Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide, with ~1.5 million new cases diagnosed each year
(1). Approximately 85% of all
cases of lung cancer is non-small cell lung cancer (NSCLC) which is
responsible for >1.2 million deaths worldwide each year
(1,2). Current outcomes of NSCLC treatment
are disappointing with dismal prognosis, and the overall 5-year
survival rates remain ~15% for the past few decades (3). This is because ~55% of patients
present with advanced stage at time of diagnosis (4). Platinum-based chemotherapy, is
currently the mainstay of the advanced NSCLC treatment (5).
Cisplatin [Cis-diammine-dichloroplatinum (II), DDP]
and its analogue carboplatin are the most commonly used platinum
agents in the platinum-based chemotherapy of NSCLC (6,7).
They are heavy metal complexes, and present their therapeutic
cytotoxic properties by covalently binding to DNA to form intra-
and interstrand cross-links which interfere with normal DNA
replication and transcription (8,9).
Without efficient DNA repair, the platinum-induced DNA damage
eventually leads to cell apoptosis (5). Although the platinum containing
agents greatly increase the effectiveness of NSCLC treatment
compared with other drugs, platinum-based chemotherapy has
apparently reached a therapeutic plateau with limited response to
treatment as well as little improvement of survival for patients
(10,11), due to the tumors rapidly developing
acquired drug resistance which has not yet been overcome (12). Many published reports suggest that
the emergence of platinum resistance, not only intrinsic, but also
acquired primarily attribute to the nucleotide excision repair
(NER) system (6,13).
NER system, a highly versatile and sophisticated
system for DNA damage removal, is the primary repair pathway for
DNA helix-distorting lesions induced by platinum agents (2,14).
High DNA repair capacity of NER pathway certainly contributes
greatly to drug resistance in the platinum-based chemotherapy of
NSCLC. DNA repair capacity of NER pathway can be represented by the
expression level of excision repair cross-complementation group 1
(ERCC1) which is one of the most important members of the pathway
(15). It has been reported that
ERCC1 together with xeroderma pigmentosum group F (XPF) forms the
ERCC1-XPF complex to nick the damaged DNA strand at the 5′ sit of
the helix-distorting platinum lesion, which is considered as the
essential rate-limiting step in the NER process (14,16).
Recently, accumulated preclinical and clinical
studies have demonstrated that ERCC1 could be as a prognostic
factor (information on the patients overall cancer outcome,
regardless of therapy), predictive (information on the effect of a
therapeutic intervention) and a potential therapeutic target in
cancer, including NSCLC (13,17).
In these studies, among patients who did not receive chemotherapy,
those with ERCC1-positive tumors had longer survival than those
with ERCC1-negative tumors (18),
however, for the patients with ERCC1-negative tumors, they had a
distinctly higher response rate (19) and attained benefit from
platinum-based chemotherapy with significantly prolonged survival,
but patients with ERCC1-positive tumors did not (18,20,21).
In some other studies, inconsistently, ERCC1 expression was not
associated with platinum response (22) or survival (23) in NSCLC patients.
It is still controversial whether ERCC1 should be
used as a predictor to guide clinical personalized chemotherapy or
not, due to the inconsistence of clinical outcomes (24). Multicenter clinical trails with
larger sample size, such as the International Adjuvant Lung Cancer
Trial Biologic Program (IALT-Bio) and the Lung Adjuvant Cisplatin
Evaluation Biologic Program (LACE-Bio) (25–27),
should be conducted for the application of ERCC1 in clinic.
Nevertheless, studies on biomarkers like ERCC1 provides an onset
for overcoming drug resistance and tailoring chemotherapy in NSCLC
treatment. Our previous retrospective studies have indicated that
ERCC1 is a potential candidate biomarker to predict tailoring
chemotherapy of NSCLC (28). In
the present study, we aim to further reveal the biological
functions of ERCC1 in cell proliferation, cell cycle, invasion and
cisplatin resistance in NSCLC cells by gene cloning, cell
transfection and RNAi techniques, to provide more evidence for the
utilization of ERCC1 as a biomarker in the customized chemotherapy
of NSCLC.
Materials and methods
Material, vectors and reagents
Tissue sample used for the cloning of ERCC1 gene was
from the redundant tissue of surgical operation performed on a
patient with brain injury. The patient gave written informed
consent before enrolment. The present study was carried out
following international and national regulation and approved by the
Ethics Committee of the Affiliated Tumor Hospital of Guangxi
Medical University. PMD18-T vector used for TA cloning, PCDNA3.1
vector for cell transfection and pSilencer 4.1-CMV-neo vector for
RNAi were obtained from Takara (Tokyo, Japan), Invitrogen
(Carlsbad, CA, USA) and Ambion (Austin, TX, USA), respectively.
TRIzol reagents, RevertAid™ First Strand cDNA Synthesis kit and
Lipofectamine 2000 were from Invitrogen. Ex Taq DNA polymerase,
restriction endonucleases BamH1 and EcoR1, and T4 DNA
ligase were from Takara. Plasmid Mini kit was from Qiagen
(Duesseldorf, Germany). M-per Mammalian Protein Extraction kit,
Bradford protein assay kit and fluorescein isothiocyanate (FITC)
reagents were purchased from Pierce (Rockford, IL, USA). Goat
anti-human antibody ERCC1 and goat anti-human antibody β-actin used
for western blot assays were from NeoMarkers (Fremont, CA, USA),
and the secondary rabbit anti-goat antibody was from Fuzhou Maixin
Biotech. Co., Ltd. (Fuzhou, China). Giemsa stain for colony-forming
assays, propidium lodide (PI) fluorescence dye and RNase A used in
the cell cycle assays were from Sigma (St. Louis, MO, USA) and
Sango (Shanghai, China), respectively. All the reagents for high
performance liquid chromatography (HPLC) assays were obtained from
Merck (Darmstadt, Germany). Mouse anti-human monoclonal antibody
(McAb) of P-glycoprotein (P-gp) and multidrug resistance-associated
protein (MRP), as well as the corresponding secondary goat
anti-mouse antibody used in the flow cytometry (FCM) were from
Invitrogen.
Cell lines, cell culture and drugs
Human NSCLC cell line H1299 was presented by Dr Zhe
Zhang from Karolinska Institutet of Sweden. Human NSCLC cell line
A549 was purchased from the cell bank of Peking Union Medical
College (Beijing, China). NSCLC cells were grown in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 μg/ml streptomycin at the final concentration
and maintained in a humidified atmosphere containing 5%
CO2 at 37°C. Cell culture reagents and FBS were obtained
from Gibco (BRL, Long Island, NY, USA), antibiotics of penicillin
and streptomycin were from Shandong Qilu Pharmaceutical Factory
(Shandong, China), and Geneticin (G418) was from Sigma. Cisplatin
and adriamycin (ADM) were purchased from Shandong Qilu
Pharmaceutical Factory and Zhejiang Haimen Pharmaceutical Factory
(Zhejiang, China), respectively.
Acquisition of the ERCC1 gene
ERCC1 gene was obtained from the tissue sample by
reverse transcription-PCR (RT-PCR) technique. In brief, the tissue
sample was incised and immediately put into liquid nitrogen. Total
RNA was isolated from the samples using TRIzol reagent according to
the manufacturer’s instructions. Approximately 0.5–5 μg of total
RNA together with random hexamers were used for the cDNA synthesis
through RT-PCR with RevertAid™ First Strand cDNA Synthesis kit. The
final cDNA was used for the following PCRs. ERCC1 primers (forward,
5′-CGCGGATTCATGGA CCCTGGGAAGGACAAAG-3′; reverse, 5′-CCGGAATTCT
CAGGGTACTTTCAAGAAGGG-3′) were designed with Primer 5 software and
synthesized in Sangon Biotech Co., Ltd. PCR was performed in a
25-μl solution including 200 nM of the primers, 200 nM of converted
cDNA, 10 mM deoxynucleotide triphosphates (dNTPs), 25 mM
MgCl2, 1.0 U Ex Taq DNA polymerase and the PCR buffer.
The amplification thermal-cycling program consisted of 30 cycles of
94°C for 30 sec, 55°C for 30 sec and 72°C for 45 sec. The obtained
PCR products were separated by the 1% agarose gel electrophoresis
and the correct one was purified to be used in the following vector
construction.
Vector construction
To obtain accurate and adequate target gene for
vector construction, TA cloning was performed in a system with a
total volume of 10 μl, including the purified PCR product 4 μl,
PMD18-T vector 1 μl, salt solution 1 μl and ddH2O 4 μl.
After 5-min incubation at room temperature, it was transformed into
the competent cell TOP 10. Positive stains were selected with
blue-white selection, and cultured. Then, the plasmid DNA was
extracted with the Plasmid Mini kit according to the manufacturer’s
instructions. Target fragment in the plasmid DNA was isolated and
inserted into the BamHI and EcoRI sites of PCDNA3.1
vector (5.5 kb) with the T4 DNA ligase. Thus, the connection
product PCDNA3.1-ERCC1 plasmid was obtained, and then was
transfected into the competent cell TOP 10 again. Positive stains
of TOP 10 were selected by the blue-white selection and identified
by PCR to exclude false clones, and further confirmed by sequencing
identification. Then, the PCDNA3.1-ERCC1 plasmid obtained through
the above vector construction processes was used in the following
cell transfection experiments.
ERCC1 transfection into the H1299 cell
line
Parental H1299 cells 0.5 ml at the density of
0.6–1×105/ml were seeded in 6-well plates per well and
incubated at 37°C for 24 h until 80% confluent at the time of
transfection. The transfection was performed using Lipofectamine
2000 according to the manufacturer’s instructions with slight
changes. Plasmid PCDNA3.1-ERCC1 obtained above was transfected into
H1299 cells; PCDNA3.1 vehicle and PEGFP-N1 plasmid with the green
fluorescent protein (GFP) gene were transfected into the H1299
cells as parallel controls; and H1299 cells treated with
transfection reagents without any plasmids or vehicles were used as
the negative control. The transfection procedures were as follows:
firstly, DNA-Lipofectamine complexes were prepared for each
transfection sample. DNA [3.0 μl (1.0 μg)] and 5 μl Lipofectamine
were mixed with 250 μl antibiotic- and FBS-free medium
respectively. After 20 min at room temperature the mixtures were
combined for 15–20 min. The final mixture was the DNA-Lipofectamine
complexes. Then, cells in each well were washed with the
antibiotic- and FBS-free medium and added with the above
DNA-Lipofectamine complexes to incubate for 5 h at 37°C, then, 1 ml
medium containing FBS were added into each well and incubated for
further 24 h. The medium was then replaced by fresh normal medium
and cells were cultured for 24–48 h to be used in the following
screening and verification tests.
G418 cytotoxicity assays
G418 was used to select the positive cells
transfected with PCDNA3.1-ERCC1 and PCDNA3.1 vehicle, as the
PCDNA3.1 vector contains the anti-G418 gene. The selecting
concentration of G418 was determined as follows: briefly, H1299
cells (1×103) grown in the antibiotics-free complete
medium were seeded in 24-well plates and cultured till the cells
attached. Then, they were treated with different concentrations of
G418 (100–1,100 μg/ml with 100 μg/ml for one gradient) and
continuously incubated at 37°C in a humidified atmosphere
containing 5% CO2 for 10–14 days. The minimum G418
concentration at which all the cells were killed was used as the
selection concentration in the transfection assays.
Reverse transcription-quantitative PCR
(RT-qPCR) assays
Total RNA was extracted with TRIzol reagent from the
positive cells that had been screened by the G418 assays. cDNA was
synthesized using the oligo(dt) primer according to the
instructions of the RevertAid™ First Strand cDNA Synthesis kit.
Subsequently, ERCC1 (441 bp) and PCDNA3.1 were amplified with the
β-actin as an internal control. The primers were: ERCC1:
5′-GATGACCCAGATCATGTTTG-3′ (forward), 5′-TGGAGTTGAAGGTAGTTTCG-3′
(reverse); PCDNA3.1: 5′-CTGCTTACTGGCTTATC-3′ (forward) and
5′-GAAAGGACAGTGGGAGTG-3′ (reverse); β-actin: 5′-CAC
CAACTGGGACGACAT-3′ (forward) and 5′-A CAGCCTGG ATAGCAACG-3′
(reverse). The amplification was performed in a 25-μl solution
containing 200 nM of each of the primers, 200 nM the converted
cDNA, 10 mM dNTPs, 25 mM MgCl2, 1.0 U Ex Taq DNA
polymerase and the PCR buffer, in conjunction with a thermal-cycle
program consisting of 30 cycles of 94°C for 30 sec, 55°C for 30 sec
and 72°C for 45 sec. Finally, the PCR products were separated on a
1% agarose gel and investigated in the Gel doc 2000 imaging and
analysis system (Bio-Rad, Hercules, CA, USA).
Western blot assays
Cells were washed with chilled PBS for three times
and added to lysis buffer M-per mammalian protein extraction to
incubate on ice for 30 min. The lysate was then centrifuged at
13,000 g for 5 min and the supernatant was collected. Concentration
of total proteins was determined with the Bradford protein assay
kit. Proteins were separated by SDS-PAGE and were transferred to a
PVDF membrane. Primary antibodies goat anti-human antibody ERCC1
(1:200) and goat anti-human antibody β-actin (1:200) were applied
to bond with target proteins on the PVDF membrane, and followed by
the combination with the secondary antibody rabbit anti-goat
(1:500). At last, the bands were identified by the enhanced FITC
reagents.
Cell growth curve
Cells (1×104) were seeded in 24-well
plates with 18 duplications for each cell sample: H1299 cells
(parental cells as control), H1299/E cells (transfected with
PCDNA3.1-ERCC1) and H1299/V cells (transfected with PCDNA3.1
vehicle only). Survival cell of each cell sample were counted in
three duplications every day. The procedures were as follows:
briefly, culture solution in each well was removed with a straw.
Cells left in the well were trypsinized with 100 μl 0.25% trypsin
for 3–5 min, and then suspended uniformly with 800 μl RPMI-1640
medium and 100 μl trypan blue staining. Survival of the cells were
evaluated under the inverted microscope (Olympus, Tokyo, Japan).
Cell number was calculated according to the formula
N=(n/4)x104xV. Finally, cell growth curves were drawn
with cell number as the y-axis and time as the x-axis.
Colony-forming assays
Cells were uniformly seeded in 24-well plates at the
density of 50, 100, 200 in triplicate, and cultured at 37°C in a
humidified atmosphere containing 5% CO2 for 5–6 days.
The culture medium was replaced by fresh medium every 3 days. When
colony contained >50 cells, it was washed with PBS, fixed with
methanol for 15 min and stained with Giemsa for 30 min, then washed
with water and dried in an airing chamber. Then, the colony was
counted under the Gel doc 2000 imaging and analysis system. Colony
forming rate was determined according to the equation: colony
forming rate = (number of colonies/number of seeded cells) ×
100%.
Cell cycle analysis
Cell cycle distribution was determined by flow
cytometry (FCM). Cells were washed twice with PBS and fixed with 1
ml chilled 70% ethanol at 4°C overnight, then washed twice again
with PBS and suspended at the density of 1×106/ml with
the staining solution containing 0.05 mg/ml PI and 0.1 mg/ml RNase
A and incubated in the dark at 4°C for 30 min. At the end of the
incubation period, the cell suspension was filtered with a 300 mesh
screen to remove adhered cells. Finally, DNA content of each cell
sample was detected on EPICS XL (Beckman, Urbana, IL, USA) and cell
cycle distribution was determined with the muticycle software.
Matrigel invasion assays
Matrigel invasion assays were performed to evaluate
the ability of cell invasion in vitro. The lower surface of
filter membrane (8 μm) of transwells was treated with 50 μl PBS
supplemented with 5 μg fibronectin (Sigma) and dried at room
temperature overnight. Matrigel (50 μl) that had been diluted to
the density of 1.25 mg/ml with the cold serum-free RPMI-1640 medium
was added to the upper surface of the filter membrane, incubating
for 4–5 h. Cells, after three washes were trypsinized with 0.25%
trypsin, and resuspended at the density of 1×106/ml in
the RPMI-1640 medium supplemented with 1% FBS. Cell suspension
solution (100 μl) was added to the upper chamber of the transwells
and 600 μl of the medium containing 1% FBS was added to the lower
chamber, incubating at 37°C in a humidified atmosphere containing
5% CO2 for 20–24 h. At the end of incubation period,
cells in both chambers were treated with 10 and 60 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
Sigma), followed by 10 min dissolution with DMSO. Matrigel invasion
ability of cells was determined through detecting the OD value of
the dissolved solution at 450 nm.
Evaluation of cisplatin response
Cisplatin response of cells was determined by MTT
assays. Cells (1×105/ml) were trypsinized with 0.25%
EDTA and seeded into 96-well plates in triplicate. After incubated
for 24 h, they were treated with different concentrations of
cisplatin (0–400 μg/ml with 50 μg/ml for a gradient) for 24 h. At
the end of the treatment period, 20 μl of MTT was added into each
well and incubated for 4 h. The cells were then collected by the
centrifugation and treated with 150 μl of DMSO for 10 min. The
plates were scanned at 570 nm in a 96-well plate reader (DG3201,
Nanjing, China). Each experiment was conducted three times in
triplicate. Rate of inhibition (IR) were calculated by the formula
IR (%) = (Acontrol −
Atreatment)/Acontrol × 100%.
Measurement of intercellular drug
concentration
Drug accumulation in cells was assessed by detecting
intercellular concentration of drugs with HPLC assays. After
treated with 14 μg/ml cisplatin or 2 μg/ml adriamycin, cells
(1×106) were trypsinized and collected by
centrifugation. Then cells were suspended with ddH2O and
lysed through the repeated freeze-thaw cycles. The lysate was
centrifuged at 12,000 rpm for 5 min and the supernatant was
collected to be analyzed with HPLC-10A VP (Shimadzu, Kyoto, Japan).
The HPLC conditions for cisplatin analysis were as follows:
chromatographic column: Shim-pack CLC-ODS (250×4.6 mm, 5 μm,
Shimadzu, Kyoto, Japan); column temperature, 35°C; mobile phase,
distilled water; test wavelength, 230 nm; flow-rate, 1.0 ml/min;
and sample size, 10 μl. For ADM analysis, the HPLC conditions were
chromatographic column: Shim-pack CLC-ODS (250×4.6 4.6 mm, 5 μm);
column temperature, 40°C; mobile phase, the mixture containing
methanol-0.01 M phosphate and 0.01% sodium dodecyl sulfate
(v/v=85/15), adjusted to pH 2.5 with phosphoric acid; wavelength,
545 nm; flow-rate, 1.3 ml/min and sample size, 20 μl.
Detection of the expression of cell
membrane proteins
Expression of two membrane proteins, P-gp and MRP,
were detected by the FCM. Cells were adjusted to the density of
5×106–1×107/ml with RPMI-1640 medium. Then,
100 μl of the cell suspension was mixed with 50 μl of the
inactivated normal rabbit serum followed by the 10-min incubation
at 4°C. At the end of incubation period, it was washed twice and
centrifuged at 1,000 rpm for 5 min, and the supernatant was
discarded. Then 5–50 μl of the chilled mouse antihuman McAb was
added and incubated at 4°C for 30 min. After washed twice and
centrifuged at 1,000 rpm for 5 min, the cells were suspended with
50 μl of the goat anti-mouse secondary antibody with fluorescent
marker and incubated at 4°C for 30 min. After washing and
centrifugation, the cells were resuspended with 0.5 ml of PBS,
filtered with the 300 mesh screen and finally detected utilizing
the FCM.
ERCC1 knockdown in A549 cell line
Human NSCLC cell line A549 with an intrinsic ERCC1
expression was selected as the parental cell in the RNAi
experiments. Four small interfering RNAs (siRNAs) targeting to
ERCC1 gene were designed according to the principles described
previously (29). The sensestrand
sequences of siRNA duplexes were as follows: siRNA897:
5′-CAGACCCTCCTGACCACATTTGG-3′; siRNA255:
5′-AAGCCCTTATTCCGATCTACACA-3′; siRNA783:
5′-AAGGCCTATGAGCAGAAACCAGC-3′; siRNA693:
5′-AAGGAGCTGGCTAAGATGTGAT-3′. In addition, one siRNA for the GAPDH
gene was designed as a positive control:
5′-GUAUGACAACAGCCUCAAGTT-3′, and another siRNA not associated with
ERCC1 gene but marked with flourescent indicator was the negative
control: 5′-TTCTCCGAACGTGTCACGT-3′. They were synthesized and
inserted into the BamH1 and HindIII sites of a
pSilencer 4.1-CMV-neo vector, respectively, and followed by the
transfection into the A549 cells utilizing Lipofectamine 2000. For
ERCC1 gene, the gene-specific siRNA with optimal efficiency of
interference was selected to be used in the following experiments.
mRNA and protein expression of ERCC1 in the three cell samples,
A549/E cells transfected with the siRNA of ERCC1, A549/V cells
transfected only with the vehicle vector (as a positive control)
and the parental A549 cells (as a negative control), was then
detected by RT-qPCR and western blot assays, respectively. In
addition, cisplatin response of A549/E cells and A549/V cells was
also detected according to the above description in ‘Evaluation of
cisplatin response’. The treatment concentration of cisplatin was
0–80 μg/ml with 10 μg/ml for a gradient.
Statistical analysis
The difference among three groups was calculated by
one-way ANOVA, and the difference between two groups was analyzed
by Student’s t-test. All of the analysis was performed on SPSS 19.0
software. A value of P<0.05 was considered as statistically
significant.
Results
ERCC1 was successfully transfected into
the NSCLC H1299 cells
To investigate the biological functions of ERCC1 on
NSCLC cells, ERCC1 gene was cloned and transfected into the NSCLC
H1299 cells with no intrinsic ERCC1 expression. ERCC1 gene was
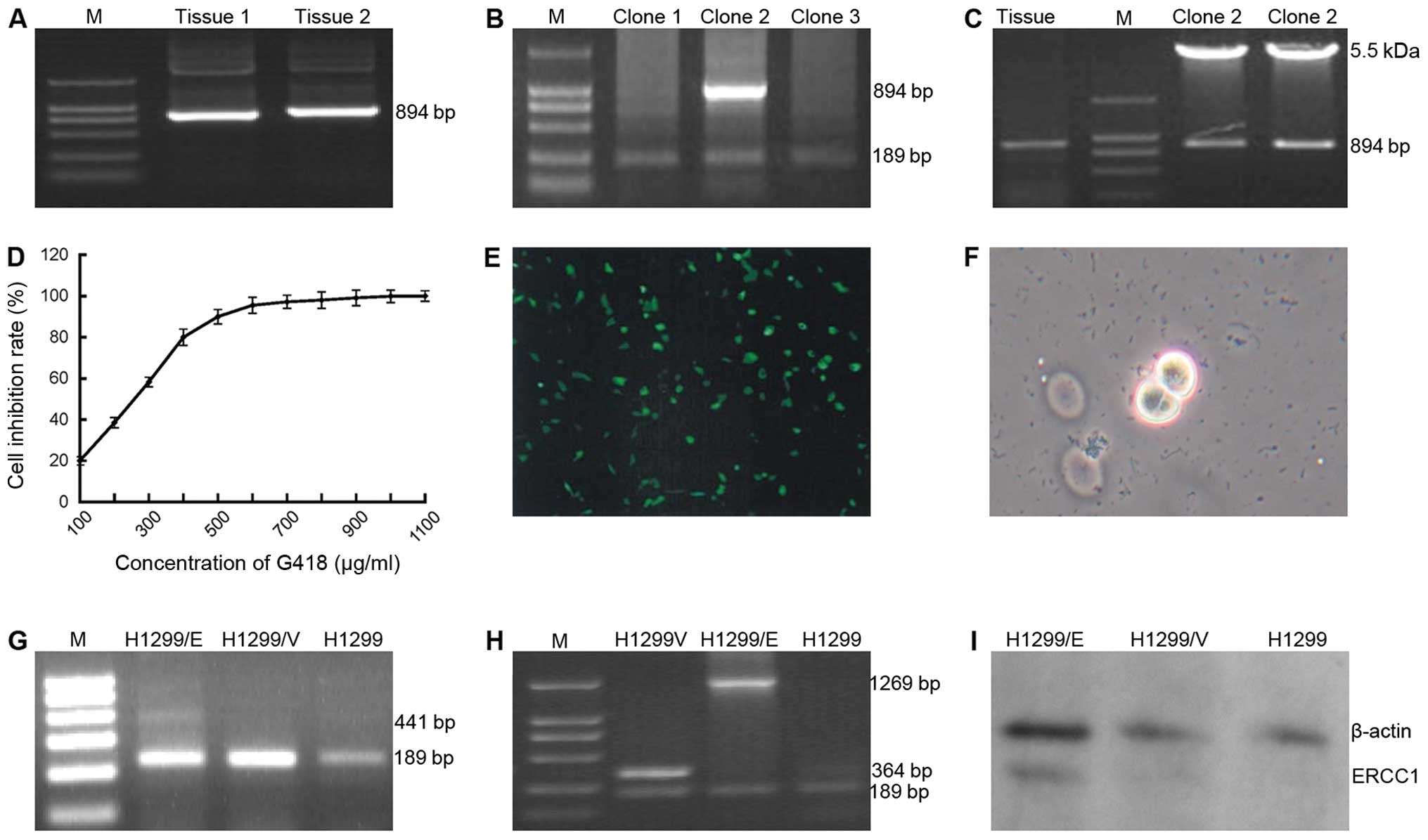
attained by RT-PCR technique (Fig.
1A) and linked to the PCDNA3.1 vector, to compose the
recombinant plasmid PCDNA3.1-ERCC1 which was transfected into the
competent cell TOP 10 treated with chilled CaCl2
solution. Positive TOP 10 clones were screened by blue-white
selection and further subject to PCR identification with the
primers of ERCC1 gene. As a result, a band of 894 bp coincided with
the target gene ERCC1 (Fig. 1B)
was successfully amplified in clone 2 which was then selected to
the following confirmation assays in which PCR identification was
conducted again with the primers of PCDNA3.1. Two bands 894 bp
(ERCC1) and 5.5 kb (PCDNA3.1) were indicated in the result of the
restriction enzyme cleave identification (Fig. 1C), which demonstrated that the
recombinant plasmid of PCDNA3.1-ERCC1 had been successfully
cloned.
G418 was used as the screening regent in the
following eukaryotic transfection experiments. The screening
concentration was determined as 600 μg/ml by the assays of
cytotoxic reaction on H1299 cells (Fig. 1D). The recombinant plasmid
PCDNA3.1-ERCC1 and PCDNA3.1 vehicle were, respectively, transfected
into the H1299 cells. They were named H1299/E and H1299/V,
respectively. H1299 cells transfected with the GFP gene pEGFP-N1
(H1299/GFP cells) and the parental H1299 cells without DNA
transfection were the positive and negative controls, respectively.
The expression of GFP detected in the H1299/GFP cells (Fig. 1E) indicated that the processes of
transfection had been accurately carried out. Following G418
screening with the concentration of 600 μg/ml, the living clones
(Fig. 1F) were selected and
cultured to an extended scale.
Further identification for the transfection was
conducted with PCR technique. ERCC1 expression (441 bp) appeared in
H1299/E cells not in H1299/V and H1299 cells when the primers of
ERCC1 were used in the PCR process (Fig. 1G), by contrast, when the primers of
PCDNA3.1 were applied, the expression of ERCC1-PCDNA3.1 (1,269 bp)
and PCDNA3.1 (364 bp) were detected in H1299/E and H1299/V cells,
respectively, and no expression of either was found in H1299 cells
(Fig. 1H). Correspondingly, the
protein expression of ERCC1 was also demonstrated in H1299/E cells,
not in H1299/V and H1299 cells by the western blot analysis
(Fig. 1I). These findings
indicated that ERCC1 gene had been successfully transfected into
H1299 cells.
ERCC1 expression had no effect on cell
proliferation, colonyforming, cell cycle and the ability of
invasion in NSCLC H1299 cells
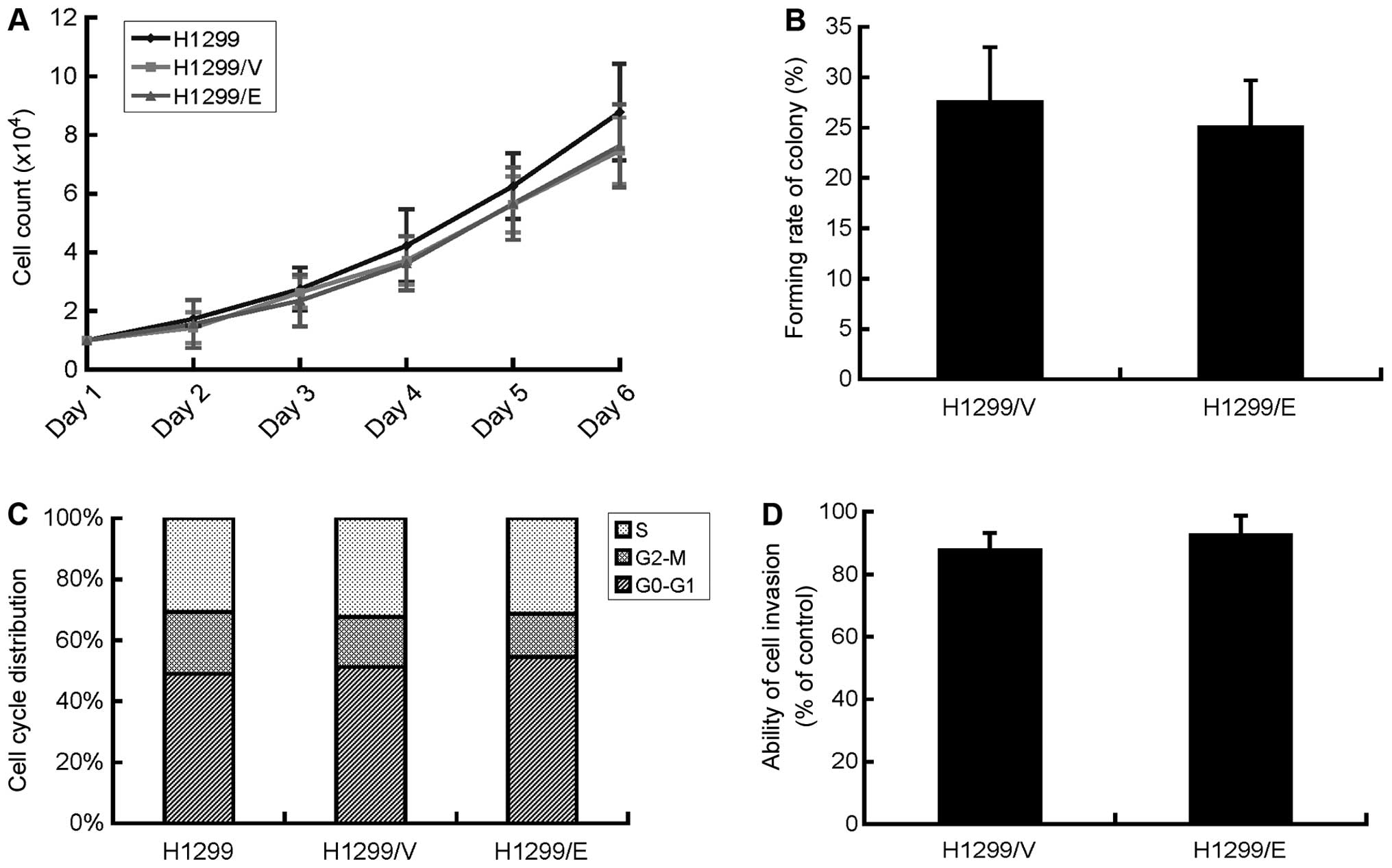
Cell proliferation of H1299/E, H1299/V and H1299
cells was evaluated by survival cell count assays. H1299/E and
H1299/V cells displayed a similar tendency in the growth curve,
they showed no significant difference with their parental cells
H1299 in growth rate with P>0.05, although they had a slightly
slower growth compared with the parental cells (Fig. 2A). Therefore, cell proliferation of
NSCLC H1299 cells in the present study was not affected by the
expression of ERCC1.
Colony-forming assays were also employed to examine
the ability of cell proliferation as well as the population
dependence of H1299/E and H1299/V cells. Consequently, the two cell
lines exhibited no significant difference in the colony-forming
rate with 25.1±4.6 and 27.6±5.4%, respectively and P>0.05
(Fig. 2B). The finding
demonstrated that ERCC1 expression had no effect on the
deterioration of the H1299 cells.
Cell cycle distribution in each cell lines of
H1299/E, H1299/V and 1299 was examined by FCM technique. The
results suggested that there was no significant difference in cell
cycle distribution among the three cell lines (Fig. 2C), indicating that the expression
of ERCC1 had no effect on the cell cycle of the H1299 cells.
Cell invasion is a common biological behavior of
cancer cells and was investigated by Matrigel invasion assays in
the present study. The invasive ability of the cells was determined
by the ratio of the cells in the upper and lower surface of the
transwell. Then, the relatively invasive ability of H1299/E and
H1299/V cells compared with their parental H1299 cell was
calculated, respectively. There was no significant difference in
the invasive ability between H1299/E and H1299/V cells (Fig. 2D). The finding indicated that the
expression of ERCC1 did not affect the invasive ability of H1299
cells in vitro.
Acquired cisplatin resistance of H1299
cells was associated with ERCC1 expression, but not correlated with
the expression of P-gp and MRP proteins
MTT assays were performed to detect the response of
H1299/E and H1299/V cells to cisplatin, respectively. The
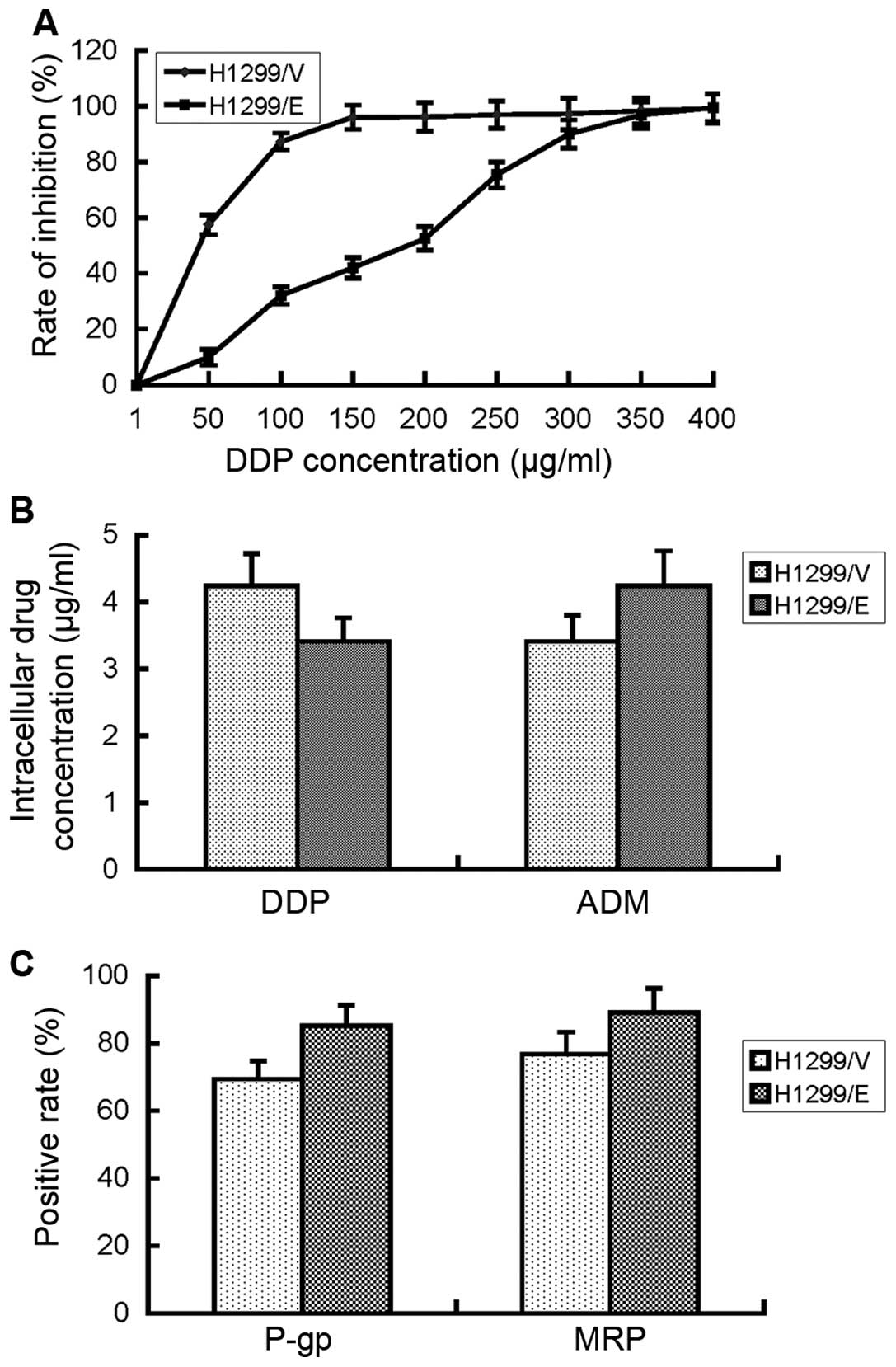
IC50 of cisplatin in the H1299/E cells was 186 μg/ml,
4.23 times higher than that of H1299/V cells which was only 44
μg/ml (Fig. 3A). The results
suggested that H1299 cells developed an evident cisplatin
resistance after transfection with ERCC1 gene.
To clarify whether the acquired cisplatin resistance
of H1299/E cells was correlated with drug influx or efflux,
intercellular accumulation of cisplatin in H1299/E and H1299/V
cells was detected by means of HPLC. Firstly, the standard curve
for cisplatin was established with the correlation coefficient of
0.9999, suggesting that the detection system of HPLC with a high
confidence was available. Then, intercellular cisplatin
concentration in H1299/E and H1299/V cells was tested and
calculated based on the established standard curve. They were 3.41
and 4.24 μg/ml, respectively, with no significant difference
between the two cell lines (P>0.05, Fig. 3B). By contrast, intercellular
concentration of ADM in the two cell lines was determined as well,
and the same result was obtained, which indicated that the results
produced by HPLC for cisplatin was credible. Consequently, the
resistance of H1299/E cells to cisplatin in the present study may
be not associated with drug influx or efflux.
As further confirmation of the above, we detected
the expression of the associated membrane proteins, P-gp and MRP,
which have been reported to participate in drug resistance
(12). The FCM result showed that
there was no significant difference in the expression of P-gp and
MRP proteins between H1299/E and H1299/V cells. Therefore,
cisplatin resistance of H1299/E cells in the present study may be
not associated with the expression of P-gp and MRP proteins.
ERCC1 expression was interfered with by
siRNA in NSCLC A549 cells
To further confirm the effect of ERCC1 expression on
the cisplatin response of NSCLC cells, RNAi technique was employed
to silence the ERCC1 expression of A549 cells. Four siRNAs were
designed, synthesized, and transfected into A549 cells to interfere
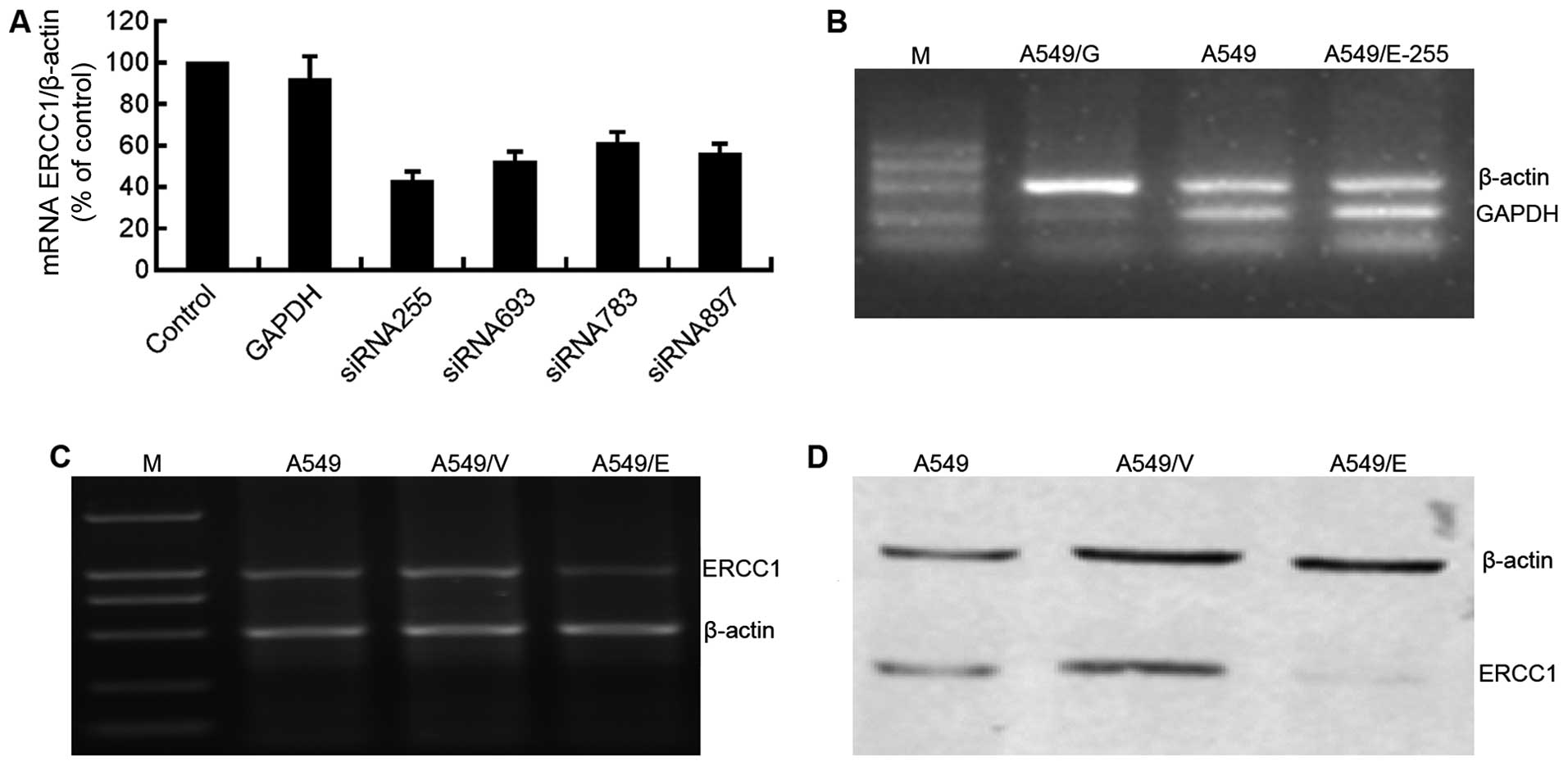
with the ERCC1 expression, siRNA255 was selected for use in the
following experiments (Fig. 4A).
As the parallel positive control, GAPDH was silenced in A549/G
cells transfected by the siRNA to GAPDH, and normally expressed in
A549/E-255 and parental A549 cells (Fig. 4B). The results demonstrated the
effectiveness of the siRNA transfection processes utilized in the
present study.
Following the transfection, RT-PCR and western blot
assays were conducted to detect the mRNA and protein expression of
ERCC1, respectively, in A549, A549/V and A549/E cells. Compared
with parental A549 and A549/V cells, mRNA (Fig. 4C) and protein (Fig. 4D) expression of ERCC1 in A549/E
cells was significantly suppressed, suggesting that NSCLC cell
model with the silenced ERCC1 (A549/E) was successfully established
and could be used in the following cytotoxicity assays.
The siRNA-induced suppression of ERCC1
enhanced the sensitivity of A549 cells to cisplatin
Cell proliferation of A549/E and A549/V cells
treated with cisplatin was detected by MTT assays. The
IC50 of cisplatin in A549/E cells was 18.2 μg/ml, much
less than that in A549/V cells at 53.4 μg/ml. The result indicated
that the suppression of ERCC1 expression by siRNA significantly
enhanced the sensitivity of A549 cells to cisplatin, thus
demonstrating that ERCC1 is implicated in the processes of
cisplatin resistance of NSCLC cells.
Discussion
ERCC1 was first discovered in the 1990s as one of
the foremost members of NER pathway which is responsible for the
repair of most platinum-induced DNA damage (6). Since then a number of studies were
conducted on the association of mRNA and protein expression of
ERCC1 as well as its genetic variations with platinum-based
chemotherapy response and resistance (30–33).
Although an exact conclusion has not been made due to the
differences of these studies in trial design and laboratory tests
(24,34), ERCC1 has attracted great attention
of the NSCLC investigators. Studies are underway to determine
further correlations between ERCC1 and platinum-based chemotherapy,
as well as the underlying mechanisms of platinum resistance in
NSCLC treatment (12).
Previous (30,31)
including our studies (28)
indicated that mRNA and protein expression of ERCC1 in NSCLC tumors
was higher than that in adjacent normal tissues, so it was reported
that ERCC1 may be associated with the development of NSCLC. As
further confirmation, in the present study, ERCC1 gene was cloned
and transfected into the NSCLC H1299 cells with no intrinsic ERCC1
expression. Then, H1299/E cells with a stable ERCC1 expression was
established (Fig. 1). The effect
of ERCC1 expression on cell proliferation, colony-forming rate,
cell cycle distribution and the invasion ability of cells were
assessed, respectively. However, no significant influence was seen
on these aspects through the comparison among H1299/E, H1299/V and
their parental H1299 cells (Fig.
2), suggesting that ERCC1 might not directly participate in the
processes of cell proliferation, cell cycle and invasion, or
correlated with the NSCLC development.
Furthermore, in previous studies (28,30),
patients with high ERCC1 expression were considered to obtain less
benefit from the platinum-based chemotherapy than those with low
ERCC1 expression, which indicated that ERCC1 may be involved in the
mechanisms of platinum resistance in NSCLC treatment. In the
present study, the IC50 of cisplatin in H1299/E cells
was 4.23 times higher than that of H1299/V cells (Fig. 3A), suggesting that the expression
of ERCC1 in H1299/E cells significantly enhanced the cisplatin
resistance of the H1299 cells.
Drug resistance of cancer cells primarily attribute
to two mechanisms: one is the decreased effective concentration of
drugs in the cell which is induced by the altered cell membrane
proteins that work as the drug pumps to decrease drug influx into
cells and/or increase drug efflux out of cells (12); the other is the decreased
sensitivity of the cancer cell to drugs which is mediated by the
increased capacity of DNA repair pathways (35,36).
In the present study, no significant difference was found in the
accumulation of cisplatin between H1299/E and H1299/V cells
(Fig. 3B), furthermore, the
expression of P-gp and MRP (12,35),
considered as the crucial drug efflux pumps, had no significant
difference between the two cell lines (Fig. 3C). The findings demonstrated that
the acquired cisplatin resistance of H1299/E cells predominately
attributed to the expression of ERCC1.
To further confirm the impact of ERCC1 on the
sensitivity of NSCLC cells to cisplatin, another NSCLC cell line
A549 with intrinsic expression of ERCC1 was selected, and the ERCC1
gene was interfered with utilizing RNAi technique. A549/E cell line
with the silenced ERCC1 expression was established (Fig. 4). IC50 of cisplatin in
A549/E cells decreased by two-thirds compared with A549/V cells in
which ERCC1 expression was not silenced, suggesting that the
interference of ERCC1 expression increased the sensitivity to
cisplatin for NSCLC cells. This agreed with the results of Chang
et al that the siRNA-induced suppression of ERCC1 enhancing
sensitivity of HeLa S3, MCF-7, and HCT116 cells to cisplatin
(37). These findings confirmed
that ERCC1 is a chemotherapy-tolerating gene in the cisplatin-based
treatment for human cancer cells.
In conclusion, based on the above and previous
findings that mRNA and protein expression of ERCC1 correlates with
survival of NSCLC patients as well as outcomes of platinum-based
treatment, in the present study, we further investigated the
biological functions of ERCC1 in the physiological processes of
NSCLC cells by cloning, transfection and RNAi technique. ERCC1 had
no effect on cell proliferation, cell cycle and invasion but
exhibited significant impact on cisplatin response of NSCLC cells.
The results demonstrated that ERCC1 might not directly participate
in the physiological processes but its high expression enhanced the
resistance of NSCLC cells to cisplatin. Therefore, we concluded
that ERCC1 is a chemotherapy-tolerating gene in cisplatin-based
treatment of NSCLC. Although ERCC1 is a promising biomarker for
NSCLC treatment, a wide range of preclinical and clinical studies
should be conducted before its clinical use.
Acknowledgements
This study was supported by grant from the project
of Guangxi Science Foundation (no. 0832077).
References
|
1
|
Le Chevalier T: Adjuvant chemotherapy for
resectable non-small-cell lung cancer: where is it going? Ann
Oncol. 21(Suppl 7): vii196–vii198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Azzoli CG, Park BJ, Pao W, Zakowski M and
Kris MG: Molecularly tailored adjuvant chemotherapy for resected
non-small cell lung cancer: a time for excitement and equipoise. J
Thorac Oncol. 3:84–93. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dziadziuszko R and Hirsch FR: Advances in
genomic and proteomic studies of non-small-cell lung cancer:
clinical and translational research perspective. Clin Lung Cancer.
9:78–84. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taillade L, Penault-Llorca F, Boulet T, et
al: Immunohistochemichal expression of biomarkers: a comparative
study between diagnostic bronchial biopsies and surgical specimens
of non-small cell lung cancer. Ann Oncol. 18:1043–1050. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Toschi L and Cappuzzo F: Impact of
biomarkers on non-small cell lung cancer treatment. Target Oncol.
5:5–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bowden NA: Nucleotide excision repair: why
is it not used to predict response to platinum-based chemotherapy?
Cancer Lett. 346:163–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simon GR, Ismail-Khan R and Bepler G:
Nuclear excision repair-based personalized therapy for non-small
cell lung cancer: from hypothesis to reality. Int J Biochem Cell
Biol. 39:1318–1328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosell R, Lord RV, Taron M and Reguart N:
DNA repair and cisplatin resistance in non-small-cell lung cancer.
Lung Cancer. 38:217–227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friboulet L, Barrios-Gonzales D, Commo F,
et al: Molecular characteristics of ERCC1-negative versus
ERCC1-positive tumors in resected NSCLC. Clin Cancer Res.
17:5562–5572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bepler G: Pharmacogenomics: a reality or
still a promise? Lung Cancer. 54(Suppl 2): S3–S7. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Custodio A, Mendez M and Provencio M:
Targeted therapies for advanced non-small-cell lung cancer: current
status and future implications. Cancer Treat Rev. 38:36–53. 2012.
View Article : Google Scholar
|
|
12
|
Stewart DJ: Tumor and host factors that
may limit efficacy of chemotherapy in non-small cell and small cell
lung cancer. Crit Rev Oncol Hematol. 75:173–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gossage L and Madhusudan S: Current status
of excision repair cross complementing-group 1 (ERCC1) in cancer.
Cancer Treat Rev. 33:565–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosell R, Vergnenegre A, Liu B, et al:
Biomarkers in lung oncology. Pulm Pharmacol Ther. 23:508–514. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen J, Emara N, Solomides C, Parekh H and
Simpkins H: Resistance to platinum-based chemotherapy in lung
cancer cell lines. Cancer Chemother Pharmacol. 66:1103–1111. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosell R, Cobo M, Isla D, Camps C and
Massuti B: Pharmacogenomics and gemcitabine. Ann Oncol. 17(Suppl
5): v13–v16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O’Brien CP, Taylor SE, O’Leary JJ and Finn
SP: Molecular testing in oncology: problems, pitfalls and progress.
Lung Cancer. 83:309–315. 2014. View Article : Google Scholar
|
|
18
|
Olaussen KA, Dunant A, Fouret P, et al:
DNA repair by ERCC1 in non-small-cell lung cancer and
cisplatin-based adjuvant chemotherapy. N Engl J Med. 355:983–991.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takenaka T, Yoshino I, Kouso H, et al:
Combined evaluation of Rad51 and ERCC1 expressions for sensitivity
to platinum agents in non-small cell lung cancer. Int J Cancer.
121:895–900. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tiseo M, Bordi P, Bortesi B, et al: A
ERCC1/BRCA1 expression and gene polymorphisms as prognostic and
predictive factors in advanced NSCLC treated with or without
cisplatin. Br J Cancer. 108:1695–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosell R, Cecere F, Santarpia M, Reguart N
and Taron M: Predicting the outcome of chemotherapy for lung
cancer. Curr Opin Pharmacol. 6:323–331. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ota S, Ishii G, Goto K, et al:
Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen
predicts survival in advanced non-small-cell lung cancer treated
with cisplatin-based chemotherapy. Lung Cancer. 64:98–104. 2009.
View Article : Google Scholar
|
|
23
|
Wachters FM, Wong LS, Timens W, Kampinga
HH and Groen HJ: ERCC1, hRad51 and BRCA1 protein expression in
relation to tumour response and survival of stage III/IV NSCLC
patients treated with chemotherapy. Lung Cancer. 50:211–219. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Filipits M and Pirker R: Predictive
markers in the adjuvant therapy of non-small cell lung cancer. Lung
Cancer. 74:355–363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Filipits M, Haddad V, Schmid K, et al:
Multidrug resistance proteins do not predict benefit of adjuvant
chemotherapy in patients with completely resected non-small cell
lung cancer: International Adjuvant Lung Cancer Trial Biologic
Program. Clin Cancer Res. 13:3892–3898. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Voortman J, Goto A, Mendiboure J, et al:
MicroRNA expression and clinical outcomes in patients treated with
adjuvant chemotherapy after complete resection of non-small cell
lung carcinoma. Cancer Res. 70:8288–8298. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Filipits M, Pirker R, Dunant A, et al:
Cell cycle regulators and outcome of adjuvant cisplatin-based
chemotherapy in completely resected non-small-cell lung cancer: the
International Adjuvant Lung Cancer Trial Biologic Program. J Clin
Oncol. 25:2735–2740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan H, Li L, Zuo CT, Mao NQ, Chen FL,
Zhang W and Tang BJ: Relationship between combined multigene
detection and response to adjuvant chemotherapy in early-stage
non-small cell lung cancer. Zhonghua Zhong Liu Za Zhi. 30:528–531.
2008.PubMed/NCBI
|
|
29
|
Elbashir SM, Lendeckel W and Tuschl T: RNA
interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev.
15:188–200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang J, Liang X, Zhou X, Huang R, Chu Z
and Zhan Q: ERCC1 expression as a prognostic and predictive factor
in patients with non-small cell lung cancer: a meta-analysis. Mol
Biol Rep. 39:6933–6942. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tepeli E, Caner V, Buyukpinarbasili N,
Cetin GO, Duzcan F, Elmas L and Bagci G: Expression of ERCC1 and
its clinicopathological correlations in non-small cell lung cancer.
Mol Biol Rep. 39:335–341. 2012. View Article : Google Scholar
|
|
32
|
Koc E, Caner V, Buyukpinarbasili N, Tepeli
E, Turk NS, Ozan Cetin G and Bagci G: The determination of
relationship between ‘excision repair cross-complementing group 1’
(ERCC1) gene T19007C and C8092A single nucleotide polymorphisms and
clinicopathological parameters in non-small cell lung cancer. Mol
Biol Rep. 39:375–380. 2012. View Article : Google Scholar
|
|
33
|
Yang Y and Xian L: The association between
the ERCC1/2 polymorphisms and the clinical outcomes of the
platinum-based chemotherapy in non-small cell lung cancer (NSCLC):
a systematic review and meta-analysis. Tumour Biol. 35:2905–2921.
2014. View Article : Google Scholar
|
|
34
|
Friboulet L, Olaussen KA, Pignon JP, et
al: ERCC1 isoform expression and DNA repair in non-small-cell lung
cancer. N Engl J Med. 368:1101–1110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar :
|
|
36
|
Altaha R, Liang X, Yu JJ and Reed E:
Excision repair cross complementing-group 1: gene expression and
platinum resistance. Int J Mol Med. 14:959–970. 2004.PubMed/NCBI
|
|
37
|
Chang IY, Kim MH, Kim HB, Lee DY, Kim SH,
Kim HY and You HJ: Small interfering RNA-induced suppression of
ERCC1 enhances sensitivity of human cancer cells to cisplatin.
Biochem Biophys Res Commun. 327:225–233. 2005. View Article : Google Scholar : PubMed/NCBI
|