stem
cells. Cell. 133:704–715. 2008. View Article : Google Scholar
|
|
18
|
Hwang-Verslues WW, Kuo WH, Chang PH, Pan
CC, Wang HH, Tsai ST, Jeng YM, Shew JY, Kung JT, Chen CH, Lee EY,
Chang KJ and Lee WH: Multiple lineages of human breast cancer
stem/progenitor cells identified by profiling with stem cell
markers. PLoS One. 4:83772009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shipitsin M, Campbell LL, Argani P,
Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T,
Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker
LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ,
Nikolsky Y, Gelman RS and Polyak K: Molecular definition of breast
tumor heterogeneity. Cancer Cell. 11:259–273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
D’Assoro AB, Liu T, Quatraro C, Amato A,
Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V,
Ecsedy J, Iankov I, Di Leonardo A, Ayers-Inglers J, Degnim A,
Billadeau D, McCubrey J, Ingle J, Salisbury JL and Galanis E: The
mitotic kinase Aurora-A promotes distant metastases by inducing
epithelial-to-mesenchymal transition in ERα(+) breast cancer cells.
Oncogene. 33:599–610. 2014.PubMed/NCBI
|
|
21
|
Lee HE, Kim JH, Kim YJ, Choi SY, Kim SW,
Kang E, Chung IY, Kim IA, Kim EJ, Choi Y, Ryu HS and Park SY: An
increase in cancer stem cell population after primary systemic
therapy is a poor prognostic factor in breast cancer. Br J Cancer.
104:1730–1738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arima Y, Hayashi N, Hayashi H, Sasaki M,
Kai K, Sugihara E, Abe E, Yoshida A, Mikami S, Nakamura S and Saya
H: Loss of p16 expression is associated with the stem cell
characteristics of surface markers and therapeutic resistance in
estrogen receptor-negative breast cancer. Int J Cancer.
130:2568–2579. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prat A, Karginova O, Parker JS, Fan C, He
X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M and Perou CM:
Characterization of cell lines derived from breast cancers and
normal mammary tissues for the study of the intrinsic molecular
subtypes. Breast Cancer Res Treat. 142:237–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
D’Assoro AB, Leontovich A, Amato A,
Ayers-Ringler JR, Quatraro C, Hafner K, Jenkins RB, Libra M, Ingle
J, Stivala F, Galanis E and Salisbury JL: Abrogation of p53
function leads to metastatic transcriptome networks that typify
tumor progression in human breast cancer xenografts. Int J Oncol.
37:1167–1176. 2010.PubMed/NCBI
|
|
25
|
D’Assoro AB, Barrett SL, Folk C, Negron
VC, Boeneman K, Busby R, Whitehead C, Stivala F, Lingle WL and
Salisbury JL: Amplified centrosomes in breast cancer: a potential
indicator of tumor aggressiveness. Breast Cancer Res Treat.
75:25–34. 2002.PubMed/NCBI
|
|
26
|
Iovino F, Lentini L, Amato A and Di
Leonardo A: RB acute loss induces centrosome amplification and
aneuploidy in murine primary fibroblasts. Mol Cancer. 5:382006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mayer IA, Abramson VG, Lehmann BD and
Pietenpol JA: New strategies for triple-negative breast cancer -
deciphering the heterogeneity. Clin Cancer Res. 20:782–790. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mehta RS: Dose-dense and/or metronomic
schedules of specific chemotherapies consolidate the
chemosensitivity of triple-negative breast cancer: a step toward
reversing triple-negative paradox. J Clin Oncol. 26:3286–3288.
2008. View Article : Google Scholar
|
|
29
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, Cristofanilli M, Hortobagyi GN and Pusztai L: Response to
neoadjuvant therapy and long-term survival in patients with
triple-negative breast cancer. J Clin Oncol. 26:1275–1281. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cristofanilli M, Buzdar AU, Sneige N,
Smith T, Wasaff B, Ibrahim N, Booser D, Rivera E, Murray JL, Valero
V, Ueno N, Singletary ES, Hunt K, Strom E, McNeese M, Stelling C
and Hortobagyi GN: Paclitaxel in the multimodality treatment for
inflammatory breast carcinoma. Cancer. 92:1775–1782. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takahashi T, Akashi-Tanaka S, Fukutomi T,
Watanabe T, Katsumata N, Miyakawa K, Hasegawa T and Tsuda H: Two
special types of breast cancer presenting as progressive disease
after neoadjuvant chemotherapy with docetaxel plus doxorubicin.
Breast Cancer. 8:234–237. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leopold PL, Vincent J and Wang H: A
comparison of epithelial-to-mesenchymal transition and
re-epithelialization. Semin Cancer Biol. 22:471–483. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rosen EM and Pishvaian MJ: Targeting the
BRCA1/2 tumor suppressors. Curr Drug Targets. 15:17–31. 2014.
View Article : Google Scholar
|
|
34
|
Khoury-Haddad H, Guttmann-Raviv N,
Ipenberg I, Huggins D, Jeyasekharan AD and Ayoub N: PARP1-dependent
recruitment of KDM4D histone demethylase to DNA damage sites
promotes double-strand break repair. Proc Natl Acad Sci USA.
111:E728–E737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Węsierska-Gądek J, Zulehner N, Ferk F,
Składanowski A, Komina O and Maurer M: PARP inhibition potentiates
the cytotoxic activity of C-1305, a selective inhibitor of
topoisomerase II, in human BRCA1-positive breast cancer cells.
Biochem Pharmacol. 84:1318–1331. 2012.PubMed/NCBI
|
|
36
|
Shen Y, Rehman FL, Feng Y, Boshuizen J,
Bajrami I, Elliott R, Wang B, Lord CJ, Post LE and Ashworth A: BMN
673, a novel and highly potent PARP1/2 inhibitor for the treatment
of human cancers with DNA repair deficiency. Clin Cancer Res.
19:5003–5015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fojo T and Bates S: Mechanisms of
resistance to PARP inhibitors - three and counting. Cancer Discov.
3:20–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yin S, Xu L, Bandyopadhyay S, Sethi S and
Reddy KB: Cisplatin and TRAIL enhance breast cancer stem cell
death. Int J Oncol. 39:891–898. 2011.PubMed/NCBI
|
|
39
|
Potemski P, Kusińska R, Pasz-Walczak G,
Piekarski JH, Watała C, Płuciennik E, Bednarek AK and Kordek R:
Prognostic relevance of cyclin E expression in operable breast
cancer. Med Sci Monit. 15:MT34–MT40. 2009.PubMed/NCBI
|
|
40
|
Willmarth NE, Albertson DG and Ethier SP:
Chromosomal instability and lack of cyclin E regulation in hCdc4
mutant human breast cancer cells. Breast Cancer Res. 6:R531–R539.
2004. View
Article : Google Scholar : PubMed/NCBI
|